

Summary
Zymoseptoria tritici causes Septoria tritici blotch (STB) on wheat. The disease interaction is characterized by clearly defined temporal phases of infection, ultimately resulting in the death of host tissue. Zymoseptoria tritici is a highly polymorphic species with significant intraspecific variation in virulence profiles. We generated a deep transcriptomic sequencing dataset spanning the entire time course of an infection using a previously uncharacterized, highly virulent Z. tritici strain isolated from a Swiss wheat field. We found that seven clusters of gene transcription profiles explained the progression of the infection. The earliest highly up‐regulated genes included chloroperoxidases, which may help the fungus cope with plant defences. The onset of necrotrophy was characterized by a concerted up‐regulation of proteases, plant cell wall‐degrading enzymes and lipases. Functions related to nutrition and growth characterized late necrotrophy and the transition to saprotrophic growth on dead plant tissue. We found that the peak up‐regulation of genes essential for mating coincided with the necrotrophic phase. We performed an intraspecies comparative transcriptomics analysis using a comparable time course infection experiment of the genome reference isolate IPO323. Major components of the fungal infection transcriptome were conserved between the two strains. However, individual small, secreted proteins, proteases and cell wall‐degrading enzymes showed strongly differentiated transcriptional profiles between isolates. Our analyses illustrate that successful STB infections involve complex transcriptomic remodelling to up‐regulate distinct gene functions. Heterogeneity in transcriptomes among isolates may explain some of the considerable variation in virulence and host specialization found within the species.
Keywords: comparative transcriptomics, hemibiotroph, RNA sequencing, Septoria leaf blotch, Zymoseptoria tritici
Introduction
Septoria tritici blotch (STB) caused by Zymoseptoria tritici (formerly Mycosphaerella graminicola) is a major disease on wheat (O'Driscoll et al., 2014). As a consequence of frequent sexual reproduction, large population sizes and significant gene flow among populations, Z. tritici field populations are phenotypically and genetically highly diverse globally (Zhan et al., 2003, 2005). Host resistance and pathogen virulence are mostly quantitative (Brown et al., 2015; Stewart and McDonald, 2014; Zhan et al., 2007). In Europe, approximately 70% of all fungicide applications in wheat production are targeted at the control of STB (Torriani et al., 2015). Resistance to several classes of fungicides has emerged rapidly in European and North American populations (Brunner et al., 2008; Cools and Fraaije, 2013; Estep et al., 2015; Torriani et al., 2009) and major gene resistance has failed quickly (Cowger et al., 2000), illustrating the high evolutionary potential of this pathogen.
The progression of Z. tritici infection on wheat is complex (O'Driscoll et al., 2014; Rudd et al., 2015). Germinating conidia and ascospores produce hyphae that penetrate stomata to initiate the infection of leaves (Kema et al., 1996). The fungus grows initially in the apoplast without penetrating host cells. Visible disease symptoms develop after a prolonged latent period, which is called biotrophic (Kema et al., 2000), although little evidence has been found for biotrophy (Sanchez‐Vallet et al., 2015). Despite the prolonged absence of visible symptoms, the fungus is probably suppressing host defence mechanisms during the latent phase (Hammond‐Kosack and Rudd, 2008; Rudd et al., 2015), including the secretion of LysM effectors that prevent chitin recognition (Lee et al., 2014; Marshall et al., 2011).
After the latent phase, the fungus switches to a necrotrophic phase initiated by unknown triggers. The death of host cells strongly resembles programmed cell death (Keon et al., 2007). Soon after the onset of necrosis, the pathogen produces pycnidia containing asexual spores in substomatal cavities. Later stages of infection are characterized by a prolonged period of saprotrophic growth during which the pathogen feeds on the dead host tissue and produces sexual fruiting bodies, called pseudothecia. The saprotrophic phase makes important contributions to STB epidemiology through the production of wind‐dispersed ascospores that infect neighbouring fields, as well as enabling the survival of the pathogen between growing seasons.
Recent transcriptomic and metabolomic studies have provided significant insights into the complex interaction between wheat and Z. tritici during the infection process (Kellner et al., 2014; Rudd et al., 2015). Rudd et al. (2015) have shown that the fungus probably benefits from increased production of fructan and other plant carbohydrates during the onset of necrotrophy. This stage is also characterized by a strong up‐regulation of plant defence genes, coinciding with the activation of pathogen genes encoding secondary metabolism and small secreted proteins. Although small secreted effector proteins have been shown to play a major role in the suppression of defence responses in other plant–pathogen interactions, the targeted disruption of genes encoding small secreted proteins specifically up‐regulated at the onset of the necrotrophic phase in Z. tritici does not affect disease progression (Mirzadi Gohari et al., 2015; Poppe et al., 2015; Rudd et al., 2015).
The significant genetic and phenotypic diversity found among natural Z. tritici isolates suggests that focusing on a single interaction between one isolate and one wheat cultivar will provide an incomplete understanding of the infection process. To better understand conserved and variable elements of the infection process, we compared the transcriptomes of a highly virulent Swiss strain of Z. tritici and the IPO323 genome reference isolate during different phases of the infection cycle. Previous studies on the comparative transcriptomics of fungal plant pathogen infections have been restricted to the host transcriptome of different rice cultivars in response to Magnaporthae oryzae (Bagnaresi et al., 2012) and the comparison of transcriptomes of related fungal species of Colletotrichum (O'Connell et al., 2012). We characterized different phases of the infection process and life cycle, including previously uncharacterized late stages of saprotrophic growth. We describe the transcription profiles of gene categories relevant for virulence, including secreted hydrolytic enzymes and peroxidases, at the different stages of the fungal infection cycle. We also present the first transcriptional profile of the genes involved in sexual reproduction. Finally, we performed comparative transcriptomics analyses between the previously published infection transcriptome of IPO323 (Rudd et al., 2015) and the Swiss strain 3D7. We identified putatively conserved and variable elements of the transcriptomic remodelling to obtain potential genes implicated in the differences in the virulence profiles of these strains.
Results and Discussion
Time course fungal transcriptomic analyses of wheat leaf infection
We investigated the infection cycle transcriptome of the Swiss Z. tritici isolate 3D7, which was collected from a Swiss wheat field in 1999 and was found to be highly virulent on several wheat cultivars (Zhan et al., 2002, 2005). Early stages of the infection were symptomless up to approximately 7 days post‐infection (dpi). The first signs of necrosis were observed at 9 dpi and, by 14 dpi, the leaves were entirely covered by lesions (Fig. 1A). The late stages of saprotrophic growth, which included the presence of pycnidiospores on the leaves, were monitored until 56 dpi.
Figure 1.

Overview of the infection transcriptomic sequence analyses. (A) Photographs of representative infected wheat leaves for the entire time course of the experiment (3–56 days post‐infection, dpi). (B) Total RNAseq reads originating from both fungal and wheat transcripts were aligned against the IPO323 reference genome of Zymoseptoria tritici. The percentage of aligned reads is indicative of the fungal biomass accumulation in infected leaves (Rudd et al., 2015). Two RNAseq datasets were generated: a six‐time‐point experiment spanning 3–56 dpi, termed ‘Time course’, and a replicated three‐time‐point experiment comprising 7, 13 and 56 dpi, termed ‘Replicated dataset’. Variation among replicates is expressed as the standard error. (C) Normalized transcription profiles of all Z. tritici genes based on the six‐time‐point experiment were clustered using self‐organizing maps. A robust clustering algorithm was used to identify seven main transcription profile clusters. The transcription profile clusters were characterized according to peak transcription and assigned to the different stages of the infection. Standard errors show variation among the genes included in each cluster. (D) For each of the seven main transcription profile clusters, the number of genes and the percentage of genes for which a secretory function was predicted (Morais do Amaral et al., 2012) are shown.
We analysed two distinct collections of infected plant material over the time course of the infection cycle. The first collection comprised six time points, including 3, 7, 11, 14, 21 and 56 dpi, and aimed to identify temporal patterns in the transcriptomic remodelling of the fungus during a complete infection cycle (‘Time course’ collection; Fig. 1B). A second collection at 7, 13 and 56 dpi utilized three independent biological replicates and covered the biotrophic, necrotrophic and saprotrophic stages of infection (‘Replicated dataset’ collection; Fig. 1B). For the time course experiment, we collected a total of 650.3 million reads (Table 1). The percentage of reads mapped to the fungal genome increased dramatically over the course of the infection, beginning at 0.8% and finishing at 74.2% (Fig. 1B). The increase in the proportion of pathogen transcripts relative to host transcripts correlated with an increase in fungal biomass in the leaves (Rudd et al., 2015). The appearance of the first necrotic lesions at 11 dpi was accompanied by a strong increase in aligned reads to the fungal genome, consistent with a rapid increase in fungal biomass in the plant tissue (Fig. 1B). For the replicated dataset experiment, we collected a total of 895 million reads, 1.1–75.4% of which were of fungal origin (Fig. 1B).
Table 1.
Overview of the transcriptomic sequencing data collected for the time course and replicated dataset experiments performed on the Swiss Zymoseptoria tritici isolate 3D7.
| Infection stage (dpi) | Library ID | Replicate | Library strategy | Total reads (millions) | Mapped reads (millions) | Alignment rate (%) |
|---|---|---|---|---|---|---|
| Time course experiment (all raw data available under the NCBI SRA experiment SRX1116288) | ||||||
| 3 | D3_2 | 1 | Single‐end | 95.30 | 0.73 | 0.80 |
| 7 | D7_5 | 1 | Single‐end | 109.46 | 1.45 | 1.30 |
| 11 | D11_1 | 1 | Single‐end | 136.81 | 7.45 | 5.40 |
| 14 | D14_7 | 1 | Single‐end | 119.07 | 68.69 | 57.70 |
| 21 | D21_9 | 1 | Single‐end | 116.65 | 86.00 | 73.70 |
| 56 | W8_5 | 1 | Single‐end | 73.03 | 54.20 | 74.20 |
| Total | 650.32 | 218.53 | ||||
| Replicated dataset experiment (all raw data available under the NCBI SRA experiment SRX1116289) | ||||||
| 7 | D7_2 | 1 | Paired‐end | 169.29 | 2.05 | 1.20 |
| 7 | D7_5 | 2 | Paired‐end | 160.27 | 2.22 | 1.40 |
| 7 | D7_8 | 3 | Paired‐end | 335.33 | 3.69 | 1.10 |
| 13 | D13_2 | 1 | Paired‐end | 40.63 | 5.09 | 12.50 |
| 13 | D13_4 | 2 | Paired‐end | 41.80 | 8.45 | 20.20 |
| 13 | D13_5 | 3 | Paired‐end | 46.49 | 11.25 | 24.20 |
| 56 | W8_1 | 1 | Paired‐end | 39.20 | 29.49 | 75.20 |
| 56 | W8_4 | 2 | Paired‐end | 34.22 | 25.79 | 75.40 |
| 56 | W8_7 | 3 | Paired‐end | 27.77 | 20.79 | 74.90 |
| Total | 894.99 | 108.82 | ||||
The genome of the Z. tritici reference isolate IPO323 comprises 10 933 annotated gene models (Goodwin et al., 2011). However, whole‐genome resequencing of the Swiss strain 3D7 showed that only 10 428 (95.4%) of the gene models were found in the genome (Croll et al., 2013). The majority of the missing gene models in 3D7 are explained by the fact that the Swiss isolate lacks four accessory chromosomes found in IPO323 (Croll et al., 2013). By combining transcriptomics data over all infection time points, we found evidence for 9457 gene models (90.7% of the predicted 3D7 transcriptome).
Clustering of transcription profiles using self‐organizing maps (SOMs) reveals distinct phases of wheat infection
We investigated the transcriptomic remodelling over the time course of an infection (Fig. 1A). For this, we first categorized all gene transcription profiles over the six time points into a matrix of 64 SOM cells. Second, we grouped the 64 SOM cells using robust k‐means clustering. We identified seven distinct expression clusters grouping genes that were up‐regulated at specific stages of the infection (total n = 8875 genes, 93.8% of all transcribed genes; Fig. 1C). We used the replicated dataset that comprised three replicates for each of the collections at 7, 13 and 56 dpi to assess the robustness of the clustering based on six time points. We performed a robust k‐means clustering and identified six well‐supported expression clusters (Fig. S1, see Supporting Information). A comparison between the expression clusters identified for the six‐time‐point and replicated three‐time‐point datasets showed that the assignment of genes to clusters was highly reproducible between the two independent datasets (Fig. S1).
The earliest expression cluster showed a peak up‐regulation at 3 dpi (1551 genes) corresponding to the initial colonization of the host (Fig. 1). At 7–11 dpi, there were two distinct clusters of 853 and 1166 up‐regulated genes, respectively, corresponding to late stages of symptomless growth in the host tissue. We identified three transcription profile clusters with 1335, 1078 and 1158 genes, respectively, corresponding to the early, middle and late stages of necrotrophic growth. The largest transcription profile cluster was found to be specific to the late saprotrophic growth phase with 1734 genes (Fig. 1D).
Transcripts encoding predicted secretion functions comprise 8.3% of the Z. tritici transcriptome (Morais do Amaral et al., 2012). During the different infection stages, the percentage of up‐regulated genes encoding secreted proteins changed according to the infection phase. The highest percentage of secreted proteins was found during the transition to the necrotrophic stage of infection (Fig. 1D). The transcription profile clusters for the biotroph–necrotroph transition and early necrotrophic phases contained 135 and 138 genes encoding secreted proteins, respectively. The lower percentage of secreted proteins (n = 51 secreted proteins) found during late biotrophic growth may reflect the fact that the secretion of proteins increases the risk of pathogen detection by host resistance proteins. The lowest percentage of secreted proteins was observed for genes up‐regulated during necrotrophic growth (11–14 dpi; n = 57 secreted proteins). Although protein secretion related to feeding (i.e. hydrolytic enzymes) is important during this stage, the transcriptional peak of many genes encoding short secreted peptides was immediately prior to the 11–14‐dpi necrotrophic period (see below).
Lifestyle transitions during infection are reflected in the distinct expression of gene categories
The lifestyle transitions during a Z. tritici infection probably require different categories of genes to be expressed at each stage of the infection. Gene ontology (GO) enrichment analyses showed that the earliest time point of 3 dpi was characterized by an over‐representation of up‐regulated genes involved in lipid metabolism, stress response and response to stimulus (Fig. 2). Encoded proteins associated with the membrane were similarly over‐represented. Later stages of biotrophy were characterized by an up‐regulation of proteins involved in cell differentiation, growth and regulatory functions. Interestingly, proteins related to sexual reproduction, including meiosis and sporulation (sexual or asexual), were also over‐represented, although the teleomorph is not expected to begin to form until approximately 30 dpi. The onset of necrotrophy was characterized by the initiation of metabolic processes, as well as processes localizing to the nuclear lumen, including transcription activation. During the late stages of necrotrophy, gene expression, cell cycle activation, protein maturation and intracellular transport dominated. Saprotrophy was dominated by cellular growth (e.g. cytoskeleton organization) and the production of secondary metabolites. The enrichment in the secondary metabolite GO terms was driven mainly by non‐ribosomal peptide synthases and components of polyketide synthase gene clusters.
Figure 2.

Gene ontology (GO) enrichment analyses for the seven transcription profile clusters against the genomic background. Significantly enriched terms are shown as a red colour gradient of hypergeometric test P values of < 0.05. (A) GO terms for significantly enriched biological processes. (B) GO terms for significantly enriched cellular compartments.
Stages of the infection cycle are characterized by distinct groups of highly up‐regulated genes
We examined the lists of the top up‐regulated genes for each cluster, and discuss notable characteristics of each stage of the infection process. Among the top 100 genes showing peak transcription at 3 dpi (early biotrophic cluster, Table S1, see Supporting Information), the most prevalent annotation was for chloroperoxidases, with a total of six genes with this annotation showing high expression at this stage. Chloroperoxidases comprise a peroxidase and catalase activity involved in the catalysis of halogenation reactions. Chloroperoxidases are known to play a role in secondary metabolite production and modification (Neumann et al., 2008). The high expression of these proteins at this early time point suggests that they play a role in the facilitation of the establishment of the fungus in the plant during the early stage of infection. Two genes encoding hydrophobins appeared near the top of this list. Hydrophobins may be involved in spore adhesion to the leaf surface (Tucker and Talbot, 2001). Two genes encoding homologues of GPR34 proteins were also highly expressed. GPR34 proteins constitute integral membrane proteins that sense molecules outside the cell and activate signal transduction pathways and, ultimately, cellular responses (Makide and Aoki, 2013). A number of genes encoding enzymes involved in lipid and fatty acid metabolism were also highly expressed at this stage. The encoded proteins include a lipid transfer protein, a fatty acid hydrolase and a secreted lipase. These findings indicate that the fungus is using its own lipid reserves during spore germination, as suggested previously in the transcriptomic study of the IPO323 strain (Rudd et al., 2015). However, the high expression of a secreted lipase suggests that the fungus may also be feeding from lipids present on the leaf surface or apoplast (Pollard et al., 2008; Reina‐Pinto and Yephremov, 2009). The high expression of genes encoding enzymes involved in alcohol metabolism, including alcohol dehydrogenase, aldehyde dehydrogenase and aldehyde reductase, suggests that the fungus needs to metabolize the alcohols generated during lipid metabolism.
The most abundant up‐regulated genes at 7 dpi encode oxidoreductases (late biotrophic cluster, Table S2, see Supporting Information). Oxidoreductases play a major role in basic metabolism and oxidative phosphorylation, but may also play an important role in the protection from reactive oxygen species and secondary metabolite production and degradation (Keller et al., 2005). Fatty acid desaturases and sterol desaturases were also highly up‐regulated at the late biotrophic stage, together with alcohol dehydrogenases, suggesting that the fungus may still be using its own lipid reserves 1 week after entering the plant. Interestingly, genes encoding proteins involved in post‐transcriptional modifications were highly represented in this list, including tRNA splicing proteins (responsible for intron splicing), RNA helicase (essential for most processes of RNA metabolism, such as ribosome biogenesis, pre‐mRNA splicing and translation initiation), Ribonucleoprotein (RNP) domain protein (processing and transport of mRNA precursors) and rRNA processing protein (efficient processing of rRNA), suggesting high post‐transcriptional activity in the pathogen at this pre‐symptomatic stage of infection.
The largest fraction (23%) of the top up‐regulated genes between 7 and 11 dpi, coinciding with the transition from biotrophic to early necrotrophic growth, consists of uncharacterized secreted proteins. Three putative effectors, MgECP2 (Stergiopoulos et al., 2010), MgTrp18 and MgTrp21 (Rudd et al., 2010), were among the top 100 genes in this list (the biotroph–necrotroph cluster, Table S3, see Supporting Information). Heat shock proteins and chaperones were also highly expressed at this time point, suggesting that these proteins play a role in protecting the fungus from antifungal compounds produced by the plant (Panaretou and Zhai, 2008). A large number of genes associated with fungal growth and cell proliferation were also highly up‐regulated at the biotroph–necrotroph transition. This group of genes included elongation factors involved in protein synthesis during cell division, CDC48 involved in cell division, a gene involved in ribosome biogenesis and a translation initiation factor, consistent with high levels of protein translation at this stage of infection. Genes encoding three carboxypeptidases and one aspartyl protease were among the most highly transcribed. A high protease activity during this stage of infection was also found in a separate analysis focused on genes encoding proteases (Fig. 3A). Proteases may be involved in obtaining nutrients from the degradation of plant proteins present in the apoplast, as suggested previously (Goodwin et al., 2011).
Figure 3.

Heatmap of the normalized expression values (top) and graphical representation of the averaged normalized expression values per time point (bottom) for secreted proteases (A) and secreted peroxidases (B). Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
Among the top 100 genes showing a peak of transcription at 11 dpi (early necrotrophic stage), we found the well‐characterized LysM effectors (Mg1LysM and Mg3LysM). At this stage, we found the largest proportion of secreted proteins of unknown function among the top 100 up‐regulated genes (26%, early necrotrophic cluster, Table S4, see Supporting Information). Many ribosomal proteins and an elongation factor were among the most highly expressed genes at this time point, consistent with active fungal growth. The high transcription of genes encoding cytochrome p450 and cytochrome mitochondrial precursors, peroxidases and a catalase suggests that the encoded proteins may be involved in the detoxification of the reactive oxygen species produced by the plant at this stage. The production of reactive oxygen species by the plant during the susceptible interaction has been reported to resemble programmed cell death (Keon et al., 2007). Genes encoding sugar, amino acid, ammonium, oligopeptide and major facilitator superfamily (MFS) transporters were also highly up‐regulated, suggesting an increase in nutrient uptake by the fungus during the transition to necrotrophy.
Interestingly, we did not find any uncharacterized secreted proteins among the top 100 up‐regulated genes between 11 and 14 dpi (necrotrophic cluster, Table S5, see Supporting Information). Most up‐regulated genes encoding uncharacterized secreted proteins showed a transcriptional peak earlier in the infection process. Genes encoding several ribosomal proteins, elongation factors, translation initiation factors and a CDC42 protein were highly up‐regulated, consistent with the high levels of protein translation at this stage of the infection cycle. The up‐regulation of a gene encoding β‐tubulin and a gene encoding a T‐complex 1 protein involved in tubulin folding suggests that the fungus accelerated mycelial growth during this phase of the infection, consistent with the predicted rapid increase in fungal biomass estimated from the increase in fungal read alignment rates (Fig. 1). Genes encoding a superoxide dismutase, which catalyses the conversion of the toxic superoxide ( ) radical into either ordinary molecular oxygen (O2) or hydrogen peroxide (H2O2), and a cytochrome p450 monooxygenase involved in oxygen reduction were among the top up‐regulated genes, suggesting that the fungus was defending itself against active oxygen species produced by the host.
Genes encoding ribosomal proteins, elongation factors and actin were highly up‐regulated at 14 dpi (late necrotrophic cluster, Table S6, see Supporting Information). This indicates high levels of translation and active fungal growth at this stage. Genes encoding enzymes involved in carbohydrate degradation and peptidases were also highly up‐regulated at this time point, consistent with the pathogen actively feeding on dead plant tissue.
During the late saprotrophic stage (saprotrophic cluster, Table S7, see Supporting Information), we found genes encoding ribosomal proteins, elongation factors and translation initiation factors to be highly up‐regulated, indicating active fungal growth. Genes coding for glycoside hydrolases and proteases probably play a role in obtaining nutrients from dead plant tissue at this late stage of infection. Different dehydrogenase genes were also highly up‐regulated, including a mannitol dehydrogenase, an enzyme that has been reported to play a role in asexual sporulation in the wheat pathogen Parastagonospora nodorum (Solomon et al., 2006). These results are consistent with those obtained by Rudd et al. (2015), which included transcriptomic data up to 21 dpi. The final stage of the infection cycle was characterized by asexual reproduction (formation of pycnidia) on dead leaf tissue.
Concerted transcriptional regulation of genes encoding proteases, peroxidases, lipases and cell wall‐degrading enzymes during biotrophic and necrotrophic stages
We conducted more detailed analyses on different gene categories associated with fungal virulence. The first group of genes analysed encodes secreted hydrolytic enzymes. These enzymes play many important roles in plant‐pathogenic fungi during the infection process. Our analyses included proteases, peroxidases, plant cell wall‐degrading enzymes (PCWDEs) and lipases.
Proteases constitute a group of enzymes that is expanded in the Z. tritici genome compared with other fungal plant pathogens (Goodwin et al., 2011). Proteases have been proposed to play a role in nutrition during the asymptomatic phase of infection by allowing the fungus to feed on plant proteins available in the apoplast. We found that the expression of proteases was specific to different stages of the infection. By plotting the average normalized expression values for all proteases for each time point, we observed that the peak of expression was at 11 dpi, at the initiation of the necrotrophic stage, suggesting an important role in host protein degradation during the attack on plant cells (Fig. 3A). Interestingly, many aspartic proteases showed high expression during the asymptomatic phase (7 dpi). These aspartic proteases may play a role in nutrient acquisition from apoplastic proteins during the biotrophic phase, as proposed previously (Goodwin et al., 2011). An alternative explanation is that these proteins play a defensive role by inactivating pathogenesis‐related (PR) proteins, which are part of the plant's basal defences (Edreva, 2005). The use of pathogen proteases to inactivate host basal defences would fit the model of latent necrotrophy recently proposed for Z. tritici (Sanchez‐Vallet et al., 2015). This model is also supported by the fact that none of the other groups of secreted hydrolytic enzymes considered in our analyses are highly expressed during the biotrophic phase, suggesting that nutrient acquisition is not very active during the asymptomatic phase. Proteases showed generally lower expression during the saprotrophic stage, indicating that they are unlikely to play an important role during the late stages of the infection.
We also examined the transcription profiles of genes encoding peroxidases. Our analyses showed that these were highly up‐regulated during the biotrophic phase between 3 and 7 dpi, with the most abundant class being chloroperoxidases (Fig. 3B). Chloroperoxidases constitute an expanded protein family in the Mycosphaerellaceae (Morais do Amaral et al., 2012). Five of the chloroperoxidase genes showed transcription values of more than 5000 reads per kilobase of transcript per million mapped reads (RPKM), indicating a strong induction of these genes during biotrophy. We hypothesize that these chloroperoxidases protect the fungus from reactive oxygen species or contribute to secondary metabolite production (Neumann et al., 2008).
In contrast with proteases and peroxidases, genes encoding PCWDEs are much reduced in the Z. tritici genome compared with the genomes of other fungal pathogens (Goodwin et al., 2011; Morais do Amaral et al., 2012). The expression of PCWDEs during the Z. tritici infection cycle is complex and has been characterized with respect to life cycle specialization (Brunner et al., 2013). Genes encoding PCWDEs were generally down‐regulated during the biotrophic stage, with the lowest average normalized transcription value at 7 dpi, followed by a peak of expression at 11 dpi (Fig. S2). The peak of expression at 11 dpi coincides with the initiation of the necrotrophic stage during which PCWDEs play an important role. α‐Amylases, a protein family expanded in the Z. tritici genome which has also been proposed to play a role in nutrition during the asymptomatic phase (Goodwin et al., 2011), showed peaks of transcription during the necrotrophic and saprotrophic stages, consistent with a major role in starch degradation.
Lipases are involved in lipid and fatty acid degradation and may contribute to virulence in addition to their role in nutrition (Subramoni et al., 2010). We found that the 29 secreted lipases of Z. tritici (Morais do Amaral et al., 2012) showed expression patterns that were specific to different infection stages (Fig. 4). The highest average transcription values for genes encoding secreted lipases were observed during the early biotrophic (3 dpi) and necrotrophic (11–14 dpi) stages of infection. This suggests that these proteins may be involved in nutrient acquisition during spore germination on the plant surface (3 dpi) and in lipid degradation during the necrotrophic phase. Genes encoding cutinases showed weak transcription during the early biotrophic stage, which fits with the model of plant penetration via stomata instead of direct penetration through the cuticle (Kema et al., 1996).
Figure 4.
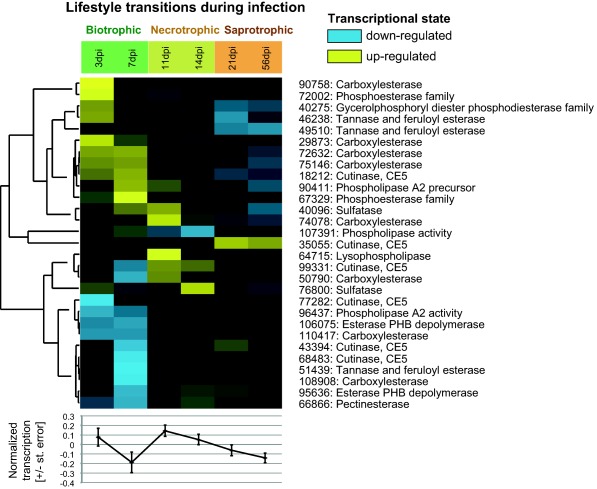
Heatmap of the normalized expression values (top) and graphical representation of the averaged normalized expression values per time point (bottom) for secreted lipases. Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
In summary, our transcriptomic analyses showed that a concerted action of proteases, PCWDEs and lipases was initiated between 7 and 11 dpi, coinciding with the onset of necrotrophy and the attack on plant cells. However, each group of enzymes showed distinct transcriptional profiles during the infection process, indicating a complex interplay between pathogen virulence and nutrient acquisition functions at each of the infection stages.
Synchronicity of genes involved in sexual reproduction and virulence progression
Sexual fruiting bodies of Z. tritici, known as pseudothecia, are produced within wheat leaf lesions (Kema et al., 1996), but have not been observed until 30–35 dpi, suggesting that the mating process may be synchronized with the local depletion of nutrients that follows the formation of the more numerous asexual pycnidia fruiting bodies. To study the synchronicity in more detail, we analysed the transcriptional profile of the genes involved in various stages of sexual reproduction. The repertoire of genes involved in mating has not yet been studied in detail in Z. tritici; hence, we identified homologues in Z. tritici based on the well‐characterized mating gene repertoire of the model fungus Aspergillus. Although a number of these genes are not specific to sexual reproduction in Aspergillus, deletion of these genes leads to an impairment of sexual reproduction (Dyer and O'Gorman, 2012). The genes involved in sexual reproduction in Aspergillus are divided into five categories: perception of environmental signals, mating processes and signal transduction, transcription factors and other regulatory proteins, endogenous physiological processes, and ascospore production and maturation (Figs 5 and S3, see Supporting Information). Most genes involved in the perception of environmental signals, including response to light, nutrients and different stresses, showed a peak of transcription during the early biotrophic stage (Fig. S3). Most of the genes in the other four categories showed a transcription peak late in the necrotrophic stage at 14 dpi, coinciding with the formation of the first asexual fruiting bodies (Figs 5 and S3). Interestingly, genes responsible for the activation of the mating process, such as the pheromone receptor gprA and several proteins in the mitogen‐activated protein (MAP) kinase cascade triggering sexual development (STE7, SteA, SteC/steB and ste50), were most strongly transcribed during the late necrotrophic stage (Fig. 5). These results strongly suggest that the activation of mating is synchronized with the activation of the necrotrophic stage, rather than with the depletion of nutrients that occurs during the saprotrophic stage. The relatively early activation of genes involved in sexual reproduction suggests that compatible Z. tritici strains co‐infecting a leaf could engage in mating soon after the onset of necrosis.
Figure 5.

Heatmap of the normalized expression values of genes predicted to be involved in mating processes and signal transduction (Dyer and O'Gorman, 2012). Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
Intraspecific variability in transcription profiles during infection
Zymoseptoria tritici exhibits significant intraspecies variability in the severity of disease symptoms caused on the host (Stewart and McDonald, 2014; Zhan et al., 2007), probably reflecting the high degree of genetic polymorphism found among isolates (Zhan et al., 2003). To obtain a first measure of the degree of intraspecific variation in the transcriptome, we performed comparative transcriptomics of 3D7 against the previously reported infection transcriptome of the reference genome isolate IPO323 (Rudd et al., 2015). The infection experiments were sampled at overlapping time points. Isolate 3D7 (this study) was sampled at 3, 7, 11, 14, 21 and 56 dpi on the winter wheat cultivar Drifter. Isolate IPO323 was sampled at 1, 3, 9, 14 and 21 dpi on the winter wheat cultivar Riband. To compare similar stages during the infection, we considered only time points from 3 to 21 dpi. We used the read alignment rate against the reference genome as a proxy to track the stages of the infection, as the alignment rate was shown to be a good predictor of fungal biomass accumulation (Fig. 6A) (Rudd et al., 2015). Both 3D7 and IPO323 infection time courses showed initial increases in read alignment rates at 7 and 9 dpi, respectively. Both isolates showed a strong increase in the read alignment rates at 14 dpi, and a levelling off at 21 dpi. Therefore, the progression in infection is largely comparable between the two experiments.
Figure 6.
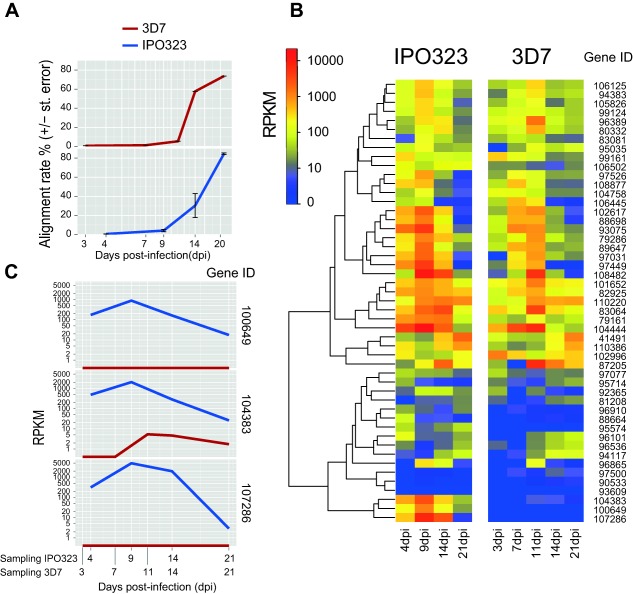
Comparative infection transcriptomic analyses of the Zymoseptoria tritici reference genome isolate IPO323 (Rudd et al., 2015) and the Swiss isolate 3D7. As isolate IPO323 was assayed at 1, 4, 9, 14 and 21 dpi and 3D7 was assayed at 3, 7, 11, 14, 21 and 56 dpi, we included infection time points spanning 3 to 21 dpi for robust transcriptomic comparisons. (A) Analysis of the synchronicity between the infection assays performed for each of the two isolates. RNAseq reads originating from both fungal and wheat transcripts were aligned against the IPO323 reference genome of Z. tritici. The percentage of aligned reads is indicative of the fungal biomass accumulation in infected leaves (Rudd et al., 2015). (B) Transcription profile comparisons of short secreted peptide (SSP) effector candidates comprising at least 5% cysteine (Morais do Amaral et al., 2012). The log‐scaled colour gradient shows reads per kilobase of transcript per million mapped reads (RPKM) values for each SSP at different time points. (C) Comparisons of transcription levels between the four SSPs that showed the most strongly differentiated transcription profiles between IPO323 and 3D7.
We compared the transcription profiles between isolates for 49 genes encoding short secreted proteins (SSPs) containing at least 5% cysteine (Morais do Amaral et al., 2012). Overall, the SSP transcription profiles were highly correlated and peak transcription most often occurred between 7 and 11 dpi (Fig. 6B). Both 3D7 and IPO323 shared a cluster of non‐transcribed SSPs, suggesting that members of this cluster may be pseudogenes or may not be involved in the infection process (Fig. 6B). Three SSPs showed very distinct transcription profiles between 3D7 and IPO323 (difference in peak RPKM of greater than 1000; Fig. 6C). The SSP‐encoding gene 104383 showed strong up‐regulation at 9 dpi in IPO323, but only weak up‐regulation (<5 RPKM) in 3D7. Using the replicated dataset available for 3D7, we confirmed that RPKM values for 104383 were <13 in all replicates of all assayed time points. For SSPs 100649 and 107286, isolate IPO323 showed strong up‐regulation at 9 dpi, but no transcription was detected for isolate 3D7. We investigated whether the lack of transcription was caused by a deletion of the SSPs 100649 and 107286 in 3D7. In the analysis of whole‐genome sequencing data for 3D7 (Croll et al., 2013), we found no reads unambiguously aligning to either gene 100649 or 107286. Hence, both of these SSPs are valuable candidates to identify differences in pathogenicity among the strains caused by deletions of genes encoding secreted proteins.
We investigated correlations in transcription profiles between 3D7 and IPO323 for two important categories of genes related to pathogenicity. We aimed to reduce biases caused by two factors, namely the asynchronicity between the infection course of the 3D7 and IPO323 experiments and biological variation not captured in the 3D7 time course experiment. Therefore, we only considered the peak RPKM detected in either experiment. We found that PCWDEs, including α‐amylases, cellulases, hemicellulases and pectinases, were strongly correlated (Fig. 7A) between the two analysed isolates. This suggests that a core set of PCWDEs is essential for successful infection, and transcription profiles are largely conserved among members of the species. A notable exception was the hemicellulase 96505, which was found to be missing from the transcriptome of isolate 3D7, but was up‐regulated in IPO323. Analyses of genomic sequences showed that 96505 was deleted in the genome of 3D7. The hemicellulase 111130 showed weak transcription (maximum of 6 RPKM) in 3D7, but stronger transcription in IPO323 (89 RPKM). Similar to the pattern for PCWDEs, transcription profiles of proteases were largely conserved between isolates 3D7 and IPO323 (Fig. 7B). Proteases with high peak transcription profiles were shared between the two isolates. However, the subtilisin‐like protease/serine protease 72659 showed a notable difference between 3D7 (879 RPKM) and IPO323 (69 RPKM). We confirmed the transcriptional patterns of 72659 and 111130 by analysing their RPKM profiles in the replicated dataset available for 3D7.
Figure 7.
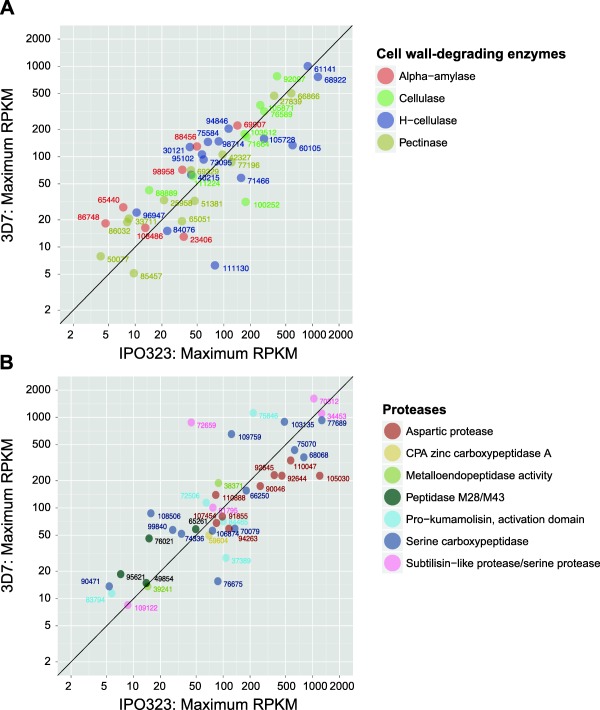
Comparative transcriptomics of specific gene categories between the Zymoseptoria tritici reference genome isolate IPO323 (Rudd et al., 2015) and the Swiss isolate 3D7. Infection time points spanning 3 to 21 dpi were included in the comparisons, and the peak transcription (in reads per kilobase of transcript per million mapped reads (RPKM)) was recorded for each gene and compared between the two isolates. (A) Peak transcription comparison of cell wall‐degrading enzymes (CWDEs). (B) Peak transcription comparison of proteases.
Conclusions
In this study, we have demonstrated the dynamic nature of a fungal transcriptome during the infection process. We characterized the transcriptional remodelling occurring during the infection cycle of a highly virulent strain of Z. tritici. We identified gene transcription profiles characterizing early biotrophy to late stages of saprotrophic growth. Each transcription profile cluster contained a set of genes indicative of the complex remodelling during the transitions from the latent period (or biotrophy) to necrotrophy and saprotrophy. Genes encoding secreted hydrolytic enzymes and peroxidases showed transcriptional profiles consistent with important roles in tolerating plant defence mechanisms, initiating necrosis and feeding on host tissue. We identified orthologues to genes involved in sexual reproduction and found that their transcriptional activity was synchronized with the onset of necrotrophy. Our intraspecific comparative transcriptomics analyses showed the conservation of major virulence components among different pathogen genotypes infecting different winter wheat cultivars.
We also identified SSPs, proteases and PCWDEs with strongly differentiated transcriptional profiles between genotypes, which were partially caused by gene deletion polymorphism segregating within the species. Our study showed that differences in the infection transcriptome among distinct isolates were caused both by variation in gene content and regulatory differences. Future large‐scale comparative transcriptomics studies will disentangle the effects caused by fungal and host genotype by comparing infections by genetically different isolates on hosts differing in resistance loci complements.
Experimental Procedures
Plant infection and RNA extraction
The Swiss Z. tritici strain ST99CH_3D7 (abbreviated 3D7) was collected from a Swiss wheat field in 1999. The genome of 3D7 was resequenced and analysed for gene and chromosomal polymorphism (Croll et al., 2013; Torriani et al., 2011), as well as for the transcriptional regulation of cell wall‐degrading enzymes (Brunner et al., 2013). For this study, the strain 3D7 was used to infect the susceptible wheat cultivar Drifter. Pathogen inoculum was prepared by growth in 50 mL of yeast sucrose broth for 6 days. After centrifugation at 2830 g for 10 min at room temperature, the spore pellet was washed to remove traces of broth, resuspended in 20 mL of double‐distilled H2O and the spore concentration was determined using a Thoma haemocytometer. Three‐week‐old Drifter plants grown in a glasshouse under 16 h of light, 70% humidity, a day temperature of 18 °C and a night temperature of 15 °C were inoculated with 120 mL of a spore suspension (106 spores/mL) containing 50 µL Tween. The second leaf on three different inoculated plants was harvested at 3, 7, 11, 13, 14, 21 and 56 dpi (Fig. 1A). Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) following the manufacturer's recommendations.
Library preparation and sequencing
RNA quality was checked using a Qubit Fluorometer (Life Technologies, Carlsbad, CA, USA) and a Bioanalyzer 2100 (Agilent, Waldbronn, Germany). All samples showed a 260 : 280 ratio between 1.8 and 2.1 and a 28S : 18S ratio of 1.5–2.0. The TruSeq RNA Sample Prep Kit v2 (Illumina Inc., San Diego, CA, USA) was used for library preparation. Samples were sequenced on an Illumina HiSeq 2000 paired‐end at 2 × 101 bp using a TruSeq SBS Kit v3‐HS (Illumina, Inc. San Diego, CA, USA).
All raw sequencing data were deposited at the National Center for Biotechnology Information (NCBI) Short Read Archive under the project SRP061444. The time course and replicated sets are accessible under experiments SRX1116288 and SRX1116289, respectively.
Transcriptomic mapping and quantification
Raw Illumina reads were quality trimmed and filtered for adapter contamination using Trimmomatic v. 0.32 (Bolger et al., 2014) with the following settings: ‘ILLUMINACLIP:TruSeq3‐PE.fa:2:30:10 LEADING:10 TRAILING:10 SLIDINGWINDOW:5:10 MINLEN:50’. Filtered reads were aligned to the IPO323 reference genome using the transcriptomic aligner tophat v. 2.0.13 (Trapnell et al., 2009). The reference genome sequence and annotation were retrieved from Ensembl release 25 (accessed January 2015). Tophat was run using default settings, except for minimum and maximum intron sizes (–min‐intron‐length 10; –max‐intron‐length 5000). We quantified reads overlapping annotated transcripts of IPO323 using HTSeq‐count, requiring a minimum alignment quality of 10 and setting the matching mode to ‘union’ (Anders et al., 2015).
Read counts for each transcript were normalized across replicates and time points using edgeR (Robinson et al., 2010). For time points at which three biological replicates were sequenced (7, 13 and 56 dpi), sets of differentially expressed genes were identified using the exactTest function, which tests for mean differences between sets of negative binomially distributed counts. We used the Benjamin–Hochberg false discovery rate (FDR) correction to account for multiple comparisons. The expression values obtained by HTSeq‐count were normalized and clustered using Cluster3 (de Hoon et al., 2004). The data were log transformed, centred and normalized (mean equals 0 and sum of squares equals 1). Java Treeview (Saldanha, 2004) was used to visualize the results from cluster analyses and to generate the final heatmaps.
For comparative transcriptomics analyses, raw RNAseq data generated for the transcriptomics study of the reference genome isolate IPO323 (Rudd et al. 2015) were accessed from the NCBI Short Read Archive (SRA study accession ERP009837). We included only RNAseq data from the wheat infection assay on cultivar Riband and excluded replicate number 3 from further analyses following the same procedure as Rudd et al. (2015), because replicate 3 tracked significantly faster than replicates 1 and 2. We processed all IPO323 RNAseq data identically as described above for isolate 3D7. However, read normalization and RPKM calculations were performed on all 3D7 and IPO323 data jointly.
Transcription profile clustering
We used SOMs to cluster transcription profiles during the six time points of the time course experiment. RPKM values were normalized (mean equals 0 and variance equals 1) and clustered using the som function in the R package som Jun Yan (2010). som: Self‐Organizing Map. R package version 0.3–5. http://CRAN.R-project.org/package=som. We predefined an 8 × 8 grid to group transcription profiles. The resulting 64 transcription profile cells were clustered using the Calinski criterion to evaluate the optimal cluster number of SOM cells. We found that nine SOM cell clusters optimally predict the different transcription profile patterns. Two SOM cell clusters were discarded because of low numbers of transcripts and weak consistency among the transcription profiles.
GO enrichment analyses
We used a local install of a Blast2GO database to annotate IPO323 reference genes according to BlastX results. BlastX was performed on the NCBI non‐redundant protein database subset for fungal sequences (accessed January 2015). The latest GO databases were downloaded in January 2015 and run using the Blast2GO command‐line interface using default parameters (Conesa et al., 2005). Enrichment of GO terms was assessed for each of the transcription profile clusters using hypergeometric tests with a false discovery rate set to 0.05. GO term sizes were set to contain at least five members in the IPO323 genome to be included in the analyses. All enrichment analyses were performed using the R packages GSEABase and GOstats (Falcon and Gentleman, 2007).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Assessment of the reproducibility of the transcription profile clustering. An identical robust k‐means clustering was performed on both the replicated three‐time‐point and six‐time‐point experiments. The transcription profile clustering for the three replicated time points identified six robust clusters that were ordered according to their peak transcription, and is shown on the y‐axis. Transcription profile clustering for the six time points is shown on the x‐axis. The heatmap shows the gene counts in overlaps according to the cluster assignments.
Fig. S2 Heatmap of the normalized expression values and graphical representation of the averaged normalized expression values per time point for secreted plant cell wall‐degrading enzymes. Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
Fig. S3 Heatmap of the normalized expression values of additional gene categories predicted to be involved in mating and sexual reproduction. Four categories (Dyer and O'Gorman, 2012) are shown: perception of environmental signals, transcription factors and other regulatory proteins, endogenous physiological processes, and ascospore production and maturation. Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
Table S1 Top 100 genes most highly expressed at the early biotrophic stage.
Table S2 Top 100 genes most highly expressed at the late biotrophic stage.
Table S3 Top 100 genes most highly expressed during the transition from the biotrophic stage to the necrotrophic stage.
Table S4 Top 100 genes most highly expressed at the early necrotrophic stage.
Table S5 Top 100 genes most highly expressed during the transition from early to late necrotrophy.
Table S6 Top 100 genes most highly expressed at the late necrotrophic stage.
Table S7 Top 100 genes most highly expressed at the saprotrophic stage.
Acknowledgements
We gratefully acknowledge funding by the Velux Foundation to DC. JPG was recipient of a fellowship from the Post doc Fellowship Programme in Plant Sciences (EU Seventh Framework Programme GA‐2010‐267243). Laboratory facilities were provided by the Genetic Diversity Centre (GDC) of ETH Zurich. Sequencing was performed at the Functional Genomics Centre Zurich (FGCZ). Parvathy Krishnan and Igy Pang provided technical assistance. JJR was supported by the Biotechnology and Biological Sciences Research Council of the UK (BBSRC) through the Institute Strategic Programme grant ‘20:20 Wheat ®’ awarded to Rothamsted Research. Andrea Sánchez Vallet provided helpful feedback on an earlier version of the manuscript.
References
- Anders, S. , Pyl, P.T. and Huber, W. (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnaresi, P. , Biselli, C. , Orrù, L. , Urso, S. , Crispino, L. , Abbruscato, P. , Piffanelli, P. , Lupotto, E. , Cattivelli, L. and Valè, G. (2012) Comparative transcriptome profiling of the early response to Magnaporthe oryzae in durable resistant vs susceptible rice (Oryza sativa L.) genotypes. PLoS One, 7, e51609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.K. , Chartrain, L. , Lasserre‐Zuber, P. and Saintenac, C. (2015) Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genet. Biol. 79, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, P.C. , Stefanato, F.L. and McDonald, B.A. (2008) Evolution of the CYP51 gene in Mycosphaerella graminicola: evidence for intragenic recombination and selective replacement. Mol. Plant Pathol. 9, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, P.C. , Torriani, S.F. , Croll, D. , Stukenbrock, E.H. and McDonald, B.A. (2013) Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol. Biol. Evol. 30, 1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. , Götz, S. , García‐Gómez, J.M. , Terol, J. , Talón, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Cools, H.J. and Fraaije, B.A. (2013) Update on mechanisms of azole resistance in Mycosphaerella graminicola and implications for future control. Pest Manag. Sci. 69, 150–155. [DOI] [PubMed] [Google Scholar]
- Cowger, C. , Hoffer, M. and Mundt, C. (2000) Specific adaptation by Mycosphaerella graminicola to a resistant wheat cultivar. Plant Pathol. 49, 445–451. [Google Scholar]
- Croll, D. , Zala, M. and McDonald, B.A. (2013) Breakage‐fusion‐bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet. 9, e1003567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer, P.S. and O'Gorman, C.M. (2012) Sexual development and cryptic sexuality in fungi: insights from Aspergillus species. FEMS Microbiol. Rev. 36, 165–192. [DOI] [PubMed] [Google Scholar]
- Edreva, A . (2005) Pathogenesis‐related proteins. Research progress in the last 15 years. Gen. Appl. Plant Physiol. 31, 105–124. [Google Scholar]
- Estep, L.K. , Torriani, S.F.F. , Zala, M. , Anderson, N.P. , Flowers, M.D. , McDonald, B.A. , Mundt, C.C. and Brunner, P.C. (2015) Emergence and early evolution of fungicide resistance in North American populations of Zymoseptoria tritici . Plant Pathol. 64, 961–971. [Google Scholar]
- Falcon, S. and Gentleman, R. (2007) Using GOstats to test gene lists for GO term association. Bioinformatics, 23, 257–258. [DOI] [PubMed] [Google Scholar]
- Goodwin, S.B. , M'barek, S.B. , Wittenberg, A.H.J. , Goodwin, S.B., M'barek, S.B., Wittenberg, A.H.J., Crane, C.F., Hane, J.K., van der Lee, T.A.J., Grimwood, J., Aerts, A., Antoniw, J., Bowler, J., van der Burgt, A., Coutinho, P.M., Csukai, M., Dehal, P., Hammond‐Kosack, K.E., Henrissat, B., Kilian, A., Lindquist, E., Mehrabi, R., Rudd, J.J., Salamov, A., Schmutz, J., Schouten, H.J., Shapiro, H., Stergiopoulos, I., Torriani, S.F.F., de Vries, R.P., Waalwijk, C., Ware, S.B., Zwiers, L.‐H., Oliver, R.P., Grigoriev, I.V. and Kema, G.H.J. (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 7, e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond‐Kosack, K.E. and Rudd, J.J. (2008) Plant resistance signalling hijacked by a necrotrophic fungal pathogen. Plant Signal. Behav. 3, 993–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon, M.J. , Imoto, S. , Nolan, J. and Miyano, S. (2004) Open source clustering software. Bioinformatics, 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Keller, N.P. , Turner, G. and Bennett, J.W. (2005) Fungal secondary metabolism – from biochemistry to genomics. Nat. Rev. Microbiol. 3, 937–947. [DOI] [PubMed] [Google Scholar]
- Kellner, R. , Bhattacharyya, A. , Poppe, S. , Hsu, T.Y. , Brem, R.B. and Stukenbrock, E.H. (2014) Expression profiling of the wheat pathogen Zymoseptoria tritici reveals genomic patterns of transcription and host‐specific regulatory programs. Genome Biol. Evol. 6, 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kema, G. , Yu, D. , Rijkenberg, F. , Shaw, M.W. and Baayen, R.P. (1996) Histology of the pathogenesis of Mycosphaerella graminicola in wheat. Phytopathology, 86, 777–786. [Google Scholar]
- Kema, G.H. , Verstappen, E.C. and Waalwijk, C. (2000) Avirulence in the wheat septoria tritici leaf blotch fungus Mycosphaerella graminicola is controlled by a single locus. Mol. Plant–Microbe Interact. 13, 1375–1379. [DOI] [PubMed] [Google Scholar]
- Keon, J. , Antoniw, J. , Carzaniga, R. , Deller, S. , Ward, J.L. , Baker, J.M. , Beale, M.H. , Hammond‐Kosack, K. and Rudd, J.J. (2007) Transcriptional adaptation of Mycosphaerella graminicola to programmed cell death (PCD) of its susceptible wheat host. Mol. Plant–Microbe Interact. 20, 178–193. [DOI] [PubMed] [Google Scholar]
- Lee, W.‐S. , Rudd, J.J. , Hammond‐Kosack, K.E. and Kanyuka, K. (2014) Mycosphaerella graminicola LysM effector‐mediated stealth pathogenesis subverts recognition through both CERK1 and CEBiP homologues in wheat. Mol. Plant–Microbe Interact. 27, 236–243. [DOI] [PubMed] [Google Scholar]
- Makide, K. and Aoki, J. (2013) GPR34 as a lysophosphatidylserine receptor. J. Biochem. 153, 327–329. [DOI] [PubMed] [Google Scholar]
- Marshall, R. , Kombrink, A. , Motteram, J. , Loza‐Reyes, E. , Lucas, J. , Hammond‐Kosack, K. , Thomma, B. and Rudd, J. (2011) Analysis of two in planta expressed LysM effector homologues from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 156, 756–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadi Gohari, A. , Ware, S.B. , Wittenberg, A.H.J. , Mehrabi, R. , Ben M'barek, S. , Verstappen, E.C.P. , van der Lee, T.A.J. , Robert, O. , Schouten, H.J. , de Wit, P.P.J.G.M. and Kema, G.H.J. (2015) Effector discovery in the fungal wheat pathogen Zymoseptoria tritici . Mol. Plant Pathol. 16, 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais do Amaral, A. , Antoniw, J. , Rudd, J.J. and Hammond‐Kosack, K.E. (2012) Defining the predicted protein secretome of the fungal wheat leaf pathogen Mycosphaerella graminicola . PLoS One, 7, e49904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann, C.S. , Fujimori, D.G. and Walsh, C.T. (2008) Halogenation strategies in natural product biosynthesis. Chem. Biol. 15, 99–109. [DOI] [PubMed] [Google Scholar]
- O'Connell, R.J., Thon, M.R., Hacquard, S., Amyotte, S.G., Kleemann, J., Torres, M.F., Damm, U., Buiate, E.A., Epstein, L., Alkan, N., Altmüller, J., Alvarado‐Balderrama, L., Bauser, C.A., Becker, C., Birren, B.W., Chen, Z., Choi, J., Crouch, J.A., Duvick, J.P., Farman, M.A., Gan, P., Heiman, D., Henrissat, B., Howard, R.J., Kabbage, M., Koch, C., Kracher, B., Kubo, Y., Law, A.D., Lebrun, M.‐H., Lee, Y.‐H., Miyara, I., Moore, N., Neumann, U., Nordström, K., Panaccione, D.G., Panstruga, R., Place, M., Proctor, R.H., Prusky, D., Rech, G., Reinhardt, R., Rollins, J.A., Rounsley, S., Schardl, C.L., Schwartz, D.C., Shenoy, N., Shirasu, K., Sikhakolli, U.R., Stüber, K., Sukno, S.A., Sweigard, J.A., Takano, Y., Takahara, H., Trail, F., van der Does, H.C., Voll, L.M., Will, I., Young, S., Zeng, Q., Zhang, J., Zhou, S., Dickman, M.B., Schulze‐Lefert, P., Ver Loren van Themaat, E., Ma, L.‐J. and Vaillancourt, L.J. (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 44, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll, A. , Kildea, S. , Doohan, F. , Spink, J. and Mullins, E. (2014) The wheat–Septoria conflict: a new front opening up? Trends Plant Sci. 19, 602–610. [DOI] [PubMed] [Google Scholar]
- Panaretou, B. and Zhai, C. (2008) The heat shock proteins: their roles as multi‐component machines for protein folding. Fungal Biol. Rev. 22, 110–119. [Google Scholar]
- Pollard, M. , Beisson, F. , Li, Y. and Ohlrogge, J.B. (2008) Building lipid barriers: biosynthesis of cutin and suberin. Trends Plant Sci. 13, 236–246. [DOI] [PubMed] [Google Scholar]
- Poppe, S. , Dorsheimer, L. , Happel, P. and Stukenbrock, E.H. (2015) Rapidly evolving genes are key players in host specialization and virulence of the fungal wheat pathogen Zymoseptoria tritici (Mycosphaerella graminicola). PLoS Pathog. 11, e1005055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina‐Pinto, J.J. and Yephremov, A. (2009) Surface lipids and plant defenses. Plant Physiol. Biochem. 47, 540–549. [DOI] [PubMed] [Google Scholar]
- Robinson, M.D. , McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd, J.J. , Antoniw, J. , Marshall, R. , Motteram, J. , Fraaije, B. and Hammond‐Kosack, K. (2010) Identification and characterisation of Mycosphaerella graminicola secreted or surface‐associated proteins with variable intragenic coding repeats. Fungal Genet. Biol. 47, 19–32. [DOI] [PubMed] [Google Scholar]
- Rudd, J.J. , Kanyuka, K. , Hassani‐Pak, K. , Rudd, J.J., Kanyuka, K., Hassani‐Pak, K., Derbyshire, M., andongabo, A., Devonshire, J., lysenko, A., Saqi, M., Desai, N.M., Powers, S.J., Hooper, J., Ambroso, L., Bharti, A., Farmer, A., Hammond‐Kosack, K.E., Dietrich, R.A. and Courbot, M. (2015) Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 167, 1158–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha, A.J. (2004) Java Treeview—extensible visualization of microarray data. Bioinformatics, 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Vallet, A. , McDonald, M.C. , Solomon, P.S. and McDonald, B.A. (2015) Is Zymoseptoria tritici a hemibiotroph? Fungal Genet. Biol. 79, 29–32. [DOI] [PubMed] [Google Scholar]
- Solomon, P.S. , Lowe, R.G. , Tan, K.‐C. , Waters, O.D. and Oliver, R.P. (2006) Stagonospora nodorum: cause of stagonospora nodorum blotch of wheat. Mol. Plant Pathol. 7, 147–156. [DOI] [PubMed] [Google Scholar]
- Stergiopoulos, I. , Van den Burg, H.A. , Okmen, B. , Beenen, H.G. , van Liere, S. , Kema, G.H. and de Wit, P.J. (2010) Tomato Cf resistance proteins mediate recognition of cognate homologous effectors from fungi pathogenic on dicots and monocots. Proc. Natl. Acad. Sci. USA, 107, 7610–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, E.L. and McDonald, B.A. (2014) Measuring quantitative virulence in the wheat pathogen Zymoseptoria tritici using high‐throughput automated image analysis. Phytopathology, 104, 985–992. [DOI] [PubMed] [Google Scholar]
- Subramoni, S. , Suárez‐Moreno, Z.R. and Venturi, V. (2010) Lipases as pathogenicity factors of plant pathogens In: Handbook of Hydrocarbon and Lipid Microbiology (Timmis K.N., ed.), pp. 3269–3277. Berlin: Handbook of Hydrocarbon and Lipid Microbiology. [Google Scholar]
- Torriani, S.F. , Brunner, P.C. , McDonald, B.A. and Sierotzki, H. (2009) QoI resistance emerged independently at least 4 times in European populations of Mycosphaerella graminicola . Pest Manag. Sci. 65, 155–162. [DOI] [PubMed] [Google Scholar]
- Torriani, S.F. , Stukenbrock, E.H. , Brunner, P.C. , McDonald, B.A. and Croll, D. (2011) Evidence for extensive recent intron transposition in closely related fungi. Curr. Biol. 21, 2017–2022. [DOI] [PubMed] [Google Scholar]
- Torriani, S.F. , Melichar, J.P. , Mills, C. , Pain, N. , Sierotzki, H. and Courbot, M. (2015) Zymoseptoria tritici: a major threat to wheat production, integrated approaches to control. Fungal Genet. Biol. 79, 8–12. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S.L. and Talbot, N.J. (2001) Surface attachment and pre‐penetration stage development by plant pathogenic fungi. Annu. Rev. Phytopathol. 39, 385–417. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Kema, G. , Waalwijk, C. and McDonald, B.A. (2002) Distribution of mating type alleles in the wheat pathogen Mycosphaerella graminicola over spatial scales from lesions to continents. Fungal Genet. Biol. 36, 128–136. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Pettway, R.E. and McDonald, B.A. (2003) The global genetic structure of the wheat pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low mitochondrial diversity, regular recombination, and gene flow. Fungal Genet. Biol. 38, 286–297. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Linde, C.C. , Jürgens, T. , Merz, U. , Steinebrunner, F. and McDonald, B.A. (2005) Variation for neutral markers is correlated with variation for quantitative traits in the plant pathogenic fungus Mycosphaerella graminicola . Mol. Ecol. 14, 2683–2693. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Torriani, S.F. and McDonald, B.A. (2007) Significant difference in pathogenicity between MAT1‐1 and MAT1‐2 isolates in the wheat pathogen Mycosphaerella graminicola . Fungal Genet. Biol. 44, 339–346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Assessment of the reproducibility of the transcription profile clustering. An identical robust k‐means clustering was performed on both the replicated three‐time‐point and six‐time‐point experiments. The transcription profile clustering for the three replicated time points identified six robust clusters that were ordered according to their peak transcription, and is shown on the y‐axis. Transcription profile clustering for the six time points is shown on the x‐axis. The heatmap shows the gene counts in overlaps according to the cluster assignments.
Fig. S2 Heatmap of the normalized expression values and graphical representation of the averaged normalized expression values per time point for secreted plant cell wall‐degrading enzymes. Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
Fig. S3 Heatmap of the normalized expression values of additional gene categories predicted to be involved in mating and sexual reproduction. Four categories (Dyer and O'Gorman, 2012) are shown: perception of environmental signals, transcription factors and other regulatory proteins, endogenous physiological processes, and ascospore production and maturation. Yellow indicates up‐regulation, blue indicates down‐regulation and black indicates the background expression level for a gene.
Table S1 Top 100 genes most highly expressed at the early biotrophic stage.
Table S2 Top 100 genes most highly expressed at the late biotrophic stage.
Table S3 Top 100 genes most highly expressed during the transition from the biotrophic stage to the necrotrophic stage.
Table S4 Top 100 genes most highly expressed at the early necrotrophic stage.
Table S5 Top 100 genes most highly expressed during the transition from early to late necrotrophy.
Table S6 Top 100 genes most highly expressed at the late necrotrophic stage.
Table S7 Top 100 genes most highly expressed at the saprotrophic stage.