

Summary
Differences in gene expression were studied after Plum pox virus (PPV, sharka disease) infection in peach GF305 leaves with and without sharka symptoms using RNA‐Seq. For each sample, more than 80% of 100‐nucleotide paired‐end (PE) Illumina reads were aligned on the peach reference genome. In the symptomatic sample, a significant proportion of reads were mapped to PPV reference genomes (1.04% compared with 0.00002% in non‐symptomatic leaves), allowing for the ultra‐deep assembly of the complete genome of the PPV isolate used (9775 nucleotides, missing only 11 nucleotides at the 5′ genome end). In addition, significant alternative splicing events were detected in 359 genes and 12 990 single nucleotide polymorphisms (SNPs) were identified, 425 of which could be annotated. Gene ontology annotation revealed that the high‐ranking mRNA target genes associated with the expression of sharka symptoms are mainly related to the response to biotic stimuli, to lipid and carbohydrate metabolism and to the negative regulation of catalytic activity. A greater number of differentially expressed genes were observed in the early asymptomatic phase of PPV infection in comparison with the symptomatic phase. These early infection events were associated with the induction of genes related to pathogen resistance, such as jasmonic acid, chitinases, cytokinin glucosyl transferases and Lys‐M proteins. Once the virus had accumulated, the overexpression of Dicer protein 2a genes suggested a gene silencing plant response that was suppressed by the virus HCPro and P1 proteins. These results illustrate the dynamic nature of the peach–PPV interaction at the transcriptome level and confirm that sharka symptom expression is a complex process that can be understood on the basis of changes in plant gene expression.
Keywords: plant–virus interaction, PPV, Prunus, RNA‐Seq, sharka disease
Introduction
Sharka disease is one of the most destructive diseases amongst stone fruits (Prunus) worldwide. It is caused by Plum pox virus (PPV), a member of the Potyvirus genus in the Potyviridae family, which encompasses a group of plant viruses with a significant impact on agronomy and economics (Scholthof et al., 2011). The estimated costs associated with the disease in Prunus spp. around the world over a period of 30 years (1975–2005) exceed 10 000 million euros (Cambra et al., 2006). This economic impact explains why PPV is amongst the most studied potyviruses, despite the technical constraints imposed by the woody nature of its natural hosts (García et al., 2014; Scholthof et al., 2011). Within Prunus, peach [P. persica (L.) Batsch] is the most important crop, with a production of 21.08 million tons in 2012 (http://faostat.fao.org/). This species is characterized by its susceptibility to PPV (Van Oosten, 1975), and no source of resistance has been reported so far to the different PPV isolates assayed (García et al., 2014; Rubio et al., 2012). In addition, many studies have demonstrated the irregular distribution of sharka symptoms in Prunus species, including peach (Albrechtova, 1986; Martínez‐Gómez and Dicenta, 2001). Host PPV symptoms vary by type and severity, depending on the strain of the virus, the timing of infection, the cultivar and environmental factors (García et al., 2014).
Genetic and physiological plasticity is a prerequisite for life in a changing environment and requires elaborate regulatory mechanisms to simultaneously alter the expression of groups of genes (Maleck et al., 2000). Transcriptome analysis is therefore essential for the interpretation of the functional elements of the genome and for understanding the cell, organisms and disease development. Host gene expression analyses in the context of PPV infection have been performed in Prunus and in model species, such as Arabidopsis thaliana L. and Nicotiana benthamiana (Domin.). Studies were first performed on peach and apricot (P. armeniaca L.) using expressed sequence tag (EST) sequencing (Wang et al., 2005) and cDNA‐amplified fragment length polymorphism (cDNA‐AFLP) (Schurdi‐Levraud et al., 2006), respectively. These studies described global changes in gene expression in the inoculated leaves, identifying dozens of differentially expressed genes (DEGs). Differences in gene expression have also been characterized using microarray technology in herbaceous Nicotiana (Dardick, 2007) and Arabidopsis (Babu et al., 2008) hosts, resulting in the identification of hundreds of genes affected by PPV infection in leaves and sharka symptom development.
High‐throughput gene expression analysis using massive RNA (cDNA) sequencing (RNA‐Seq) represents, to date, the most powerful tool for the characterization of transcriptomes in comparison with EST sequencing, cDNA‐AFLP or microarray analysis (Martínez‐Gómez et al., 2011; Wang et al., 2009). This new RNA‐Seq technology has been used to study plant–pathogen interactions in the case of fungi (Bagnaresi et al., 2012; Barakat et al., 2012; Fernández et al., 2012; Gao et al., 2013; Kunjeti et al., 2012; Li et al., 2012), viruses (Lu et al., 2012; Zhang et al., 2012) and bacteria (Socquet‐Juglard et al., 2013). It could be very useful for the clarification of gene expression changes in the PPV–Prunus interaction. In Prunus species, RNA‐Seq has been applied in peach to facilitate the identification and isolation of genes controlling horticultural traits (Chen et al., 2014; Wang et al., 2013) and of genes affected by Xanthomonas arboricola infection (Socquet‐Juglard et al., 2013). The technology has also been used to analyse expression profiles of seasonal bud dormancy in Japanese apricot (P. mume Sieb. et Zucc.) (Zhong et al., 2013) and fruit development in sweet cherry (Alkio et al., 2014). Peach presents several agronomical and molecular advantages, such as self‐compatibility, a short juvenile phase and a small genome size (224.6 Mb), which makes it suitable as a model species within the Prunus genus and the Rosaceae family. Furthermore, a high‐quality draft genome has recently been published (http://www.rosaceae.org), which is now the reference genome for RNA‐Seq studies in Prunus (Verde et al., 2013).
In the present study, high‐throughput Illumina sequencing was used to perform gene expression analysis (RNA‐Seq) during PPV infection in the GF305 peach indicator. Together with confirmatory quantitative polymerase chain reaction (qPCR) experiments, the results obtained provide a global picture of the peach leaf transcriptome after PPV infection and sharka symptom development.
Results and Discussion
RNA‐Seq transcriptome profiles from peach samples
GF305 peach buds were grafted onto PPV‐inoculated GF305 seedlings used as rootstocks. Both grafted clonal GF305 and GF305 seedling rootstocks were analysed for PPV infection using enzyme‐linked immunosorbent assay (ELISA) and reverse transcription‐polymerase chain reaction (RT‐PCR). Total RNA was extracted from the leaves of symptomless but infected (ELISA‐ and RT‐PCR‐positive) peach trees (INS), symptomatic and infected (ELISA‐ and RT‐PCR‐positive) peach trees (IWS) and control non‐infected (ELISA‐ and RT‐PCR‐negative) peach trees (NI). The total RNA obtained was subjected to RNA Illumina sequencing (Fig. 1). The RNA samples were fragmented and ligated with adaptors prior to cDNA synthesis and PCR amplification. Totals of 122, 127.5 and 131.9 million 100‐bp paired‐end reads were generated from INS, IWS and NI, respectively (Table 1).
Figure 1.
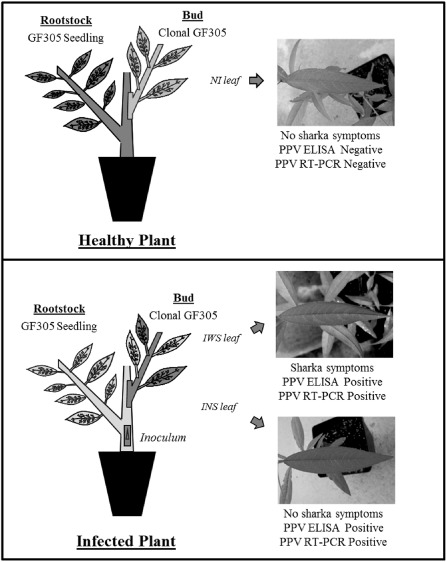
Schematic representation of the plant models tested for resistance to Plum pox virus (PPV) and RNA‐Seq. Healthy Plant: GF305 clonal peach grafted onto healthy peach GF305 rootstock seedlings. Detail of non‐inoculated leaf (NI) and reverse transcription‐polymerase chain reaction (RT‐PCR). Infected Plant: GF305 clonal peach grafted onto infected peach GF305 rootstock seedlings. Details of infected leaf with symptoms (IWS) and symptomless leaf (INS) and RT‐PCR.
Table 1.
Mapping characteristics of the GF305 peach and Plum pox virus (PPV) reads to the reference genomes in the three samples assayed: non‐infected (control) peach leaves (NI); PPV‐infected leaves without sharka symptoms (asymptomatic) (INS); and PPV‐infected leaves with sharka symptoms (symptomatic) (IWS)
| Sample | Raw reads | Clean reads | Reads mapped P. persica v 1.0 | Reads mapped PPV genomes |
|---|---|---|---|---|
| NI | 131 945 461 | 95 805 431 (72%) | 81 173 368 (85.0%) | 0 (0.0%) |
| INS | 122 016 326 | 85 138 600 (69.6%) | 71 173 787 (83.6%) | 147 (0.00002%) |
| IWS | 127 541 851 | 91 166 685 (71.6%) | 76 522 953 (84.0%) | 946 121 (1.04%) |
| Total | 381 503 638 | 272 110 716 (71.4%) | 228 870 108 (83.8%) | 946 268 (0.34%) |
Raw reads were trimmed by removing adaptor sequences, empty reads and low‐quality sequences. As a result, 272.11 million high‐quality reads (71.4%), designated as clean reads, were generated for the three treatments. Of the clean reads, 97.6% (265.57 million) were paired‐end reads and 2.4% (6.53 million) were single‐end reads. By iterative alignment, an average of 83.8% of the clean reads were successfully mapped to the v1.0 peach reference genome, whereas 16.2% of the clean reads did not map onto any of the eight mega‐scaffolds of the peach genome. Of the unmapped reads mapping to the PPV genome, 147 (0.00002% of the total reads for the sample) were obtained for the INS samples and 946 121 (1.04% of the total reads) for the IWS samples.
Percentages of unmapped reads of between 5% and 40% are typically obtained during high‐throughput RNA sequencing or DNA re‐sequencing projects (Cao et al., 2011; Wang et al., 2013). It is assumed that these unmapped reads could contain novel splice junctions and sequences belonging to other sources of contamination (Philippe et al., 2013). The percentages obtained in the present work are similar to the 11% of unmapped reads recently reported after the exploration of the peach transcriptome performed by Wang et al. (2013). Our results confirm that the irregular distribution of sharka symptoms described in natural PPV host plants, particularly in Prunus species, including peach (Albrechtova, 1986; Martínez‐Gómez and Dicenta, 2001), is associated with differences in virus concentration, evaluated by RNA‐Seq as the frequency of PPV reads in the symptomatic sample, which is orders of magnitude higher than that in asymptomatic infected leaves.
RNA‐seq reads from the three different samples (NI, INS, IWS) mapped to the peach genome resulted in the identification of a total of 54 016 protein‐coding transcripts, 43 752 of which were multi‐exon transcripts (Table 2). These 54 016 transcripts were generated from 31 716 loci, with a total of 3864 additional genes, compared with the 27 852 genes predicted for the peach reference genome (Verde et al., 2013). These results are in keeping with those of Wang et al. (2013), who reported a total of 2140 new genes not included in the peach reference gene set following an RNA‐seq analysis.
Table 2.
Elements found in the GF305 peach transcriptome samples assayed by RNA‐Seq
| Transcriptome elements | Experiment peach mRNA | Reference peach mRNA |
|---|---|---|
| Total transcripts | 54 016 | 28 689 |
| Multi‐exon transcripts | 43 752 | 23 410 |
| Total loci | 31 716 | 27 852 |
| Multi‐transcript loci | 11 012 | 26 552 |
| Transcripts per locus | 1.70 | 1.03 |
| Novel/total exons (%) | 20 048/191 174 (10.5%) | — |
| Novel/total introns (%) | 11 662/134 800 (8.7%) | — |
| Novel/total loci (%) | 4755/31 716 (15.0%) | — |
| Novel/total isoforms (%) | 16 704/54 016 (31.0%) | — |
In total, the present RNA‐seq analysis revealed 20 048 novel exons (10.5%), 11 662 novel introns (8.7%) and 4755 new loci (15%), in comparison with the peach reference genome (Table 2).
The large number of PPV reads in the symptomatically infected sample made it possible to assemble the complete genome of the PPV isolate used, with a length of 9786 nucleotides and missing only 11 nucleotides at the extreme 5′ genome end. The complete genome sequence of the PPV 3.30 RB/GF‐IVIA isolate thus reconstructed has been deposited in the GenBank database under accession number KJ849228. The mapping of reads on the PPV genome assembly provided limited evidence for sequence heterogeneity and confirmed the presence of a single PPV isolate. A single high‐frequency polymorphism was detected at genome position 2995, and both G (57%) and A (43%) were observed. Comparison with the 990 nucleotide fragments deposited in GenBank for the 3.30 RB/GF‐IVIA original isolate and its variants (accession numbers AF172346–50) showed a similarity of 99.7%, with only three to four mutations. Considering the complete genomic sequence, the highest level of identity was observed with typical PPV‐D isolates and, in particular, with the complete PPV‐Ou1 and PPV‐Ou6 sequences, with 99.1% nucleotide identity (PPV‐Ou1, AB545926) and 99.5% amino acid identity in the encoded protein (PPV‐Ou6, AB576062). These results confirm the suitability of high‐throughput RNA sequencing for the determination of the complete genome sequences of PPV isolates (Sheveleva et al., 2013) and the slow accumulation of mutations in the PPV genome during long‐time maintenance and the passage of PPV in Prunus host plants (Predajňa et al., 2012; Vozárová et al., 2013).
Identification of new single nucleotide polymorphisms (SNPs), INDELs and alternative splicing (AS) variants
RNA‐Seq clean reads from the three treatments were used for SNP and INDEL identification in transcribed regions employing the peach genome as a reference. A total of 13 679 variations was identified, 12 990 of which were SNPs (95%) and 689 of which were INDELs (5%). Of these identified SNPs, 425 could be annotated to the reference SNP database [see Table S1 (Supporting Information), which also contains detailed information concerning the scaffold and the position in which the SNP variant is found (reference/variant), the quality score of the variant, the total number of reads covering the variant and, finally, the estimated variant allele frequency]. Table S1 also contains detailed information regarding the SNPs that were identified and not annotated. The SNP density was one SNP per 4.15 kb (0.24 SNPs/kb). The highest density of SNPs was founded at scaffold 1, with 2946 SNPs, followed by scaffolds 4 and 6, with 2669 and 1833 SNPs, respectively. Of all the SNPs identified, 58.5% were transitions and 41.5% were transversions. The transitions A/G and T/C were the two most abundant SNPs, accounting for 15.5% and 15.1%, respectively, of all SNPs.
SNPs can occur in both coding and non‐coding regions of genes and may have functional consequences in terms of gene transcription or gene function. These functional consequences are the biological cause of the association of SNPs with different agronomic traits in plants. Our data agree with results described by Wang et al. (2013), who also analysed different peach genotypes by RNA‐Seq. These authors identified 9587 different, mainly unannotated, SNPs. In addition, Koepke et al. (2012) identified 2243 SNPs in cherry with RNA‐Seq. Verde et al. (2012), however, described an overall density of 4.4 SNPs/kb using whole genome sequencing (WGS) on 56 peach genotypes. The frequency of SNPs in genomic DNA sequences is much higher than that observed in the transcribed regions. An average of one SNP every 598 bp has been described in Prunus, and this variability is higher in non‐coding regions (one SNP every 390 non‐coding bp) than in coding regions (one SNP per 1850 coding bp) (Aranzana et al., 2012). With regard to the functional consequences of SNPs, one unannotated SNP (5595) has been located (scaffold 1 position 8 188 815) in the peach genome region in which resistance to PPV was described by Zuriaga et al. (2013) in apricot. These authors described the presence of different SNPs identified in the PPVres region by WGS of nine different resistant and susceptible apricot cultivars.
We also detected significant AS events in 359 genes. Of these, 10 located on scaffold 1 showed AS forms in the inoculated without symptoms vs. inoculated with symptoms comparison, 181 located on scaffold 1–9 corresponded to AS in the control vs. inoculated without symptoms samples and another 168 located on scaffold 1–9 were AS in control leaves vs. inoculated leaves with symptoms (Table S2, see Supporting Information). This table contains the new isoforms detected and the associated genes, as well as genes that show differential AS for each condition. Table S2 also shows the identifiers of isoforms or genes, the site of transcription initiation for that isoform, the value of the statistical test and its significance before and after correction for multiple tests.
AS events are widely accepted to play an important role in the regulation of gene expression in plants and are one of the most critical mechanisms for the generation of protein diversity (Lozada, 2007). Halterman et al. (2003) described, in barley, how the powdery mildew resistance gene is alternatively spliced to produce five different transcript regions. Verde et al. (2013) described 838 AS processes in a total of 26 689 protein‐coding genes identified in the peach reference genome. Wang et al. (2013), analysing a larger number of peach genotypes, described a total of 10 835 AS events in 5520 transcribed regions. However, none of the AS events identified in the comparison between the control and PPV‐inoculated peach transcriptomes was located in the peach genome region in which the gene responsible for resistance to PPV was described in apricot (between 8 050 804 and 8 244 925 of scaffold 1) (Zuriaga et al., 2013).
Host transcriptional changes in symptomatic and asymptomatic peach leaves infected with PPV
The normalized expression levels from uninfected control and PPV‐infected plants (symptomatic and asymptomatic) were compared to detect differentially expressed transcripts. Read counts obtained for each of the samples were FPKM (fragments per kilobase of exon per million fragments mapped) normalized prior to DEG analysis. DEGs selected by a fold change of ≥2 or ≤−2 and a statistical value of q = 0.05 were compared in three different ways. A total of 761 DEGs was obtained after the analysis of the non‐infected peach leaves against the symptomless infected leaves (NI vs. INS). In contrast, only 222 DEGs were identified after comparing non‐infected peach leaves against symptomatic leaves (NI vs. IWS). In both cases, 63 of the 761 and 222 DEGs corresponded to novel or unannotated genes. A total of 571 DEGs was identified after comparison between symptomless and symptomatic infected peach leaves (INS vs. IWS). Forty‐five genes of this total were novel or unannotated genes [Table 3; see also Table S3 (Supporting Information)]. Totals of 322, 95 and 149 DEGs were specific for NI vs. INS, NI vs. IWS and INS vs. IWS, respectively (Fig. 2).
Table 3.
Differentially expressed genes (DEGs) from the three GF305 peach samples assayed [non‐infected control peach leaves (NI), PPV infected but without sharka symptoms (INS) and PPV infected with sharka symptoms] in the three comparisons performed (NI vs. INS, NI vs. IWS, INS vs. IWS)
| NI vs. INS | NI vs. IWS | INS vs. IWS | |
|---|---|---|---|
| Total DEGs | 761 | 222 | 571 |
| Annotated DEGs | 698 | 159 | 526 |
| Novel DEGs | 63 | 63 | 45 |
| Shared DEGs | 439 | 127 | 422 |
| Specific DEGs | 322 | 95 | 149 |
| Up‐regulated | 299 | 52 | 91 |
| Down‐regulated | 23 | 43 | 58 |
Figure 2.
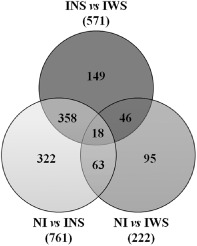
Venn diagram of differentially expressed genes among the three comparisons performed (NI vs. INS, NI vs. IWS and INS vs. IWS). INS, inoculated leaves without symptoms; IWS, inoculated leaves with symptoms; NI, non‐inoculated GF305 peach leaves.
Curiously, NI vs. INS showed a greater number of total and specific deregulated genes in comparison with the NI vs. IWS situation. This difference could indicate that the gene expression profile on NI vs. INS leads to a complex and extensive transcriptome remodelling in peach leaves, suggesting the existence of an active reaction limiting the appearance of symptoms. These results also confirm the dynamic nature of the PPV–peach interaction at the transcriptome level, in agreement with previous results obtained at the protein level (Díaz‐Vivancos et al., 2006). Using two‐dimensional polyacrylamide gel electrophoresis (2D‐PAGE), these authors observed dozens of protein spots that were differentially expressed in control and PPV‐inoculated GF305 peach plants. However, after analysis by matrix‐assisted laser desorption/ionization‐time‐of‐flight mass spectrometry (MALDI‐TOF‐MS), most of these spots had not been identified, with the exception of thaumatin‐like proteins associated with PPV infection. These results agree with ours, in that it was possible to observe a clear expression of thaumatin‐like protein genes (ppa010479m, ppa010522m, ppa025773m) in the sample showing sharka symptoms after PPV inoculation (NI vs. IWS, Table S3). Similarly, in Arabidopsis, Baerenfaller et al. (2008) have described a high correlation between protein and the abundance of transcripts (transcriptome analysis) of between 0.50 and 0.68.
In addition, in non‐symptomatic leaves, 299 (92%) and 23 (8%) of the 322 genes were up‐ and down‐regulated, respectively, relative to non‐infected leaves (NI vs. INS). However, in the symptomatic leaves, of the 95 genes, 52 (54%) were up‐regulated and 43 (46%) were down‐regulated relative to control leaves (Table 3; Tables S3 and S4, see Supporting Information).
These results confirm the large amount of data generated by RNA‐Seq in the study of PPV–host interactions in comparison with EST sequencing (Wang et al., 2005) and cDNA‐AFLP analysis (Schurdi‐Levraud et al., 2006). The results obtained with microarray technology in herbaceous species are of similar magnitude. In agreement with our results, Babu et al. (2008), using microarray analysis, identified 2013 Arabidopsis genes that were significantly induced and 1457 that were significantly repressed by systemically PPV‐infected leaf tissues. Sicard et al. (2008) identified different quantitative trait loci (QTL) controlling symptom development during viral infection in A. thaliana. Dardick (2007) also described gene expression changes concomitant with PPV symptom severity using microarrays in Nicotiana leaves. These authors identified up to 744 DEGs in severely infected leaves.
In our study, PPV infection and sharka symptom development were associated with gene expression changes in PPV‐infected peach leaves. The differentially expressed regulated genes linked with the appearance of sharka symptoms may be critical to virus infection, particularly the early response genes (control leaves and PPV‐inoculated leaves without sharka symptoms), with a higher differential response and a larger number of genes involved. This critical early response in PPV infection has also been highlighted in Arabidopsis (Babu et al., 2008). Furthermore, Dardick (2007) has described gene expression changes concomitant with PPV symptom severity in Nicotiana leaves.
The differential expression changes observed in this study correlate very well with the irregular distribution of the virus that has been described throughout its infected natural Prunus host (Albrechtova, 1986), particularly in peach (Martínez‐Gómez and Dicenta, 2001). Indeed, peach is characterized as a natural host with specific pathogenicity properties dependent on the different isolates (Dallot et al., 2001; García et al., 2014). Furthermore, these DEGs are candidates for future functional characterization in order to shed light on molecular virus–host interactions.
Functional analysis of DEGs in symptomatic and non‐symptomatic peach leaves
Of the 761 total DEGs identified in the NI vs. INS pair‐wise analysis and the 222 identified in the NI vs. IWS analysis, totals of 718 and 172 DEGs, respectively, were assigned with one or more gene ontology (GO) terms. In both cases, the entire GO assignments fell into broad categories for two of the three major GO functional domains (Biological Processes and Molecular Function). Sixteen significantly different GO annotations were obtained for the NI vs. INS pair, 10 belonging to Molecular Function and six to Biological Processes. By contrast, nine significantly different GO annotations were obtained for the NI vs. IWS pair, four belonging to Molecular Function and five to Biological Processes. With the exception of the ‘cellular process’, eight of the nine secondary GO terms were over‐represented for the NI vs. IWS pair (Fig. 3). For NI vs. INS, only the secondary GO terms belonging to ‘signalling’ and ‘cellular process’ were under‐represented from the 16 significant annotated groups. Although the number of secondary GO terms was more abundant in the NI vs. INS pair‐wise analysis, the differences from the background/reference were always more significant in the case of NI vs. IWS (Fig. 3).
Figure 3.
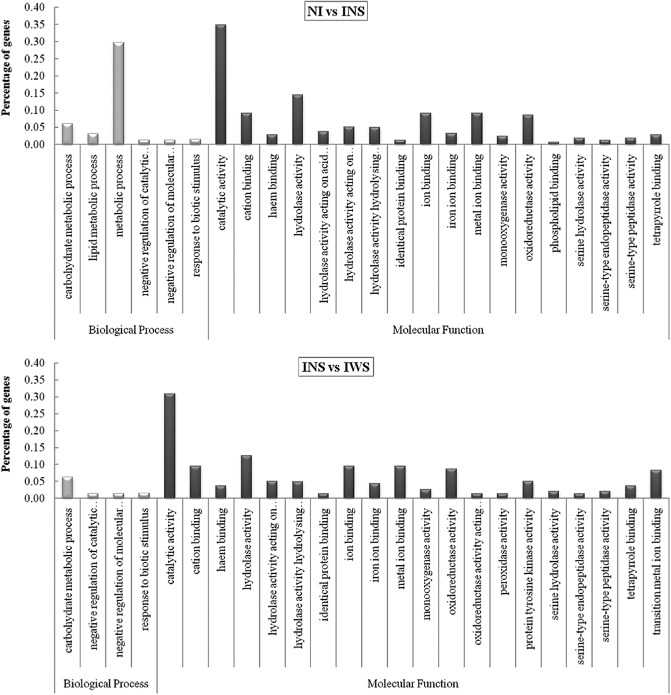
Significant gene ontology (GO) annotations (False Discovery Rate, FDR < 0.05) of differentially expressed genes compared with total analysed transcripts from the three assayed samples [non‐infected control peach leaves (NI); PPV infected but without sharka symptoms (INS); and PPV infected with sharka symptoms (IWS)] in the two comparisons performed [NI vs. INS (A); and INS vs. IWS (C)]. Annotations are grouped by biological process or molecular function. Percentage of genes is listed for each category.
The GO results are consistent with the hypothesis that biotic stress marks a transition from growth and reproduction to physiology and metabolism tailored to the defence response (Bilgin et al., 2010). In general, peach leaves in which the virus has accumulated and symptom development has appeared seem to have a low representation of genes involved in the ‘cellular process’ and a high representation of genes implicated in ‘catalytic activities’ and ‘regulation of metabolic’ and ‘biological processes’ (Fig. 3). Curiously, one of the DEGs with high statistical significance involved in ‘metabolic processes’ was an endoribonuclease Dicer protein 2a (ppa020875), which was significantly up‐regulated in the INS vs. IWS pair (Table S3). HC‐Pro and P1 PPV proteins have been widely reported to counteract antiviral defence through RNA silencing, and play a key role in the recognition of the double‐stranded RNA originating during virus replication (Valli et al., 2006).
In contrast, non‐symptomatic peach leaves showed a normal abundance of genes belonging to ‘cellular processes’ and a higher abundance of genes belonging to ‘death’, ‘cell wall organization’, ‘transporter activity’ and ‘electron carrier activity’. These genes include, for example, several disease resistance proteins (ppa015274m, ppa015499m, ppa014872m, ppb017543m), chitinases and Lys‐M proteins (ppa010947m, ppa010952m, ppa017855m, ppa006147m) and proteins involved in jasmonate (JA) biosynthesis (allene oxide synthase, ppa004133m and allene oxide cyclase 4 chloroplastic, ppa012079m) and signalling (some basic bHLH transcription factors: ppa014004m, ppa016030m, ppa015634m, ppa016514m) (NI vs. INS, Table S3). The increase in JA levels leads to a degradation of JAZ proteins (Staswick, 2008) and to the depression of MYC2 (and its redundant homologues MYC3 and MYC4), a bHLH transcription factor that plays a central role in JA signalling, resulting in the transcriptional activation of downstream target genes (Katsir et al., 2008). In addition, in inoculated asymptomatic leaves, an induction of JA‐responsive genes involved in defence, as well as of jasmonates, was observed, as has already been described by different authors (Devoto et al., 2005; Shan et al., 2009). Higher patterns of expression (FPKM values) were also observed in genes related to the defence response and the response to biotic stimuli in PPV‐inoculated leaves without visible symptoms relative to control non‐infected leaves and leaves infected with PPV with symptoms. These genes include pectate lyase (ppa00665m), endoglucanases (ppa004653m and ppa004644m) and glucan endo‐1,3‐β‐glucosidase (ppa005142m) (NI vs. INS, Table S3).
Recently, a PPV‐resistant locus (PPVres) has been delineated in apricot via syntenic analysis with the peach genome (Zuriaga et al., 2013). This PPVres locus in peach contains a total of 31 predicted transcripts, some of which have been reported previously to confer resistance against potyviruses, such as S‐adenosyl‐methionine (SAM) synthetase (Uzarowska et al., 2009). Curiously, both SAM synthetase (ppa006841m) and ubiquitin carboxyl‐terminal hydrolase (ppa008951m), localized in the PPVres locus, were only over‐expressed in the non‐symptomatic leaves in which virus accumulation had not occurred (NI vs. INS, Table S3). The transient induction of the accumulation of transcripts corresponding to ppa006841m in inoculated asymptomatic leaves only also suggests that processes occurring during the first phase of PPV infection are accompanied by the production of SAM, which is a general donor of methyl groups in the transmethylation reactions in the cytosol, chloroplasts and mitochondria. A high level of SAM is needed for phenylpropanoid synthesis as it is the main methyl group donor utilized by o‐diphenol‐O‐methyltransferases (OMTs) in mono‐lignol biosynthesis. Shen et al. (2002) reported that SAM genes were strongly expressed in highly lignified tissues and suggested that the lignification might consume large amounts of SAM. In INS leaves, SAMs showed the same pattern as observed for 4‐coumarate‐CoA ligase‐like 9 (ppa000359m), which could be associated with an increase in lignin‐like materials and/or phenylpropanoids esterified to cell wall materials in response to PPV infection. These cell wall modifications have already been linked to resistance mechanisms activated following fungal and viral attacks (Boevink and Oparka, 2005).
In addition, some cytokinin glucosyl transferases (ppa004968m and ppa005447m) were also over‐expressed in the non‐symptomatic leaves in which virus accumulation had not occurred (NI vs. INS, Table S3). In this regard, some authors have recently described the role of cytokinins in resistance to Pseudomonas syringae in Arabidopsis (Choi et al., 2010) and Nicotiana (Großkinsky et al., 2011).
In addition to GO term assignment, KEGG pathway mapping based on KEGG orthology (KO) terms for assignments was also carried out with the total list of DEGs. Of the 222 and 761 total DEGs, 141 and 649 transcripts were matched with previously annotated Arabidopsis genes for NI vs. IWS and NI vs. INS, respectively. Among the transcripts with KEGG annotation, 29 and 92 significant annotated genes were distributed in a total of nine groups in both cases. For symptomatic (seven genes) and asymptomatic (29 genes) peach leaves, biosynthesis of secondary metabolites was the most represented functional annotated KEGG group. Nevertheless, ‘starch and sucrose metabolism’ (nine genes) and ‘plant–pathogen interaction’ (eight genes) were only represented in the NI vs. INS pair‐wise analysis (Fig. 4). These results support the idea that leaves in which the virus is unable to accumulate could activate a defence response that prevents virus accumulation.
Figure 4.
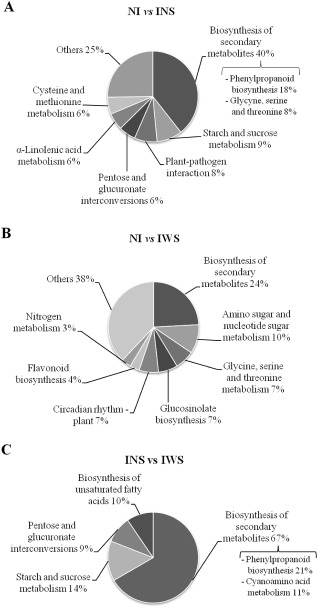
KEGG annotations of transcripts from the three assayed samples [non‐infected control peach leaves (NI); PPV infected but without sharka symptoms (INS); and PPV infected with sharka symptoms (IWS)] in the three comparisons performed [NI vs. INS (A); NI vs. IWS (B); and INS vs. IWS (C)]. Percentage of genes per current KEGG annotation is expressed between comparisons.
In accordance with our results, Wang et al. (2005) found an altered expression of genes in peach leaves involved in defence, cellular transport and protein synthesis and of proteins with binding function after infection with PPV. In addition, Schurdi‐Levraud et al. (2006) described a differential expression of genes coding for proteins involved in metabolism, signal transduction, defence, stress and intra/intercellular connections after the resistant apricot cultivar Goldrich had been infected with PPV.
In addition, Babu et al. (2008) identified Arabidopsis genes that were significantly induced (mainly associated with the metabolism of soluble sugar, starch and amino acid, intracellular membrane/membrane‐bound organelles, chloroplast and protein fate) and repressed (genes related to development/storage proteins, protein synthesis and translation, and cell wall‐associated components) by systemically PPV‐infected leaf tissues. Some of these genes showed high sequence similarity to peach uniESTs corresponding to orthologues related to defence, metabolism and protein synthesis. Finally, Dardick (2007) described DEGs mainly associated with stress and the pathogenesis process in Nicotiana leaves that were severely infected with PPV. PPV‐infected leaves also showed widespread repression of genes associated with plastid functions in Nicotiana.
Validation of gene expression profiles using RT‐qPCR
The expression trends of 12 representative genes were confirmed by real‐time RT‐qPCR in a separate experiment. Expression trends of the selected genes related to the defence response and response to biotic stimulus using RT‐qPCR were similar to the results obtained with RNA‐Seq analysis (Fig. 5). However, differences between treatments in the RNA‐Seq analysis were greater than those observed in the RT‐qPCR analysis.
Figure 5.
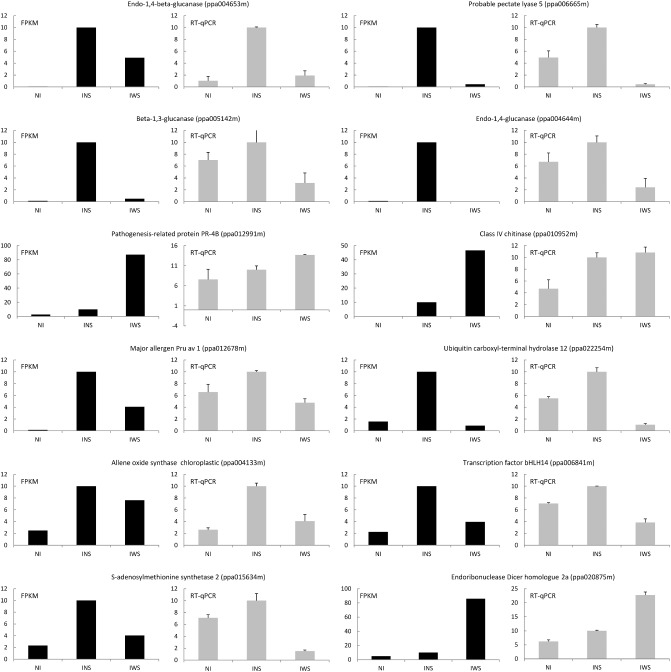
Validation of RNA‐Seq analysis by reverse transcription‐quantitative polymerase chain reaction (RT‐qPCR). FPKM (fragments per kilobase of exon per million fragments mapped) values obtained with RNA‐Seq and qPCR values in the analysis of selected genes in the three assayed samples: non‐inoculated control peach leaves (NI); PPV infected without sharka symptoms but ELISA and RT‐PCR positive (INS); and PPV infected with sharka symptoms and ELISA and RT‐PCR positive (IWS). Error bars represent the standard error for three independent experimental replicates.
On average, the expression of the following encoding genes was higher in leaves inoculated with PPV but without visible symptoms than in the non‐infected control leaves and the leaves infected with PPV with symptoms: pectate lyase (ppa006665m); the two endoglucanases (ppa004653m and ppa004644m); the allene oxide synthase (ppa004133m); the transcription factor bHLH14 (ppa006841m); the SAM synthetase 2 (ppa015634m); the ubiquitin carboxyl‐terminal hydrolase (ppa020875m); the major allergen Pu av1 (ppa012678m); and the glucan endo‐1,3‐β‐glucosidase (ppa005142m). However, the expression of genes encoding the two pathogenesis‐related proteins, a chitinase (ppa010952m) and a PR‐4B (ppa012991m), and the endoribonuclease Dicer homologue 2a (ppa02875m) was, on average, higher in the symptomatic leaves (Fig. 5).
The different expression levels between RNA‐Seq and RT‐qPCR could be caused by the bioinformatics process performed in the RNA‐Seq analysis, which includes several factors that can affect the reproducibility of quantitative expression profiles, including alignment choices, estimation of transcript expression, etc. (Labaj et al., 2011). In addition, other factors affecting this discrepancy include the dynamic nature of the transcriptome (Martínez‐Gómez et al., 2012) and the fact that, in the RNA‐Seq analysis, we used a pool of biological samples which were assayed separately in the RT‐qPCR assays. Mochizuki et al. (2014), for example, established a correlation between the levels of mRNA expressed from some genes and the severity of leaf symptoms in tobacco after infection with Cucumber mosaic virus (CMV).
The single composite sample analysis assayed (Auer and Doerge, 2010) using RNA‐Seq has also been applied in the study of plant–pathogen interactions in fungi, including chestnut resistance to blight disease (Barakat et al., 2012), rust fungus infection in coffee (Fernández et al., 2012) and Fusarium infection in banana (Li et al., 2012). The single composite sample analysis has also been used to study the susceptibility of Chenopodium to different viruses (Zhang et al., 2012). In all of these studies, as in ours, the composite sample consisted of a pool of leaves. In the same vein, partial validation of RNA‐Seq data (read number) with the application of qPCR (expression level) in biological samples has been described in analyses of plant–pathogen interactions in the case of fungi (Bagnaresi et al., 2012; Fernández et al., 2012; Gao et al., 2013) and viruses and viroids (Lu et al., 2012; Zhang et al., 2012). In these assays, a selected number of genes were evaluated in different biological replications in order to validate the RNA‐Seq results obtained.
In our study, RNA‐Seq and qPCR data confirmed that sharka symptom expression in Prunus woody hosts is a complex process involving different genes that can be understood only on the basis of changes in gene expression. The irregular distribution of PPV in woody natural hosts could be a result of different responses at the cellular level, with a greater level of differential expression of genes than described in herbaceous hosts at the genomic (Sicard et al., 2008) and transcriptomic (Babu et al., 2008; Dardick, 2007) levels, as sharka symptom expression is much more uniform in herbaceous hosts. Indeed, the authors studying these hosts have only found differences during different infection times, whereas, in woody hosts, we observed differences throughout the plant at the same infection time.
Conclusions
RNA‐Seq results showed the dynamic nature of the peach–PPV interaction at the transcriptomic level and confirmed that sharka symptom expression is a complex process that can be understood only on the basis of changes in gene expression. Furthermore, the comparison between control leaves and PPV‐inoculated leaves with symptoms reflects the whole process of sharka disease development. The DEGs, particularly the more numerous early response genes, may be critical for virus infection and are suitable candidates for future functional characterization in order to shed light on molecular virus–host interactions. Early PPV infection in peach leaves without symptoms is associated with an induction of genes related to pathogen resistance, such as jasmonic acid, resistance proteins, chitinases and Lys‐M proteins. In addition, processes occurring during the first phase of PPV infection are accompanied by the production of SAM, which is a general donor of methyl groups in transmethylation reactions. Further targeted functional studies of these early defence‐related genes will help to unravel their biological function during the course of PPV infection. When the virus is fully installed, the overexpression of Dicer protein 2a genes could be representative of a gene silencing response suppressed by the HCPro and P1 PPV proteins. Furthermore, significant AS events were detected in 359 genes and a total of 12 990 SNPs were identified in the transcribed peach region, 425 of which could be annotated.
Experimental Procedures
Plant material and sharka evaluation procedures
Plant inoculations were carried out in GF305, a peach cultivar used as a biological indicator of PPV (Bernhard et al., 1969), using the PPV strain 3.30 RB/GF‐IVIA belonging to the PPV‐D strain. This isolate, first obtained from Japanese plum and later propagated in GF305 peach, was introduced into the glasshouses of Centro de Edafología y Biología Aplicada del Segura (CEBAS‐CSIC) in Murcia in 1995 from the IVIA collection of Dr Mariano Cambra (Valencia, Spain). For 15 years, the cultivar was propagated by successive graft inoculation passages (one or two per year) in GF305 peach in a quarantine shelter. Two‐month‐old GF305 seedling rootstocks were inoculated by grafting a piece of bark from other previously infected GF305 plants showing strong sharka symptoms. Additional seedlings were kept as controls. Two months later, clonal GF305 buds were grafted onto the previously inoculated and control GF305 seedling rootstocks. One month after grafting, plants were subjected to an artificial period of dormancy in a cold chamber at 7 °C, in darkness, for 2 months. The plants were then transferred to an insect‐proof glasshouse. Sharka symptoms were scored using a scale from ‘0’ (no symptoms) to ‘5’ (maximum intensity). The presence of the virus was confirmed by double antibody sandwich indirect‐ELISA (DASI‐ELISA) using the specific monoclonal antibody 5B‐IVIA/AMR (Plant Print Diagnostics SL, Valencia, Spain) (Cambra et al., 1994). In addition, an RT‐PCR analysis was carried out using two specific primers for the coat protein: VP337 (5′‐CTCTGTGTCCTCTTCTTGTG‐3′) and VP338 (5′‐CAATAAAGCCATTGTTGGATC‐3′) (Martínez‐Gómez et al., 2003).
RNA preparation and Illumina sequencing
Three different leaf pools (single composite samples) (Auer and Doerge, 2010) were collected for RNA‐Seq: one pool containing leaves from non‐inoculated GF305 (NI); one pool of leaves from PPV‐inoculated GF305 showing sharka symptoms (IWS) and ELISA and RT‐PCR positive; and one pool of leaves from PPV‐inoculated GF305 without sharka symptoms but ELISA and RT‐PCR positive (INS) (Fig. 1). Total RNA was extracted using the Rneasy Plant Mini Kit (Qiagen, Hilden, Germany). The quality and quantity of total RNA samples were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and samples were normalized at the same concentration (5 μg, 200 ng/μL). Later, samples were sent to the Centre for Genomic Regulation (CRG, Barcelona, Spain) for library preparation and RNA sequencing. mRNA was obtained using Sera‐mag (Thermo Scientific). The cDNA libraries were prepared according to the Illumina protocols. Fragments of about 300 bp were excised from agarose and enriched by PCR for 16 cycles. Finally, the cDNA libraries were sequenced using an Illumina HiSeq2000 machine to perform 100 paired‐end sequencing.
Mapping of RNA‐Seq reads, DEGs, AS events and SNPs
A double quality control was performed for RNA‐Seq reads obtained by HiSeq2000. First, we aligned 10% of randomly selected sequenced reads against the peach genome (v1.0) (http://www.rosaceae.org) using the GEM MAP algorithm (http://sourceforge.net/apps/mediawiki/gemlibrary), allowing for up to two mismatches in the first 100 bases. Then, a secondary quality control was performed with FastQC software. Pre‐processing of the reads was performed with the fastx‐toolkit, a specific aScidea tool, in order to filter low‐quality regions. High‐quality reads that passed the quality filter threshold were mapped to the P. persica genome v1.0 obtained from the Genome database for Rosaceae (GDR, http://www.rosaceae.org/peach/genome) using Tophat 1.4.0 (Trapnell et al., 2009) and Bowtie 0.12.7 (Lindner and Friedel, 2012). In addition, both of these programs were selected to analyse and identify annotated and new exon–exon splice junctions and to quantify transcript abundance in order to find the isoforms over‐represented on the samples, as no databases of AS are available as yet for P. persica. The presence of PPV reads was quantified by mapping the reads on a PPV strain D reference genome (X16415) using the CLC Genomics workbench version 7.0 (http://www.clcbio.com/blog/clc‐genomics‐workbench‐7‐0/). The same strategy allowed for the complete reconstruction of the genome of the PPV 3.30 RB/GF‐IVIA isolate, whilst the parsing of read mapping made it possible to search for potential sites of sequence variability. Comparisons between the assembled PPV 3.30 RB/GF sequence and other PPV isolates were performed using blast (Altschul et al., 1990) against the GenBank database. Differential gene expression and transcript abundance were calculated with the program Cufflinks 1.3.0 (Trapnell et al., 2010). The resulting lists of differentially expressed isoforms were filtered by ln(fold_change) > 2 and ln(fold_change) < −2 and a q value of 0.05. Genes selected as being differentially expressed were clustered to identify common patterns of expression. In addition, FPKM values were used to normalize and quantify the gene expression level. The analysis of biological significance was based on GO (Ashburner et al., 2000) and KEGG enrichment (Ogata et al., 1999) using the hypergeometric statistical test (Young et al., 2010) and the Bonferroni multi‐test adjustment method, and considering a significance level cut‐off of 0.05. GO and KEGG analyses were performed using AgriGO (http://bioinfo.cau.edu.cn/agriGO/) (Du et al., 2010) and GeneCodis (http://genecodis.cnb.csic.es/) (Carmona‐Saez et al., 2007). Among the dozens of software programs developed for GO and KEGG analysis, AgriGO and GeneCodis are considered to be the most suitable for horticultural crops (http://neurolex.org/wiki/Category:Resource:Gene_Ontology_Tools). Variant calling analysis from read mapping files was performed using Samtools 0.1.18 (Li et al., 2009). High‐quality variants were obtained after filtering SNPs with quality scores >20 and INDELs with scores >50.
Validation of RNA‐Seq analysis by RT‐qPCR
To validate the RNA‐Seq analysis, RT‐qPCR was performed in a new experiment with new plant materials. Total RNAs extracted using the Rneasy Plant Mini Kit from two inoculated replications [PPV inoculated showing sharka symptoms (IWS) and ELISA and RT‐PCR positive; and PPV inoculated without sharka symptoms but ELISA and RT‐PCR positive (INS)] and one control replication (NI) were used to examine the expression pattern of selected genes. Reverse transcription was conducted using the PrimeScript Reverse Transcriptase Kit (Invitrogen, Applied Biosystems, Madrid, Spain). Twelve representative genes specifically expressed in the different treatments related to the defence response and response to biotic stimulus, with q < 0.05 and high FPKM values, were selected to validate RNA‐Seq data (Table S5, see Supporting Information). Gene‐specific primers were designed using Primer3Plus (Untergasser et al., 2012). Both synthesized cDNA and the primer pairs were then incubated with LightCycler 480 SYBR Green (Roche Diagnostics, Basel, Switzerland) at 95 °C for 5 min, followed by 40 cycles at 95 °C for 10 s, 58 °C for 5 s and 72 °C for 10 s. Real‐time PCR efficiencies (E) and crossing points (CP) were calculated from the slopes in the LightCycler Software. In order to normalize the qPCR data, three different reference genes were evaluated: peach 18S rRNA (Rasori et al., 2002), actin and expansin (Tong et al., 2009) (Table S5). Two Excel‐based programs, geNorm (Vandesompele et al., 2002) and Norm‐Finder (Andersen et al., 2004), were used to evaluate the stability of each reference gene. In our case, expansin was the most stable gene and was used to normalize the expression of the genes. The relative expression ratio of this target gene was then calculated based on the E and CP values of the target and reference genes according to the following equation: ratio = (E target) Δ(CPtarget in the sample) / (E ref) Δ(CPref in the sample). Each sample was measured in triplicate and the mean ratios ± standard deviations were calculated.
Supporting information
Table S1 Unannotated and annotated single nucleotide polymorphisms (SNPs) identified by RNA‐Seq in the three GF305 peach samples: control (NI), inoculated without symptoms (INS) and inoculated with symptoms (IWS).
Table S2 Alternative splicing events identified by RNA‐Seq in the three GF305 peach samples: control (NI), inoculated without symptoms (INS) and inoculated with symptoms (IWS).
Table S3 Total differentially expressed genes in non‐inoculated GF305 peach leaves (NI), inoculated leaves without symptoms (INS) and inoculated leaves with symptoms (IWS).
Table S4 Specific differentially expressed genes in non‐inoculated GF305 peach leaves (NI), inoculated leaves without symptoms (INS) and inoculated leaves with symptoms (IWS).
Table S5 Primer sequences representing differentially expressed genes in the different treatments selected and reference genes for real‐time polymerase chain reaction (PCR).
Acknowledgements
This study was supported by projects AGL2010‐16335 of the Spanish Ministry of Science and Innovation and 08672/PI/08 of the Seneca Foundation of the Region of Murcia (Spain). We thank Dr Heinz Himmelbauer of the Centre for Genomic Regulation (CRG) of Barcelona for help with the description of the RNA‐Seq protocol and Dr Mariano Cambra (IVIA, Valencia, Spain) for the generous gift of the 3.30 RB/GF‐IVIA isolate.
References
- Albrechtova, L. (1986) Investigations on the distribution of sharka virus (Plum pox) in tissue of Prunus domestica . Zeit. Pflanzenk. Pflanzenschutz, 93, 190–201. [Google Scholar]
- Alkio, M. , Jonas, U. , Declerq, M. , Van Nocker, S. and Knoche, M. (2014) Transcriptional dynamics of the developing sweet cherry (Prunus avium L.) fruit: sequencing, annotation and expression profiling of exocarp‐associated genes. Horticult. Res. 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipmann, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andersen, C.L. , Jensen, J.L. and Ørntoft, T.F. (2004) Normalization of real‐time quantitative reverse transcription‐PCR data: a model‐based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. [DOI] [PubMed] [Google Scholar]
- Aranzana, M.J. , Illa, E. , Howad, B. and Arús, P. (2012) A first insight into peach [Prunus persica (L.) Batsch] SNP variability. Tree Genet. Genomes, 8, 1359–1369. [Google Scholar]
- Ashburner, M. , Ball, C.A. , Blake, J.A. , Botstein, D. , Butler, H. , Cherry, J.M. , Davis, A.P. , Dolinski, K. , Dwight, S.S. , Eppig, J.T. , Harris, M.A. , Hill, D.P. , Issel‐Tarver, R. , Lewis, S. , Matese, J.E. , Richardson, J.E. , Ringwald, M. , Rubin, G.M. and Sherlock, G. (2000) Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer, P.L. and Doerge, R.W. (2010) Statistical design and analysis of RNA‐Seq data. Genetics, 185, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu, M. , Griffiths, J.S. , Huang, T.S. and Wang, A. (2008) Altered gene expression changes in Arabidopsis thaliana leaf tissues and protoplast in response to Plum pox virus infection. BMC Genomics, 9, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller, K. , Grossmann, J. , Grobei, M.A. , Hirsch‐Hoffmann, M. , Yalovsky, S. , Zimmermann, P. , Grossniklaus, U. , Gruissem, W. and Baginsky, S. (2008) Genomic scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science, 320, 938–941. [DOI] [PubMed] [Google Scholar]
- Bagnaresi, P. , Biselli, C. , Orrù, L. , Urso, S. , Crispino, L. , Abbruscato, P. , Piffanelli, P. , Lupotto, E. , Cattivelli, L. and Valè, G. (2012) Comparative transcriptome profiling of the early response to Magnaporthe oryzae in durable resistant vs susceptible rice (Oryza sativa L.) genotypes. PLoS ONE, 7, e51609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat, A. , Staton, M. , Cheng, C.H. , Park, J. , Mohammed, N.B. , Ficklin, S. , Yeh, C.C. , Hebard, F. , Baier, K. , Powel, W. , Schuster, S.C. , Wheeler, N. , Abbott, A. , Carlson, J.E. and Sederoff, R. (2012) Chesnut resistance to the blight disease: insights from transcriptome analysis. BMC Plant Biol. 12, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard, R. , Marénaud, C. and Sutic, D. (1969) Le pêcher GF305 indicateur polyvalent des virus des espèces a noyau. Ann. Phytopathol. 1, 603–617. [Google Scholar]
- Bilgin, D. , Zavala, J.A. , Zhu, J. , Clough, S.J. , Ort, D.R. and DeLucia, E.H. (2010) Biotic stress globally down regulates photosynthesis genes. Plant Cell Environ. 33, 1597–1613. [DOI] [PubMed] [Google Scholar]
- Boevink, P. and Oparka, K.J. (2005) Focus issue on virus–plant cell interaction and virus–host interactions during movement processes. Plant Physiol. 138, 1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambra, M. , Asensio, M. , Gorris, M.T. , Camarasa, E. , García, J.A. , Moya, J.J. , López‐Abella, D. , Vela, C. and Sanz, A. (1994) Detection of Plum pox virus using monoclonal antibodies to structural and non‐structural proteins. EPPO Bull. 24, 569–578. [Google Scholar]
- Cambra, M. , Capote, N. , Myrta, A. and Llácer, G. (2006) Plum pox virus and the estimated costs associated with sharka disease. EPPO Bull. 36, 202–204. [Google Scholar]
- Cao, J. , Schneeberger, K. , Ossowski, S. , Gunther, T. , Bender, S. , Fitz, J. , Koening, D. , Lanz, C. , Stegle, O. , Lippert, C. , Wang, X. , Ott, F. , Muller, J. , Alonso‐Blanco, C. , Borgwardt, K. , Schmid, K.J. and Weigel, D. (2011) Whole‐genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. [DOI] [PubMed] [Google Scholar]
- Carmona‐Saez, P. , Chagoyen, M. , Tirado, F. , Carazo, J.M. and Pascual‐Montado, A. (2007) GENECODIS: a web‐based tool for finding significant concurrent annotations in gene lists. Gen. Biol. 8, R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Mao, Y. , Liu, H. , Yu, F. , Li, S. and Yin, T. (2014) Transcriptome differentially expressed genes relevant to variegation in peach flowers. PLoS ONE, 9, e90842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. , Huh, S.U. , Kojima, M. , Sakakibara, H. , Paek, K.H. and Hwang, I. (2010) The cytokinin‐activated transcription factor ARR2 promotes plant immunity via TGA3/NPR1‐dependent salicylic acid signaling in Arabidopsis. Dev. Cell, 19, 284–295. [DOI] [PubMed] [Google Scholar]
- Dallot, S. , Quiot‐Douine, L. , Sáenz, P. , Cervera, M.T. , García, J.A. and Quiot, J.B. (2001) Identification of Plum pox virus determinants implicated in specific interactions with different Prunus spp. Phytopathology, 91, 159–164. [DOI] [PubMed] [Google Scholar]
- Dardick, C. (2007) Comparative expression profiling of Nicotiana benthamiana leaves systemically infected with three tree viruses. Mol. Plant–Microbe Interact. 20, 1004–1007. [DOI] [PubMed] [Google Scholar]
- Devoto, A. , Ellis, C. , Magusin, A. , Chang, H.S. , Chilcott, C. , Zhu, T. and Turner, J.G. (2005) Expression profiling reveals COI1 to be a key regulator of genes involved in wound‐ and methyl jasmonate‐induced secondary metabolism, defence, and hormone interactions. Plant Mol. Biol. 58, 497–513. [DOI] [PubMed] [Google Scholar]
- Díaz‐Vivancos, P. , Rubio, M. , Mesonero, V. , Periago, P.M. , Ros‐Barceló, A. , Martínez‐Gómez, P. and Hernández, J.A. (2006) The apoplastic antioxidant system in Prunus: response against long‐term Plum pox virus infection. J. Exp. Bot. 57, 3813–3824. [DOI] [PubMed] [Google Scholar]
- Du, Z. , Zhou, X. , Ling, Y. , Zhang, Z. and Su, Z. (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38 (Suppl 2), W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández, D. , Tisserant, E. , Talhinhas, P. , Azinheira, H. , Vieira, A. , Petitot, A.S. , Loureiro, A. , Poulain, J. , Da Silva, C. and Duplessis, S. (2012) 454‐pyrosequencing of Coffea arabica leaves infected by rust fungus Hemileia vastatrix: in planta‐expressed pathogen‐secreted proteins and plant functions in a late compatible‐rust interaction. Mol. Plant Pathol. 12, 17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L. , Tu, Z.J. , Millet, B.P. and Bradeen, J.M. (2013) Insights into organ‐specific pathogen defence responses in plants: RNA‐seq analysis of potato tuber–Phytophthora infestans interactions. BMC Genomics, 14, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García, J.A. , Glasa, M. , Cambra, C. and Candresse, T. (2014) Plum pox virus and sharka: a model potyvirus and a major disease. Mol. Plant Pathol. 15, 226–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Großkinsky, D.K. , Naseem, M. , Abdelmohsen, U.R. , Plickert, N. , Engelke, T. , Griebel, T. , Zeier, J. , Novák, O. , Strnad, M. , Pfeifhofer, H. , van der Graaff, E. , Simon, U. and Roitsch, T. (2011) Cytokinins mediate resistance against Pseudomonas syringae in tobacco through increased antimicrobial phytoalexin synthesis independent of salicylic acid signaling. Plant Physiol. 157, 815–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman, D.A. , Wei, F. and Wise, D. (2003) Powdery mildew‐induced Mla mRNAs are alternatively spliced and contain multiple upstream open reading frames. Plant Physiol. 131, 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsir, L. , Schilmiller, A.L. , Staswick, P.E. , He, S.Y. and Howe, G.A. (2008) COI1 is a critical component of a receptor for jasmonate and the bacterial virulence factor coronatine. Proc. Natl. Acad. Sci. USA, 105, 7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepke, T. , Schaeffer, S. , Krishnan, V. , Harper, A. , Whiting, M. , Oraguzie, N. and Dhingra, A. (2012) Rapid gene‐based SNP and haplotype marker development in non‐model eukaryotes using 3′UTR sequencing. BMC Genomics, 13, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunjeti, S.H. , Evans, T.A. , Marsh, A.G. , Gregory, N.F. , Kunjeti, S. , Meyers, B.C. , Kalavacharls, V.S. and Donofrio, N.M. (2012) RNA‐Seq reveals infection‐related global gene changes in Phytophthora phaseoli, the causal agent of lima bean downy mildew. Mol. Plant Pathol. 13, 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labaj, P.P. , Leparc, G.G. , Linggi, B.L. , Markille, L.M. , Wiley, H.S. and Kreil, D.P. (2011) Characterization and improvement of RNA‐Seq precision in quantitative transcript expression profiling. Bioinformatics, 27, i383–i391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C.Y. , Geng, G. , Yang, J. , Viljoen, A. , Jin, Y. , Kuang, R.B. , Zuo, C. , Lv, Z.C. , Yang, Q. , Sheng, O. , Wei, Y. , Hu, C. , Dong, T. and Yi, G. (2012) Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum sp. cubense tropical race 4. BMC Genomics, 13, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. and 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner, R. and Friedel, C.C. (2012) A comprehensive evaluation of alignment algorithms in the context of RNA‐Seq. PLoS ONE, 7, e52403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada, E.S. (2007) Alternative processing as a mechanism for regulating gene expression In: Regulation of Gene Expression in Plants (Basset C.L., ed.), pp. 67–100. New York: Springer. [Google Scholar]
- Lu, J. , Du, Z.X. , Kong, J. , Chen, L.N. , Qiu, Y.H. , Li, G.F. , Meng, X.H. and Zhu, S.F. (2012) Transcriptome analysis of Nicotiana tabacum infected by Cucumber mosaic virus during systemic symptom development. PLoS ONE, 7, e43447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleck, K. , Levine, A. , Eulgem, T. , Morgan, A. , Schimid, J. , Lawton, K.A. , Dangl, J.L. and Dietrich, R.A. (2000) The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat. Genet. 26, 403–410. [DOI] [PubMed] [Google Scholar]
- Martínez‐Gómez, P. and Dicenta, F. (2001) Distribution of coat protein and nucleic acid of Plum pox virus (PPV) in seedlings of peach rootstock GF305 and apricot cv. Real Fino. Phytopathol. Mediterr. 40, 157–164. [Google Scholar]
- Martínez‐Gómez, P. , Rubio, M. , Dicenta, F. , Aparicio, F. and Pallás, V. (2003) Comparative analysis of three diagnostic methods for the evaluation of Plum pox virus (PPV) resistance in apricot breeding programs. Acta Hortic. 622, 353–357. [Google Scholar]
- Martínez‐Gómez, P. , Crisosoto, C. , Bonghi, C. and Rubio, M. (2011) New approaches to Prunus transcriptome analysis. Genetica, 139, 755–769. [DOI] [PubMed] [Google Scholar]
- Martínez‐Gómez, P. , Sánchez‐Pérez, R. and Rubio, M. (2012) Clarifying omics concepts, challenges and opportunities for Prunus breeding in the post‐genomic era. OMICS: J. Integrative Biol. 16, 268–283. [DOI] [PubMed] [Google Scholar]
- Mochizuki, T. , Ogata, Y. , Hirata, Y. and Ohki, S.T. (2014) Quantitative transcriptional changes associated with chlorosis severity in mosaic leaves of tobacco plants infected with Cucumber mosaic virus . Mol. Plant Pathol. 15, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata, H. , Goto, S. , Sato, K. , Fujibuchi, W. , Bono, H. and Kanehisa, M. (1999) KEGG: kyoto encyclopaedia of genes and genomes. Nucleic Acids Res. 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe, N. , Salson, M. , Commes, T. and Rivals, E. (2013) CRAC: an integrated approach to the analysis of RNA‐Seq. Genome Biol. 14, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predajňa, L. , Šubr, Z. , Candresse, T. and Glasa, M. (2012) Evaluation of the genetic diversity of Plum pox virus in a single plum tree. Virus Res. 167, 112–117. [DOI] [PubMed] [Google Scholar]
- Rasori, A. , Ruperti, B. , Bonghi, C. , Tonutti, P. and Ramina, A. (2002) Characterization of two putative ethylene receptor genes expressed during peach fruit development and abscission. J. Exp. Bot. 53, 2333–2339. [DOI] [PubMed] [Google Scholar]
- Rubio, M. , Martínez‐Gómez, P. , Pascal, T. and Dicenta, F. (2012) Sensitivity of peach cultivars against a Dideron isolate of Plum pox virus. Sci. Hortic. 144, 81–86. [Google Scholar]
- Scholthof, K.B.G. , Adkins, S. , Czosnek, H. , Paulukaitis, P. , Jacquot, E. , Hohn, T. , Hohn, B. , Saunders, K. , Candresse, T. , Ahlquist, P. and Foster, G.D. (2011) Top 10 plant viruses in molecular plant pathology. Mol. Plant Pathol. 12, 938–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurdi‐Levraud, V. , Hullot, C. , Wawrzy'nczak, D. , Mathieu, E. , Eyquard, J.P. , le Gall, O. and Decroocq, V. (2006) Plum pox virus induces differential gene expression in the partially resistant stone fruit tree Prunus armeniaca cv. Goldrich. Gene, 374, 96–103. [DOI] [PubMed] [Google Scholar]
- Shan, X. , Zhang, Y. , Peng, W. , Wang, Z. and Xie, D. (2009) Molecular mechanism for jasmonate‐induction of anthocyanin accumulation in Arabidopsis . J. Exp. Bot. 60, 3849–3860. [DOI] [PubMed] [Google Scholar]
- Shen, B. , Li, C. and Tarczynski, M.C. (2002) High free‐methionine and decreased lignin content result from a mutation in the Arabidopsis S‐adenosyl‐L‐methionine synthetase 3 gene. Plant J. 29, 371–380. [DOI] [PubMed] [Google Scholar]
- Sheveleva, A. , Kudryavtseva, A. , Speranskaya, A. , Belenikin, M. , Melnikova, N. and Chirkov, S. (2013) Complete genome sequence of a novel Plum pox virus strain W isolate determined by 454 pyrosequencing. Virus Genes, 47, 385–388. [DOI] [PubMed] [Google Scholar]
- Sicard, O. , Loudet, O. , Keurentjes, J.J. , Candresse, T. , Le Gall, O. , Revers, F. and Decroocq, V. (2008) Identification of quantitative trait loci controlling symptom development during viral infection in Arabidopsis thaliana . Mol. Plant–Microbe Interact. 21, 198–207. [DOI] [PubMed] [Google Scholar]
- Socquet‐Juglard, D. , Kamber, T. , Pothier, J.F. , Christen, D. , Gessler, C. , Duffy, B. and Patochi, A. (2013) Comparative RNA‐Seq analysis of early‐infected peach leaves by the invasive phytopathogen Xanthomonas arboricola pv. pruni . PLoS ONE, 8, e54196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staswick, P.E. (2008) JAZing up jasmonate signalling. Trends Plant Sci. 13, 66–71. [DOI] [PubMed] [Google Scholar]
- Tong, Z. , Gao, Z. , Wang, F. , Zhou, J. and Zang, Z. (2009) Selection of reliable reference genes for gene expression studies in peach using real‐time PCR. BMC Mol. Biol. 10, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Barem, M.J. , Salzberg, S.L. , Wold, B.J. and Pachter, L. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser, A. , Cutcutache, I. , Koressaar, T. , Ye, J. , Faircloth, B.C. , Remm, M. and Rozen, S.G. (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzarowska, A. , Dionisio, G. , Sarholz, B. , Piepho, H.P. , Xu, M. , Ingvardsen, C.R. , Wenzel, G. and Lübberstedt, T. (2009) Validation of candidate genes putatively associated with resistance to SCMV and MDMV in maize (Zea mays L.) by expression profiling. BMC Plant Biol. 9, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli, A. , Martín‐Hernández, A.M. , López‐Moya, J.J. and García, J.A. (2006) RNA silencing suppression by a second copy of the P1 serine protease of cucumber vein yellowing Ipovirus, a member of the family Potiviridae that lacks the cysteine protease HCPro. J. Virol. 80, 10 055–10 063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele, J. , De Preter, K. , Pattyn, F. , Poppe, B. , Van Roy, N. , De Paepe, A. and Speleman, F. (2002) Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1–0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oosten, H.J. (1975) Susceptibility of some woody plant species, mainly Prunus spp., to sharka (plum pox) virus. Neth. J. Plant Pathol. 81, 199–203. [Google Scholar]
- Verde, I. , Bassil, N. , Scalabrin, S. , Gilmore, B. , Lawley, C.T. , Gasic, K. , Micheletti, D. , Rosyara, U.R. , Cattonaro, F. , Vendramin, E. , Main, D. , Aramini, V. , Blas, A.L. , Mockler, T.C. , Bryant, D.W. , Wilhelm, L. , Troggio, M. , Sosinski, B. , Aranzana, M.J. , Arús, P. , Iezzoni, A. , Morgante, M. and Peace, C. (2012) Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS ONE, 7, e35668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde, I. , Abbott, A.G. , Scalabrin, S. , Jung, S. , Shu, S. , Marroni, F. , Zhebentyayeva, T. , Dettori, M.T. , Grimwood, J. , Cattanoro, F. , Zuccolo, A. , Rossini, L. , Jenkins, J. , Vendramin, E. , Meisel, L.A. , Decroocq, V. , Sosininski, B. , Prochnik, S. , Mitros, T. , Policriti, A. , Cipriani, G. , Dondini, L. , Ficklin, S. , Goodstein, D.M. , Xuan, P. , Del Fabbro, C. , Aramini, V. , Copeti, D. , González, S. , Horner, D. , Falchi, R. , Lucas, S. , Mica, E. , Maldonado, J. , Lazzari, B. , Bielenberg, D. , Pirona, R. , Miculan, M. , Barakat, A. , Testolin, R. , Stella, A. , Tartarin, S. , Tonutti, P. , Arús, P. , Orellana, A. , Wells, C. , Main, D. , Vizzotto, G. , Silva, H. , Salamani, F. , Schmutz, J. , Morgante, M. and Rokhsar, D. (2013) The high‐quality draft of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494. [DOI] [PubMed] [Google Scholar]
- Vozárová, Z. , Kamencayová, M. , Glasa, M. and Subr, Z. (2013) Plum pox virus accumulates mutations in different genome parts during a long‐time maintenance in Prunus host plants and passage in Nicotiana benthamiana . Acta Virol. 57, 369–372. [PubMed] [Google Scholar]
- Wang, A. , Chapman, P. , Chen, L. , Stobbs, L.W. , Brown, D.C.W. and Brandle, J.E. (2005) A comparative survey, by expressed sequence tag analysis, of genes expressed in peach leaves infected with Plum pox virus (PPV) and free from PPV. Can. J. Plant Pathol. 27, 410–419. [Google Scholar]
- Wang, L. , Zhao, S. , Gu, C. , Zhou, Y. , Zhou, H. , Ma, J. , Cheng, J. and Han, Y. (2013) Deep RNA‐Seq uncovers the peach transcriptome landscape. Plant Mol. Biol. 83, 365–377. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Gerstein, M. and Snyder, M. (2009) RNA‐Seq: a revolutionary tool for transcriptomics. Nat. Rev. 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, D.M. , Wakefield, M.J. , Smyth, G.K. and Oshlack, A. (2010) Gene ontology analysis for RNA‐seq: accounting for selection bias. Gen. Biol. 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Pei, X. , Zhang, C. , Lu, Z. , Wang, Z. , Jia, S. and Li, W. (2012) De novo foliar transcriptome of Chenopodium amaranticolor and analysis of its gene expression during virus‐induced hypersensitive response. PLoS ONE, 7, e45953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, W. , Gao, Z. , Zhuang, W. , Shi, T. , Zhang, Z. and Ni, Z. (2013) Genome‐wide expression profiles of seasonal bud dormancy at four critical stages in Japanese apricot. Plant Mol. Biol. 86, 247–264. [DOI] [PubMed] [Google Scholar]
- Zuriaga, E. , Soriano, J.M. , Zhebentyayeva, T. , Romero, C. , Dardick, C. , Cañizares, J. and Badenes, M.L. (2013) Genomic analysis reveals MATH gene(s) as candidate for Plum pox virus (PPV) resistance in apricot (Prunus armeniaca L.). Mol. Plant Pathol. 13, 663–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Unannotated and annotated single nucleotide polymorphisms (SNPs) identified by RNA‐Seq in the three GF305 peach samples: control (NI), inoculated without symptoms (INS) and inoculated with symptoms (IWS).
Table S2 Alternative splicing events identified by RNA‐Seq in the three GF305 peach samples: control (NI), inoculated without symptoms (INS) and inoculated with symptoms (IWS).
Table S3 Total differentially expressed genes in non‐inoculated GF305 peach leaves (NI), inoculated leaves without symptoms (INS) and inoculated leaves with symptoms (IWS).
Table S4 Specific differentially expressed genes in non‐inoculated GF305 peach leaves (NI), inoculated leaves without symptoms (INS) and inoculated leaves with symptoms (IWS).
Table S5 Primer sequences representing differentially expressed genes in the different treatments selected and reference genes for real‐time polymerase chain reaction (PCR).