

Abstract
Enhancers serve as critical regulatory elements in higher eukaryotic cells. The characterization of enhancer function has evolved primarily from genome-wide methodologies, including chromatin immunoprecipitation (ChIP-seq), DNase-I hypersensitivity (DNase-seq), digital genomic footprinting (DGF), and the chromosome conformation capture techniques (3C, 4C, and Hi-C). These population-based assays average signals across millions of cells and lead to enhancer models characterized by static and sequential binding. More recently, fluorescent microscopy techniques, including fluorescence recovery after photo-bleaching, fluorescence correlation spectroscopy, and single molecule tracking (SMT), reveal a highly dynamic binding behavior for these factors in live cells. Furthermore, a refined analysis of genomic footprinting suggests that many transcription factors leave minimal or no footprints in chromatin, even when present and active in a given cell type. In this study, we review the implications of these new approaches for an accurate understanding of enhancer function in real time. In vivo SMT, in particular, has recently evolved as a promising methodology to probe enhancer function in live cells. Integration of findings from the many approaches now employed in the study of enhancer function suggest a highly dynamic view for the action of enhancer activating factors, viewed on a time scale of milliseconds to seconds, rather than minutes to hours.
INTRODUCTION
A primary effort in current biology concerns the identification of genomic elements that regulate gene expression, and the mechanisms by which they operate. This issue has been central to molecular biology for decades, and will likely remain a main line research effort for years to come. These efforts have led to many breakthrough findings, including the first identification of enhancer elements in 1981.1–3 Since their discovery, enhancer elements have been intensively investigated, both as isolated elements in model gene systems, and as widely distributed elements in the genome. Throughout this long period of investigation, the factors that act at enhancers to activate or repress their function have been studied by methods that utilize large numbers of cells; thus the results usually represent an average across millions of cells. Furthermore, the techniques used invariably involve fixation of cells, purification of subcellular fractions, or in vitro reconstitution approaches that rely on templates much less complex that the in vivo chromatin substrate. In aggregate, these studies have led to models that envisage factors binding at their genomic targets for minutes or hours at a time to form relatively long-lived macromolecular complexes.4–6 More recently, genome-wide studies have identified large numbers of enhancers in the mammalian genome.7,8 A large literature now exists on the characterization of these elements and their activities in many cell types. Again, this literature focuses almost exclusively on static methodology.
Figure 1 presents a core summary of the current paradigm for enhancer action. Transcription factors (TFs) responsible for activating the enhancer bind to localized regions (a) either depleted of nucleosomes or populated with modified nucleosomes accessible by the factors. These events trigger two separate processes. By unspecified mechanisms, including recruitment of bridging factors (b) such as mediator,9 the enhancer is brought into proximity (c) to the target promoter. This process brings additional activities that further modify chromatin architecture at the promoter and recruit the many macromolecular complexes necessary for promotor function. In some cases, transcription at the enhancer itself is activated (d), producing enhancer RNAs (eRNA).10,11 For some enhancers, these noncoding RNAs then diffuse to distant genomic sites and regulate gene expression through poorly understood mechanisms.12,13 Alternatively, the eRNAs may function directly at the enhancer site itself, perhaps recruiting unique activities to the site.
FIGURE 1.
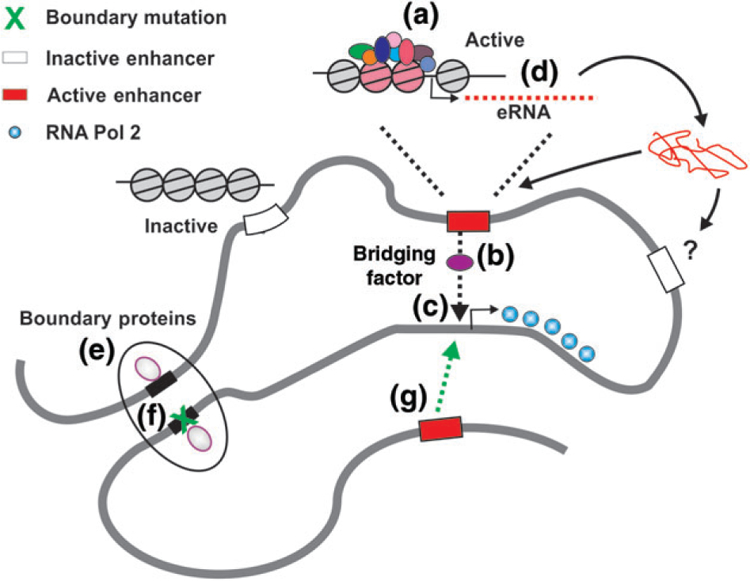
Current paradigm for enhancer function.
(a) Enhancers are regulated by transcription factors that bind and create regions of modified ‘open’ chromatin at the enhancer site. (b) During this process, the enhancer is brought into proximity of a target promoter by putative bridging factors (c), thereby activating the promoter. (d) In some cases, enhancers produce transcripts from their site, termed enhancer RNAs (eRNAs). These eRNAs either act at other targets in the genome by poorly understood mechanisms, or provide some cis function directly at the site. Enhancers are organized within large domains (designated as topologically associating domains, clusters, and other terms), that are formed by boundary proteins (e). Their activity is thought to be limited within these domains, but mutations in the boundary elements (f ) can break the boundary and allow penetration of other enhancers external to the domain (g).
Enhancers are believed to be organized within larger domains that demarcate genetically active regions (e). These regions in turn are characterized structurally as larger scale loops that appear in genome-wide domain interaction maps as ‘topologically associating domains,’ or TADs.14 While enhancer/promoter interactions are generally thought to be confined within a given TAD, mutations have been identified15,16 that appear to ‘break’ the TAD boundary (f ), thereby allowing enhancers from neighboring domains to invade (g) and activate promoters within a given domain. These long-range interactions are under intense study with the ‘chromosome conformation capture’ methods (3C, 4C, HiC) pioneered by Dekker et al.17 This static, population-oriented, view of enhancer function has been extensively reviewed18–24 in many formats. We focus here on recent developments in the real-time action of enhancers and their binding factors in live cells, moving toward a true molecular model for enhancer function.
FOOTPRINTING
TF Footprints and Chromatin Accessibility as a Proxy for TF Binding
In eukaryotic cells, transcriptional regulation at enhancers involves alteration in the chromatin land-scape to allow TF binding. A chromatin environment favoring binding is one where the DNA template is accessible and certain histone modifications/variants are present. An explosion of experimental and computational techniques in the last decade led to critical insights regarding the triumvirate of enhancer features: TF binding, DNA accessibility, and histone modifications/variants. These insights were made possible primarily due to genome-wide profiling of enhancer features by high-throughput sequencing. Binding of TFs to enhancers as well as mapping his-tone features is routinely profiled by chromatin immunoprecipitation (ChIP-seq) and have been extensively reviewed.25,26 The most prevalent method for measuring DNA accessibility is the DNase hyper-sensitivity assay followed by sequencing (DNase-seq).27 The properties of DNase have been studied for decades; it was found to resemble several key TF properties,28 thus making it a good enzyme for accessibility assays. Additionally, several other chromatin accessibility techniques that present advantages over DNase-seq in particular aspects are available (TACh,29 FAIRE-seq,30 and RED-seq31). Among these alternative techniques, assay for transposase-accessible chromatin (ATAC-seq)32 provides several critical improvements over DNase-seq, mainly in terms of reduced amount of starting material needed and a more reproducible, less cumbersome protocol. Single-cell ATAC-seq is emerging as a powerful technique and has already led to critical discoveries.33 Single-cell analysis of chromatin accessibility enables evaluation of chromatin states without averaging the signal across the population and is therefore expected to be an invaluable technique. However, in contrast to DNase, Tn5 transposase is less well characterized. It is still not clear how well Tn5-accessible sites represent true regulatory elements. Also, a systematic comparison of Tn5-accessible sites to DNase-accessible sites is lacking.
The sequences bound by TFs within enhancers are locally protected from enzymatic cleavage due to occlusion of the DNA-cleaving enzyme from the DNA substrate. This protection results in reduced cleavage rate detected as a ‘footprint’34 that can be assayed genome-wide when using high-throughput sequencing.35 Although footprints in individual sites are observed, single footprints cannot be reliably determined individually. Thus, footprints are commonly presented and quantified by aggregating DNase cut counts across all motif occurrences and the motif-flanking region36 (Figure 2(a)).
FIGURE 2.

Genomic footprinting. (a) The genome-wide accessibility of transcription factors (TFs) to their DNA motif sequences is measured via sequencing DNA liberated from the chromatin fiber by cleaving enzymes. In the resulting enhancer landscape map, regions bound by TFs can be locally protected and are thus less frequently cleaved. A motif-centered aggregate plot of cleavage events across the genome can present this reduced cleavage rate as a footprint (pink). However, some TFs do not leave a footprint although they measurably bind to DNA (grey). The zigzag pattern at the motif sequence (‘signature’) occurs regardless of TF binding and originates from enzyme cleavage bias. (b) Several scenarios arising by combining chromatin immunoprecipitation (ChIP-seq) and footprinting data. (1) The appearance of both a ChIP-seq peak as well as a footprint at the motif is the canonical case. (2) A lack of a footprint in bound sites is possible in highly dynamic TFs or in the case of a DBD structure that more readily allows DNase access. (3) A footprint at unbound sites can originate from another factor recognizing the same motif. (4) Peaks bound with no detectable motif (and therefore no footprint) can suggest indirect binding. (5) When the TF is inactive under the examined conditions, no footprint would be found. [Correction added on 10 November 2017, after first online publication: Figure 2b has been updated.]
In the early years of genomic footprinting, it was suggested the technique offers an unprecedented view into the complete TF-DNA interactome of the cell following only one accessibility experiment (as opposed to ChIP-seq profiles generated singularly for each TF). Additionally, it was suggested to serve as a gateway for detecting previously unrecognized TF motifs.7 However, several reports highlight the difficulty in direct inference of TF binding from genomic footprinting. First, DNase has a nonrandom sequence specificity,37 as do all DNA cleaving enzymes. This cut bias affects motif-centered aggregation plots and can result in a footprint-like cleavage pattern at the motif (to differentiate it from a bona fide footprint, this pattern was termed signature, Figure 2(a)). This mis-identification of cut bias-originated signatures as footprints was described in several recent studies by comparing DNase-digested chromatin to similarly digested naked DNA.38–40 In fact, from the handful of TF-bound motifs examined in these studies, more motif-centered cleavage patterns were found to stem from cut bias rather than from bona fide footprints associated with TF binding. In addition, footprint patterns suggested to represent previously unrecognized motifs were also shown to originate from cut bias.38
Inferring TF Dynamics by Footprints
As for the interpretation of ChIP-seq experiments, researchers interpret genomic footprint patterns with a static model of TF binding in mind. The two approaches produce a population-averaged output, making conclusions drawn from them harder to relate to TF dynamics. However, while ChIP-seq is performed on crosslinked material, DNA accessibility assays are performed in isolated nuclei, where factors are free to come on and off chromatin. Thus, foot-printing can potentially shed some light on dynamics. In a scenario whereby TFs bind DNA for long periods of time (minutes to hours), a factor is expected to shield DNA from cleavage resulting in a measurable footprint. The lack of a footprint in bound motifs collides with this view and could be partially explained by the dynamic model of TF action discussed below. Factors with a short residence time of a few seconds would allow frequent access of DNase to chromatin leading to increased cut rate. However, ‘slower’ TFs would dock on DNA for longer periods and protect it from cleavage. Thus, the possibility arises that lack of footprints at bound sites (as measured by ChIP-seq) is due to short residence time. Interestingly, many footprint-lacking motifs bind nuclear receptors, a class of TFs repeatedly shown to only transiently reside on DNA.41–46
A complementary explanation to the lack of a footprint might be found in the structure of the DNA binding domain (DBD) of different TFs. Some DBDs may bind DNA in a manner more permissible to DNase access while others block it altogether. A systemic appreciation of footprinting capacity as a function of DBD structure is warranted to examine the effect of different DBD classes on DNase occlusion.
In summary, the genomic footprinting field has evolved to more faithfully represent different TF-DNA binding patterns in combination with ChIP-seq data. A more wholesome view on TF binding patterns can be divided to several plausible scenarios: (1) The canonical, straight forward case would be a motif bound by a TF as measured by ChIP-seq, this motif would present a footprint pattern in cleavage aggregate plots; (2) A ChIP-seq peak is found at the motif but a footprint pattern is lacking. This can be explained either by short residence time or a DBD structure that more readily allows DNase access; (3) A footprint is found at the motif but a peak is absent. This is most probably due to a different TF binding the same motif; (4) A peak is found but a motif is absent (and thus a motif-centered footprint pattern cannot be measured) would suggest indirect binding either by tethering to other factors or chromatin looping. ChIP-seq artifacts47 represent another possibility; and (5) Only a motif is found but no peak or footprint would suggest that the motif is unbound in most cells in the population (Figure 2(b)).
ENHANCER FACTOR ACTION IN LIVING CELLS
Results from in vivo population assays such as ChIP are interpreted under paradigms that visualize the recruitment of multiple molecular partners into stable and long-lived complexes in a well-ordered, sequential manner.48 When TF dynamics in live cells was first visualized through the use of fluorescently tagged proteins,41 this view came under attack. In the classic fluorescence recovery after photobleaching (FRAP) experiment, a green fluorescent protein (GFP)-labeled enhancer-binding factor (glucocorticoid receptor-GR) was observed to bind directly to a tandem array of responsive genes (Figure 3(a)). The accumulated pool of GFP-GR was bleached, and then observed after turning off the bleach beam. The bound, bleached molecules were found to be rapidly replaced by unbleached molecules present in the nucleoplasm (Figure 3(b)). Thus, it appeared that the actual residence time for this enhancer factor was in the range of a few seconds, in marked contrast to the widely accepted view of TF occupancy at the time. In the decade following this experiment, many factors were found to manifest similar rapid exchange properties.48–50 Most factors that have now been studied by live-cell microscopy exhibit dwell times on chromatin on the order of seconds.51
FIGURE 3.

Tracking enhancer-binding factors in live cells. Transcription factor (TF) action at enhancers has been studied in live cells primarily by two methodologies. (a) In fluorescence recovery after photobleaching (FRAP), GFP-labeled factors are visualized either throughout the nucleus, or at gene arrays containing specific binding sites for a given factor. (b) Bleaching of the bound factors followed by observation during recovery provides direct information regarding the residence time of the factors. (c) In single-molecule tracking (SMT), movements of individual molecules are observed within a thin slice of the nucleus. In this example, the zone of observation is provided by highly inclined laminated optical sheet microscopy. (d) Single molecules are visualized as bright diffraction-limited spots, and their movement is tracked in real time. (e) Representation of SMT data. Collected tracks (black) showing the single-molecule residence times fitted to a single-(blue) or double-exponential (red) decay model. Expanded view represents the same data and fits with y-axis plotted as a log 10. (f ) Results from SMT from several laboratories suggest that factors search the genome via many very short binding events, but eventually find authentic response elements, where they reside for longer binding times on the order of 5–15 seconds.
The many FRAP studies carried out following the GR observations in 2000 firmly established the concept of highly mobile TFs. However, the FRAP technology is itself a molecular population method. Furthermore, robust quantitative interpretation of FRAP experiments is exceedingly difficult52,53; in fact, the estimates of molecular mobility with this technique are often within considerable error range. Fortunately, an alternate methodology has recently evolved that shows great promise in tackling this important question. Single molecule tracking (SMT) of individual fluorescently tagged molecules is not a new technique. However, the photobleaching problems associated with the classic family of fluorescent proteins, green (GFP), red (RFP), cyan (CFP), and yellow (YFP) chromofluors prevents tracking of molecules labeled with these tags over the necessary time periods. Development of the HALO and Snap-tag protein tagging methods,54,55 coupled with synthesis of the bright and photoresistant organic JF-fluors,56 now allows relatively long-term tracking of individual TFs.
In the SMT methodology, the movement of fluorescently tagged molecules is observed within a thin plane in the nucleus of live cells (Figure 3(c)). This can be achieved with alternate microscopy approaches; one method (depicted in this panel) is highly inclined laminated optical sheet microscopy (HILO).57 Using this approach, single molecule tracks can be observed over extended periods. The factors are observed to periodically stop, and then diffuse quickly to alternate sites (Figure 3(d)). Large data sets can be collected involving thousands of tracks. This approach has now been applied for approximately two-dozen TFs in a variety of cellular systems.52,57–69
In light of its obvious advantages, considerable effort is devoted to overcome the current limitations of SMT. One concern is how to deduce ‘productive’ binding, namely binding to DNA at the cognate motif, leading to transcriptional regulation (Figure 1). This is separated from ‘scanning’ binding events (i.e., nonspecific DNA–protein encounters). This issue was resolved by the observation that a single component model was insufficient to describe the data collected. Rather, a two-component model fits the data with high precision (Figure 3(e)). This indicates that the DNA-bound population of molecules includes two mathematically distinguished subpopulations, or two fractions: a short-lived (‘fast stops’) and a longer-lived (‘slow stops’) fraction. Previous reports suggest that the slow fraction of molecules represents specific binding events associated with enhancers or promoters, while the fast fraction defines nonspecific binding to chromatin or a DNA scanning mechanism.60,62,70 In addition, technical issues and standard practices are actively debated in the field. Some of the debated issues are the tradeoff between long acquisition time and bleaching as well as labeling enough of the TF population versus avoiding too much labeled molecules that will render single molecule resolution impossible. The methodology has been discussed in detail in recent technical reviews.70,71
With the implementation of these methods, an alternate view of TF/chromatin interactions is evolving (Figure 3(e)). According to this new view, TFs search the genome by rapid sampling of DNA sites available on accessible nucleosomes. When an authentic recognition element (RE, or response element) for a given factor is encountered, the factor will engage multiple chromatin modifying activities (histone modifiers, ATP-dependent remodeling systems, etc.) and initiate enhancer activation. This process, how-ever, is itself highly dynamic, that is, even at these regulatory sites, the factors are only briefly resident. Current estimates suggest scanning stops are in the range of 3–500 milliseconds, while residence times at true REs range from 5 to 15 seconds (Figure 3(f)).
This model sheds new light on enhancer activity and TF cooperativity on the chromatin template. Indeed, a study examining two TFs that bind the same motif found these TFs do not compete but rather enhance each other’s binding.72 This phenomenon, termed dynamic assisted loading, was since described in various systems.68,69,73–78 Thus, data have accumulated to suggest a dynamic mode of action for TFs whereby cooperativity can occur in a manner not necessitating physical interaction between TFs and is mediated by changes in chromatin availability.
In addition to the emergence of TF binding SMT, single molecule analysis of transcripts is rapidly evolving. Single molecule-level information enables evaluation of different behaviors for various subpopulations of transcripts originating from the same promoter. Additionally, temporal information is easier to obtain compared to population-based techniques. These advantages in transcript SMT lead to a shift in how we envisage almost every aspect of transcriptional regulation; including initiation, elongation, and splicing.79
CONCLUSION
Two mutually exclusive views have dominated the recent literature concerning the action of TFs at enhancer elements. The static view, based on bio-chemistry with naked DNA and a variety of cell population approaches, argues that TFs are sequentially recruited to DNA and reside on response elements over time periods of minutes or hours. In contrast, a highly dynamic model is evolving from the two approaches discussed in this review. Live cell microscopy, including the fluorescence techniques of fluorescence correlation spectroscopy (FCS), FRAP, and SMT, reveals that many, perhaps most, factors are only transiently bound to the template, even at authentic response elements.
The introduction of digital genomic footprinting in 200980 provided a potent new method to examine factor interaction with the chromatin land-scape. Elimination of the need for a new dataset for each factor (ChIP-seq) allows in principle the opportunity to examine all known TF-binding events by detecting DNA protection at their motifs in the genome. However, a careful examination of these datasets now argues that many TFs do not leave significant footprints at their binding sites.35,38,39,81 We suggest here that the ‘missing footprints’ result from the highly dynamic behavior of TFs on genome sites revealed in the live cell experiments. For a comprehensive understanding of enhancer function, these conflicting perspectives must eventually be resolved. SMT is a rapidly growing field, with constant improvement in tools and technique from a growing number of investigators. It is anticipated that newer approaches will eventually allow the development of a consensus for true TF dwell times at authentic response elements. These studies must also be extended to an analysis of the linkage between TF binding at enhancers and the real-time transcriptional output for promoter(s) dependent on those sites.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), the National Cancer Institute (NCI), the Center for Cancer Research (CCR).
Footnotes
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1.Gruss P, Dhar R, Khoury G. Simian virus 40 tandem repeated sequences as an element of the early promoter. Proc Natl Acad Sci USA 1981, 78:943–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benoist C, Chambon P. In vivo sequence requirements of the SV40 early promotor region. Nature 1981, 290:304–10. [DOI] [PubMed] [Google Scholar]
- 3.Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 1981, 27:299–308. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 2014, 159:358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller F, Stasevich TJ, Mazza D, McNally JG. Quantifying transcription factor kinetics: at work or at play? Crit Rev Biochem Mol Biol 2013, 48:492–514. [DOI] [PubMed] [Google Scholar]
- 6.Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995, 83:1091–1100. [DOI] [PubMed] [Google Scholar]
- 7.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orom UA, Shiekhattar R. Noncoding RNAs and enhancers: complications of a long-distance relationship. Trends Genet 2011, 27:433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mousavi K, Zare H, Dell’orso S, Grontved L, Gutierrez-Cruz G, Derfoul A, Hager GL, Sartorelli V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell 2013, 51:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161:1012–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupianez DG, Spielmann M, Mundlos S. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet 2016, 32:225–37. [DOI] [PubMed] [Google Scholar]
- 17.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science 2002, 295: 1306–1311. [DOI] [PubMed] [Google Scholar]
- 18.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 2015, 16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van BK, Corces VG. Lost in transition: dynamic enhancer organization across naive and primed stem cell states. Cell Stem Cell 2014, 14:693–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plank JL, Dean A. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell 2014, 55:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiekhattar R Opening the chromatin by eRNAs. Mol Cell 2013, 51:557–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell 2013, 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denker A, de Laat W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev 2016, 30:1357–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinn JL, Chang HY. Genome regulation by long non-coding RNAs. Annu Rev Biochem 2012, 81:145–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivera CM, Ren B. Mapping human epigenomes. Cell 2013, 155:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 2014, 15:272–286. [DOI] [PubMed] [Google Scholar]
- 27.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oliveri M, Daga A, Lunardi C, Navone R, Millo R, Puccetti A. DNase I behaves as a transcription factor which modulates Fas expression in human cells. Eur J Immunol 2004, 34:273–9. [DOI] [PubMed] [Google Scholar]
- 29.Grøntved L, Bandle R, John S, Baek S, Chung HJ, Liu Y, Aguilera G, Oberholtzer C, Hager GL, Levens D. Rapid genome-scale mapping of chromatin accessibility in tissue. Epigenetics Chromatin 2012, 5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, et al. A map of open chromatin in human pancreatic islets. Nat Genet 2010, 42:255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen PB, Zhu LJ, Hainer SJ, McCannell KN, Fazzio TG. Unbiased chromatin accessibility profiling by RED-seq uncovers unique features of nucleosome variants in vivo. BMC Genomics 2014, 15:1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013, 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 23:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galas DJ, Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 1978, 5:3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung MH, Baek S, Hager GL. Genome-wide footprinting: ready for prime time? Nat Methods 2016, 13:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baek S, Sung MH. Genome-scale analysis of cell-specific regulatory codes using nuclear enzymes. Methods Mol Biol 2016, 1418:225–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazarovici A, Zhou T, Shafer A, Dantas Machado AC, Riley TR, Sandstrom R, Sabo PJ, Lu Y, Rohs R, Stamatoyannopoulos JA. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci USA 2013, 110:6376–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods 2014, 11:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sung MH, Guertin MJ, Baek S, Hager GL. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell 2014, 56:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yardimci GG, Frank CL, Crawford GE, Ohler U. Explicit DNase sequence bias modeling enables high-resolution transcription factor footprint detection. Nucleic Acids Res 2014, 42:11865–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNally JG, Mueller WG, Walker D, Wolford RG, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 2000, 287:1262–1265. [DOI] [PubMed] [Google Scholar]
- 42.Groeneweg FL, van Royen ME, Fenz S, Keizer VI, Geverts B, Prins J, de Kloet ER, Houtsmuller AB, Schmidt TS, Schaaf MJ. Quantitation of glucocorticoid receptor DNA-binding dynamics by single-molecule microscopy and FRAP. PLoS One 2014, 9:e90532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu Y, Stavreva DA, Luo Y, Indrawan A, Chang M, Hager GL. Dynamic interaction of HDAC1 with a glucocorticoid receptor regulated gene is modulated by the activity state of the promoter. J Biol Chem 2011, 286:7641–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meijsing SH, Elbi C, Luecke HF, Hager GL, Yamamoto KR. The ligand binding domain controls glucocorticoid receptor dynamics independent of ligand release. Mol Cell Biol 2007, 27:2442–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rayasam GV, Elbi C, Walker DA, Wolford R, Fletcher TM, Edwards DP, Hager GL. Ligand specific dynamics of the progesterone receptor in living cells and during chromatin remodeling in vitro. Mol Cell Biol 2005, 25:2406–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agresti A, Scaffidi P, Riva A, Caiolfa VR, Bianchi ME. GR and HMGB1 interact only within chromatin and influence each other’s residence time. Mol Cell 2005, 18:109–121. [DOI] [PubMed] [Google Scholar]
- 47.Jain D, Baldi S, Zabel A, Straub T, Becker PB. Active promoters give rise to false positive ‘Phantom Peaks’ in ChIP-seq experiments. Nucleic Acids Res 2015, 43:6959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coulon A, Chow CC, Singer RH, Larson DR. Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet 2013, 14:572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 2014, 15:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hager GL, McNally JG, Misteli T. Transcription dynamics. Mol Cell 2009, 35:741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Royen ME, Zotter A, Ibrahim SM, Geverts B, Houtsmuller AB. Nuclear proteins: finding and binding target sites in chromatin. Chromosome Res 2011, 19:83–98. [DOI] [PubMed] [Google Scholar]
- 52.Mazza D, Abernathy A, Golob N, Morisaki T, McNally JG. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res 2012, 40:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazza D, Mueller F, Stasevich TJ, McNally JG. Convergence of chromatin binding estimates in live cells. Nat Methods 2013, 10:691–692. [DOI] [PubMed] [Google Scholar]
- 54.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 2008, 3:373–382. [DOI] [PubMed] [Google Scholar]
- 55.Gautier A, Juillerat A, Heinis C, Corrêa IR Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem Biol 2008, 15:128–136. [DOI] [PubMed] [Google Scholar]
- 56.Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 2015, 12:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tokunaga M, Imamoto N, Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 2008, 5:159–161. [DOI] [PubMed] [Google Scholar]
- 58.Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science 2007, 316:1191–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gebhardt JCM, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, Maniatis T, Xie XS. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods 2013, 10:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morisaki T, Muller WG, Golob N, Mazza D, McNally JG. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat Commun 2014, 5:4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Z, Legant WR, Chen B-C, Li L, Grimm JB, Lavis LD, Betzig E, Tjian R. 3D imaging of Sox2 enhancer clusters in embryonic stem cells. Elife 2014, 3:e04236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 2014, 156:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Izeddin I, Récamier V, Bosanac L, Cissé II, Boudarene L, Dugast-Darzacq C, Proux F, Bénichou O, Voituriez R, Bensaude O. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife 2014, 3: e02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Royen ME, van Cappellen WA, Geverts B, Schmidt T, Houtsmuller AB, Schaaf MJ. Androgen receptor complexes probe DNA for recognition sequences by short random interactions. J Cell Sci 2014, 127:1406–1416. [DOI] [PubMed] [Google Scholar]
- 65.Sugo N, Morimatsu M, Arai Y, Kousoku Y, Ohkuni A, Nomura T, Yanagida T, Yamamoto N. Single-molecule imaging reveals dynamics of CREB transcription factor bound to its target sequence. Sci Rep 2015, 5:10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Speil J, Baumgart E, Siebrasse J-P, Veith R, Vinkemeier U, Kubitscheck U. Activated STAT1 transcription factors conduct distinct saltatory movements in the cell nucleus. Biophys J 2011, 101:2592–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coleman RA, Liu Z, Darzacq X, Tjian R, Singer RH, Lionnet T. Imaging transcription: past, present, and future. Cold Spring Harb Symp Quant Biol 2015, 80:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD. Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell 2016, 165:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goldstein I, Baek S, Presman DM, Paakinaho V, Swinstead EE, Hager GL. Transcription factor assisted loading and enhancer dynamics dictate the hepatic fasting response. Genome Res 2017, 27:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Presman DM, Ball DA, Paakinaho V, Grimm JB, Lavis LD, Karpova TS, Hager GL. Quantifying transcription factor dynamics at the single-molecule level in live cells. Methods 2017, 116:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Z, Lavis LD, Betzig E. Imaging live-cell dynamics and structure at the single-molecule level. Mol Cell 2015, 58:644–659. [DOI] [PubMed] [Google Scholar]
- 72.Voss TC, Schiltz RL, Sung M-H, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, Hager GL. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 2011, 146:544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, Vinson C, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 2011, 43:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 2013, 32:1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grøntved L, Schiltz RL, Hager GL. Reprogramming of the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res 2013, 73:5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madsen MS, Siersbaek R, Boergesen M, Nielsen R, Mandrup S. Peroxisome proliferator-activated receptor gamma and C/EBPalpha synergistically activate key metabolic adipocyte genes by assisted loading. Mol Cell Biol 2014, 34:939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, et al. Genetic variation determines PPAR-gamma function and anti-diabetic drug response in vivo. Cell 2015, 162:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu B, Gates LA, Stashi E, Dasgupta S, Gonzales N, Dean A, Dacso CC, York B, O’Malley BW. Coactivator-dependent oscillation of chromatin accessibility dictates circadian gene amplitude via REV-ERB loading. Mol Cell 2015, 60:769–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen H, Larson DR. What have single-molecule studies taught us about gene expression? Genes Dev 2016, 30:1796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 2009, 6:283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gusmao EG, Allhoff M, Zenke M, Costa IG. Analysis of computational footprinting methods for DNase sequencing experiments. Nat Methods 2016, 13:303–309. [DOI] [PubMed] [Google Scholar]
FURTHER READINGS
- Coulon A, Chow CC, Singer RH, Larson DR. Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet 2013, 14:572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 2014, 15:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 2013, 14:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinstead EE, Paakinaho V, Presman DM, Hager GL. Pioneer factors and ATP-dependent chromatin remodeling factors interact dynamically: a new perspective. Multiple transcription factors can effect chromatin pioneer functions through dynamic interactions with ATP-dependent chromatin remodeling factors. Bioessays 2016, 38:1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez DG, Spielmann M, Mundlos S. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet 2016, 32:225–237. [DOI] [PubMed] [Google Scholar]