

Summary
RNA interference (RNAi), a conserved RNA‐mediated gene regulatory mechanism in eukaryotes, plays an important role in plant growth and development, and as an antiviral defence system in plants. As a counter‐strategy, plant viruses encode RNAi suppressors to suppress the RNAi pathways and consequently down‐regulate plant defence. In geminiviruses, the proteins AC2, AC4 and AV2 are known to act as RNAi suppressors. In this study, we have designed a gene silencing vector using the features of trans‐acting small interfering RNA (tasiRNA), which is simple and can be used to target multiple genes at a time employing a single‐step cloning procedure. This vector was used to target two RNAi suppressor proteins (AC2 and AC4) of the geminivirus, Tomato leaf curl New Delhi virus (ToLCNDV). The vector containing fragments of ToLCNDV AC2 and AC4 genes, on agro‐infiltration, produced copious quantities of AC2 and AC4 specific siRNA in both tobacco and tomato plants. On challenge inoculation of the agro‐infiltrated plants with ToLCNDV, most plants showed an absence of symptoms and low accumulation of viral DNA. Transgenic tobacco plants were raised using the AC2 and AC4 tasiRNA‐generating constructs, and T1 plants, obtained from the primary transgenic plants, were tested for resistance separately against ToLCNDV and Tomato leaf curl Gujarat virus. Most plants showed an absence of symptoms and low accumulation of the corresponding viruses, the resistance being generally proportional to the amounts of siRNA produced against AC2 and AC4 genes. This is the first report of the use of artificial tasiRNA to generate resistance against an important plant virus.
Keywords: artificial tasiRNA, geminivirus, microRNA, tobacco, tomato, virus resistance
Introduction
RNA interference (RNAi) is an important antiviral system, which presumably evolved even before the divergence of plants and animals (Sharp, 2001). Although many classes of small RNAs are known, these have been classified into three main categories, small interfering RNAs (siRNAs), microRNAs (miRNAs) and piwi‐interacting RNAs (piRNAs), based on their origins, structure, associated effector proteins and biological roles (Ketting, 2011). In plant genomes, a novel group of siRNAs has been discovered, called trans‐acting siRNAs (tasiRNAs), which are endogenous siRNAs, directing the cleavage of non‐identical transcripts. Each tasiRNA locus (known as TAS gene) produces a non‐coding transcript, which, in turn, is targeted by a specific miRNA. It has been postulated that the miRNA cleavage product is subsequently converted into double‐stranded RNA (dsRNA) by the RNA‐dependent RNA polymerase (RdRp), RDR6, and the dsRNA, in turn, is further successively cleaved mostly into 21‐nucleotide siRNAs in accurate phases by a Dicer‐like enzyme, DCL4 (Howell et al., 2007). Arabidopsis contains four families of TAS genes, three of which (TAS1/2/4) require one miRNA binding site (BS), whereas TAS3 requires two miRNA BSs for the maturation of tasiRNA (Yoshikawa et al., 2005). The tasiRNAs can also be produced artificially (atasiRNA) from any non‐coding gene when the miRNA binding/processing site(s) is tethered at the appropriate end of the gene. Such atasiRNAs could be put to use to tweak many biological processes, including virus control.
Begomoviruses (family, Geminiviridae; genus, Begomovirus) are single‐stranded circular DNA viruses, which can have either a monopartite (single‐component) or bipartite (two‐component) genome (King et al., 2011). Leaf curl diseases in tomato, caused by begomoviruses, can result in crop losses of up to 100% in many tropical and subtropical regions from the Old to the New World (Moriones and Navas‐Castillo, 2000). More than 15 species of begomovirus have been reported to be associated with leaf curl disease of tomato in India (Yadava and Mukherjee, 2012), the most prevalent and notorious being Tomato leaf curl New Delhi virus (ToLCNDV). The DNA‐A of begomoviruses contains five (sometimes six) open reading frames (ORFs), one (AV1) or two (AV1 and AV2) in the viral sense strand, which code for coat and pre‐coat protein, respectively, and four (AC1–AC4) in the complementary sense strand, where AC1 codes for replication‐associated protein (Rep), AC2 for transcriptional activator (TrAP) and AC3 is the replication enhancer (Ren). Together with the above proteins, AC2/C2 (Voinnet et al., 1999; van Wezel et al., 2002), AC4/C4 (Vanitharani et al., 2004) and AV2/V2 (Sharma and Ikegami, 2010; Tien et al., 2013; Zrachya et al., 2007) are known to encode RNAi suppressor proteins. The viral RNAi suppressor proteins provide important handles for virus control.
To control viral infection, the application of insecticide for whitefly, which acts as a vector for begomoviruses, can be used. However, several insecticide‐resistant varieties of whitefly have already emerged and most insecticides are environmental pollutants and carcinogens (Tien et al., 2013). The lack of well‐characterized resistance genes against most begomoviruses precludes the otherwise effective breeding‐based approach of the development of begomovirus‐resistant plants. The hairpin constructs (Bonfim et al., 2007; Sunitha et al., 2013) and synthetic or artificial miRNA expression constructs (Niu et al., 2006; Qu et al., 2007; Tien et al., 2013), utilizing the principles of RNAi and the targeting of viral promoters (transcriptonal gene silencing, TGS) or viral coding sequences (post‐transcriptional gene silencing, PTGS), have been employed to achieve viral resistance. These approaches can be very useful for virus control, especially when viral RNAi suppressors are targeted.
Recently, vectors for gene silencing induced by miRNA (De Felippes et al., 2012) and synthetic tasiRNA (Carbonell et al., 2014) have been developed that can be employed in different species to silence multiple unrelated genes using a single vector. To date, no study has reported the silencing of any viral gene employing the atasiRNA approach. Here, we report the construction of a vector that gives rise to atasiRNA from a DNA sequence of interest. These vectors with a single‐step cloning process provide an alternative to difficult multistep cloning of transgenes in vectors producing hairpin RNA and, unlike miRNA, which produces a single effector molecule for silencing, atasiRNA can produce more than one siRNA to target one or multiple gene (s) using only one construct. In this article, we report the design and construction of a gene silencing binary vector that uses a single‐step cloning process and produces atasiRNAs to target two begomoviral gene suppressors separately, namely AC2 and AC4. These constructs, when agro‐infiltrated into tomato (cultivar Pusa Ruby), followed by challenge with the agro‐infectious clone of ToLCNDV after 6 days, conferred tolerance against the virus. We further raised transgenic tobacco plants producing atasiRNA against AC2 and AC4 genes of ToLCNDV, and these transgenics showed a high degree of resistance against the challenge virus.
Results
Design of the vector generating atasiRNAs
The tasiRNA/atasiRNAs are known to silence targets in the normal siRNA pathway (de la Luz Gutierrez‐Nava et al., 2008). We designed an atasiRNAi vector (TRiV) containing two BSs of miR390, which flank a multiple cloning site (MCS with 10 restriction enzyme recognition sites) in which a non‐coding gene of interest, the source of tasiRNAs, could be cloned (Fig. 1A). The 5′‐BS of miR390 was placed just downstream of the 35S promoter and the 3′‐BS preceded the nopaline synthase (NOS) transcription termination site. The pCambia2300 backbone provided both the promoter and transcription termination sites (Fig. 1A). In the MCS region, we cloned the portions of the ToLCNDV‐AC2 and ToLCNDV‐AC4 genes conserved in tomato‐infecting begomoviruses, separately, thus producing two independent vectors, namely TRiV‐AC2 and TRiV‐AC4. The AC2 and AC4 genes have been demonstrated to encode the RNAi suppressors (Yadava and Mukherjee, 2012; Yadava et al., 2010), and thus were chosen as the targets of the silencing processes. As both the AC2 and AC4 genes overlap with the Rep gene (Fig. 1B), which controls the initiation of replication of viral DNA, the plant‐generated atasiRNAs will also target the Rep mRNA of the invading virus simultaneously for silencing, making the resistance more effective. On the basis of sequence homology within tomato‐infecting begomoviral sequences from the Indian subcontinent, determined by clustal‐w alignment, the AC2 segment of 278 nucleotide residues, spanning the overlapping region of AC1/AC2 (TRiV‐AC2), was selected. Similarly, the AC4 region of around 206 nucleotides, spanning the overlapping region of AC1/AC4 (TRiV‐AC4), was also selected. The exact AC1/AC2 and AC1/AC4 sequences were taken from the DNA‐A genome of ToLCNDV (Pandey et al., 2009; GenBank DQ629101). As the 3′‐BS region is very important for the processing of tasiRNA sequences, we introduced site‐specific mutations at different positions in this region to generate the TRiV‐M1 and TRiV‐M2 vectors (Fig. 1C). The locations and nature of the mutations are highlighted with bold and underlined nucleotides. These vectors were made to impair the supposed formation of atasiRNAs, so that these could serve as the negative controls. In each of the mutant vectors, the same conserved regions of AC2 and AC4 were cloned further, thus generating the TRiVM1‐AC2 and TRiVM2‐AC2, as well as TRiVM1‐AC4 and TRiVM2‐AC4, vectors.
Figure 1.
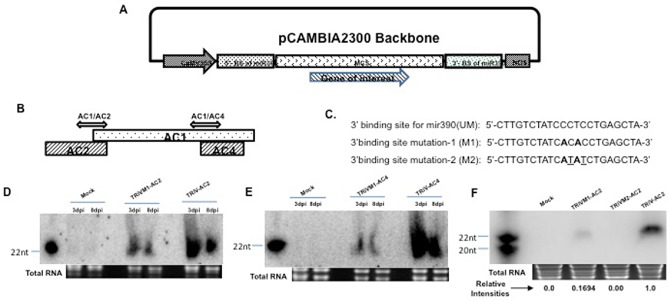
Cloning and expression of TRiV‐AC2 and TRiV‐AC4 in tobacco. (A) Map showing the TRiV construct containing 5′‐ and 3′‐binding sites of miR390 and the multiple cloning site (MCS) in which the sequence of interest can be cloned. (B) Schematic representation of DNA‐A showing the positions of AC1, AC2 and AC4 open reading frames (ORFs). Double‐headed arrows represent the fragments used for cloning in TRiV. (C) Sequences of wild‐type (UM) and mutated 3′‐binding sites of miR390. The mutated nucleotides are denoted in bold letters in M1 and additional mutated nucleotides are underlined in M2. (D) Northern blot analyses performed at 3 and 8 days post‐infiltration (dpi) of small interfering RNA (siRNA) from tobacco leaves infiltrated with Mock (empty vector), mutated (TRiVM1‐AC2) and TRiV‐AC2. (E) Same analyses with Mock, TRiVM1‐AC4 and TRiV‐AC4, as indicated above the lanes. Polymerase chain reaction (PCR)‐amplified AC1/AC2 and AC1/AC4 fragments (C) were taken as probes for TRiV‐AC2 and TRiV‐AC4, respectively. AC2SM22 and AC4SM22 were used as size markers for (D) and (E), respectively. Loading controls are indicated by total RNA below each panel. (F) Northern blot of siRNA at 8 dpi in plants infiltrated with TRiV‐AC2 with its mutants (TRiVM1 and TRiVM2). The relative intensities of the bands are presented below. AC2SM20 and AC2SM22 were used as size markers for 20 and 22 nucleotides (nt), respectively.
Transient expression of atasiRNA in tobacco and tomato plants
As mentioned earlier, miR390 guides cleavage at the 3′‐BS for the initiation of processing of the functional tasiRNA (Allen and Howell, 2010). Hence, we mutated base pairs 10 and 12 at the 3′‐BS of miR390 (TRiVM1) in the TRiV backbone to generate the negative control with impaired ability to produce small RNAs. The TRiV‐AC2 and TRiV‐AC4 vectors, together with the mutants (TRiVM1‐AC2 and TRiVM1‐AC4), were infiltrated individually in tobacco plants. The total RNAs were isolated from the systemically inoculated leaves of the agro‐infiltrated plants and analysed using Northern blotting to detect the production of small RNA at 3 and 8 days post‐infiltration (dpi) (Fig. 1D, E). TRiVM1‐AC2 produced a lower expression level of small RNA relative to TRiV‐AC2 when observed at 3 dpi (Fig. 1D, compare lanes 5 and 3) and 8 dpi (Fig. 1D, compare lanes 6 and 4). Similar results were obtained when TRiVM1‐AC4 was compared with TRiV‐AC4 (Fig. 1E). As these mutations did not block siRNA formation completely, additional mutations were introduced in the 3′‐BS. The additional mutations at the ninth and 11th positions (de la Luz Gutierrez‐Nava et al., 2008) in the 3′‐BS (TRiVM2; Fig. 1C) led to the complete loss of expression of siRNA (Fig. 1F). Using TRiVM2 as a control, TRiV‐AC2 and TRiV‐AC4 were also checked in a manner similar to that mentioned above in tomato plants for the production of siRNA. The accumulation of small RNA was observed in tomato plants (Fig. 2A, B). These results validate the construction of atasiRNA vectors that successfully give rise to the siRNAs in both tobacco and tomato.
Figure 2.
Expression of TRiV‐AC2 and TRiV‐AC4 in tomato plants. Northern blot analysis for the expression of small interfering RNA (siRNA) from TRiV‐AC2 (A) and TRiV‐AC4 (B) and their respective mutant derivatives (TRiVM2) in tomato plants at 8 days post‐infiltration (dpi). Samples are indicated on the top of each lane. Mock represents the empty vector. Lower panel shows total RNA as a loading control. The probes and markers used were the same as in Fig. 1. nt, nucleotides.
Ability of the construct to induce resistance against virus in transient assays in tomato plants
To examine the effects of atasiRNA on the growth and spread of ToLCNDV, transient expression assays were carried out in tomato plants. Tomato plants were infiltrated with agrobacteria containing TRiV‐AC2 and TRiV‐AC4 plasmids, followed by challenge with the agro‐infectious ToLCNDV clone at 3 dpi (Pratap et al., 2011). Virus infection was carried out in leaves just above the agro‐infiltrated leaf. For TRiV‐AC2, of nine plants, only two (plants 1 and 5) showed disease symptoms consisting of upward curling of the leaflet margins, reduction in the leaflet area, swelling of the vein and stunting of the plant at 21 days post‐virus challenge (Fig. 3B), whereas, for TRiV‐AC4, only three of nine plants showed similar symptoms (Fig. 3C, plants 4, 6 and 8). All plants were found to show severe symptoms in the case of TRiM2‐AC2 (Fig. 3A). Similarly, all plants, agro‐infiltrated first with TRiVM2‐AC4, showed severe disease symptoms when challenged with the agro‐infectious clones (data not shown). Rolling circle amplification (RCA) was carried out with genomic DNA isolated from all infiltrated plants, followed by restriction digestion with EcoRI, and the products showed amplification of viral DNA in plants 1 and 5 for TRiV‐AC2 and plants 4, 6 and 8 for TRiV‐AC4 (Fig. 3A, C). The experiment was repeated thrice and the results are shown in Table 1.
Figure 3.

Validation of the construct by transient expression assay in planta. Symptoms of leaf curl in the leaves of tomato plants infiltrated with TRiVM2‐AC2 as a control (A), TRiV‐AC2 (B) and TRiV‐AC4 (C), followed by challenge of the systemically inoculated leaves at 6 days post‐infiltration (dpi) with the agro‐infectious clone of Tomato leaf curl New Delhi virus (ToLCNDV). The third leaf from the top of the inoculated site in the challenged plants is shown at 21 dpi. The gels below each panel (A–C) show respective rolling circle amplification (RCA) products of the third leaf from the top, digested with EcoRI, from systemically inoculated leaves at 30 dpi. Polymerase chain reaction (PCR) amplification with actin‐specific primers is shown below for normalization. L indicates 1‐kb ladder, and P and n represent positive control (pUC19 plasmid) and number of plants used for the experiment, respectively.
Table 1.
Transient virus resistance assay, with the tomato plants infiltrated with artificial trans‐acting small interfering RNA (atasi‐RNA) constructs, followed by challenge with the agro‐infectious Tomato leaf curl N ew D elhi virus (ToLCNDV) construct in three independent experiments for each assay. i and n indicate the number of plants showing viral amplification at 30 days post‐infiltration (dpi) and the number of plants challenged with ToLCNDV, respectively, for each atasi‐RNA construct used
| Experiment | TRiV‐AC2 (i/n) | TRiV‐AC4 (i/n) | TRiVM2‐AC2 (i/n) |
|---|---|---|---|
| 1 | 1/4 | 0/4 | 4/4 |
| 2 | 2/9 | 2/10 | 9/9 |
| 3 | 1/10 | 3/9 | 10/10 |
Analysis of transgenic tobacco plants harbouring atasiRNA constructs
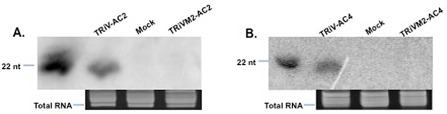
As the TRiV‐AC2 and TRiV‐AC4 constructs were validated in tobacco using transient inoculation conditions, we used the same constructs to raise transgenic tobacco (Nicotiana tabacum) plants. Fifteen lines of T0 plants for each construct were screened with polymerase chain reaction (PCR) and found to harbour the transgene. Five lines amplifying the expected band of TRiV‐AC2 (lines 1, 2, 3, 6 and 21) and six lines amplifying the expected band of TRiV‐AC4 (lines 4, 14, 15, 19, 21 and 23) were analysed further for the expression of atasiRNA by Northern blotting using an equal amount of total RNA from the leaf samples of transgenic plants. The AC1/AC2 and AC1/AC4 fragments were used as probes in the case of TRiV‐AC2 and TRiV‐AC4 plants, respectively. The results revealed that all five plants tested in the case of TRiV‐AC2 (Fig. 4A) and five of the six tested plants in the case of TRiV‐AC4 (Fig. 4B) accumulated the desired siRNAs. The intensity of the RNA bands revealed that the expression of siRNA was at a maximum in line 2, followed by lines 3, 21, 6 and 1, in the case of TRiV‐AC2 (Fig. 4A), and line 4, followed by lines 14, 21, 15 and 23, in the case of TRiV‐AC4 (Fig. 4B). As a control, the TRiVM2‐AC2 construct was also used to generate transgenic tobacco, which showed normal growth with no detectable (with the AC2 probe) siRNAs (data not shown).
Figure 4.

Northern blots showing the artificial trans‐acting small interfering RNA (atasi‐RNA) production in T0 transgenic tobacco plants. Northern blot of siRNA isolated from the top leaf of six‐leaf stage T0 transgenic tobacco plants harbouring TRiV‐AC2 (A) and TRiV‐AC4 (B). The numbers on the lanes represent the plant identifier numbers, and UT indicates untransformed plants as controls. Sizes of standard marker RNAs are indicated at the sides and relative band intensities of siRNAs are indicated at the bottom. nt, nucleotides.
Assessment of resistance to ToLCNDV in transgenic tobacco plants
To examine the effects of atasiRNA on the growth and spread of ToLCNDV, five and six lines expressing atasiRNA in transgenic tobacco plants harbouring the TRiV‐AC2 and TRiV‐AC4 transgenes, respectively, were challenged with the agro‐infectious clone of ToLCNDV. No symptoms developed in four (1, 2, 3 and 6) of the five lines tested with the TRiV‐AC2 transgenics at 30 dpi. For the sake of representation, only line 3 is illustrated in Fig. 5A. All five untransformed plants challenged with the virus exhibited severe symptoms at 15 dpi and were dwarfed. Further, RCA reactions were carried out with 100 ng of genomic DNA isolated from all lines, followed by restriction digestion of the RCA products with EcoRI to linearize the products. The products revealed that, at 30 dpi, lines 2, 3 and 6 did not contain viral DNA in the upper non‐inoculated leaves (Fig. 5C). Similarly, for the TRiV‐AC4 plants, of the six lines challenged, at 30 dpi, three lines (4, 14 and 21) showed no symptoms (Fig. 5B) and two lines (15 and 23) showed mild symptoms. RCA analysis indicated that two lines (4 and 21) showed no viral DNA accumulation (Fig. 5D).
Figure 5.

Resistance assay of transgenic tobacco plants. T0 transgenic TRiV‐AC2 line 3 (A) and TRiV‐AC4 line 4 (B) plants. Plants with the third leaf from the bottom challenged with the agro‐infectious clone of Tomato leaf curl New Delhi virus (ToLCNDV) are shown on the left with untransformed plants shown on the right. (C, D) EcoRI‐digested rolling circle amplification (RCA) products representing viral DNA in the fifth leaf from the bottom at 30 days post‐infiltration (dpi). UT represents the untransformed plant challenged with the agro‐infectious clone. H indicates plant not challenged with ToLCNDV; numbers indicate transgenic plant line numbers. Polymerase chain reaction (PCR) amplification with actin‐specific primers is shown below for normalization. L indicates 1‐kb size marker.
Quantification of ToLCNDV DNA in virus‐inoculated transgenic tobacco lines
The level of viral DNA relative to the internal β‐actin standard was calculated in the challenged plants expressing siRNA following real‐time PCR. Total RNA from infected leaf samples was collected at 30 dpi and the viral nucleic acid was estimated using the coat protein primers together with the internal β‐actin standard. The ΔΔCt value for each real‐time PCR was derived by taking into account the values of the internal standard, and these values were used to calculate the relative levels of viral DNA. As shown in Fig. 6, for TRiV‐AC2, line 3 exhibited the lowest virus accumulation (around 15‐fold decrease compared with the untransformed plant), followed by lines 6 and 2 (Fig. 6A), which were high siRNA‐expressing lines, whereas line 21, which also expressed siRNA, albeit weakly, showed high viral accumulation. For TRiV‐AC4, lines 4, 14 and 21 showed around a six‐fold decrease in virus accumulation compared with the wild‐type, and these lines expressed siRNA at high levels. In all cases, there was an inverse relationship between siRNA expression level and viral DNA accumulation.
Figure 6.
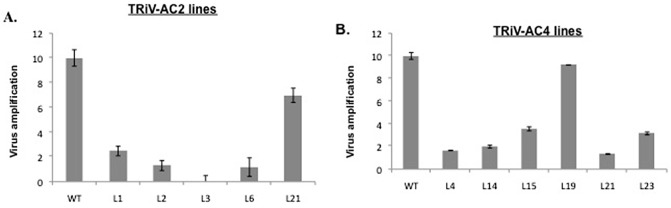
Quantification of virus in transgenics by quantitative polymerase chain reaction (qPCR). Column graph showing virus accumulation in different T0 transgenic lines [TRiV‐AC2 (A) and TRiV‐AC4 (B)] at 30 days post‐infiltration (dpi), calculated by real‐time PCR, using coat protein primers and β‐actin as an internal control. WT represents wild‐type plant inoculated with virus. The fifth leaf from the bottom was taken for the study. Standard deviations are shown as bars.
Assessment of resistance of T1 transgenic tobacco plants to ToLCNDV and Tomato leaf curl Gujarat virus (ToLCGV)
To test whether the viral resistance observed in the high‐siRNA‐producing T0 plants was retained in the T1 generation, three and two lines for TRiV‐AC2 and TRiV‐AC4, respectively, selected on the basis of their high siRNA levels, were selfed, and seeds were used to obtain T1 plants to test their viral resistance. The T0 lines selected were 2, 3 and 6 from TRiV‐AC2 and 4 and 21 from TRiV‐AC4. T1 plant lines growing on kanamycin and tested to be PCR positive were challenged with the infectious clone of ToLCNDV. In addition, to test whether the same T1 plant lines also showed resistance to another tomato‐infecting begomovirus from India, ToLCGV, the same plant lines were separately challenged with the infectious clone of ToLCGV (Chakraborty et al., 2003). Viral resistance was assessed at 21 dpi by detecting the presence of viral DNA using RCA, followed by digestion with EcoRI. As shown in Table 2, an average of 80% of plants challenged with either ToLCNDV or ToLCGV were resistant, assessed by a lack of RCA amplification of viral DNA. Control untransformed plants were all infected.
Table 2.
Virus resistance assay performed for T1 transgenic tobacco plants challenged separately with infectious clones representing T omato leaf curl N ew D elhi virus (ToLCNDV) and Tomato leaf curl Gujarat virus (ToLCGV)
| Constructs | Progeny of T0 transgenic lines | Number of non‐symptomatic plants and plants not amplifying viral DNA by rolling circle amplification/number of plants challenged | |
|---|---|---|---|
| ToLCNDV | ToLCGV | ||
| TRiV‐AC2 | Line 2 | 4/6 | 3/5 |
| Line 3 | 4/5 | 4/5 | |
| Line 6 | 5/6 | 3/4 | |
| TRiV‐AC4 | Line 4 | 5/8 | 5/7 |
| Line 21 | 3/5 | 2/5 | |
| Untransformed control | — | 0/5 | 0/5 |
To determine whether there is any relationship between viral resistance and the accumulation of tasiRNA in transgenic plant lines (T1), small RNAs extracted from two TiRV‐AC2 plants showing resistance against ToLCNDV and two plants showing resistance against ToLCGV were compared with small RNAs extracted from two plants not resistant to ToLCNDV and two plants not resistant to ToLCGV. A similar analysis was performed for TiRV‐AC4 plant lines. As shown in Fig. 7, there is a good correlation between strong accumulation of siRNAs and viral resistance to both ToLCNDV and ToLCGV in both TiRV‐AC2 and TiRV‐AC4 lines. In Fig. 7A, lanes 1–4 represent siRNAs from a resistant plant, which showed a stronger hybridizing band, indicating greater siRNA accumulation, relative to lanes 5–8, representing siRNAs from a susceptible plant.
Figure 7.
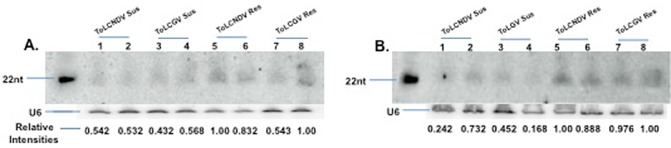
Northern blot to compare small interfering RNA (siRNA) production in resistant and susceptible plants in the T1 generation. Northern blot of T1 transgenic TRiV‐AC2 line 2 (A) and TRiV‐AC4 line 21 (B) to compare siRNA expression in susceptible and resistant plants at 30 days post‐infiltration (dpi). The top leaf was taken for the experiment. ToLCNDV Res and ToLCNDV Sus represent Tomato leaf curl New Delhi virus (ToLCNDV)‐resistant and ToLCNDV‐susceptible plants, respectively, and ToLCGV Res and ToLCGV Sus represent Tomato leaf curl Gujarat virus (ToLCGV)‐resistant and ToLCGV‐susceptible plants, respectively. The relative intensities of the bands are represented below, where the resistant line showing the highest band intensity was taken as unity for comparison for each ToLCNDV and ToLCGV separately. AC2SM22 and AC4SM22 were used as size markers in (A) and (B), respectively. nt, nucleotides.
Discussion
In this article, we have described the construction of a gene silencing vector using the features of tasiRNAs, employing a single‐step cloning process. The production of tasiRNA from TAS1 and TAS2 is dependent on miR173, and from TAS3 and TAS4 on miR390 and miR828, respectively (Allen and Howell, 2010). To date, miR173 is known to be present only in two members of the Brassicaceae, whereas miR390 is conserved among plant species. Therefore, we planned to design an atasiRNA vector using the feature of the TAS3 locus. Recently, Felippes and Weigel (2009) have demonstrated that the miRNA target site in TAS1 is not only necessary, but also sufficient, to trigger the formation of tasiRNA, whereas other sequences in the TAS1 backbone play only a minor role in the biogenesis of tasiRNA.
Working on this idea, the specific features of tasiRNA can be employed to carry out the silencing of any gene of interest. The advantages in designing atasiRNA vectors are as follows: (i) no requirement for secondary structure as in miRNA vectors; and (ii) multiple target sequences can be incorporated (Ossowski et al., 2008). Based on this idea, an atasiRNA vector was designed for the first time to target a virus of agronomic importance. A binary vector was designed that produced atasiRNA for the silencing of important geminivirus gene(s). The conserved regions of the overlapping AC2/AC1 and AC4/AC1 were chosen as targets because, under field conditions, infection normally involves a mixture of viruses (Méndez‐Lozano et al., 2003; Rentería‐Canett et al., 2011; Saunders and Stanley, 1995), some of which can be synergistic and increase the severity of the disease (Chakraborty et al., 2008). By selecting these regions, we speculated that siRNAs generated from TRiV‐AC2 or TRiV‐AC4 vectors would confer resistance against a number of tomato‐infecting begomoviruses.
After the design of the vectors, the expression of small RNAs was checked by Northern analysis in both tomato and tobacco plants. Two mutant controls were designed by mutating the nucleotides at different positions in the 3′‐BS of miR390. As, during the biogenesis of TAS3, cleavage occurs at the canonical position between bases 10 and 11, the first mutation (M1) was created at the 10th and 12th positions (Fig. 1C). This mutation resulted in reduced expression of siRNA (Fig. 1D, E), which may be a result of the reduced cleavage activity of miR390. Further mutations at positions 9 and 11 (M2, Fig. 1C) led to the complete loss of expression of siRNA (Fig. 1F), implying the additional importance of positions 9 and/or 11 for miRNA‐mediated cleavage.
Transgenic tobacco plants harbouring the atasiRNA‐generating transgenes were produced following Agrobacterium‐mediated transformation. All the phenotypic characters were observed to be normal relative to those of the untransformed plants, indicating the absence of off‐target effects of the designed atasiRNAs on the host plants, and the transgenes were stably introduced (Fig. 3A). The integration of T‐DNA into the host genome during Agrobacterium‐mediated transformation is believed to be random (Gelvin, 2003; Tzfira and Citovsky, 2006), and its mechanism is still not fully understood. In the light of this, observations that some transgenic plants could not express mature atasiRNAs and some expressed higher levels of atasiRNAs relative to others were not surprising, and could be a result of variations in the integration sites of the transgenes.
When T0 TRiV‐AC2 transgenic tobacco plants were challenged with ToLCNDV, lines 2, 3 and 6 showed complete resistance to the virus, together with high accumulation of siRNAs (Figs 4A, 5C). Line 21 developed symptoms despite expressing siRNAs. However, symptom development was delayed by 8 days relative to the control and showed recovery at 45 dpi. All the other challenged lines showed the presence of viral DNA. For TRiV‐AC4, lines 4, 14 and 21 showed resistance to the virus, revealed by both the absence of symptoms following virus challenge and the lack of amplification of viral DNA using RCA, and high siRNA expression was also observed in these lines (Figs 4B, 5D). Although untransformed plants were taken as a negative control for the transgenic study, based on the results of a transient assay, we assume that transgenic plants containing mutated constructs would have given similar results. These observations are in agreement with the results derived from real‐time PCR. The viral resistance of T0 plant lines was retained in the T1 lines, indicating that the resistance is genetically stable. The T1 lines were also resistant to ToLCGV, which is 96% similar to ToLCNDV in the target region, indicating that the resistance is applicable to related viruses, an important feature with regard to field resistance, where infection with multiple related viruses is a common occurrence.
The vectors reported here with a single‐step cloning process will provide an alternative source to the difficult multistep cloning vectors involved in either hairpin siRNA‐mediated or artificial miRNA‐mediated silencing. Many reports in which an artificial miRNA approach has been used to target a virus or viruses have shown a delay in symptom development (Ali et al., 2013; Tien et al., 2013); in some cases, a significant amount of resistance has been reported (Qu et al., 2007). Although the atasiRNA‐mediated resistance reported here is also dependent on miRNA, the levels of resistance cannot be compared between atasiRNA‐mediated and artificial miRNA‐mediated approaches. The efficiency of atasiRNA vectors needs to be analysed in different model crop plants using different target genes for silencing. However, it must be kept in mind that many viruses are also known to suppress miRNA pathways (Baulcombe, 2004) and, during mixed infection with such viruses, miRNA pathways may be down‐regulated and thus tasiRNA production could be affected.
In addition to providing an excellent tool for tasiRNA production, the reported vectors can easily provide a valuable system for the targeting of the virus for silencing. The sources of the observed siRNAs were the designed vectors, but these siRNAs silenced the viral ORFs in the infected plants. Thus, these siRNAs truly function as tasiRNAs. Working with tasiRNA or TAS genes will further increase our understanding of this newly discovered, less studied RNAi pathway, providing an insight into the complex developmental regulatory mechanisms in plants.
Experimental Procedures
Construction of gene silencing vector
Determination of the sequence of miR390 BS
Sequences of miR390 from different plant species were downloaded from miRbase (Griffiths‐Jones et al., 2006), and sequences of TAS3 were downloaded from the National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov). In silico sequence homology analysis of these miR390 sequences and the sequences of the 5′‐ and 3′‐BSs of miR390 on TAS genes was performed. Based on previous reports (Allen et al., 2005; Williams et al., 2005; Yifhar et al., 2012) and our in silico analysis, the sequences of the 5′‐ and 3′‐BSs of miR390 for the proposed vector were determined. The 5′‐BS of miR390 had the sequence 5′‐GGTGCTATCCTACCTGAGCTT‐3′ and the 3′‐BS for miR390 had the sequence 5′‐CTTGTCTATCCCTCCTGAGCTA‐3′.
Design of the MCS for the vector
The strategy to synthesize the proposed vector involved the use of the backbone of pCambia2300 (CAMBIA, Canberra, Australia) and pRT100 (Topfer et al., 1987). pCambia2300 is a binary vector used for plant transformation, which harbours the pUC18 MCS in its backbone. pUC18 MCS was removed from the vector backbone using EcoRI and SalI. pCambia2300 plasmid DNA was digested with EcoRI and SalI, which created 3′ overhangs on both ends of the linearized vector. The linearized vector was gel purified and 3′ overhangs were filled to create blunt ends by treatment with Klenow enzyme (New England Biolabs, USA) at 37 °C for 2 h. The linearized, end‐filled DNA of pCambia2300 was purified by phenol–chloroform treatment and precipitated with absolute ethanol. Purified vector was self‐ligated using ligase (5 U/μL) from MBI‐Fermentas (Amherst, NY, USA) at 16 °C overnight and transformed into DH5α cells made competent using standard methods (Sambrook and Russell, 2001). Transformed bacterial colonies were screened by colony PCR using the primers pCamF/pCamR (Table 3) flanking pUC18 MCS. One positive clone (named as pCambia2300‐pUC) was checked by restriction endonuclease and sequencing for the absence of the respective restriction sites. Ten restriction sites absent in the vector were used in MCS (SacI, KpnI, SmaI, SalI, HincII, AccI, EcoRI, SpeI, PmlI, ApaI) of the proposed vector. A complete 48‐bp MCS sequence containing sites for 10 restriction enzymes was 5′‐GAGCTCGGTACCCCCGGGGTCGACGAATTCACTAGTCACGTGGGGCCC‐3′. On the basis of the above analysis, double‐stranded, 103‐bp, tasi‐RNAi cassettes (XhoI‐5′miR390 BS‐MCS‐3′miR390 BS‐BamH1) were chemically synthesized from GENEART AG, Germany. The clone was received in the pMA vector (GENEART AG, Germany) in the form of lyophilized DNA, which was transformed in DH5α competent cells. Plasmid DNA from a positive clone was isolated for further use.
Table 3.
Sequences of primers used for the study
| Construct name or purpose | Primer name | Sequence (5′ → 3′) |
|---|---|---|
| pRT‐100 | pRTF | CAAATGCCATCATTGCGATA |
| pRTR | GCAGGTCACTGGATTTTGGT | |
| ToLCNDV‐AC2 | AC2REPF | GAGCTCTTCCCACTACCTTCCTCTGC |
| AC2REPR | ACTAGTTAAACACGCCATTCTCTGCTT | |
| ToLCNDV‐AC4 | AC4REPF | GGGGTACCAGCTGATCGTCCATCGACTT |
| AC4REPR | GGAATTCCTCCTCACTTGCATGTGCTC | |
| For real‐time polymerase chain reaction (PCR) | β‐actinFP | TGGTCGTACCACCGGTATTGTGTT |
| β‐actinRP | CCACGCTCGGTAAGGATCTTCATC | |
| AV2 FP | TCTAGAATGTGGGATCCATTATTGC | |
| AV2 RP | CCCGGGCTATACATTCTGTACATTCTGG | |
| To mutate miR390 binding sites | tasi‐m1‐3′BS | GGATCCTAGCTCAGGTGTGATAGACAAG |
| tasi‐m2‐3′BS | GGATCCTAGCTCAGAGATGATAGACAAG | |
| pCambia2300 multiple cloning site (MCS) flanking primers | pCamF | CATGTTGGCAAGCTGCTCTA |
| pCamR | GTTTTCCCAGTCACGACGTT | |
| Primers for rolling circle amplification (RCA) | β‐actin(TB)F | ACAATGAGCTCCGAGTTGCT |
| β‐actin(TB)R | TTGATCTTCATGCTGCTTGG | |
| β‐actin(TM)F | AGGCTGTGCTTTCTCTGTATG | |
| β‐actin(TM)R | GCTTTTCCTTCATGTCACGTAC | |
| For screening transgenic plants | 35S‐(TB) | TGACGCACAATCCCACTATC |
| TRiV‐AC2, size marker for Northern blotting | AC2SM20 | TTCCCACTACCTTCCTCTGC |
| AC2SM22 | TCTTCCCACTACCTTCCTCTGC | |
| TRiV‐AC4, size marker for Northern blotting | AC4SM20 | AGCTGATCGTCCATCGACTT |
| AC4SM22 | CCAGCTGATCGTCCATCGACTT |
Cloning of tasi‐RNAi cassette in the binary vector
Plasmid DNA of pRT100 was digested with XhoI and BamHI restriction enzymes to linearize the vector. Linearized vector was purified by phenol–chloroform treatment. pMA vector was also digested with BamHI and XhoI to release a 100‐bp miR390‐MCS‐miR390 cassette. The 100‐bp fragment was gel purified and cloned in linearized pRT100 vector. Clones were checked by colony PCR using pRT100 vector‐specific primers pRTF/pRTR (Table 3). Recombinant pRT100 plasmid was digested with HindIII, and ∼760‐bp fallout containing the 35S promoter, tasi‐RNAi cassette and terminator was gel purified. Similarly, pCambia2300‐pUC18 was digested with HindIII to linearize the vector. The linear vector was treated with alkaline phosphatase enzyme at 37 °C for 10 min to remove 5′‐ and 3′‐phosphate groups to avoid self‐ligation of the vector. After treatment with alkaline phosphatase, vector DNA was gel purified and ligated to HindIII‐digested pRT100 fallout. The ligation reaction was kept at 4 °C overnight and further transformed in DH5α ultracompetent cells. Colonies were screened by PCR and restriction digestion analysis. The prepared vector was named as the TRiV vector (tasiRNAi vector).
Cloning of the target gene to be silenced in the TRiV vector
The target genes were selected from ToLCNDV. Sequences of AC1/AC2 (302 bp, nucleotide positions 1417–1719) and AC1/AC4 (207 bp, nucleotide positions 2238–2445) of ToLCNDV (accession number DQ629101) were used for restriction mapping for non‐cutters. Primers were designed using the appropriate restriction sites at their 5′‐end for cloning in the MCS of the TRiV vector. The primers used to amplify the respective AC2/AC1 genes were AC2REPF/AC2REPR and, for the AC2/AC1 genes, AC4REPF/AC4REPR (Table 3). PCR amplification was carried out in a 50‐μL reaction mixture containing 50 ng of DNA template, 1 μL of 10 μm of each primer, 1 μL of 2.5 mm of deoxynucleotide triphosphates (dNTPs), 1 × Taq buffer and 2 U of Taq DNA polymerase. PCR amplification conditions involved an initial denaturation at 95 °C for 3 min, followed by 35 cycles of denaturation for 20 s at 95 °C, annealing for 30 s at 60 °C and extension for 30 s at 72 °C. Final extension for 10 min at 72 °C was carried out, after which the reaction was terminated at 4 °C. Amplified DNA was gel purified and T/A cloning was performed in pGEM‐T Easy T/A cloning vector following the manufacturer's protocol. DNA fragments representing AC1/AC2 and AC1/AC4 were further mobilized from the pGEM‐T to the TRiV vector using SacI/SpeI (AC1/AC2) and KpnI/EcoRI (AC1/AC4) restriction sites to obtain the recombinant clones TRiV‐AC2 and TRiV‐AC4, respectively. To determine the gene silencing efficiency of these vectors, empty vector TRiV, TRiV‐AC2 and TRiV‐AC4 were transformed in Agrobacterium tumefaciens strain EHA‐105. The preparation of competent cells of EHA‐105 and the transformation of competent cells with the desired vector construct were performed following the freeze–thaw method (Jyothishwaran et al., 2007).
Agro‐infiltration
To check the molecular expression of the TRiV‐AC2 and TRiV‐AC4 constructs, Agrobacterium cells containing the plasmids were grown to an optical density at 600 nm (OD600) of 1.0 and collected by centrifugation, and the cells were resuspended in an equal volume of 2‐(N‐morpholino)ethanesulfonic acid (MES) buffer containing 200 μm acetosyringone. The mixture was kept at 28 °C for 1 h before infiltration. Around 4 mL of the mixture was infiltrated on the abaxial surface of each leaf in two leaves. Agrobacterium‐mediated transient expression in tomato (Pusa Ruby) leaves was achieved through pressure infiltration at the six‐leaf stage, as described previously (Karjee et al., 2008) with minor modifications.
Tobacco transformation
The constructs TRiV‐AC2 and TRiV‐AC4 were used to transform N. tabacum employing the modified A. tumefaciens‐mediated transformation protocol (Lloyd et al., 1986), followed by selection with 100 mg/L kanamycin. The genomic DNA of the regenerated seedlings was isolated using a DNeasy Plant Mini Kit (Qiagen, USA) and was screened via PCR using 35S‐(TB)/AC2REPR for TRiV‐AC2 and 35S‐(TB)/AC4REPR for TRiV‐AC4 (Table 3).
Northern blotting
Total RNA was isolated using TRIZOL (Sigma, USA) from the systemically inoculated leaves of wild‐type and transgenic plants at 3 and 8 dpi. Total RNA containing siRNA was resolved in 12% denaturing urea gel. RNAs were transferred to a nylon membrane (Hybond N, Amersham, UK) by electroblotting at 0.8 A for 40 min (BIO‐RAD, USA). The membranes were cross‐linked by a UV cross‐linker (UVP). Northern hybridization was carried out using the standard protocol at 37 °C. AC1/AC2 and AC1/AC4 sequences (as described above) were used as probes for TRiV‐AC2 and TRiV‐AC4, respectively, and the probes were labelled using 3000 Ci/mmol of [α‐32P]dCTP (PerkinElmer Life Sciences, USA) and purified in a G25 column (GE Healthcare Life Sciences, UK) according to the supplier's protocol. Oligonucleotides of 20 (AC2SM20, AC2SM22) and 22 (AC4SM20, AC2SM22) residues, complementary for both TRiV‐AC2 and TRiV‐AC4, were synthesized and used as markers (Table 3). The membranes were autoradiographed using a TYPHOON phosphor imager (GE Healthcare Life Sciences). The siRNA band intensities were quantified using ImageJ software (http://rsbweb.nih.gov/ij/index.html).
Site‐directed mutagenesis
In order to insert mutations, in vitro mutagenesis was carried out at the 3′‐BS of miR390 in TRiV. The mutations were performed in two steps. In the first step, site‐directed mutagenesis was performed using the primer tasi‐m1‐3′BS (Table 3), with the tasi‐RNAi cassette in the pMA vector used as a template. The PCR product was cloned into pGEM‐T and was confirmed for mutation through sequencing. Plasmid DNA of pRT100 was digested with XhoI and BamHI restriction enzymes to linearize the vector. The linearized vector was purified by phenol–chloroform treatment. The pGEM‐T vector was also digested with BamHI and XhoI to release a 100‐bp mutated‐miR390‐MCS‐miR390 cassette, having mutations at positions 10 and 12 on the 3′‐BS of miR390 from the 3′‐end. The 100‐bp fragment was gel purified and cloned into the linearized pRT100 vector. The positive clone was checked and mobilized to pCambia2300 in the same way and using the same primers as performed for the TRiV construct, and named as TRiVM1. AC1/AC2 and AC1/AC4 were further mobilized from the pGEM‐T to the TRiVM1 vector using SacI/SpeI and KpnI/EcoRI restriction sites, respectively (as described above), to obtain the recombinant clones TRiVM1‐AC2 and TRiVM1‐AC4, respectively. Further mutation was performed at positions 9 and 11 from the 3′‐end using the first mutated pGEM‐T construct as a template for the second step, employing the tasi‐m2‐3′BS (Table 3), and was named TRiVM2; moreover, the recombinant clones were made with the AC1/AC2 and AC1/AC4 sequences, and named TRiVM2‐AC2 and TRiVM2‐AC4, respectively.
RCA of genomic DNA
RCA was performed according to the protocol of the manufacturer of the Templiphi 500 amplification kit (GE Healthcare Life Sciences) using 100 ng of genomic DNA from virus‐challenged tomato/tobacco plants (described earlier). The RCA products were digested with EcoRI and resolved on 1.2% agarose gels. PCR of the same DNA sample was conducted for β‐actin using the primers β‐actin (TB)F/β‐actin(TB)R for tobacco and β‐actin(TM)F/β‐actin(TM)R for tomato (Table 3) to quantify the viral DNA present. PCR band intensities were quantified using ImageJ software.
Real‐time PCR
Primer design
Primers for real‐time PCR‐based detection of ToLCNDV were synthesized from the coat protein region using the primer Express™ software (Applied Biosystems, USA). Primer pairs AV2FP/AV2RP and β‐actinFP/β‐actinRP (Table 3) were used.
First‐strand cDNA synthesis
The first‐strand cDNA was generated using a high‐capacity cDNA archive kit (Applied Biosystems). Two reaction mixtures were prepared and mixed later. Reaction 1 contained 1 μg of RNA, and RNAse‐free MQ water was added to make a total volume of 10 μL. Reaction 2 contained 2 μL of 10 × buffer, 0.8 μL of 100 mm dNTPs, 2 μL of 10 × random primers, 1 μL of reverse transcriptase and 4.2 μL of RNAse‐free MQ water for each reaction. The reverse transcriptase reaction was set at 25 °C for 10 min, followed by 37 °C for 2 h and then 85 °C for 5 min. After this, the samples were incubated at 4 °C. The cDNA thus synthesized was used for real‐time PCR.
PCR amplification
For real‐time PCR, 2 μL each of 5′‐ and 3′‐primers (5 mm each), 1 μL of cDNA and 25 μL of PCR SYBER GREEN reaction mix (Applied Biosystems) were mixed with 20 μL of RNAse‐free water, and 15 μL of this reaction mix were loaded in triplicate in 96‐well plates. β‐Actin was used as an endogenous control for all samples tested employing the β‐actin‐specific forward and reverse primers, β‐actinFP/β‐actinRP (Table 3). Amplification was carried out at 50 °C (2 min)/95 °C (10 min)/[95 °C (15 s)/60 °C (1 min) × 40 cycles]. At the end of the cycles, the Ct values were obtained. The values obtained for the desired genes were normalized to the β‐actin reference values, ΔΔCt values were calculated and the relative mRNA level was obtained from the formula: relative mRNA level = 2–(ΔΔCt). The results thus obtained were plotted as relative mRNA level versus sample identity.
Acknowledgements
AS would like to gratefully acknowledge the University Grants Commission, New Delhi for a Research Fellowship. This work was funded by a research grant to SKM from the Department of Biotechnology, Government of India. The gift of an infectious clone of ToLCGV from Professor S. Chakraborty is gratefully acknowledged. There are no associated conflicts of interest for any author with regard to the manuscript.
References
- Ali, I. , Amin, I. , Briddon, R.W. and Mansoor, S. (2013) Artificial microRNA‐mediated resistance against the monopartite begomovirus Cotton leaf curl Burewala virus . Virol. J. 10, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, E. and Howell, M.D. (2010) miRNAs in the biogenesis of trans‐acting siRNAs in higher plants. Semin. Cell Dev. Biol. 21, 798–804. [DOI] [PubMed] [Google Scholar]
- Allen, E. , Xie, Z. , Gustafson, A.M. and Carrington, J.C. (2005) microRNA‐directed phasing during trans‐acting siRNA biogenesis in plants. Cell, 12, 207–221. [DOI] [PubMed] [Google Scholar]
- Baulcombe, D. (2004) RNA silencing in plants. Nature, 431, 356–363. [DOI] [PubMed] [Google Scholar]
- Bonfim, K. , Faria, J.C. , Nogueira, E.O. , Mendes, E.A. and Aragão, F.J.L. (2007) RNAi‐mediated resistance to Bean golden mosaic virus in genetically engineered common bean (Phaseolus vulgaris). Mol. Plant–Microbe Interact. 20, 717–726. [DOI] [PubMed] [Google Scholar]
- Carbonell, A. , Takeda, A. , Fahlgren, N. , Johnson, S.C. , Cuperus, J.T. and Carrington, J.C. (2014) New generation of artificial microRNA and synthetic trans‐acting small interfering RNA vectors for efficient gene silencing in Arabidopsis. Plant Physiol. 165, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, S. , Pandey, P.K. , Banerjee, M.K. , Kalloo, G. and Fauquet, C.M. (2003) Tomato leaf curl Gujarat virus, a new Begomovirus species causing a severe leaf curl disease of tomato in Varanasi, India. Phytopathology, 93, 1485–1495. [DOI] [PubMed] [Google Scholar]
- Chakraborty, S. , Vanitharani, R. , Chattopadhyay, B. and Fauquet, C.M. (2008) Supervirulent pseudo‐recombination and asymmetric synergism between genomic components of two distinct species of begomovirus associated with severe tomato leaf curl disease in India. J. Gen. Virol. 89, 818–828. [DOI] [PubMed] [Google Scholar]
- De Felippes, F.F. , Wang, J. and Weigel, D. (2012) MIGS: miRNA‐induced gene silencing. Plant J. 70, 541–547. [DOI] [PubMed] [Google Scholar]
- Felippes, F.F. and Weigel, D. (2009) Triggering the formation of tasiRNAs in Arabidopsis thaliana: the role of microRNA miR173. EMBO Rep. 10, 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelvin, S.B. (2003) Agrobacterium‐mediated plant transformation: the biology behind the gene‐jockeying tool. Microbiol. Mol. Biol. Rev. 67, 16–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths‐Jones, S. , Grocock, R.J. , Dongen, S.V. , Bateman, A. and Enright, A.J. (2006) miRBase: microRNA sequences, targets, and gene nomenclature. Nucleic Acids Res. 34, D140–D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, M.D. , Fahlgren, N. , Chapman, E.J. , Cumbie, J.S. , Sullivan, C.M. , Givan, S.A. , Kasschau, K.D. and Carrington, J.C. (2007) Genome‐wide analysis of RNA dependent RNA Polymerase6/Dicerlike4 pathway in Arabidopsis reveals dependency on miRNA and tasiRNA directed targeting. Plant Cell, 19, 926–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyothishwaran, G. , Kotresha, D. , Selvaraj, T. , Srideshikan, S.M. , Rajvanshi, P.K. and Jayabaskaran, C. (2007) A modified freeze–thaw method for efficient transformation of Agrobacterium tumefaciens . Curr. Sci. 93, 770–772. [Google Scholar]
- Karjee, S. , Islam, M.N. and Mukherjee, S.K. (2008) Screening and identification of virus‐encoded RNA silencing suppressors. Methods Mol. Biol. 442, 187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting, R.F. (2011) The many faces of RNAi. Dev. Cell, 20, 148–161. [DOI] [PubMed] [Google Scholar]
- King, A.M.Q. , Adams, M.J. , Carstens, E.B. and Lefkowitz, E.J. (2011) Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego, CA: Elsevier Academic Press. [Google Scholar]
- Lloyd, A.M. , Barnason, A.R. , Rogers, S.G. , Byrne, M.C. , Fraley, R.T. and Horsch, R.B. (1986) Transformation of Arabidopsis thaliana with Agrobacterium tumefaciens . Science, 234, 464–466. [DOI] [PubMed] [Google Scholar]
- de la Luz Gutierrez‐Nava, M. , Aukerman, M.J. , Sakai, H. , Tingey, S.V. and Williams, R.W. (2008) Artificial trans‐acting siRNAs confer consistent and effective gene silencing. Plant Physiol. 147, 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez‐Lozano, J. , Torres‐Pacheco, I. , Fauquet, C.M. and Rivera‐Bustamante, R.F. (2003) Interactions between geminiviruses in a naturally occurring mixture: Pepper huasteco virus and Pepper golden mosaic virus. Phytopathology, 93, 270–277. [DOI] [PubMed] [Google Scholar]
- Moriones, E. and Navas‐Castillo, J. (2000) Tomato yellow leaf curl virus, an emerging virus complex causing epidemics worldwide. Phytopathology, 71, 123–134. [DOI] [PubMed] [Google Scholar]
- Niu, Q.W. , Lin, S.S. , Reyes, J.L. , Chen, K.S. , Wu, H.W. , Yeh, S.D. and Chua, N.H. (2006) Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat. Biotechnol. 24, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Ossowski, S. , Schwab, R. and Weigel, D. (2008) Gene silencing in plants using artificial microRNAs and other small RNAs. Plant J. 53, 674–690. [DOI] [PubMed] [Google Scholar]
- Pandey, P. , Choudhury, N.R. and Mukherjee, S.K. (2009) A geminiviral amplicon (VA) derived from Tomato leaf curl virus (ToLCV) can replicate in a wide variety of plant species and also acts as a VIGS vector. Virol. J. 6, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap, D. , Kashikar, A.R. and Mukherjee, S.K. (2011) Molecular characterization and infectivity of a Tomato leaf curl New Delhi virus variant associated with newly emerging yellow mosaic disease of eggplant in India. Virol. J. 8, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, J. , Ye, J. and Fang, R. (2007) Artificial microRNA‐mediated virus resistance in plants. J. Virol. 81, 6690–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentería‐Canett, I. , Xoconostle‐Cázares, B. , Ruiz‐Medrano, R. and Rivera‐Bustamante, R.F. (2011) Geminivirus mixed infection on pepper plants: synergistic interaction between PHYVV and PepGMV. Virol. J. 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. and Russell, D.W. (2001) The Inoue method for preparation and transformation of competent E. coli: ‘Ultra‐competent’ cells In: Molecular Cloning: A Laboratory Manual, Vol. 1, 3rd edn. pp. 1.112–1.115. New York: Cold Spring Harbor Press. [Google Scholar]
- Saunders, K. and Stanley, J. (1995) Complementation of African cassava mosaic virus AC2 gene function in a mixed bipartite geminivirus infection. J. Gen. Virol. 76, 2287–2292. [DOI] [PubMed] [Google Scholar]
- Sharma, P. and Ikegami, M. (2010) Tomato leaf curl Java virus V2 protein is a determinant of virulence, hypersensitive response and suppression of posttranscriptional gene silencing. Virology, 396, 85–93. [DOI] [PubMed] [Google Scholar]
- Sharp, P.A. (2001) RNA interference. Genes Dev. 15, 485–490. [DOI] [PubMed] [Google Scholar]
- Sunitha, S. , Shanmugapriya, G. , Balamani, V. and Veluthambi, K. (2013) Mungbean yellow mosaic virus (MYMV) AC4 suppresses post‐transcriptional gene silencing and an AC4 hairpin RNA gene reduces MYMV DNA accumulation in transgenic tobacco. Virus Genes, 46, 496–504. [DOI] [PubMed] [Google Scholar]
- Tien, V.V. , Choudhury, N.R. and Mukherjee, S.K. (2013) Transgenic tomato plants expressing artificial microRNAs for silencing the pre‐coat and coat proteins of a begomovirus, Tomato leaf curl New Delhi virus, show tolerance to virus infection. Virus Res. 172, 35–45. [DOI] [PubMed] [Google Scholar]
- Topfer, R. , Matzeit, V. , Gronenborn, B. , Schell, J. and Steinbiss, H.H. (1987) A set of plant expression vectors for transcriptional and translational fusions. Nucleic Acids Res. 15, 5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzfira, T. and Citovsky, V. (2006) Agrobacterium‐mediated genetic transformation of plants: biology and biotechnology. Curr. Opin. Biotechnol. 17, 147–154. [DOI] [PubMed] [Google Scholar]
- Vanitharani, R. , Chellappan, P. , Pita, J.S. and Fauquet, C.M. (2004) Differential roles of AC2 and AC4 of cassava geminiviruses in mediating synergism and suppression of posttranscriptional gene silencing. J. Virol. 78, 9487–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinnet, O. , Pinto, Y.M. and Baulcombe, D.C. (1999) Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses. Proc. Natl. Acad. Sci. USA, 96, 14 147–14 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wezel, V.R. , Dong, X. , Liu, H. , Tien, P. , Stanley, J. and Hong, Y. (2002) Mutation of three cysteine residues in Tomato yellow leaf curl virus‐China C2 protein causes dysfunction in pathogenesis and posttranscriptional gene‐silencing suppression. Mol. Plant–Microbe Interact. 15, 203–208. [DOI] [PubMed] [Google Scholar]
- Williams, L. , Carles, C.C. , Osmont, K.S. and Fletcher, J.C. (2005) A database analysis method identifies an endogenous trans‐acting short‐interfering RNA that targets the Arabidopsis ARF2, ARF3, and ARF4 genes. Proc. Natl. Acad. Sci. USA, 102, 9703–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava, P. and Mukherjee, S.K. (2012) Artificial microRNA and its applications In: Regulatory RNAs: Basics, Methods and Applications (Bibekanand M. and Zhumur G., eds), pp. 505–521. Berlin and Heidelberg: Springer‐Verlag. [Google Scholar]
- Yadava, P. , Suyal, G. and Mukherjee, S.K. (2010) Begomovirus DNA replication and pathogenicity. Curr. Sci. 98, 360–368. [Google Scholar]
- Yifhar, T. , Pekker, I. , Peled, D. , Friedlander, G. , Pistunov, A. , Sabban, M. , Wachsman, G. , Alvarez, J.P. , Amsellem, Z. and Eshed, Y. (2012) Failure of the tomato trans‐acting short interfering RNA program to regulate AUXIN RESPONSE FACTOR3 and ARF4 underlies the wiry leaf syndrome. Plant Cell, 24, 3575–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa, M. , Peragine, A. , Park, M.Y. and Poethig, R.S. (2005) A pathway for the biogenesis of trans‐acting siRNAs in Arabidopsis. Genes Dev. 19, 2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrachya, A. , Glick, E. , Levy, Y. , Arazi, T. , Citovsky, V. and Gafni, Y. (2007) Suppressor of RNA silencing encoded by Tomato yellow leaf curl virus‐Israel. Virology, 358, 159–165. [DOI] [PubMed] [Google Scholar]