

Summary
P hytophthora sojae is an oomycete pathogen of soybean. As a result of its economic importance, P . sojae has become a model for the study of oomycete genetics, physiology and pathology. The lack of efficient techniques for targeted mutagenesis and gene replacement have long hampered genetic studies of pathogenicity in Phytophthora species. Here, we describe a CRISPR/Cas9 system enabling rapid and efficient genome editing in P . sojae. Using the RXLR effector gene Avr4/6 as a target, we observed that, in the absence of a homologous template, the repair of Cas9‐induced DNA double‐strand breaks (DSBs) in P . sojae was mediated by non‐homologous end‐joining (NHEJ), primarily resulting in short indels. Most mutants were homozygous, presumably as a result of gene conversion triggered by Cas9‐mediated cleavage of non‐mutant alleles. When donor DNA was present, homology‐directed repair (HDR) was observed, which resulted in the replacement of Avr4/6 with the NPT II gene. By testing the specific virulence of several NHEJ mutants and HDR‐mediated gene replacements in soybean, we have validated the contribution of Avr4/6 to recognition by soybean R gene loci, Rps4 and Rps6, but also uncovered additional contributions to resistance by these two loci. Our results establish a powerful tool for the study of functional genomics in Phytophthora, which provides new avenues for better control of this pathogen.
Keywords: Avr4/6, CRISPR/Cas9, gene replacement, genome editing, oomycetes, Phytophthora sojae, RXLR effector
Introduction
Phytophthora sojae causes ‘damping off’ of soybean seedlings as well as stem and root rot of established plants (Tyler, 2007). Morphologically and physiologically, oomycetes such as P. sojae resemble filamentous fungi, but evolutionarily they are classified in the kingdom Stramenopila (Tyler, 2001). Most Phytophthora species are plant pathogens that can damage a huge range of agriculturally and ornamentally important plants (Erwin and Ribeiro, 1996). Because of its economic impact, P. sojae, together with P. infestans, has been developed as a model species for the study of oomycete plant pathogens (Tyler, 2007).
The first two genome sequences of oomycetes (P. sojae and P. ramorum) were published approximately 9 years ago (Tyler et al., 2006), but functional genomics studies have been hampered by the lack of efficient strategies for genome engineering. DNA transformation procedures have been developed (Judelson et al., 1993a, 1993b), but gene knockouts and gene replacements have never been possible because insertion of transgenes occurs exclusively by non‐homologous end‐joining (NHEJ) (Judelson, 1997; Tyler and Gijzen, 2014). Alternative approaches for functional analysis have included TILLING (Targeting Induced Local Lesions in Genomes) (Lamour et al., 2006) and gene silencing (Ah‐Fong et al., 2008; Judelson et al., 1993b; Wang et al., 2011; Whisson et al., 2005). However, TILLING, which is based on random mutagenesis, is very laborious, requires long‐term storage of large pools of mutants and has not been proven to be very useful in oomycetes. Gene silencing (RNAi, RNA interference), triggered using hairpin, antisense and sense RNA constructs (Ah‐Fong et al., 2008) or using double‐stranded RNA (dsRNA) directly (Wang et al., 2011; Whisson et al., 2005), has been proven to be useful. However, knockdown of genes in oomycetes by RNAi is incomplete, and varies among gene targets, experiments and laboratories. Also, selective silencing of closely related genes is difficult.
Recent advances in engineered nucleases that specifically cleave genomic sequences in living cells have provided valuable tools to create targeted mutations in numerous organisms, from vertebrates, insects and plants (reviewed in Gaj et al., 2013) to microbes, including parasites (Peng et al., 2015; Shen et al., 2014; Wagner et al., 2014; Zhang et al., 2014) and fungi (Jacobs et al., 2014; Liu et al., 2015; Vyas et al., 2015). These nucleases, which include zinc finger nucleases (ZFNs), transcription activator‐like effector nucleases (TALENs) and CRISPR/Cas (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated), can generate a double‐strand break (DSB) at specific sites. By triggering repair of the DSB, by either error‐prone NHEJ or homology‐directed repair (HDR), such methods can increase the rate of gene editing to levels that enable the ready isolation of cells or organisms bearing a desired genetic change (Miller et al., 2011). ZFNs and TALENs are engineered proteins containing a modular DNA recognition domain and a DNA cleavage domain.
Like ZFNs and TALENs, the type II CRISPR/Cas9 system derived from the adaptive immune system of Streptococcus pyogenes also has DNA recognition and cleavage functions (Cong et al., 2013; Mali et al., 2013). However, DNA recognition is mediated by a single guide RNA (sgRNA) rather than a fused DNA recognition protein domain. The specificity of this system relies on the sgRNA, which can direct the nuclease Cas9 to the target DNA sequence (Cong et al., 2013; Mali et al., 2013).
Here, we have implemented the CRISPR/Cas9 system in P. sojae, using the RXLR effector gene Avr4/6 (Dou et al., 2010) as a target. RXLR effectors are a large superfamily of virulence proteins secreted by many oomycetes that have the ability to enter host cells in order to promote host susceptibility (Jiang and Tyler, 2012). The presence of some RXLR effectors, such as Avr4/6, can be recognized by intracellular receptors encoded by plant resistance genes (R genes), triggering vigorous defence responses (Jiang and Tyler, 2012). The presence of Avr4/6 is recognized by soybean R genes Rps4 and Rps6 (Dou et al., 2010; Gijzen et al., 1996; Whisson et al., 1994); recognition by Rps4 requires the N‐terminus of Avr4/6, whereas recognition by Rps6 requires the C‐terminus (Dou et al., 2010). Our results demonstrate that CRISPR/Cas9‐mediated gene disruption and gene replacement is an efficient and useful strategy for the testing of the function of specific genes in P. sojae, such as Avr4/6, which should be useful for all oomycetes.
Results
Establishment of the CRISPR/Cas9 system for P . sojae
To establish a CRISPR/Cas9 system for P. sojae, we had to establish efficient expression of Cas9, efficient expression of guide RNAs and targeting of the Cas9 enzyme to the nucleus.
For expression in P. sojae, we selected the Streptococcus pyogenes Cas9 encoded by a gene with human‐optimized codons (hSpCas9), because this Cas9 version has been used in diverse organisms (Cong et al., 2013; Peng et al., 2015; Zhang et al., 2014) and matches P. sojae codon usage relatively well. To test whether this protein could be efficiently expressed in P. sojae, we fused green fluorescent protein (GFP) to the C‐terminus of hSpCas9. Furthermore, to direct hSpCas9‐GFP into the P. sojae nucleus, we used a strong synthetic nuclear localization sequence (NLS) derived from a P. sojae bZIP transcription factor (Fang and Tyler, 2015), which we fused to the N‐terminus of SpCas9 (Fig. 1A). Preliminary experiments had shown that commonly used mammalian NLS signals did not work efficiently in P. sojae. Expression of the P. sojae NLS (PsNLS) fused hSpCas9‐GFP construct in P. sojae transformants resulted in a bright GFP signal strongly localized within the nuclei of P. sojae hyphae (Fig. 1A). These results indicated that hSpCas9 was strongly expressed in P. sojae without further codon optimization, and that the bZIP‐derived NLS efficiently targeted the fusion protein to P. sojae nuclei.
Figure 1.
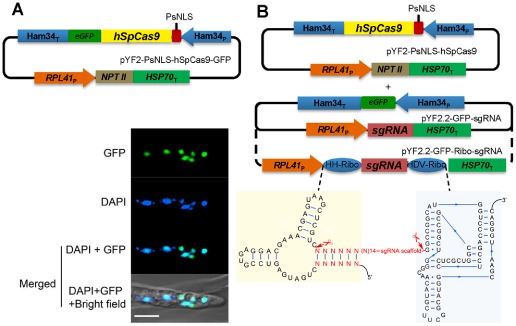
Cas9 and guide RNA constructs for P hytophthora sojae genome editing. (A) Top: plasmid for expression of hSpCas9 fused to eGFP and a nuclear localization sequence (NLS) in P . sojae. PsNLS, a strong synthetic NLS derived from a P. sojae bZIP transcription factor; eGFP, enhanced green fluorescent protein. Bottom: P . sojae hyphae expressing PsNLS‐hSpCas9‐GFP from pYF2, counter‐stained with 4′,6‐diamidino‐2‐phenylindole (DAPI); scale bars, 10 μm. (B) Top: plasmids for expression of CRISPR constructs in P . sojae. Cas9 expression is driven by the Ham34 promoter, on a plasmid with the selectable marker NPT II driven by the P . sojae RPL41 promoter. Transcription of single guide RNA (sgRNA) (including flanking ribozymes) is driven by the RPL41 promoter on a plasmid with an eGFP expression cassette (used as a screening marker). Bottom: double ribozyme construct for release of sgRNAs from the primary RNA polymerase II transcript.
In most systems, sgRNAs are synthesized by RNA polymerase III (RNA pol III), typically using a U6 small nuclear RNA (snRNA) promoter (Cong et al., 2013; Hwang et al., 2013; Mali et al., 2013; Shen et al., 2014; Zhang et al., 2014). However, no RNA pol III promoters have yet been functionally defined in oomycetes. U6 gene sequences are highly conserved among different oomycetes (Fig. S1A, see Supporting Information), and so we cloned the full‐length P. sojae and P. infestans U6 gene regions (Fig. S1B,C) and inserted a 150‐bp fragment of the eGFP (enhanced green fluorescent protein) gene near the 3′ end as a polymerase chain reaction (PCR) reporter sequence (Fig. S1D). Surprisingly, however, we did not detect any transcripts spanning the GFP reporter fragment by reverse transcription‐polymerase chain reaction (RT‐PCR) (data not shown).
Recently, the generation of sgRNAs from RNA pol II promoters has been demonstrated in wheat (Upadhyay et al., 2013), yeast (Gao and Zhao, 2014) and Arabidopsis (Gao et al., 2015). The yeast and Arabidopsis systems used cis‐acting ribozymes to trim flanking sequences from the sgRNAs, whereas the wheat system did not. Thus, we employed the constitutive P. sojae RPL41 promoter (Dou et al., 2008a, 2008b) to direct the transcription of sgRNAs, and evaluated sgRNA constructs that either were or were not flanked on the 5′ side by a hammerhead (HH) ribozyme and on the 3′ side by a HDV ribozyme (Gao and Zhao, 2014) (Fig. 1B).
To simplify the generation and screening of P. sojae transformants, the hSpCas9 gene and a resistance selection marker (NPT II) were placed in one plasmid, whereas the sgRNA gene together with a GFP marker gene were placed in a second plasmid (Figs 1B and S4, see Supporting Information).
Cas9‐mediated mutagenesis of Avr4/6
To test the P. sojae sgRNA:Cas9 system, we selected as a target a P. sojae gene encoding an RXLR avirulence effector, Avr4/6 (GenBank: GU214064.1). Avr4/6 is a single copy gene with no close paralogues. Furthermore, loss of Avr4/6 function was expected to confer a phenotype that would not affect in vitro growth, namely the ability to successfully infect soybean cultivars containing resistance genes Rps4 or Rps6 (Dou et al., 2010). sgRNAs targeting Avr4/6 were designed using the web tool sgRNA Designer (Doench et al., 2014). sgRNA candidates rated highly by the tool were further filtered by off‐target analysis. Finally, two sgRNAs (sgRNA versions A and B) were selected in which the respective Cas9 cleavage sites overlapped unique restriction enzyme sites (Bst UI and Tsp 45I, respectively) that could be used to rapidly screen for mutations (Fig. 2A).
Figure 2.
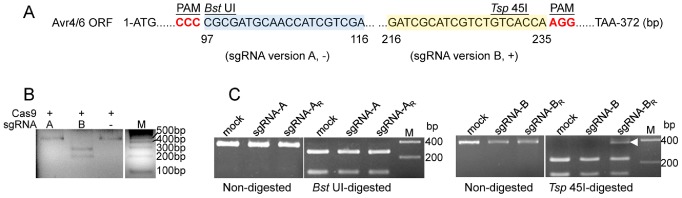
Single guide RNAs (sgRNAs) for targeting of Avr4/6. (A) Two sgRNA target sites within the Avr4/6 open reading frame (ORF). Target sites of sgRNA A (sgRNA‐A) and B (sgRNA‐B) are highlighted in blue and yellow, respectively. sgRNAs A and B target Avr4/6 on the negative (−) and positive (+) DNA strand, respectively. The sgRNA‐A site overlaps with a Bst UI restriction enzyme site (CGCG) and sgRNA‐B with a Tsp 45I site (GTG/CAC). PAM, Protospacer Adjacent Motif (bold red). (B) In vitro cleavage assay indicating that Avr4/6 sgRNA‐B can direct Cas9 cleavage of target polymerase chain reaction (PCR) products, but sgRNA‐A cannot. The DNA template was amplified from pBS‐Avr4/6 using M13F and M13R [supplemental sequences in Appendix S1 (see Supporting Information)]. The colours of the original gel are inverted for clarity. (C) PCR and restriction enzyme analysis of P hytophthora sojae pooled transient expression transformants, indicating that only sgRNA‐B flanked by ribozymes (sgRNA‐BR) produced amplicons resistant to restriction enzyme cleavage (arrowhead). Approximately 25%–30% of the amplicon was resistant to Tsp 45I digestion. The experiment was performed in triplicate. sgRNA‐A, ‐B, sgRNA lacking ribozymes; sgRNA‐AR, ‐BR, sgRNAs flanked byribozymes; mock, P . sojae transformants only receiving Cas9 plasmid. In (B) and (C), all gel panels placed together were from the same gel; white dividers indicate lanes not adjacent in these gels.
To examine the activity of the designed sgRNAs, Avr4/6‐A and Avr4/6‐B, we carried out sgRNA‐mediated in vitro cleavage assay of target DNA. In these assays, the sgRNAs were synthesized using T7 RNA polymerase and purchased SpCas9 protein. Cas9/sgRNA‐B completely cleaved the target DNA, whereas Cas9/sgRNA‐A showed no activity in vitro (Fig. 2B).
In parallel with the in vitro assays, we used transient expression in P. sojae protoplasts to test whether the hSpCas9 and sgRNAs produced in vivo could modify the endogenous Avr4/6 gene. The two Avr4/6‐specific sgRNAs were assembled into the P. sojae expression plasmid under the control of the RPL41 promoter, either flanked with (Avr4/6‐sgRNA‐AR, ‐BR) or without (Avr4/6‐sgRNA‐A, ‐B) ribozymes. The sgRNA constructs were co‐transformed with the hSpCas9 expression plasmid into P. sojae strain P6497. Transformants were enriched by G418 selection 12 h after transformation when hyphae had regenerated. After 24 h, DNA was extracted from the culture containing the pooled transformants. Avr4/6 sequences were amplified from the pool of genomic DNAs (gDNAs) and screened for mutants resistant to the relevant restriction enzymes (Bst UI for A and Tsp 45I for B). We found that the Avr4/6 amplicons from the two sgRNA‐A transformations (constructs with and without ribozymes) were still fully subject to restriction enzyme cleavage, indicating failure of Cas9‐mediated mutagenesis (Fig. 2C). In contrast, the transformation utilizing sgRNA version B flanked by ribozymes yielded restriction enzyme‐resistant amplicons (Fig. 2C). However, the transformation utilizing sgRNA version B without ribozymes did not yield restriction enzyme‐resistant amplicons (Fig. 2C). To validate the enzyme cleavage results, we sequenced the nested PCR products amplified from the enzyme digestion products from the sgRNA‐BR transformants. The sequence chromatograms showed pure sequences proximal to the target site and mixed sequences distal to the target site (data not shown), suggesting the presence of mutations at the target site. These observations indicate that, in P. sojae, RNA pol II can be successfully used for the generation of sgRNA, provided that ribozymes are employed to remove the surrounding sequences from the transcripts. The failure of sgRNA version A may result from strong self‐complementarity that was subsequently discovered in its sequence, which could block its binding to target DNA.
To characterize CRISPR/Cas9‐generated Avr4/6 mutations in detail, the transformation with the ribozyme‐containing Avr4/6‐sgRNA‐BR construct was repeated. Individual G418‐resistant transformants were isolated and screened for the presence of GFP, indicating the presence of the sgRNA construct. Of 50 primary transformants screened, six exhibited green fluorescence. Of these, four yielded Avr4/6 amplicons that were partially or fully resistant to Tsp 45I digestion (Fig. 3A), indicating the presence of Avr4/6 mutations. As P. sojae protoplasts and hyphae are multinucleate, and hence might be expected to harbour nuclei with a diversity of Avr4/6 mutations, we isolated zoospores (which are mononucleate) from three of the primary transformants (T11, T18 and T32; the fourth, T30, did not produce zoospores). Ten single zoospore lines were isolated from each transformant, and the Avr4/6 amplicons were screened by restriction enzyme digestion and Sanger sequencing. T11, T18 and T32 yielded seven, 10 and 10 pure mutant lines, respectively (summarized in Table 1). All 27 pure lines showed a homogeneous sequence profile (Fig. S2, see Supporting Information), indicating that all were already homozygous, and carried the same mutation in both alleles. All 10 of the T32 lines were homozygous for the same mutation. The lines derived from T18 included nine lines homozygous for one Avr4/6 mutation and one homozygous for a different mutation. The lines derived from T11 included two homozygous for one mutation (mut1) and five homozygous for a second mutation (mut2). The three remaining lines were heterozygous and biallelic, containing a third mutation paired with mut1 (Figs 3B and S2). No lines retained any wild‐type alleles, either homozygous or heterozygous.
Figure 3.
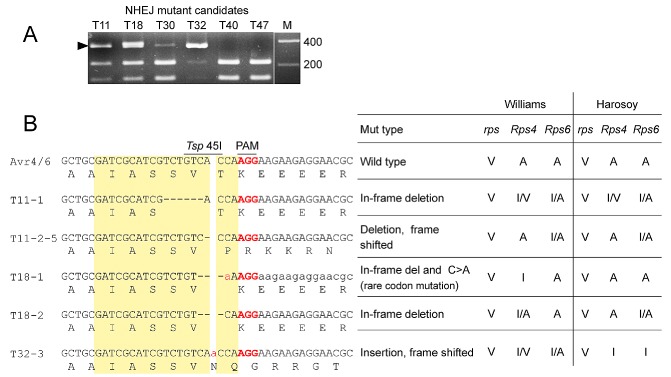
Characterization of individual non‐homologous end‐joining (NHEJ)‐mediated mutants. (A) Tsp 45I screening of Avr4/6 amplicons from six individual transformants carrying hSpCas9 and sgRNA‐BR plasmids. Arrowhead indicates Tsp 45I‐resistant amplicons. Transformants are expected to carry a mixture of modified and non‐modified Avr4/6 genes. (B) Left: sequences of Avr4/6 mutant amplicons from single zoospore lines derived from transformants T11, T18 and T32 (T30 produced no zoospores). Target sites are highlighted in yellow and the PAM sequences are shown in bold red. Right: summary of virulence assays of Avr4/6 mutants on Rps4‐ and Rps6‐containing cultivars. V, virulent; A, avirulent; I, intermediate; I/A intermediate to avirulent; I/V intermediate to virulent.
Table 1.
Avr4/6 CRISPR/Cas‐induced non‐homologous end‐joining (NHEJ) mutations
| Mutants | Mutant patterns | |||||
|---|---|---|---|---|---|---|
| Homozygotes | Heterozygotes | Biallele | ||||
| WT/WTa | mut1/mut1 | mut2/mut2 | WT/mut1 | WT/mut2 | mut1/mut2,3 | |
| T11b | 0 | 2 | 5 | 0 | 0 | 3c |
| T18d | 0 | 9 | 1 | 0 | 0 | 0 |
| T32e | 0 | 10 | 0 | 0 | 0 | 0 |
WT, wild‐type sequence.
T11, mut1, 6‐bp deletion; mut2, 1‐bp insertion.
Heterozygous mut1/mut3; mut3 is a 3‐bp deletion. The sequencing profiles of the heterozygotes were disambiguated using the web‐tool TIDE (Brinkman et al., 2014).
T18, mut1, 3‐bp deletion; mut2, 3‐bp deletion plus 1‐bp substitution.
T32, mut1, 1‐bp insertion.
Each of the mutations consisted of a short indel, located specifically at the Cas9 cleavage site, i.e. between the 17th and 18th nucleotides of the sgRNA target site. Deletions of 1, 3 and 6 bp were observed, one contained a 1‐bp insertion, and one combined a 3‐bp deletion with a 1‐bp replacement (Fig. 3B and Table 1).
We also tested the stability of the mutants by subculturing each of the single zoospore lines for at least three generations on medium without G418 selection. All of the mutated sites examined remained the same as in the first generation, based on the sequencing of the Avr4/6 amplicons. Interestingly, one transformant, T47, which did not show obvious mutations in the first generation, acquired the same single adenine insertion as T32‐3 after subculture of the unpurified transformant for one generation (Fig. S3, see Supporting Information), presumably because the sgRNA:Cas9 constructs were integrated into the genome and continued to actively cleave the target in each generation. Collectively, these results indicate that our CRISPR/Cas9 system can efficiently and specifically trigger the introduction of NHEJ mutations, typically short indels, into the P. sojae genome.
Homologous gene replacement stimulated by the CRISPR/Cas9 system
Donor DNA‐mediated repair of sgRNA‐guided Cas9 cuts has been proven to be an efficient method to facilitate gene replacements via HDR (Cong et al., 2013; Mali et al., 2013). To determine whether sgRNA:Cas9‐mediated DSB could stimulate homologous recombination in P. sojae, we co‐transformed the CRISPR constructs that were successfully used for mutation of Avr4/6, together with uncut donor DNA plasmids that contained the entire NPT II open reading frame (ORF) flanked by different lengths of the sequences surrounding the Avr4/6 gene. An equimolar ratio of the three plasmids was used (Fig. 4A). As preliminary experiments had shown that expression of the NPT II gene from the Avr4/6 promoter was insufficient for G418 selection, the NPT II gene was included in the Cas9 plasmid for the selection of transformants. We used homology arms consisting of three different lengths of 5′ and 3′ flanking sequences, namely 250 bp, 500 bp and 1 kb, to assess which would enable the highest recombination efficiency (Fig. 4B). The NPT II gene in the Cas9 expression plasmid served as a negative control, because it lacked any Avr4/6 flanking sequences.
Figure 4.
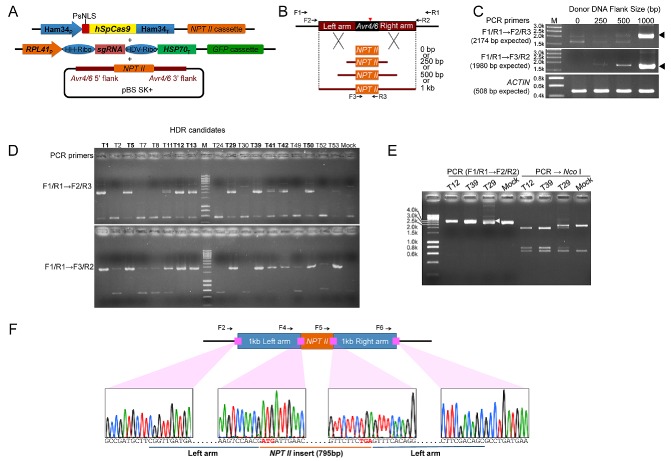
Homology‐directed repair (HDR)‐mediated replacement of the Avr4/6 open reading frame (ORF) with an NPT II ORF. (A) Strategy used for gene replacement. Plasmids containing a homologous donor DNA (NPT II with Avr4/6 flanking sequences) were co‐transformed with the Avr4/6 sgRNA‐BR and hSpCas9 constructs. (B) Three different sizes of the homology arms, 250 bp, 500 bp and 1 kb, flanking the Avr4/6 locus, were used. The NPT II gene in the hSpCas9 expression plasmid served as the control (0‐bp homologous arm, mock). Primers used to screen the HDR mutants and to validate the replaced region are shown as arrows. Primer pair F1/R1 and nested primer pairs F2/R3, F3/R2 and F2/R2 were used for HDR mutant screening. Red arrowhead indicates the CRISPR/Cas9 cleavage site (between 232 and 233 bp of Avr4/6 ORF). (C) Analysis of genomic DNA from the pooled transformants produced using the four sizes of flanking sequences, employing nested PCR. Arrowheads indicate sizes expected if HDR has occurred. ACTIN, actin control for DNA quality. (D) Screening of individual HDR transformants generated with the 1‐kb flanking sequence plasmid. The nine positive HDR mutants are highlighted in bold. (E) PCR analysis of representative zoospore‐purified lines of HDR mutants, demonstrating that T12 and T39 are homozygotes, whereas T29 is a heterozygote. DNA sizes (bp) before restriction enzyme cleavage: WT, 2936; HDR mutant, 3359; after Nco I digestion: WT, 2273 + 663; HDR mutant, 1945 + 751 + 663. The arrowhead indicates the fragment amplified from the NPT II‐replaced allele. Primer F4, Avr4/6_up500bp_PhusF (Table S1). (F) Sanger sequencing traces of junction regions confirming that the Avr4/6 ORF was cleanly replaced by the NPT II ORF in a representative zoospore‐purified clone (HDR‐T12‐1). Start and stop codons are indicated in bold red.
Following co‐transformation and G418 enrichment, the bulk transformants were subjected to gDNA extraction and PCR analysis. PCR amplifications using primers located outside the Avr4/6 homology arms and within the NPT II gene were used to detect homologous recombination events. The results suggested that HDR had occurred, but the frequency was variable depending on the length of the flanking sequences in the donor DNA plasmids. The transformant population generated with the 1‐kb flanking sequences showed the highest frequency of gene replacement. The population generated with the 500‐bp flanking sequences showed a much lower frequency compared with the 1‐kb population. The population from the 250‐bp flanking sequences showed very low recombination frequencies (Fig. 4C).
Next, to characterize HDR events in detail, we generated single P. sojae transformants derived from the 1‐kb arm donor. After screening 68 individual G418 resistant transformants for GFP production, we identified 18 transformants bearing the two CRISPR components. Then, using nested primers specific for HDR events (Fig. 4B), we found evidence for gene replacement events in nine of the transformants (Fig. 4D). Sanger sequencing across the junctions of the flanking sequences and NPT II in the nested PCR products was also consistent with replacement of the Avr4/6 ORF with the NPT II gene (data not shown). Three HDR mutants, namely HDR‐T12, HDR‐T29 and HDR‐T39, that readily produced zoospores were selected for functional tests. After zoospore isolation, the Avr4/6 region of each single zoospore line was examined by PCR amplification using primers flanking the two homologous arms and cleavage of the amplicon by the restriction enzyme Nco I. We found that all of the 11 single zoospore lines obtained from HDR‐T12 and all eight obtained from HDR‐T39 were homozygotes (Fig. 4E); this was further validated by Sanger sequencing (Fig. 4F). In contrast, all 20 of the single zoospore lines of T29 appeared to be heterozygotes that contained an HDR event in just one of the two Avr4/6 alleles (Fig. 4E). This was further verified by PCR amplification using primers outside of the homology arms and in the NPT II gene. More detailed analysis of three of the HDR‐T29 lines revealed that the non‐HDR alleles possessed the same mutation, an adenine deletion, in every case (Fig. S3), presumably caused by NHEJ.
Modified recognition of Avr4/6 mutants by soybean carrying the Rps4 and Rps6 loci
In order to test the effects of the CRISPR/Cas9‐induced Avr4/6 mutations on P. sojae recognition by plants containing the Rps4 and Rps6 loci, the five homozygous NHEJ mutants and two homozygous HDR mutants were inoculated onto hypocotyls of soybean isolines containing Rps4 (L85‐2352) or Rps6 (L89‐1581) in a Williams background, as well as isolines containing Rps4 (HARO4272) or Rps6 (HARO6272) in a Harosoy background. At 4 days post‐inoculation (dpi), the specific virulence of the different mutants was scored and analysed by Fisher's exact test (Table 2). We observed that the frame‐shifted mutants T32‐3 and T11‐2‐5 both showed increased killing of Rps4‐ or Rps6‐containing soybean seedlings in both Williams and Harosoy backgrounds (Table 2). The increased killing of both Rps4 and Rps6 plants by T32‐3 was statistically significant (P < 0.01), whereas the increased killing by T11‐2‐5 of Rps4, but not Rps6, plants was significant (P < 0.05). However, the increased killing in every case was still significantly (P < 0.05) less than the killing of rps plants lacking Rps4 or Rps6 (Table 2). Thus, T32‐3 was scored as intermediate on Rps4 and Rps6 plants, whereas T11‐2‐5 was scored as avirulent and intermediate, respectively. The other NHEJ mutants, containing in‐frame deletions, also showed increased killing of Rps4‐ and Rps6‐containing plants, but significantly (P < 0.03) less killing than observed with rps plants. The Avr4/6 mutant having a two‐amino‐acid deletion (T11‐1) showed an intermediate phenotype that was close to fully virulent on Rps4 plants, whereas the two mutants with a single amino acid deletion (18‐1 and 18‐2) showed intermediate to avirulent phenotypes (Table 2).
Table 2.
Characterization of the virulence of P hytophthora sojae Avr4/6 non‐homologous end‐joining (NHEJ) and homology‐directed repair (HDR) mutants on soybean
| Williams | Ws (rps) | L85‐2352 (Rps4) | L89‐1581 (Rps6) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strains | sva | virb | sv | P value 1c | P value 2d | vir | sv | P value 1 | P value 2 | vir |
| WT | 0/60 | V | 39/58 | – | – | A | 30/46 | – | – | A |
| T11‐1 | 0/40 | V | 7/47 | 0.014 | <0.0001 | I/V | 12/35 | <0.0001 | 0.0074 | I/A |
| T11‐2‐5 fse | 0/52 | V | 41/70 | <0.0001 | 0.36 | A | 18/53 | <0.0001 | 0.034 | I/A |
| T18‐1 | 1/40 | V | 18/53 | 0.0001 | 0.0006 | I | 15/32 | <0.0001 | 0.16 | A |
| T18‐2 | 0/39 | V | 16/45 | <0.0001 | 0.0016 | I/A | 12/26 | <0.0001 | 0.21 | A |
| T32‐3 fs | 0/54 | V | 6/60 | 0.029 | <0.0001 | I/V | 18/51 | <0.0001 | 0.0044 | I/A |
| HDR‐T12‐1 | 0/20 | V | 9/28 | 0.0063 | 0.0027 | I | 8/32 | 0.0174 | 0.0006 | I/V |
| HDR‐T39‐1 | 0/19 | V | 11/31 | 0.0035 | 0.0067 | I | 7/33 | 0.0390 | 0.0002 | I/V |
| Harosoy | (1‐7)1 (rps) | 4272 (Rps4) | 6272 (Rps6) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strains | sv | vir | sv | P value 1 | P value 2 | vir | sv | P value 1 | P value 2 | vir |
| WT | 0/40 | V | 60/85 | – | – | A | 53/76 | – | – | A |
| T11‐1 | 0/38 | V | 5/37 | 0.025 | <0.0001 | I/V | 15/39 | <0.0001 | 0.0024 | I/A |
| T11‐2‐5 fs | 0/40 | V | 47/76 | <0.0001 | 0.25 | A | 34/79 | <0.0001 | 0.0011 | I/A |
| T18‐1 | 0/40 | V | 19/36 | <0.0001 | 0.094 | A | 15/28 | <0.0001 | 0.16 | A |
| T18‐2 | 0/39 | V | 20/35 | <0.0001 | 0.20 | A | 16/40 | <0.0001 | 0.0027 | I/A |
| T32‐3 fs | 0/42 | V | 15/64 | 0.0004 | <0.0001 | I | 25/64 | <0.0002 | 0.0003 | I |
| HDR‐12‐1 | 0/20 | V | 6/28 | 0.034 | <0.0001 | I/V | 10/35 | 0.0090 | <0.0001 | I/V |
| HDR‐39‐1 | 0/21 | V | 10/33 | 0.0043 | 0.0001 | I/V | 9/31 | 0.0073 | 0.0002 | I/V |
sv, soybean seedlings surviving after infection/total seedlings.
vir, virulence of wild‐type (WT) and mutants: V, virulent; A, avirulent; I, intermediate; I/A intermediate to avirulent; I/V, intermediate to virulent.
P value 1, difference between Rps4/Rps6 and the rps control.
P value 2, difference between mutants and WT P. sojae.
fs, frame‐shift mutant.
The two homozygous HDR mutants (T12‐1 and T39‐1) both showed significantly (P < 0.01) more killing of Rps4‐ or Rps6‐containing soybean seedlings in both Williams and Harosoy backgrounds (Table 2; Fig. 5), but the killing was significantly (P < 0.05) less than on rps plants. Thus, both mutants were scored as intermediate to virulent on Rps4 and Rps6.
Figure 5.
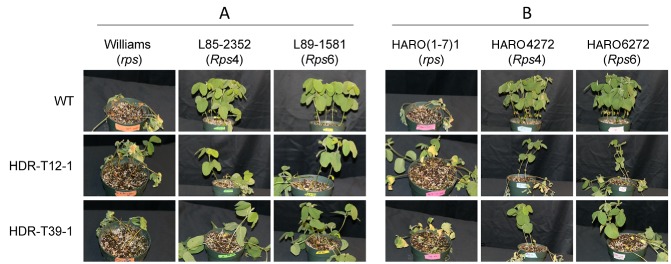
Infection phenotypes of Avr4/6 mutants. Representative photographs showing soybean seedlings inoculated on the hypocotyls with P hytophthora sojae Avr4/6 homology‐directed repair (HDR) mutants. L85‐2352 and HARO4272 contain Rps4, whereas L89‐1581 and HARO6272 contain Rps6. (A) Williams isolines. (B) Harosoy isolines. Photographs were taken at 4 days post‐inoculation.
Discussion
Substantial numbers of oomycete genomes have now been sequenced, and even larger numbers are underway (Jiang and Tyler, 2012; Jiang et al., 2013; Kamoun et al., 2015). These genomes contain 15 000–25 000 genes, approximately one‐half of which, in most species, show the rapid sequence divergence expected of infection‐related genes (Baxter et al., 2010; Haas et al., 2009; Jiang and Tyler, 2012; Jiang et al., 2013; Tyler et al., 2006). To date, however, the tools available for the assessment of the functions of these genes have been limited to RNAi‐mediated gene silencing and to the expression of ectopic transgenes.
Here, we have adapted the CRISPR/Cas9 sequence‐specific nuclease technology for use in P. sojae. This involved overcoming several technical hurdles. One of these was the fact that commonly used mammalian NLSs do not function well in P. sojae and that P. sojae nuclear proteins use dispersed NLSs (Y. Fang and B. M. Tyler, unpublished data). The NLSs from several P. sojae nuclear proteins were delineated to the point that a small highly efficient NLS tag could be constructed (Y. Fang and B. M. Tyler, unpublished data). A second hurdle was that no RNA pol III promoters had been characterized in oomycetes, and that the U6 genes we attempted to use did not appear to be well transcribed under our transformation conditions. We solved this by using a strong RNA pol II promoter (RPL41) in conjunction with self‐cleaving ribozymes to release the sgRNA from the RNA pol II transcript. A third hurdle was effective expression of the nuclease protein. Our initial attempts to use TALENs were blocked because the TALEN constructs were silenced extremely strongly in P. sojae, presumably because of their highly repetitive nature. The humanized SpCas9 protein was readily expressed, but, unexpectedly, the hSpCas9‐GFP fusion protein used to validate expression was not effective in generating mutations in P. sojae, despite the fact that Cas9‐GFP fusions have been used in other organisms, such as Toxoplasma gondii (Shen et al., 2014). We were successful only after we used the non‐fused hSpCas9. A fourth hurdle was to identify effective sgRNAs. One of the two sgRNAs predicted using the sgRNA Designer web tool proved to be ineffective both in vitro and in vivo. This sgRNA, sgRNA‐A, was found to be strongly self‐complementary, potentially preventing hybridization with the target DNA. This observation, which is in agreement with the sgRNA design guideline proposed by Peng et al. (2015), underlines that sgRNAs with strong secondary structure predictions should be eliminated. We also observed a discrepancy between in vitro and in vivo assays of Cas9/sgRNA activity, namely that the sgRNA lacking ribozymes was functional in the in vitro assay, whereas only the sgRNA flanked by ribozymes was effective in vivo. There clearly is room for further optimization of sgRNA design.
Using a single sgRNA, we were able to generate small indels at the site of the Cas9 cleavage. Although these were useful, future attempts to disrupt genes with large deletions will probably require a pair of sgRNAs, or the use of HDR to introduce specific mutations or to replace the gene entirely, as we did when replacing Avr4/6 with NPT II. Of interest is the fact that, from two of our transformant lines, we were able to recover two or three different mutations. Several of these mutations were segregated into different single zoospore lines, indicating that the mutations had occurred in different nuclei of the regenerating protoplasts. One set of lines was biallelic, indicating that two different mutations had occurred in the same nucleus. Also of interest is the observation that most of the mutations were recovered as homozygotes in this diploid organism. We speculate that, once a mutation occurred in one allele that made it resistant to further Cas9 cleavage, cleavage of the remaining unmodified allele by Cas9 in most cases led to gene conversion of that allele to match the first allele. During subculture and infection assays of all of our Cas9‐expressing mutants, we did not observe negative effects on P. sojae growth or overall virulence. Future work, including meiotic analysis, careful examination of possible off‐target effects and targeting of additional genes, will expand our understanding of the utility and any limitations of the technology in P. sojae and other oomycetes.
CRISPR‐mediated gene disruptions and gene replacements will find numerous applications in P. sojae and other oomycetes. With careful design of sgRNAs, single members of closely related gene families could be eliminated, or tags for transcriptional measurements could be introduced, to determine their individual contributions. Alternatively, entire gene clusters could be eliminated with a pair of sgRNAs. Gene replacements will enable mutations of all kinds, including promoter mutations and epitope and fluorescent protein tags, to be introduced into the endogenous gene, where expression and phenotypes will not be confounded by position effects, overexpression artefacts or contributions from the unmodified native gene. Targeted gene insertions are also expected to solve a long‐standing problem in some oomycetes, such as P. sojae, in which ectopic transgenes are invariably poorly expressed, even from the strongest promoter. Gene disruptions will also be useful for the creation of a much wider choice of selectable markers for transformation through the creation of auxotrophic mutants. Gene deletions could also be used to remove integrated transgenes, so that selectable markers, such as NPT II, can be recycled for repeated transformation experiments.
Some of the advantages of these CRISPR‐enabled approaches are illustrated by insights gained from our manipulation of the Avr4/6 gene. The elimination of the gene by replacement with NPT II confirms that Avr4/6 makes a major contribution to recognition of the pathogen by plants containing the Rps4 and Rps6 loci, consistent with the findings of Dou et al. (2010). However, the mutants did not kill the Rps4‐ and Rps6‐containing plants as completely as they killed rps plants lacking Rps4 and Rps6. The Rps4‐ and Rps6‐containing isolines were produced by introgression, not by transformation with individual R genes (Sandhu et al., 2004). As the Rps4 and Rps6 loci, which are allelic, both contain many nucleotide‐binding and leucine‐rich repeat (NB‐LRR) genes (Sandhu et al., 2004), we speculate that additional NB‐LRR genes at these loci (or even the Rps4 and Rps6 genes themselves) can recognize additional effectors produced by the P. sojae strain used in these studies (P6497). Dou et al. (2010) also observed that P. sojae strains silenced for Avr4/6 did not completely kill Rps4‐ and Rps6‐containing lines, but these observations were ascribed to incomplete silencing of Avr4/6. With the availability of complete Avr4/6 deletion mutants, we can now more confidently conclude that the Rps4 and Rps6 loci make additional contributions to resistance other than through the recognition of Avr4/6.
The two frame‐shift mutants killed Rps6 plants nearly as well as did the HDR mutants (62% combined killing versus 74%), indicating that these mutants were no longer recognized by Rps6‐containing plants. T32‐3 killed Rps4 plants more effectively than did the two HDR mutants (83% versus 74% killing), but T11‐2‐5 was clearly avirulent on Rps4 plants (40% versus 74%; wild‐type, 31%). The explanation for this difference may lie in the observation by Dou et al. (2010) that the N‐terminal domain of Avr4/6, up to and including the dEER motif, is sufficient for recognition by Rps4 plants, whereas recognition by Rps6 plants requires the C‐terminus. As the site of the CRISPR‐induced NHEJ mutations is immediately upstream of the dEER motif, the +1 frame‐shift in T32‐3 retained the N‐terminal domain but eliminated the dEER motif, presumably abolishing effector entry into the plant (Dou et al., 2008b). The –1 frame‐shift in T11‐2‐5 also eliminated the dEER motif, but in its place created a highly positively charged sequence (RKKRNARSR). Thus, we speculate that this positively charged sequence may act as a surrogate cell entry sequence (Dou et al., 2008b; Kale et al., 2010; Milletti, 2012; Snyder and Dowdy, 2004) delivering the N‐terminal fragment for recognition by Rps4.
Of the three in‐frame deletions, the two single‐amino‐acid deletion mutants scored as intermediate to avirulent on both Rps4 and Rps6 plants, suggesting that recognition was only slightly impaired. The two‐amino‐acid deletion mutant (T11‐1) showed a stronger loss of Avr4/6 function, possibly as a result of disruption of the structure of Avr4/6.
In summary, the adaptation of CRISPR/Cas9‐mediated gene targeting to oomycetes is expected to rapidly advance the functional analysis of these extremely destructive and important plant and animal pathogens.
Experimental Procedures
P hytophthora sojae strains and growth conditions
The reference P. sojae isolate P6497 (Race 2) used in this study was routinely grown and maintained in cleared V8 medium at 25 °C in the dark. Zoospores were induced and isolated from approximately 1‐week‐old cultures grown on clarified V8 agar, as described previously (Judelson et al., 1993a). Single P. sojae transformants were incubated in 12‐well plates containing V8 medium supplemented with 50 μg/mL G418 (Geneticin, AG Scientific, San Diego, California, USA) for 2–3 days before small‐scale genomic DNA extraction.
sgRNA design
sgRNA target sites were selected according to the web tool sgRNA Designer (http://www.broadinstitute.org/rnai/public/analysis‐tools/sgrna‐design; Doench et al., 2014). Potential off‐target sites were checked using the FungiDB (www.fungidb.org) alignment search tool (blastn) against the P. sojae genome and visual inspection of the results. Sequences that perfectly matched the final 12 nucleotides of the target sequence and NGG PAM sequence were discarded (Cong et al., 2013). Ribozymes were designed according to Gao and Zhao (2014). The first six nucleotides of the HH ribozyme were designed to be the reverse complement of the first six nucleotides of the sgRNA target sequences.
Plasmid construction
All the primers used in this study are listed in Table S1 and further details on plasmid construction are described in Methods S1 (see Supporting Information). A map and sequence file for the plasmid backbones used for the expression of Cas9 and the sgRNAs targeting the Avr4/6 locus can be found in Fig. S4 and the supplemental sequences in Appendix S1.
sgRNA:Cas9 in vitro activity assay
To test the activity of the designed sgRNAs, an in vitro cleavage assay was carried out (Gao and Zhao, 2014). Briefly, sgRNA was in vitro transcribed through run‐off reactions with T7 RNA polymerase using the MEGAshortscript™ T7 kit (Ambion, Austin, TX, USA) according to the manufacturer's manual. Templates for sgRNA synthesis were generated by PCR amplification from the sgRNA expression plasmid pYF2.2‐GFP‐sgRNAs (Table S1). The target DNA was amplified from pCR2.1‐Avr4/6 using primers M13F and M13R (Appendix S1). SpCas9 nuclease was purchased from New England Biolabs Inc., Ipswich, MA, USA, and the cleavage assay was performed according to the product manual.
Improved transformation of P . sojae
Polyethylene glycol (PEG)‐mediated protoplast transformations were conducted using a modification of the previously described methods (Dou et al., 2008a; Mcleod et al., 2008). Two‐ to four‐day‐old P. sojae mycelial mats, cultured in nutrient pea broth, were harvested and pretreated with 0.8 m mannitol for 10 min, and then digested in 20 mL of enzyme solution [0.4 m mannitol, 20 mm KCl, 20 mm 2‐(N‐morpholino)ethanesulfonic acid (MES), pH 5.7, 10 mm CaCl2, 0.5% Lysing Enzymes from Trichoderma harzianum (Sigma L1412: St Louis, MO, USA) and 0.5% CELLULYSIN® Cellulase (Calbiochem 219466: San Diego, California, USA)] for approximately 40 min at room temperature with gentle shaking. The mixture was filtered through a Falcon™ Nylon Mesh Cell Strainer (BD Biosciences: San Diego, CA, USA) and protoplasts were pelleted by centrifugation at 1200 g for 2 min in a Beckman Coulter, Miami, FL, USA benchtop centrifuge with swing buckets. After washing with 30 mL of W5 solution (5 mm KCl, 125 mm CaCl2, 154 mm NaCl and 177 mm glucose), protoplasts were resuspended in 10 mL of W5 solution and left on ice for 30 min. Protoplasts were collected by centrifugation at 1200 g for 2 min in the Beckman centrifuge and resuspended at 106/mL in MMg solution (0.4 m mannitol, 15 mm MgCl2 and 4 mm MES, pH 5.7). DNA transformation was conducted in a 50 mL Falcon tube, where 1 mL protoplasts were well mixed with 20–30 μg of DNA for single plasmid transformation. For co‐transformation experiments, 20–30 μg of the plasmid carrying the NPT II selectable marker gene were used, together with an equimolar ratio of any other DNAs included. Then, three successive aliquots of 580 μL each of freshly made PEG solution (40% PEG 4000 v/v, 0.2 m mannitol and 0.1 m CaCl2) were slowly pipetted into the protoplast suspension and gently mixed. After 20 min incubation on ice, 10 mL of pea broth containing 0.5 m mannitol were added, and the protoplasts were regenerated overnight at 18 °C in the dark. For the production of stable transformants, the regenerated protoplasts were collected by centrifugation at 2000 g for 2 min in the Beckman centrifuge, and then resuspended and evenly divided into three Falcon tubes containing 50 mL of liquid pea broth containing 1% agar (42 °C), 0.5 m mannitol and 50 μg/mL G418 (AG Scientific, San Diego, CA, USA). The resuspended protoplasts were then poured into empty 60 mm × 15 mm Petri dishes. Mycelial colonies could be observed after 2 days of incubation at 25 °C in the dark. The visible transformants were transferred to V8 liquid medium containing 50 μg/mL G418 and propagated for 2–3 days at 25 °C prior to analysis. For transient expression, 50 μg/mL G418 was usually added to the regeneration medium after overnight recovery to enrich the positive transformants. After 1 day of incubation at 25 °C in the dark, hyphae were collected for gDNA extraction.
Detection and quantification of targeted mutagenesis
To detect the results of targeted mutagenesis in transformants, total gDNA was extracted from pooled or individual P. sojae transformants. For pooled transformants, 48 h after transformation, 1 mL of the mycelial culture was pelleted, resuspended in 500 mL of lysis buffer [200 mm tris(hydroxymethyl)aminomethane (Tris), pH 8.0, 200 mm NaCl, 25 mm ethylenediaminetetraacetic acid (EDTA), pH 8.0, 2% sodium dodecylsulfate (SDS), plus 0.1 mg/mL RNaseA added prior to use] and broken by vortexing with 0.5‐mm glass beads. For individual transformants, an approximately 7‐mm‐diameter clump of P. sojae hyphae was blotted dry on Kimwipe™ paper, frozen in liquid nitrogen, ground to a powder using a polypropylene pestle, and then resuspended in 500 μL of lysis buffer. Hyphal lysates were incubated at 37 °C for 30 min for RNA digestion, and the DNA was recovered by phenol–chloroform extraction and isopropanol precipitation.
All PCR amplifications were conducted using Phusion® high‐fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA) in order to exclude the possibility of mutations caused during PCR amplification. Generally, approximately 10 ng of gDNA was used as PCR template. Nested PCR was conducted if necessary, using 1 : 1000 diluted PCR products as a DNA template for the second round.
To detect NHEJ mutations, the entire 372‐bp Avr4/6 ORF was amplified and examined by digestion with the relevant restriction enzyme. For pooled transformants, a nested PCR was performed to enrich the mutated target before sequencing; this step was not needed for individual transformants, including single zoospore lines. To detect HDR events in pooled and individual transformants (other than zoospore lines), primers located outside the Avr4/6 homology arms and in the NPT II gene were used. For screening single zoospore lines, PCR was performed by using primers only outside the homology arms. In both cases, nested PCR was carried out for efficient amplification of the targets. PCR products were sequenced directly by the Sanger dideoxy method in the Oregon State University Center for Genome Research and Biocomputing, Corvallis, OR, USA.
Confocal microscopy
Laser scanning confocal microscopy (Carl Zeiss, Oberkochen, Germany, LSM 780 NLO) was used to examine the expression and subcellular localization of hSPCas9 fused to the NLS and to GFP. Living hyphae were picked from liquid cultures after 2–3 days of growth of transformants. Samples were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) for 20 min in the dark (Talbot, 2001) before microscopy examination. Images were captured using a 63× oil objective with excitation/emission settings of 405 nm/410–490 nm for DAPI and 488 nm/510–535 nm for GFP.
Infection assays
The ability of Avr4/6 mutants to infect soybean plants carrying Rps4 and Rps6 was evaluated by hypocotyl inoculation, as described previously (Dou et al., 2010). The wild‐type and mutant P. sojae strains were grown on V8 plates without G418 selection for approximately 5 days. Soybean cultivars HARO(1‐7)1 (rps), HARO4272 (Harosoy background, Rps4, Rps7), HARO6272 (Harosoy background, Rps6, Rps7), Williams (rps), L85‐2352 (Williams background, Rps4) and L89‐1581 (Williams background, Rps6) were used. Each pathogenicity test was performed in triplicate, each replicate consisting of at least 19 seedlings. A strain was considered to be avirulent if the number of inoculated Rps4 or Rps6 seedlings surviving was significantly higher than the number of surviving seedlings lacking the Rps gene, as determined by Fisher's exact test (Sokal and Rohlf, 1995), and the number was not significantly different from the number of surviving seedlings inoculated with the unmodified control strain P6497. A strain was considered to be virulent if the number of surviving Rps4 or Rps6 seedlings was not significantly different from the number of surviving rps seedlings, and was significantly fewer than the number of seedlings surviving P6497 inoculation. A strain was considered to be intermediate if the number of surviving Rps4 or Rps6 seedlings was not significantly different from the number of surviving rps seedlings, and also was not significantly different from the number of seedlings surviving P6497 inoculation. A strain was also considered to be intermediate if the number of surviving Rps4 or Rps6 seedlings was significantly greater than the number of surviving rps seedlings, and also significantly fewer than the number of seedlings surviving P6497 inoculation. Intermediate phenotypes were further designated intermediate/virulent or intermediate/avirulent if the P values indicating a difference from virulent or avirulent controls differed by more than 10‐fold.
Supporting information
Fig. S1 Phytophthora U6 promoter evaluation. (A) Alignment of selected oomycete U6 genes, showing that U6 transcripts are highly conserved. The numbers of U6 genes were variable in different oomycete species: eight in P. sojae P6497 (PsojP6497), 127 in P. infestans (PinfT30‐4), five in P. capsici LT1534 (PcapLT1534), one in P. parasitica (Ppar INRA‐310) and one in Hyaloperonospora arabidopsidis (HaraEmoy2). Genome data were obtained from the fungidb.org website. (B) Alignment of the eight annotated P. sojae U6 genes. PsU6‐1 was used to test promoter activity. The red lines indicate the border of the upstream and downstream tRNAs. (C) One of the 127 P. infestans U6 genes cloned to test U6 promoter activity. Top: position of the PiU6 gene on P. infestans T30‐4 Supercontig 65. Bottom: PiU6 sequence used for promoter activity test. The putative U6 coding region is underlined; a putative TATA‐box is indicated in red. (D) The plasmid used for testing the functions of the PsU6‐1 and PiU6 promoters in P. sojae. Residues 1–150 bp of eGFP (enhanced green fluorescent protein) were used as a transcription detection marker. Arrows indicate the primer pair U6GFP_F and U6GFP_R used for cloning the eGFP fragment and also for the detection of U6 transcripts by reverse transcription‐polymerase chain reaction (RT‐PCR). The EcoNI restriction enzyme site used for insertion of the GFP detection marker is underlined in (A) and (B) and double underlined in (C).
Fig. S2 Representative sequencing chromatograms of the Avr4/6 mutations in the single zoospore purified mutants. Regions in the box show the single guide RNA (sgRNA) target sites within the Avr4/6 gene. The unambiguous sequencing profiles indicate that these mutant lines are all homozygous.
Fig. S3 Sanger sequencing profiles revealing that the subcultured Cas9:sgRNA transformant T47 (NHEJ‐T47) and homology‐directed repair (HDR) mutant T29 (HDR‐T29‐2) had non‐homologous end‐joining (NHEJ) mutations (1‐bp insertion and deletion, respectively). Red triangles indicate the differences between wild‐type (WT) and mutants.
Fig. S4 Plasmid backbones used for the expression of hSpCas9 and single guide RNA (sgRNA) in Phytophthora sojae. (A) pYF2‐2XGFP is used for tracking the subcellular localization and expression of PsNLS‐fused hSpCas9. PsNLS is inserted into SacII and SpeI sites. hSpCas9 is inserted into SpeI and AflII sites for subcellular localization examination, and SpeI and ApaI sites for CRISPR expression. (B) pYF2.2‐GFP is used for the expression of sgRNA (inserted into NheI and AgeI sites).
Table S1 Sequences of the oligonucleotides used in this study.
Appendix S1 Supplemental sequences. Sequences of plasmids used in this study.
Methods S1 Generation of Phytophthora sojae CRISPR/Cas9 plasmids.
Acknowledgements
We thank F. Arredondo, S. Taylor and D. Wellappili (Oregon State University, Corvallis, OR, USA) for experimental assistance, M. A. Saghai‐Maroof (Virginia Tech, Blacksburg, VA, USA) for soybean seed and H. Judelson (University of California‐Riverside, CA, USA) and members of the Tyler Laboratory for useful advice. We acknowledge the Sequencing and Confocal Microscopy Facilities of the Center for Genome Research and Biocomputing at Oregon State University. This work was supported in part by grant 2011‐68004‐30104 from the Agriculture and Food Research Initiative of the US Department of Agriculture (USDA) National Institute for Food and Agriculture.
References
- Ah‐Fong, A.M. , Bormann‐Chung, C.A. and Judelson, H.S. (2008) Optimization of transgene‐mediated silencing in Phytophthora infestans and its association with small‐interfering RNAs. Fungal Genet. Biol. 45, 1197–1205. [DOI] [PubMed] [Google Scholar]
- Baxter, L. , Tripathy, S. , Ishaque, N. , Boot, N. , Cabral, A. , Kemen, E. , Thines, M. , Ah‐Fong, A. , Anderson, R. , Badejoko, W. , Bittner‐Eddy, P. , Boore, J.L. , Chibucos, M.C. , Coates, M. , Dehal, P. , Delehaunty, K. , Dong, S. , Downton, P. , Dumas, B. , Fabro, G. , Fronick, C. , Fuerstenberg, S.I. , Fulton, L. , Gaulin, E. , Govers, F. , Hughes, L. , Humphray, S. , Jiang, R.H.Y. , Judelson, H. , Kamoun, S. , Kyung, K. , Meijer, H. , Minx, P. , Morris, P. , Nelson, J. , Phuntumart, V. , Qutob, D. , Rehmany, A. , Rougon‐Cardoso, A. , Ryden, P. , Torto‐Alalibo, T. , Studholme, D. , Wang, Y. , Win, J. , Wood, J. , Clifton, S.W. , Rogers, J. , Van den Ackerveken, G. , Jones, J.D.G. , McDowell, J.M. , Beynon, J. and Tyler, B.M. (2010) Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science, 330, 1549–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman, E.K. , Chen, T. , Amendola, M. and van Steensel, B. (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L. , Ran, F.A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , Hsu, P.D. , Wu, X. , Jiang, W. , Marraffini, L.A. and Zhang, F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench, J.G. , Hartenian, E. , Graham, D.B. , Tothova, Z. , Hegde, M. , Smith, I. , Sullender, M. , Ebert, B.L. , Xavier, R.J. and Root, D.E. (2014) Rational design of highly active sgRNAs for CRISPR‐Cas9‐mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou, D. , Kale, S.D. , Wang, X. , Chen, Y. , Wang, Q. , Wang, X. , Jiang, R.H. , Arredondo, F.D. , Anderson, R.G. , Thakur, P.B. , McDowell, J.M. , Wang, Y. and Tyler, B.M. (2008a) Conserved C‐terminal motifs required for avirulence and suppression of cell death by Phytophthora sojae effector Avr1b. Plant Cell, 20, 1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou, D. , Kale, S.D. , Wang, X. , Jiang, R.H.Y. , Bruce, N.A. , Arredondo, F.D. , Zhang, X. and Tyler, B.M. (2008b) RXLR‐mediated entry of Phytophthora sojae effector Avr1b into soybean cells does not require pathogen‐encoded machinery. Plant Cell, 20, 1930–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou, D. , Kale, S.D. , Liu, T. , Tang, Q. , Wang, X. , Arredondo, F.D. , Basnayake, S. , Whisson, S. , Drenth, A. , Maclean, D. and Tyler, B.M. (2010) Different domains of Phytophthora sojae effector Avr4/6 are recognized by soybean resistance genes Rps 4 and Rps 6. Mol. Plant–Microbe Interact. 23, 425–435. [DOI] [PubMed] [Google Scholar]
- Erwin, D.C. and Ribeiro, O.K. (1996) Phytophthora Diseases Worldwide. St. Paul, MN: American Phytopathological Society. [Google Scholar]
- Fang, Y. and Tyler, B.M. (2015) The Oomycete Phytophthora sojae uses non‐canonical nuclear localization signals to direct proteins into the nucleus. Fung. Genet. Rep. 61 (Suppl.), Abstract #165. [Google Scholar]
- Gaj, T. , Gersbach, C.A. and Barbas, C.F., 3rd (2013) ZFN, TALEN, and CRISPR/Cas‐based methods for genome engineering. Trends Biotechnol. 31, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. and Zhao, Y. (2014) Self‐processing of ribozyme‐flanked RNAs into guide RNAs in vitro and in vivo for CRISPR‐mediated genome editing. J. Integr. Plant Biol. 56, 343–349. [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Zhang, Y. , Zhang, D. , Dai, X. , Estelle, M. and Zhao, Y. (2015) Auxin binding protein 1 (ABP1) is not required for either auxin signaling or Arabidopsis development. Proc. Natl. Acad. Sci. USA, 112, 2275–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijzen, M. , Förster, H. , Coffey, M.D. and Tyler, B. (1996) Cosegregation of Avr4 and Avr6 in Phytophthora sojae . Can. J. Bot. 74, 800–802. [Google Scholar]
- Haas, B.J. , Kamoun, S. , Zody, M.C. , Jiang, R.H.Y. , Handsaker, R.E. , Cano, L.M. , Grabherr, M. , Kodira, C.D. , Raffaele, S. , Torto‐Alalibo, T. , Bozkurt, T.O. , Ah‐Fong, A.M.V. , Alvarado, L. , Anderson, V.L. , Armstrong, M.R. , Avrova, A.O. , Baxter, L. , Beynon, J.L. , Boevink, P.C. , Bollmann, S.R. , Bos, J.I.B. , Bulone, V. , Cai, G. , Cakir, C. , Carrington, J.C. , Chawner, M. , Conti, L. , Costanzo, S. , Ewan, R. , Fahlgren, N. , Fischbach, M.l.A. , Fugelstad, J. , Gilroy, E.M. , Gnerre, S. , Green, P.J. , Grenville‐Briggs, L.J. , Griffith, J.M. , Grünwald, N.J. , Horn, K. , Horner, N.R. , Hu, C.H. , Huitema, E. , Jeong, D.H. , Jones, A.M.E. , Jones, J.D.G. , Jones, R.W. , Karlsson, E.K. , Kunjeti, S.G. , Lamour, K. , Liu, Z. , Ma, L.J. , Maclean, D.J. , Chibucos, M.C. , McDonald, H. , McWalters, J. , Meijer, H.J.G. , Morgan, W. , Morris, P.F. , Munro, C.A. , O'Neill, K. , Ospina‐Giraldo, M.D. , Pinzon, A. , Pritchard, L. , Ramsahoye, B. , Ren, Q. , Restrepo, S. , Roy, S. , Sadanandom, A. , Savidor, A. , Schornack, S. , Schwartz, D.C. , Schumann, U.D. , Schwessinger, B. , Seyer, L. , Sharpe, T. , Silvar, C. , Song, J. , Studholme, D.J. , Sykes, S. , Thines, M. , van de Vondervoort, P.J.I. , Phuntumart, V. , Wawra, S. , Weide, R. , Win, J. , Young, C. , Zhou, S. , Fry, W.E. , Meyers, B.C. , van West, P. , Ristaino, J.B. , Govers, F. , Birch, P.R.J. , Whisson, S.C. , Judelson, H.S. and Nusbaum, C. (2009) Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans . Nature, 461, 393–398. [DOI] [PubMed] [Google Scholar]
- Hwang, W.Y. , Fu, Y. , Reyon, D. , Maeder, M.L. , Tsai, S.Q. , Sander, J.D. , Peterson R.T., Yeh, J.R. and Joung, J.K. (2013) Efficient genome editing in zebrafish using a CRISPR‐Cas system. Nat. Biotechnol. 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, J.Z. , Ciccaglione, K.M. , Tournier, V. and Zaratiegui, M. (2014) Implementation of the CRISPR‐Cas9 system in fission yeast. Nat. Commun. 5, 5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, R.H. and Tyler, B.M. (2012) Mechanisms and evolution of virulence in oomycetes. Annu. Rev. Phytopathol. 50, 295–318. [DOI] [PubMed] [Google Scholar]
- Jiang, R.H. , de Bruijn, I. , Haas, B.J. , Belmonte, R. , Löbach, L. , Christie, J. , van den Ackerveken, G. , Bottin, A. , Dumas, B. , Fan, L. , Gaulin, E. , Govers, F. , Grenville‐Briggs, L.J. , Horner, N.R. , Levin, J.Z. , Mammella, M. , Meijer, H.J.G. , Morris, P. , Nusbaum, C. , Oome, S. , Rooyen, D.V. , Saraiva, M. , Secombes, C.J. , Seidl, M.F. , Snel, B. , Stassen, J. , Sykes, S. , Tripathy, S. , van den Berg, H. , Vega‐Arreguin, J.C. , Wawra, S. , Young, S. , Zeng, Q. , Dieguez‐Uribeondo, J. , Russ, C. , Tyler, B.M. and van West, P. (2013) Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen Saprolegnia parasitica . PLoS Genet. 9, e1003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judelson, H.S. (1997) The genetics and biology of Phytophthora infestans: modern approaches to a historical challenge. Fungal Genet. Biol. 22, 65–76. [DOI] [PubMed] [Google Scholar]
- Judelson, H.S. , Coffey, M.D. , Arredondo, F.R. and Tyler, B.M. (1993a) Transformation of the oomycete pathogen Phytophthora‐megasperma F‐Sp glycinea occurs by DNA integration into single or multiple chromosomes. Curr. Genet. 23, 211–218. [DOI] [PubMed] [Google Scholar]
- Judelson, H.S. , Dudler, R. , Pieterse, C.J. , Unkles, S.E. and Michelmore, R.W. (1993b) Expression and antisense inhibition of transgenes in Phytophthora infestans is modulated by choice of promoter and position effects. Gene, 133, 63–69. [DOI] [PubMed] [Google Scholar]
- Kale, S.D. , Gu, B. , Capelluto, D.G.S. , Dou, D.‐L. , Feldman, E. , Rumore, A. , Arredondo, F.D. , Hanlon, R. , Fudal, I. , Rouxel, T. , Lawrence, C.B. , Shan, W. and Tyler, B.M. (2010) External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell, 142, 284–295. [DOI] [PubMed] [Google Scholar]
- Kamoun, S. , Furzer, O. , Jones, J.D.G. , Judelson, H.S. , Ali, G.S. , Dalio, R.J.D. , Roy, S.G. , Schena, L. , Zambounis, A. , Panabières, F. , Cahill, D. , Ruocco, M. , Figueiredo, A. , Chen, X.‐R. , Hulvey, J. , Stam, R. , Lamour, K. , Gijzen, M. , Tyler, B.M. , Grünwald, N.J. , Mukhtar, M.S. , Tomé, D.F.A. , Tör, M. , Van den Ackerveken, G. , McDowell, J. , Daayf, F. , Fry, W.E. , Lindqvist‐Kreuze, H. , Meijer, H.J.G. , Petre, B. , Ristaino, J. , Yoshida, K. , Birch, P.R.J. and Govers, F. (2015) The Top 10 oomycete pathogens in molecular plant pathology. Mol. Plant Pathol. 16, 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour, K.H. , Finley, L. , Hurtado‐Gonzales, O. , Gobena, D. , Tierney, M. and Meijer, H.J. (2006) Targeted gene mutation in Phytophthora spp. Mol. Plant–Microbe Interact. 19, 1359–1367. [DOI] [PubMed] [Google Scholar]
- Liu, R. , Chen, L. , Jiang, Y. , Zhou, Z. and Zou, G. (2015) Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 1, 15 007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P. , Yang, L. , Esvelt, K.M. , Aach, J. , Guell, M. , DiCarlo, J.E. et al (2013) RNA‐guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcleod, A. , Fry, B.A. , Zuluaga, A.P. , Myers, K.L. and Fry, W.E. (2008) Toward improvements of oomycete transformation protocols. J. Eukaryot. Microbiol. 55, 103–109. [DOI] [PubMed] [Google Scholar]
- Miller, J.C. , Tan, S. , Qiao, G. , Barlow, K.A. , Wang, J. , Xia, D.F. , Meng, X. , Paschon, D.E. , Leung, E. , Hinkley, S.J. , Dulay, G.P. , Hua, K.L. , Ankoudinova, I. , Cost, G.J. , Urnov, F.D. , Zhang, H.S. , Holmes, M.C. , Zhang, L. , Gregory, P.D. and Rebar, E.J. (2011) A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148. [DOI] [PubMed] [Google Scholar]
- Milletti, F. (2012) Cell‐penetrating peptides: classes, origin, and current landscape. Drug Discov. Today, 17, 850–860. [DOI] [PubMed] [Google Scholar]
- Peng, D. , Kurup, S.P. , Yao, P.Y. , Minning, T.A. and Tarleton, R.L. (2015) CRISPR‐Cas9‐mediated single‐gene and gene family disruption in Trypanosoma cruzi . MBio, 6, e2097–e2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu, D. , Gao, H. , Cianzio, S. and Bhattacharyya, M.K. (2004) Deletion of a disease resistance nucleotide‐binding‐site leucine‐rich‐repeat‐like sequence is associated with the loss of the Phytophthora resistance gene Rps4 in soybean. Genetics, 168, 2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, B. , Brown, K.M. , Lee, T.D. and Sibley, L.D. (2014) Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio, 5, e01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, E.L. and Dowdy, S.F. (2004) Cell penetrating peptides in drug delivery. Pharm. Res. 21, 389–393. [DOI] [PubMed] [Google Scholar]
- Sokal, R.R. and Rohlf, F.J. (1995) Biometry: The Principles and Practice of Statistics in Biological Research, San Francisco, CA: WH. Freeman & Co. [Google Scholar]
- Talbot, N.J. (2001) Molecular and Cellular Biology of Filamentous Fungi: A Practical Approach, New York: Oxford University Press. [Google Scholar]
- Tyler, B.M. (2001) Genetics and genomics of the oomycete–host interface. Trends Genet. 17, 611–614. [DOI] [PubMed] [Google Scholar]
- Tyler, B.M. (2007) Phytophthora sojae: root rot pathogen of soybean and model oomycete. Mol. Plant Pathol. 8, 1–8. [DOI] [PubMed] [Google Scholar]
- Tyler, B.M. and Gijzen, M. (2014) The Phytophthora sojae genome sequence: foundation for a revolution In: Genomics of Plant‐Associated Fungi and Oomycetes: Dicot Pathogens (Dean R.A., Lichens‐Park A. and Kole C., eds), pp. 133–157, New York: Springer. [Google Scholar]
- Tyler, B.M. , Tripathy, S. , Zhang, X. , Dehal, P. , Jiang, R.H.Y. , Aerts, A. , Arredondo, F.D. , Baxter, L. , Bensasson, D. , Beynon, J.L. , Chapman, J. , Damasceno, C.M.B. , Dorrance, A.E. , Dou, D. , Dickerman, A.W. , Dubchak, I.L. , Garbelotto, M. , Gijzen, M. , Gordon, S.G. , Govers, F. , Grünwald, N.J. , Huang, W. , Ivors, K.L. , Jones, R.W. , Kamoun, S. , Krampis, K. , Lamour, K.H. , Lee, M.‐K. , McDonald, W.H. , Medina, M. , Meijer, H.J.G. , Nordberg, E.K. , Maclean, D.J. , Ospina‐Giraldo, M.D. , Morris, P.F. , Phuntumart, V. , Putnam, N.H. , Rash, S. , Rose, J.K.C. , Sakihama, Y. , Salamov, A.A. , Savidor, A. , Scheuring, C.F. , Smith, B.M. , Sobral, B.W.S. , Terry, A. , Torto‐Alalibo, T.A. , Win, J. , Xu, Z. , Zhang, H. , Grigoriev, I.V. , Rokhsar, D.S. and Boore, J.L. (2006) Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science, 313, 1261–1266. [DOI] [PubMed] [Google Scholar]
- Upadhyay, S.K. , Kumar, J. , Alok, A. and Tuli, R. (2013) RNA‐guided genome editing for target gene mutations in wheat. G3 (Bethesda) 3, 2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas, V.K. , Barrasa, M.I. and Fink, G.R. (2015) A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci. Adv. 1, e1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, J.C. , Platt, R.J. , Goldfless, S.J. , Zhang, F. and Niles, J.C. (2014) Efficient CRISPR‐Cas9‐mediated genome editing in Plasmodium falciparum . Nat. Methods, 11, 915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Han, C. , Ferreira, A.O. , Yu, X. , Ye, W. , Tripathy, S. , Kale, S.D. , Gu, B. , Sheng, Y. , Sui, Y. , Wang, X. , Zhang, Z. , Cheng, B. , Dong, S. , Shan, W. , Zheng, X. , Dou, D. , Tyler, B.M. and Wang, Y. (2011) Transcriptional programming and functional interactions within the Phytophthora sojae RXLR effector repertoire. Plant Cell, 23, 2064–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisson, S. , Drenth, A. , Maclean, D. and Irwin, J. (1994) Evidence for outcrossing in Phytophthora sojae and linkage of a DNA marker to two avirulence genes. Curr. Genet. 27, 77–82. [DOI] [PubMed] [Google Scholar]
- Whisson, S.C. , Avrova, A.O. , Van West, P. and Jones, J.T. (2005) A method for double‐stranded RNA‐mediated transient gene silencing in Phytophthora infestans . Mol. Plant Pathol. 6, 153–163. [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Xiao, B. , Jiang, Y. , Zhao, Y. , Li, Z. , Gao, H. , Ling, Y. , Wei, J. , Li, S. , Lu, M. , Su, X. , Cui, H. and Yuan, J. (2014) Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. MBio, 5, e01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Phytophthora U6 promoter evaluation. (A) Alignment of selected oomycete U6 genes, showing that U6 transcripts are highly conserved. The numbers of U6 genes were variable in different oomycete species: eight in P. sojae P6497 (PsojP6497), 127 in P. infestans (PinfT30‐4), five in P. capsici LT1534 (PcapLT1534), one in P. parasitica (Ppar INRA‐310) and one in Hyaloperonospora arabidopsidis (HaraEmoy2). Genome data were obtained from the fungidb.org website. (B) Alignment of the eight annotated P. sojae U6 genes. PsU6‐1 was used to test promoter activity. The red lines indicate the border of the upstream and downstream tRNAs. (C) One of the 127 P. infestans U6 genes cloned to test U6 promoter activity. Top: position of the PiU6 gene on P. infestans T30‐4 Supercontig 65. Bottom: PiU6 sequence used for promoter activity test. The putative U6 coding region is underlined; a putative TATA‐box is indicated in red. (D) The plasmid used for testing the functions of the PsU6‐1 and PiU6 promoters in P. sojae. Residues 1–150 bp of eGFP (enhanced green fluorescent protein) were used as a transcription detection marker. Arrows indicate the primer pair U6GFP_F and U6GFP_R used for cloning the eGFP fragment and also for the detection of U6 transcripts by reverse transcription‐polymerase chain reaction (RT‐PCR). The EcoNI restriction enzyme site used for insertion of the GFP detection marker is underlined in (A) and (B) and double underlined in (C).
Fig. S2 Representative sequencing chromatograms of the Avr4/6 mutations in the single zoospore purified mutants. Regions in the box show the single guide RNA (sgRNA) target sites within the Avr4/6 gene. The unambiguous sequencing profiles indicate that these mutant lines are all homozygous.
Fig. S3 Sanger sequencing profiles revealing that the subcultured Cas9:sgRNA transformant T47 (NHEJ‐T47) and homology‐directed repair (HDR) mutant T29 (HDR‐T29‐2) had non‐homologous end‐joining (NHEJ) mutations (1‐bp insertion and deletion, respectively). Red triangles indicate the differences between wild‐type (WT) and mutants.
Fig. S4 Plasmid backbones used for the expression of hSpCas9 and single guide RNA (sgRNA) in Phytophthora sojae. (A) pYF2‐2XGFP is used for tracking the subcellular localization and expression of PsNLS‐fused hSpCas9. PsNLS is inserted into SacII and SpeI sites. hSpCas9 is inserted into SpeI and AflII sites for subcellular localization examination, and SpeI and ApaI sites for CRISPR expression. (B) pYF2.2‐GFP is used for the expression of sgRNA (inserted into NheI and AgeI sites).
Table S1 Sequences of the oligonucleotides used in this study.
Appendix S1 Supplemental sequences. Sequences of plasmids used in this study.
Methods S1 Generation of Phytophthora sojae CRISPR/Cas9 plasmids.