

Abstract
The KMT2 (lysine methyltransferase) family of histone modifying proteins play essential roles in regulating developmental pathways, and mutations in the genes encoding these proteins have been strongly linked to many blood and solid tumor cancers. The KMT2A-D proteins are histone 3 lysine 4 (H3K4) methyltransferases embedded in large COMPASS-like complexes important for RNA Polymerase II-dependent transcription. KMT2 mutations were initially associated with pediatric Mixed Lineage Leukemias (MLL) and found to be the result of rearrangements of the MLL1/KMT2A gene at 11q23. Over the past several years, large-scale tumor DNA sequencing studies have revealed the potential involvement of other KMT2 family genes, including heterozygous somatic mutations in the paralogous MLL3/KMT2C and MLL2(4)/KMT2D genes that are now among the most frequently associated with human cancer. Recent studies have provided a better understanding of the potential roles of disrupted KMT2C and KMT2D family proteins in cell growth aberrancy. These findings, together with an examination of cancer genomics databases provide new insights into the contribution of KMT2C/D proteins in epigenetic gene regulation and links to carcinogenesis.
Keywords: Lysine methyltransferase, chromatin, epigenetic, co-occurrence
1. Introduction
Post-translational histone modifications provide a vast epigenetic regulatory language that can be written and decoded by a large number of proteins and are important for controlling the timing and level of gene expression among all eucaryotes [1]. The methylation of lysine residues on N-terminal histone tails can provide both gene activation and repression signals and is one of the best-studied epigenetic modifications [2]. Methylation of histone 3 lysine 4 (H3K4) is frequently associated with active transcription, with di- and trimethylated H3K4 associated with active gene promoters and monomethylation found at gene enhancers [3]. The enzymes that place methyl modifications or marks on H3K4 are known as the KMT2 (lysine methyltransferase) family and are found embedded within enormous protein complexes, named COMPASS (COMplex of Proteins ASsociated with SET1) [4]. The discovery almost 30 years ago of recurrent translocation-associated leukemias involving MLL1/KMT2A, a homolog of the Drosophila Trithorax (Trx) protein important in Hox gene regulation, provided a critical platform for investigations into the roles of the KMT2 proteins in animal development and cancer [5–7]. More recently, genes encoding other KMT2 family proteins have been implicated in cancer [8]. Tumor genome and exome sequencing studies combined with cancer cell and model system genetic analyses over the past decade have revealed an unanticipated vital role for the KMT2 family enzymes in a diverse set of cancers, both as drivers of oncogenesis and as critical cooperating mutations in both cancer progression and post-therapy relapse.
2. KMT2 COMPASS-like complexes
KMT2 family proteins (SET1A, SET1B, MLL1/KMT2A, MLL4(2)/KMT2B, MLL3/KMT2C and MLL2(4)/KMT2D) provide the histone lysine methyltransferase activity of mammalian COMPASS complexes. Designated MLL1–5 based on relatedness to the MLL1 gene (Mixed Lineage Leukemia), KMT2 proteins comprise three subgroups, called COMPASS and COMPASS-like based homology with Drosophila Trx, Trr and dSet1 [4, 8]. Genome duplication during mammalian evolution resulted in two paralogs in each KMT2 subgroup. KMT2A/MLL1 and KMT2B/MLL4(2) are paralogous proteins within the Trx related subgroup and we refer to this group as the MLX family (MLL-TRX). KMT2C and KMT2D are paralogous proteins within the Trr related subgroup that we refer to as the MLR family (MLL-TRR). KMT2F and KMT2G are paralogous proteins within the dSET1 subgroup (KMT2F and KMT2G will not be covered in this discussion) [5]. The COMPASS complexes are large, multi-subunit assemblies [4, 9] that carry out mono-, di- and trimethylation of histone 3 lysine 4 (H3K4). These modifications are placed on H3 N-terminal tails within nucleosomes at gene promoter and enhancer regions and are universally associated with transcription activation. The SET1A/B complexes perform the majority of di- and tri-methylation of H3K4 throughout the genome [9]; whereas, the KMT2A/B (MLX) complexes are more limited in the targets of their enzymatic activities, primarily catalyzing di- and tri-methylation of H3K4 at gene promoters and nearby cis-regulatory sites where they maintain active transcription (Fig. 1). The KMT2C and KMT2D COMPASS-like (MLR) complexes monomethylate H3K4 (H3K4me1) at transcription enhancers throughout the human genome [10, 11], with estimates ranging from approximately 12,000 to over 20,000 sites depending on cell type and developmental stage [12, 13].
Figure 1. Human COMPASS-like complexes and their histone targets.
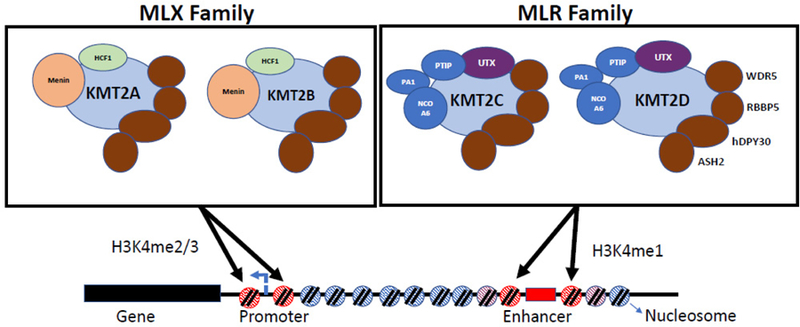
Two COMPASS-like families of complexes exist in higher eukaryotes, we indicate as MLX (MLL-TRX) and MLR (MLL-TRR) with distinct, though potentially limited overlapping target specificity. The MLX and MLR family complexes all contain catalytic subunits that methylate nucleosomes on histone 3 lysine 4 (H3K4) to produce di/trimethylation (H3K4me2/3; MLX family) or monomethylation (H3K4me1; MLR family). These histone modifications are associated with gene promoters and enhancers, respectively, and are linked to the activation of gene transcription. The MLX and MLR complexes both contain a set of core components (WDR5, RBBP5, ASH2 and hDPY30), as well as unique subunits. The MLX complexes contain Menin and HCF1 subunits, while the MLR complexes contain the histone lysine demethylase UTX, PTIP, PA1 and NCOA6.
The MLR COMPASS complexes are recruited to enhancers through interactions with sequence specific transcription factors, including ligand-associated nuclear receptors and pioneer factors that activate enhancers de novo. Although nucleosomal H3K4me1 marking is important for establishing enhancer identity, full activation appears to require acetylation of H3K27 by the histone acetyltransferase p300/CBP, recruited to enhancers through direct interactions with the COMPASS-like complexes [12, 14–16]. The presence of multiple methyl groups on H3K27, catalyzed by the Polycomb repressor PRC2 complex, can block the addition of the activating acetyl mark. In this case, the UTX/KDM6A lysine demethylase, a component of the MLR complexes, removes the methyl mark to allow for the addition of the acetyl group and subsequent enhancer activation. Through their epigenetic marking and commissioning of transcription enhancers, the MLR complexes contribute essential functions in developmental signaling [8, 17, 18].
KMT2A-D proteins are large (2715–5537aa) with multiple functional domains (see Fig. 2A). All contain plant homeodomain zinc finger structures (PHDf) that provide histone recognition and binding functions and interaction sites for other proteins. While the KMT2A/B proteins contain a single three PHD finger cluster within the N-terminal region and a single PHD finger near the C-terminus, the KMT2C/D proteins contain two closely related PHD finger clusters in the N-terminal regions (6–7 zinc-fingers) and a single PHD finger near the C-terminus. Each KMT2C/D cluster contains three to four zinc-coordinating PHD fingers in tandem. The first PHD cluster in KMT2C was recently shown to be involved in protein associations involving the BAP1 histone deubiquitinating repressor complex to control the expression of Polycomb-mediated transcriptional repression [13], while the highly conserved second cluster is thought to mediate direct chromatin interactions. A large number of cancer-associated missense mutations affecting the PHD finger clusters suggest these domains provide critical functions in controlling cellular growth and differentiation pathways through regulated binding to both modified histones and chromatin modifying complexes [13, 19].
Figure 2. Organization and mutation clustering of the KMT2A-D family proteins.
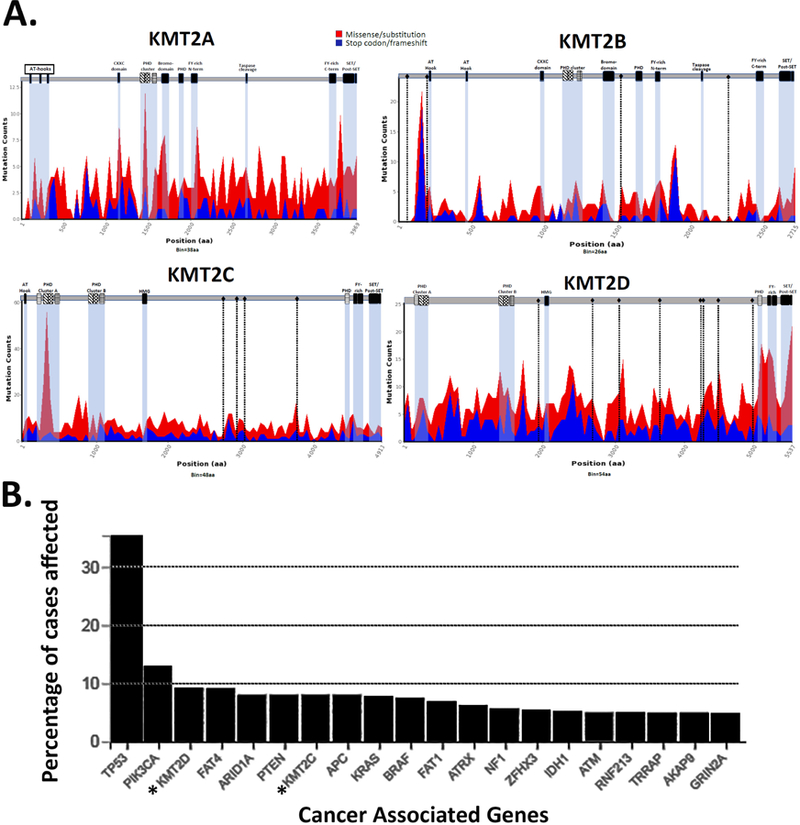
A) The KMT2A-D proteins are enormous (2715–5265aa), and each contains multiple domains, including Plant Homeodomain (PHD), SET/Post-SET methyltransferase, FY-rich (FYR) regions, as well as domains that are specific for each member or family. The KMT2A/B proteins are cleaved post-translationally by the taspase protease; although, there is no known cleavage of the mature KMT2C/D proteins. The PHD domain cluster in KMT2A/B is highly related to the PHD ‘b’ domain cluster in KMT2C/D. The black diamonds represent putative nuclear receptor (NR) binding motifs (LLXXL, LXXLL). Data obtained from The Cancer Genome Atlas (TCGA) was used to map cancer associated mutations in each protein and to identify mutational hotspots by binning based on mutation type (missense/nonsense), suggesting important functional protein domains [13, 31]. B) KMT2C and KMT2D are among the most frequently mutated cancer associated genes. Cancer mutation data was obtained from the NCI Genomic Data Commons Portal (GDC) that represents a summary of over 33,000 cases, including The Cancer Genome Atlas (TCGA) data. The top 20 most frequently mutated genes are shown together with the percent of cases affected across multiple cancer types.
The histone modifying enzyme activity of the KMT2 family resides in the SET histone methyltransferase domain (KMTase; reviewed in [20]). The SET domain of the KMT2 family catalyzes the addition of methyl groups onto H3K4 residues that subsequently serve as markers for additional regulatory control. While the precise functional roles of methylated H3K4 in nucleosomes is uncertain, three key findings suggest that the H3K4me1 histone mark catalyzed by the MLR complexes is not essential for transcriptional regulation during development but is potentially important to control precise transcription levels or timing of gene transcription in vivo, perhaps critically in cancer. First, the histone methylation activity of the Drosophila Trr and murine Kmt2c/d proteins do not appear to be required for maintaining transcription activity after genes have been initially activated [21, 22]. Second, both Kmt2c and Kmt2d are required for murine embryonic stem cell (mESC) differentiation, though not critical for self-renewal or maintenance of stem cell identity [12]. Third, while the SET methyltransferase activity of MLR complexes may be primarily important in the marking of enhancers, the SET domains are frequently mutated in cancers suggesting that they are responsible for chromatin modifications that broadly impact gene regulation. In support of this idea, there have been reported examples of SET domain cancer mutations in KMT2A and KMT2C that alter their enzymatic activity, in some cases increasing methylation activity while in other cases decreasing histone methylation [23, 24], and truncations that specifically remove the SET domain in various tumors suggest that the methyltransferase activity may be important for cellular homeostasis. The discovery of both gain and loss of SET domain activity in cancer implies that there is considerable complexity in the potential mechanisms of oncogenesis associated with altered methyltransferase function. Additional protein domains of functional biological importance in the KMT2A/B family include AT-hooks, CXXC and bromodomains that are involved in DNA binding or protein-protein interactions. The AT-hooks bind to minor groove DNA and likely help to direct the KMT2 proteins to specific genomic regions [25] and deletion of the AT hooks can impair leukemic fusion protein functions [26]. The CXXC domain can distinguish epigenetically modified DNA (CpG methylation), as it only binds to unmethylated DNA, frequently found in active gene promoters. The CXXC domain is also involved in subnuclear localization and target gene selection of the KMT2A COMPASS complexes [27] and is reported to help recruit repressive factors, such as histone deacetylases (HDAC) and Polycomb group (PcG) complexes [27, 28]. The AT hooks and CXXC domain are included in all KMT2A fusion associated leukemias and are thought to contribute to oncogenesis [26, 27]. The KMT2C/D proteins contain an HMG domain that may provide similar DNA binding as the AT-hooks, and multiple nuclear receptor (NR) interaction motifs (LLXXL/LXXLL) that are important for recruitment of the complexes to NR-regulated enhancer targets [29, 30]. However, it is unknown whether these domains in KMT2C/D play any direct role in cancer.
The establishment of several disease-associated mutation databases (e.g., TCGA, OMIM, COSMIC) has revealed that cancer-associated truncating/inactivating mutations are widely dispersed along the KMT2A-D proteins, confirming their importance as tumor suppressors (Fig. 2A). These databases also provide opportunities to gain insight into the potential biological significance of these domains in cancer. Missense mutation ‘hotspots’ are associated with known functional domains, including the PHD fingers, SET methyltransferase and regions associated with protein interactions [13, 31]. As was recently shown for the KMT2C protein using MutClustSW, a recursive Smith-Waterman based algorithm [19], mutational hotspots are not randomly distributed, but strongly associated with the first PHD finger cluster and with a second nearby region that has no identifiable domain. The first PHD finger cluster of KMT2C is involved in protein interactions with the BAP1 histone deubiquitinating complex and cancer associated mutations in this PHD domain disrupted recruitment of KMT2C to enhancers [13]. Alignment of the KMT2 proteins with mutation counts reveals additional ‘hotspots’ for missense mutations, supporting the existence of undiscovered functionally important domains that may be crucial for proper gene regulation.
3. Emerging roles for KMT2C and KMT2D in cancer
The epigenetic functions of the COMPASS complexes are vital for normal animal development and frequent mutations affecting these proteins or other components of the COMPASS complexes are associated with a large number of cancers [32]. Mutations involving KMT2 family genes are among the most commonly seen in many diverse cancer types and there is a statistically significant co-occurrence, raising the possibility that the simultaneous disabling of several KMT2 genes might be a critical step in some cancer types. However, expression of KMT2A fusion oncoproteins can produce leukemias in the absence of additional KMT2 mutations [33–35]. Similarly, murine knockout studies have shown that loss of Kmt2d can promote lymphomagenesis independent of additional mutations affecting other KMT2 family genes [18, 36]. Finally, a majority of cancers associated with KMT2 family genes are the result of somatic heterozygous loss-of-function mutations, suggesting that haploinsufficiency may underlie disease for these epigenetic regulators [8].
As mentioned earlier, the majority of KMT2A associated cancers are the result of gene fusions producing leukemic oncoproteins, linking the N-terminal portion of KMT2A to over 135 different partner proteins, including 35 that are frequently recurring and nine partners that account for over 90% of KMT2A-based leukemias [37]. While the involvement of KMT2A/MLL1 translocation fusions in cancer have been extensively reviewed [5, 23, 33, 35, 38], recent cancer exome sequencing studies have revealed a large number of missense (78%) and nonsense (22%) mutations in the gene with an overall somatic mutation frequency of 2.9%, suggesting possible loss and gain of function alterations of KMT2A in oncogenesis [23]. KMT2B is also found mutated in a variety of cancer types, with the majority (71%) being missense mutations and an overall somatic mutation frequency in cancer of 1.6%. Overexpression of KMT2B has also been observed in colorectal and breast cancer cell lines [39]. Although direct involvement in cancer is uncertain, KMT2B is important for maintaining promoter bivalency in embryonic stem cells [40] and it has been implicated in pediatric dystonia [41].
KMT2C and KMT2D were first linked to cancer through tumor sequencing studies showing frequent loss in pediatric and adult medulloblastoma [42, 43], with KMT2D among the most highly mutated genes in Non-Hodgkin lymphoma [44, 45]. More recently, the National Cancer Institute Genomic Data Commons (GDC) summary of over 33,000 cases revealed that KMT2D and KMT2C were the third and seventh most commonly mutated cancer genes (Fig. 2B). In addition, the cBio Portal database compendium [46, 47] of almost 47,000 cancer samples representing 175 studies reveals that KMT2D and KMT2C are mutated in approximately 14–16% of all cases, with the highest percentages in both melanoma (27%) and non-melanoma (~48%) skin cancers, urothelial carcinoma (40%), bladder cancer (33%), lung cancer (18–26%), head and neck and esophagogastric cancers (19%), as well as colorectal and small bowel cancers (13.5%). Surprisingly, only in prostate cancer are both genes frequently amplified (13–19%), while KMT2C is amplified in approximately 5% of ovarian cancers. However, there is no obvious strong correlation between changes in the RNA levels of these genes and cancer phenotype, suggesting that loss-of-function mutations rather than expression level changes are generally associated with oncogenesis.
Haploinsufficiency of the human 7q chromosome region in acute myeloid leukemia implicated KMT2C as a likely tumor suppressor [48], and recent tumor sequencing data (TCGA-BRCA study) has revealed a strong enrichment for KMT2C mutations in breast cancer, with approximately 8% across breast cancer types and over 10% within invasive breast cancers according to the cBio Portal database, with substantially greater frequency than any other H3K4 methyltransferase. Importantly, KMT2C was recently shown to be important for driving hormone-stimulated cell proliferation in estrogen receptor alpha positive (ERα+) HER2- breast cancer cell lines but not ERα+ HER2+ cells, while knockdown of KMT2D more broadly suppressed proliferation in breast cancer cells [49]. In this context, it appears that KMT2C is a critical co-factor in promoting ERα function and it appears to also have an important role in suppressing hormone-independent tumor expansion. This dual role of KMT2C in breast cancer cell lines is consistent with patient data, in that KMT2C loss was more frequent (30%) in patients with metastatic hormone-refractory disease and they had a shorter progression-free survival following aromatase therapy [49]. In addition, Zhang et al. [50] developed a somatic mammary stem cell based mouse organoid model to identify functionally important genes involved in tumorigenesis. They found that murine kmt2c inactivation in cells overexpressing Pik3ca blocked mammary gland differentiation and simultaneously increased cell stem cell self renewal activity through activation of the HIF pathway. These studies suggest that KMT2C loss may be an important oncogenic driver that promotes increased stem cell-like properties, especially in ERα+ breast cancers that also overexpress PIK3CA.
KMT2D is one of the most commonly mutated genes in both follicular lymphomas and diffuse large B cell lymphomas that arise from germinal center B cells with a mutation rate ranging from 35% to 85% [36, 44, 45]. The loss of KMT2D function appears to be an early cooperating event with overexpression of the BCL2 oncogene to drive lymphomagenesis. Genetic ablation of murine Kmt2d in B cells promotes lymphoma development and impedes B cell differentiation, supporting its role as a tumor suppressor in germinal center B cells [18, 36]. Conditional deletion of Kmt2d led to expansion of germinal center B-cells (GCB), while shRNA knockdown of KMT2D in human lymphoma cells was associated with increased proliferation in vitro, as well as decreased expression of tumor suppressor genes (e.g., SOCS3) that regulate B cell signaling pathways including JAK-STAT [18]. The loss of KMT2D results in reduced monomethylation marks at enhancers of multiple tumor suppressors which are known target genes of KMT2D [18, 51–54]. Thus, KMT2D appears to function as a tumor suppressor in non-Hodgkin lymphoma by regulating genes required for B-cell development, as well as other cancer types through its function in enhancer activation.
4. Lessons from KMT2C/D mutations in Lung Cancer
Lung cancers, both non small cell (NSCLC, 85%) and small cell (SCLC, 15%), are a leading cause of cancer-associated deaths worldwide with few treatment options and a high recurrence rate. SCLC is a highly aggressive neuroendocrine tumor with a very poor prognosis in which TP53 and RB1 are frequently mutated. Several whole genome or exome sequencing studies have also revealed a high frequency of KMT2D (8–24%) but not KMT2C mutations [55–61]. These studies revealed that KMT2D is not only among the most frequently mutated genes in SCLC, but there is a strong bias for the loss of KMT2D via truncating mutations in the pathogenesis of SCLC with potentially significant prognostic impact [58].
Three subtypes of NSCLC include adenocarcinoma (LUAD), squamous cell carcinoma (LUSC) and large cell carcinoma (LCC). Meta analysis of 3065 samples covering 9 studies [62–67] shows KMT2C/D alterations in over 40% of squamous cell cancers of the lung, and up to 30% of adenocarcinoma of the lung. In both LUAD and LUSC, there is a trend towards significant co-occurrence of KMT2C and KMT2D mutations, with a log odds ratios of 0.861 and 0.837, respectively. While mutations account for the vast majority of alterations, amplifications are also observed. However, many of these cancers also have reduced KMT2D or KMT2C expression independent of mutation status and reduced expression was correlated with worse overall survival. A study from Yin et al. [68] identified deleterious KMT2D mutations in 12/105 NCSLC patients as well as reduced expression of KMT2D in tumors relative to paired adjacent non tumor tissues. This study also included whole exome sequencing of nine non small cell lung cancers and found that KMT2D was the second most significantly mutated gene after TP53, showing three missense mutations and two nonsense mutations in three out of nine samples. While all tumor samples showed reduced expression of KMT2D independent of mutation status, only KMT2D mutations were significantly associated with lower recurrence free survival. Interestingly, many of the mutations in KMT2D resulted in silenced or significantly reduced expression in all of the sequenced tumor tissues compared with adjacent non-tumorous lung tissue. This study did not address the functional status of KMT2C in these mutated tissues, making it difficult to assess possible redundancy. A more recent analysis of 108 early stage (I-III) lung squamous cell carcinoma patients with matched normal tissue revealed KMT2D was mutated in 10.2% of lung tumors [69], consistent with TCGA data (178 patients) with a mutation frequency of 17.4%. In additon, it was found that patients with KMT2D mutations had a worse recurrence free survival, regardless of TP53 status. Based on univariate analysis, KMT2D mutation status was the factor most strongly associated with poor prognosis [69]. Together, these findings indicate that KMT2D mutations also have a strong association with NCSLC pathogenesis. Again, functional status of KMT2C was not assessed in this study. As might be expected, many of the identified mutations in NSCLC affected genes involved with DNA damage checkpoint and cell cycle regulation. Choi et al. [69] found that among LUSC patients with TP53 mutations, co-occurrence of KMT2D mutations was strongly correlated with poor recurrence free survival. An examination of the TCGA dataset for all NSCLC subtypes shows a significant co-occurrence between mutations in TP53 and both KMT2D (odds ratio 0.763) and KMT2C (odds ratio 0.908) with adjusted p-values <0.001 for both. A strong tendency of co-occurrence in lung squamous cell carcinomas and adenocarcinomas suggest that these three genes should be assessed in NSCLC tumors.
Expression levels of KMT2C/D genes have significant prognostic implications in some cancer types [70–73]. Significantly better survival rates are seen in adenocarcinoma of the lung with high expression levels of either KMT2C or KMT2D (Fig. 3A). However, this association is not seen in squamous cell carcinomas of the lung, with similar survival rates regardless of KMT2C/D expression levels (data not shown). Regardless of this difference, survival rates in all lung cancers are 20% higher at 10 years with high expression of KMT2C and KMT2D compared to low expression of both. The high rate of KMT2D mutations in these samples is suggestive that these alterations function as driver mutations, or mutations that are responsible for the development and progression of cancers. Data from the same meta analysis reveals that truncating mutations of KMT2C and KMT2D are present throughout all stages of the analyzed cancers, suggesting these mutations are present from early stages of cancer providing additional support for these alterations as driver mutations (Fig. 3B).
Figure 3. KMT2C/D involvement in lung cancer development and survival.
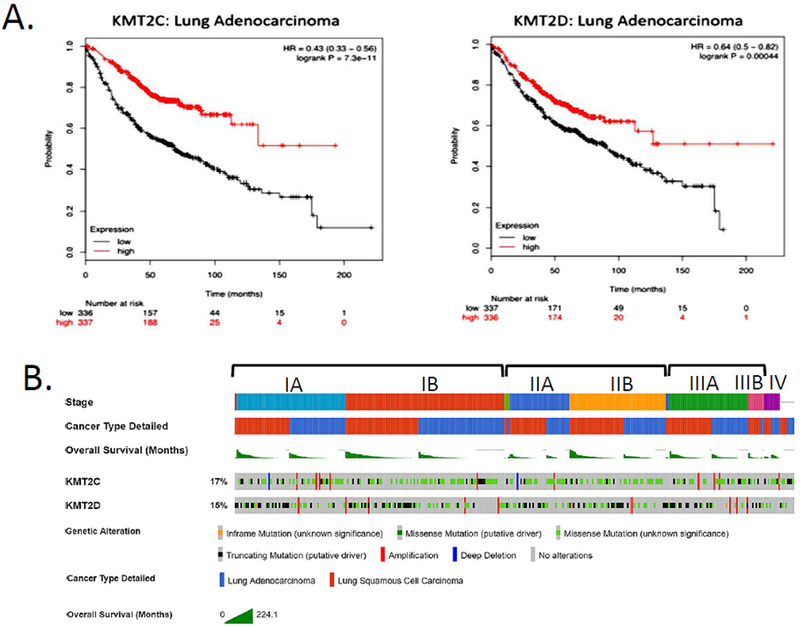
A) K-M plotter analysis of the correlation between KMT2C and KMT2D transcript expression and survival probability among lung adenocarcinoma patients. Low expression of both genes is correlated with shorter overall survival time. B) Association of KMT2C and KMT2D mutations with lung cancer type, stage of first detection and overall patient survival. Data was obtained from the cBio Portal, analyzing 1144 patients with lung adenocarcinoma or squamous cell carcinoma. Staging is according to the American Joint Committee on Cancer (AGCC). Truncating mutations of KMT2D are commonly observed in early stage lung squamous cell carcinomas, suggesting driver function; whereas, missense mutations in KMT2C are common in early stage lung adenocarcinomas.
5. Functional distinctions between KMT2C and KMT2D in cancer
Possible oncogenic mechanisms associated with KMT2C/D loss involve the dysregulation of transcriptional enhancers and transcription factor-dependent programs that drive cellular pathways required for tumor suppression and anti-tumor immune evasion [12, 21]. For example, several studies have demonstrated important direct interactions between KMT2C/D and the tumor suppressor TP53 [74–78]. The activation of TP53 target genes following doxorubicin (DNA damaging agent) treatment requires the coactivation functions of both KMT2C and KMT2D [77], and KMT2D enrichment sites overlap significantly with TP53 genomic sites [79]. These studies reveal several potential mechanisms through which KMT2C and KMT2D cooperate with TP53 in both transcription activation dependent and independent pathways to promote tumorigenesis. In human colon cancer cells harboring specific TP53 mutations, there is aberrant gene activation that is dependent on KMT2D deposition of H3K4me1 marks on enhancers [74, 78] providing a mechanistic explanation by which specific TP53 gain of function mutations contribute to tumor formation. Intriguingly, KMT2C also appears to play an important role in cellular damage responses that rely on TP53 function in maintaining genomic stability through DNA double strand break repair, a role that is independent of transcription activation [77].
Within early stage cancer cells, the KMT2C/D proteins help to maintain epithelial cell states, such that loss may contribute to a more stem-cell like state and progression to metastasis. In gastric epithelial cells, the depletion of KMT2C is associated with a transition to a more mesenchymal phenotype, which can be reversed by adding back KMT2C [73]. Further, tumor-derived organoids depleted of KMT2C were more highly invasive in mouse xenografts. One possible mechanism may involve KMT2C/D interactions with cell-type specific transcription factors that drive mesenchymal-epithelial transition, such as GRHL2, that function to activate epithelial cell phenotypes and sensitize tumor cells to Natural Killer (NK) cell activity [80]. Simultaneous removal of both KMT2C (−/−) and KMT2D (+/−) in MCF10a cells converted them from an epithelial to a more mesenchymal morphology with reduced sensitivity to NK killing, suggesting an important role for these epigenetic modifiers in anti-tumor immunity [80].
The finding of KMT2C and KMT2D in essentially identical COMPASS complexes that catalyze mono-methylation of H3K4 at gene enhancers has raised the possibility that their functions were largely overlapping, such that loss of both might be important for oncogenesis [12, 81]. Support for this theory can be seen through meta analysis of >58,000 samples from the cBio Portal dataset that demonstrates significant tendency towards co-occurrence of KMT2C and KMT2D mutations with a log odds ratio of 1.65 (p-value <0.001), perhaps indicating that loss of both proteins’ tumor suppressor functions are important to confer malignant characteristics. However, the preponderance of mutations affecting only one of the two genes in specific cancer types suggests that each may have a critical role in cell-type or tissue-specific cancer development. The high frequency of KMT2C and KMT2D mutations, together with significant co-occurrence (p < 0.001) with various driver mutations (e.g., TP53, PIK3CA, PTEN, APC) and the SWI/SNF complex component ARID1A (log odds ratio 1.73, p = <0.001) across multiple cancer types suggests that disruption of either KMT2C or KMT2D individually might serve as founder or gatekeeper mutations in early tumor cells, resulting in changes in the epigenomic landscape that are permissive for additional oncogenic changes.
Although mouse knockout studies reveal that Kmt2c can partially compensate for the absence of Kmt2d [81], both are essential genes which implies that each may have distinct functions in gene regulation. The mechanism(s) that distinguish KMT2C and KMT2D functions are not well understood as they share nearly identical domains and organization and presumably form identical MLR complexes [82]. One possible distinguishing difference is that KMT2D contains fewer zinc fingers within the first PHD domain cluster, possibly allowing for functional differences. Analysis of cancer-associated mutations reveals that there is a large clustering of lung and breast cancer missense mutations in the first KMT2C domain that are not found similarly in KMT2D [13]. This domain in KMT2C is involved in direct contacts with the BAP1 histone deubiquitinating complex that is linked to Polycomb-dependent gene silencing, suggesting that KMT2C may play a more critical role in cancers associated with Polycomb silenced gene enhancers [13]. Since KMT2D is lacking one of the conserved PHD finger domains and does not interact with the BAP1 protein, this difference may reflect how each protein is capable of distinct protein interactions that may account for differences in target gene regulation. Another difference is that KMT2D can be inactivated through phosphorylation by SGK1, a PI3K effector closely related to AKT1, and attenuate estrogen receptor (ER) activation in breast cancer cells [83, 84]. Treatment of ER+ breast cancers with a PI3K inhibitor is effective clinically, but also results in increased ER-dependent transcription that can result in therapeutic resistance [85]. ER activates its targets through interaction with KMT2D at enhancers. One of those targets is the estrogen inducible kinase SGK1, whose expression is induced upon inhibition of PI3K. Subsequently, SGK1 can inactivate KMT2D through phosphorylation at S1331 near the second PHD finger cluster implicated in chromatin binding and result in downregulation of global H3K4me1 levels at ER-regulated loci, as part of a negative feedback loop [83]. Although the mechanism is unknown, targeted phosphorylation and inactivation of KMT2D by both SGK1 and/or AKT1 can lead to downregulation of enhancers controlled by KMT2D-associated transcription factors. Notably, KMT2C does not contain the AGC kinase consensus sequence (RXRXXS/T) in a similar position relative to the PHD finger cluster. The distinctions between the KMT2C and KMT2D proteins are unlikely to be limited to these differences, but probably include varied protein interactions and differential chromatin targeting.
6. Germline roles of KMT2-family genes in cancer predisposition
The KMT2A-D genes are each essential for organismal viability, with strong depletion, homozygous loss of function mutations or deletions resulting in embryonic or perinatal lethality (KMT2A/MLL1: [86] [87]; KMT2B/MLL4(2): [88]; KMT2C/MLL3 and KMT2D/MLL2(4): [81, 89]. Somatic mutations in the KMT2A-D genes have been strongly linked to cancer development; however, it is less clear the impacts of de novo germline mutations in cancer predisposition. The KMT2A-D genes have each been associated with developmental disorders, with common phenotypic features that include intellectual disability and often skeletal abnormalities. Germline heterozygous mutations in KMT2A are associated with WeidemannSteiner syndrome [90, 91] and rarely, pediatric eosinophilia [92]. Inactivating mutations of KMT2B that include microdeletions and pathogenic variants have been linked to pediatric dystonia, a hyperkinetic movement disorder [41, 93, 94]. Heterozygous germline loss of KMT2C is associated with autism spectrum disorder and Kleefstra syndrome, a developmental disorder associated with skeletal and intellectual defects [95, 96]. Heterozygous mutations in human KMT2D are also frequently associated with developmental anomalies and malignancy. Inactivating germline mutations in KMT2D are strongly correlated (55–80%) with Kabuki Syndrome, a developmental disorder characterized by distinct facial features, mild to severe developmental delay and intellectual disability, skeletal defects and cardiac abnormalities [97].
Cancer predisposition is not a significant feature of KMT2A/B/C germline mutations. Although translocation fusions of KMT2A with a variety of partner proteins are directly linked to pediatric leukemias [33, 35], there is no clear link between germline loss of KMT2A function and oncogenesis. In contrast, heterozygous germline inactivating mutations in KMT2D have been linked to several cancers. The de novo mutations are inherited in an autosomal dominant pattern and Kabuki syndrome patients have a modest predisposition to cancer, including lymphoma, Wilms tumor (kidney), hepatoblastoma (liver), synovial sarcoma (lung) and neuroblastoma [98]. Brain-specific knockout of Kmt2d in mice is associated with spontaneous medulloblastoma, perhaps through hyperactivation of the Ras and Notch pathways and down-regulation of tumor suppressor genes [99]. In light of the significant role of somatic loss of KMT2C/D in multiple cancer types, the relative lack of pediatric cancers associated with KMT2A/B/C suggest that heterozygous inactivation of these genes is insufficient to drive epigenetic changes associated with tumor formation and likely require additional cooperating mutations for cancer development. However, reduced KMT2D germline function is associated with several pediatric cancers, perhaps allowing for epigenetic changes that in combination with other driver mutations, leads to aberrant growth [100, 101] through the misregulation of critical signaling pathways controlled by KMT2D.
During embryonic development, KMT2D and KMT2C have important roles in enhancer priming and de novo enhancer activation; thus, loss of these activities in early animal development may lead to an inability to appropriately activate critical developmental signaling pathways important for controlling growth and differentiation [79]. These functions appear to be dispensable for maintaining cell identity and self-renewal in both murine embryonic stem cells (ESCs) and somatic cells, but essential for reprogramming ESCs during differentiation and for generating induced pluripotent stem cells (iPSCs) [12]. Murine ESCs expressing KMT2D were able to form cystic embryoid bodies consisting of all three germ layers, while those with KMT2D deleted only formed poorly differentiated primitive stem cells. Somatic cells manipulated to induce the formation of pluripotent stem cells (iPSC) were able to form embryonic stem cell like colonies when KMT2D was present; whereas, cells with KMT2D deleted showed a dramatic decrease in reprogramming efficiency and showed a decreased expression of pluripotency markers. These results reveal a critical role of KMT2C/D COMPASS complexes in establishing pluripotent cell identity during somatic reprogramming, thus suggesting a role for loss of KMT2C/D function in aberrant growth and differentiation.
7. Conclusions and perspectives
Cancer exome databases reveal that the KMT2C/MLL3 and KMT2D/MLL2(4) histone lysine methyltransferases are among the most frequently mutated genes across a variety of cancer types. Most of these cancers are associated with mutations that alter the proteins through missense changes or truncations resulting in reduced functions, supporting their roles as tumor suppressors. Genetic removal of these genes in cancer cells, embryonic stem cells, as well as both vertebrate and invertebrate animal models has confirmed their requirement as transcription enhancer regulators.
While it is difficult to therapeutically target loss-of-function tumor suppressors directly, recent work has shown there is promise for targeting repressor enzymes, such as EZH2 that places a trimethyl mark on histone 3 lysine 27 (H3K27) which serves to block some enhancer activities [13]. Inhibitors that block the activities of EZH2, such as GSK126, result in the restoration of gene expression in KMT2C mutant cells. A recently discovered role for KMT2C in homology-driven DNA repair and genomic instability in bladder cancer cells has opened the possibility that some KMT2C-associated cancers may be targeted by PARP½ inhibitors [102]. Another potential target of inhibition is the COMPASS-like complex subunit WDR5, found in all COMPASS complexes. A report by Senisterra et al [103] identified a compound that would inhibit WDR5 interaction with MLL1/KMT2A and histone H3, reducing histone methyltransferase activity and H3 binding in vitro. Whether this compound would disrupt WDR5 interactions with KMT2C/D remains to be determined. The loss of kmt2d in a zebrafish model of Kabuki syndrome results in hyperactivation of MEK within the RAS/MAPK pathway and treatment with the BRAF inhibitor desmethyl dabrafenib (dmDf) can rescue the Kabuki-like phenotypes [104]. Dabrafenib has shown clinical efficacy in treating tumors (especially melanoma) with BRAF hyperactivation mutations; thus, tumors with KMT2D mutations may be candidates for treatment with MAPK pathway inhibitors.
Highlights.
KMT2C/D methyltransferases are frequently mutated in cancer
KMT2C/D mutations frequently co-occur in lung cancer
Somatic loss of KMT2C/D function as drivers of oncogenesis
KMT2C/D function to maintain epithelial states
MLR COMPASS complexes control gene transcriptional enhancer functions
Acknowledgements
We thank Claudia Zraly, David Ford and Matthew Kroll for helpful advice and comments on the manuscript. R. F. was supported by a NIH T35 award from the NHLBI (HL120835). The research in the laboratory of A.D. is supported by the National Science Foundation (MCB1716431).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement:
The authors declare that they have no conflict of interest.
References
- [1].Rothbart SB, Strahl BD, Interpreting the language of histone and DNA modifications, Biochim Biophys Acta, 1839 (2014) 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Greer EL, Shi Y, Histone methylation: a dynamic mark in health, disease and inheritance, Nat Rev Genet, 13 (2012) 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Calo E, Wysocka J, Modification of enhancer chromatin: what, how, and why?, Mol Cell, 49 (2013) 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shilatifard A, The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis, Annu Rev Biochem, 81 (2012) 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rao RC, Dou Y, Hijacked in cancer: the KMT2 (MLL) family of methyltransferases, Nat Rev Cancer, 15 (2015) 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Slany RK, When epigenetics kills: MLL fusion proteins in leukemia, Hematol Oncol, 23 (2005) 1–9. [DOI] [PubMed] [Google Scholar]
- [7].Slany RK, The molecular mechanics of mixed lineage leukemia, Oncogene, 35 (2016) 5215–5223. [DOI] [PubMed] [Google Scholar]
- [8].Ford DJ, Dingwall AK, The cancer COMPASS: navigating the functions of MLL complexes in cancer, Cancer Genet, 208 (2015) 178–191. [DOI] [PubMed] [Google Scholar]
- [9].Mohan M, Herz HM, Smith ER, Zhang Y, Jackson J, Washburn MP, Florens L, Eissenberg JC, Shilatifard A, The COMPASS family of H3K4 methylases in Drosophila, Mol Cell Biol, 31 (2011) 4310–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A, The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers, Mol Cell Biol, 33 (2013) 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Piunti A, Shilatifard A, Epigenetic balance of gene expression by Polycomb and COMPASS families, Science, 352 (2016) aad9780. [DOI] [PubMed] [Google Scholar]
- [12].Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, Ge K, Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition, Proc Natl Acad Sci U S A, 113 (2016) 11871–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, Cozzolino KA, Louis N, He X, Morgan MA, Takahashi YH, Collings CK, Smith ER, Ntziachristos P, Savas JN, Zou L, Hashizume R, Meeks JJ, Shilatifard A, Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy, Nat Med, 24 (2018) 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lai B, Lee JE, Jang Y, Wang L, Peng W, Ge K, MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis, Nucleic Acids Res, 45 (2017) 6388–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH, Nakadai T, Chramiec A, Krivtsov AV, Armstrong SA, Roeder RG, A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription, Mol Cell, 67 (2017) 308–321 e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tie F, Banerjee R, Conrad PA, Scacheri PC, Harte PJ, Histone demethylase UTX and chromatin remodeler BRM bind directly to CBP and modulate acetylation of histone H3 lysine 27, Mol Cell Biol, 32 (2012) 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Baas R, van Teeffelen H, Tjalsma SJD, Timmers HTM, The mixed lineage leukemia 4 (MLL4) methyltransferase complex is involved in transforming growth factor beta(TGF-beta)-activated gene transcription, Transcription, 9 (2018) 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, Lee JE, Chen HT, Ennishi D, Scott DW, Mottok A, Hother C, Liu S, Cao XJ, Tam W, Shaknovich R, Garcia BA, Gascoyne RD, Ge K, Shilatifard A, Elemento O, Nussenzweig A, Melnick AM, Wendel HG, The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development, Nat Med, 21 (2015) 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rhee JK, Yoo J, Kim KR, Kim J, Lee YJ, Cho BC, Kim TM, Identification of local clusters of mutation hotspots in cancer-related genes and their biological relevance, IEEE/ACM Trans Comput Biol Bioinform, (2018). [DOI] [PubMed]
- [20].Qian C, Zhou MM, SET domain protein lysine methyltransferases: Structure, specificity and catalysis, Cell Mol Life Sci, 63 (2006) 2755–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD 2nd, Garcia BA, Adelman K, Wysocka J, Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation, Mol Cell, 66 (2017) 568–576 e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rickels R, Herz HM, Sze CC, Cao K, Morgan MA, Collings CK, Gause M, Takahashi YH, Wang L, Rendleman EJ, Marshall SA, Krueger A, Bartom ET, Piunti A, Smith ER, Abshiru NA, Kelleher NL, Dorsett D, Shilatifard A, Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability, Nat Genet, 49 (2017) 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Weirich S, Kudithipudi S, Jeltsch A, Somatic cancer mutations in the MLL1 histone methyltransferase modulate its enzymatic activity and dependence on the WDR5/RBBP5/ASH2L complex, Mol Oncol, 11 (2017) 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Weirich S, Kudithipudi S, Kycia I, Jeltsch A, Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme, Clin Epigenetics, 7 (2015) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zeleznik-Le NJ, Harden AM, Rowley JD, 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene, Proc Natl Acad Sci U S A, 91 (1994) 10610–10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Slany RK, Lavau C, Cleary ML, The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX., Molecular and Cellular Biology, 18 (1998) 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bach C, Mueller D, Buhl S, Garcia-Cuellar MP, Slany RK, Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2, Oncogene, 28 (2009) 815–823. [DOI] [PubMed] [Google Scholar]
- [28].Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ, MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein, Proc Natl Acad Sci U S A, 100 (2003) 8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sedkov Y, Cho E, Petruk S, Cherbas L, Smith ST, Jones RS, Cherbas P, Canaani E, Jaynes JB, Mazo A, Methylation at lysine 4 of histone H3 in ecdysone-dependent development of Drosophila, Nature, 426 (2003) 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Froimchuk E, Jang Y, Ge K, Histone H3 lysine 4 methyltransferase KMT2D, Gene, 627 (2017) 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fantini D, Glaser AP, Rimar KJ, Wang Y, Schipma M, Varghese N, Rademaker A, Behdad A, Yellapa A, Yu Y, Sze CC, Wang L, Zhao Z, Crawford SE, Hu D, Licht JD, Collings CK, Bartom E, Theodorescu D, Shilatifard A, Meeks JJ, A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer, Oncogene, 37 (2018) 1911–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Herz HM, Hu D, Shilatifard A, Enhancer malfunction in cancer, Mol Cell, 53 (2014) 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang W, Ernst P, Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis, Curr Opin Hematol, 24 (2017) 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krivtsov AV, Armstrong SA, MLL translocations, histone modifications and leukaemia stem-cell development, Nat Rev Cancer, 7 (2007) 823–833. [DOI] [PubMed] [Google Scholar]
- [35].Krivtsov AV, Hoshii T, Armstrong SA, Mixed-Lineage Leukemia Fusions and Chromatin in Leukemia, Cold Spring Harb Perspect Med, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, Ge K, Dalla-Favera R, Pasqualucci L, Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis, Nat Med, 21 (2015) 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, Sutton R, Venn NC, Emerenciano M, Pombo-de-Oliveira MS, Barbieri Blunck C, Almeida Lopes B, Zuna J, Trka J, Ballerini P, Lapillonne H, De Braekeleer M, Cazzaniga G, Corral Abascal L, van der Velden VHJ, Delabesse E, Park TS, Oh SH, Silva MLM, Lund-Aho T, Juvonen V, Moore AS, Heidenreich O, Vormoor J, Zerkalenkova E, Olshanskaya Y, Bueno C, Menendez P, Teigler-Schlegel A, Zur Stadt U, Lentes J, Gohring G, Kustanovich A, Aleinikova O, Schafer BW, Kubetzko S, Madsen HO, Gruhn B, Duarte X, Gameiro P, Lippert E, Bidet A, Cayuela JM, Clappier E, Alonso CN, Zwaan CM, van den Heuvel-Eibrink MM, Izraeli S, Trakhtenbrot L, Archer P, Hancock J, Moricke A, Alten J, Schrappe M, Stanulla M, Strehl S, Attarbaschi A, Dworzak M, Haas OA, Panzer-Grumayer R, Sedek L, Szczepanski T, Caye A, Suarez L, Cave H, Marschalek R, The MLL recombinome of acute leukemias in 2017, Leukemia, 32 (2018) 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Winters AC, Bernt KM, MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches, Front Pediatr, 5 (2017) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Natarajan TG, Kallakury BV, Sheehan CE, Bartlett MB, Ganesan N, Preet A, Ross JS, Fitzgerald KT, Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon, Cancer Cell Int, 10 (2010) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mas G, Blanco E, Ballare C, Sanso M, Spill YG, Hu D, Aoi Y, Le Dily F, Shilatifard A, Marti-Renom MA, Di Croce L, Promoter bivalency favors an open chromatin architecture in embryonic stem cells, Nat Genet, 50 (2018) 1452–1462. [DOI] [PubMed] [Google Scholar]
- [41].Meyer E, Carss KJ, Rankin J, Nichols JM, Grozeva D, Joseph AP, Mencacci NE, Papandreou A, Ng J, Barral S, Ngoh A, Ben-Pazi H, Willemsen MA, Arkadir D, Barnicoat A, Bergman H, Bhate S, Boys A, Darin N, Foulds N, Gutowski N, Hills A, Houlden H, Hurst JA, Israel Z, Kaminska M, Limousin P, Lumsden D, McKee S, Misra S, Mohammed SS, Nakou V, Nicolai J, Nilsson M, Pall H, Peall KJ, Peters GB, Prabhakar P, Reuter MS, Rump P, Segel R, Sinnema M, Smith M, Turnpenny P, White SM, Wieczorek D, Wiethoff S, Wilson BT, Winter G, Wragg C, Pope S, Heales SJ, Morrogh D, U.K. Consortium, S. Deciphering Developmental Disorders, N.B.R.D. Consortium, Pittman A, Carr LJ, Perez-Duenas B, Lin JP, Reis A, Gahl WA, Toro C, Bhatia KP, Wood NW, Kamsteeg EJ, Chong WK, Gissen P, Topf M, Dale RC, Chubb JR, Raymond FL, Kurian MA, Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia, Nat Genet, 49 (2017) 223–237. [DOI] [PubMed] [Google Scholar]
- [42].Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, The genetic landscape of the childhood cancer medulloblastoma, Science, 331 (2011) 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ, Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations, Nature, 488 (2012) 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA, Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma, Nature, 476 (2011) 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, Dalla-Favera R, Analysis of the coding genome of diffuse large B-cell lymphoma, Nat Genet, 43 (2011) 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data, Cancer Discov, 2 (2012) 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci Signal, 6 (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, Stock W, LeBeau MM, Shannon KM, Kogan S, Zuber J, Lowe SW, MLL3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia, Cancer Cell, 25 (2014) 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gala K, Li Q, Sinha A, Razavi P, Dorso M, Sanchez-Vega F, Chung YR, Hendrickson R, Hsieh JJ, Berger M, Schultz N, Pastore A, Abdel-Wahab O, Chandarlapaty S, KMT2C mediates the estrogen dependence of breast cancer through regulation of ERalpha enhancer function, Oncogene, 37 (2018) 4692–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang Z, Christin JR, Wang C, Ge K, Oktay MH, Guo W, Mammary-Stem-Cell-Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation, Cell Rep, 16 (2016) 3146–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, Pasqualucci L, Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma, Nature, 459 (2009) 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Molavi O, Wang P, Zak Z, Gelebart P, Belch A, Lai R, Gene methylation and silencing of SOCS3 in mantle cell lymphoma, Br J Haematol, 161 (2013) 348–356. [DOI] [PubMed] [Google Scholar]
- [53].Cheung KJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, Schein J, Morin RD, Moore R, Shah SP, Qian H, Paul JE, Telenius A, Relander T, Lam W, Savage K, Connors JM, Brown C, Marra MA, Gascoyne RD, Horsman DE, Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis, Cancer Res, 70 (2010) 9166–9174. [DOI] [PubMed] [Google Scholar]
- [54].Guo C, Chen LH, Huang Y, Chang CC, Wang P, Pirozzi CJ, Qin X, Bao X, Greer PK, McLendon RE, Yan H, Keir ST, Bigner DD, He Y, KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation, Oncotarget, 4 (2013) 2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Augert A, Zhang Q, Bates B, Cui M, Wang X, Wildey G, Dowlati A, MacPherson D, Small Cell Lung Cancer Exhibits Frequent Inactivating Mutations in the Histone Methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance), J Thorac Oncol, 12 (2017) 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, Vaka D, Torres A, Wang MS, Korbel JO, Menon R, Chun SM, Kim D, Wilkerson M, Hayes N, Engelmann D, Putzer B, Bos M, Michels S, Vlasic I, Seidel D, Pinther B, Schaub P, Becker C, Altmuller J, Yokota J, Kohno T, Iwakawa R, Tsuta K, Noguchi M, Muley T, Hoffmann H, Schnabel PA, Petersen I, Chen Y, Soltermann A, Tischler V, Choi CM, Kim YH, Massion PP, Zou Y, Jovanovic D, Kontic M, Wright GM, Russell PA, Solomon B, Koch I, Lindner M, Muscarella LA, la Torre A, Field JK, Jakopovic M, Knezevic J, Castanos-Velez E, Roz L, Pastorino U, Brustugun OT, LundIversen M, Thunnissen E, Kohler J, Schuler M, Botling J, Sandelin M, Sanchez-Cespedes M, Salvesen HB, Achter V, Lang U, Bogus M, Schneider PM, Zander T, Ansen S, Hallek M, Wolf J, Vingron M, Yatabe Y, Travis WD, Nurnberg P, Reinhardt C, Perner S, Heukamp L, Buttner R, Haas SA, Brambilla E, Peifer M, Sage J, Thomas RK, Comprehensive genomic profiles of small cell lung cancer, Nature, 524 (2015) 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, Conley S, Zhong H, Liu Z, Brohawn P, Shen D, Wu S, Ge X, Jiang Y, Zhao Y, Lou Y, Morehouse C, Zhu W, Sebastian Y, Czapiga M, Oganesyan V, Fu H, Niu Y, Zhang W, Streicher K, Tice D, Zhao H, Zhu M, Xu L, Herbst R, Su X, Gu Y, Li S, Huang L, Gu J, Han B, Jallal B, Shen H, Yao Y, Genomic Landscape Survey Identifies SRSF1 as a Key Oncodriver in Small Cell Lung Cancer, PLoS Genet, 12 (2016) e1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Simbolo M, Mafficini A, Sikora KO, Fassan M, Barbi S, Corbo V, Mastracci L, Rusev B, Grillo F, Vicentini C, Ferrara R, Pilotto S, Davini F, Pelosi G, Lawlor RT, Chilosi M, Tortora G, Bria E, Fontanini G, Volante M, Scarpa A, Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D, J Pathol, 241 (2017) 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ross JS, Wang K, Elkadi OR, Tarasen A, Foulke L, Sheehan CE, Otto GA, Palmer G, Yelensky R, Lipson D, Chmielecki J, Ali SM, Elvin J, Morosini D, Miller VA, Stephens PJ, Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer, J Clin Pathol, 67 (2014) 772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dowlati A, Lipka MB, McColl K, Dabir S, Behtaj M, Kresak A, Miron A, Yang M, Sharma N, Fu P, Wildey G, Clinical correlation of extensive-stage small-cell lung cancer genomics, Ann Oncol, 27 (2016) 642–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Umemura S, Mimaki S, Makinoshima H, Tada S, Ishii G, Ohmatsu H, Niho S, Yoh K, Matsumoto S, Takahashi A, Morise M, Nakamura Y, Ochiai A, Nagai K, Iwakawa R, Kohno T, Yokota J, Ohe Y, Esumi H, Tsuchihara K, Goto K, Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis, J Thorac Oncol, 9 (2014) 1324–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, Jayakumaran G, Gao SP, Johnsen HC, Hanrahan AJ, Zehir A, Rekhtman N, Ginsberg MS, Li BT, Yu HA, Paik PK, Drilon A, Hellmann MD, Reales DN, Benayed R, Rusch VW, Kris MG, Chaft JE, Baselga J, Taylor BS, Schultz N, Rudin CM, Hyman DM, Berger MF, Solit DB, Ladanyi M, Riely GJ, Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies, Cancer Discov, 7 (2017) 596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK, Somatic mutations affect key pathways in lung adenocarcinoma, Nature, 455 (2008) 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer, Science, 348 (2015) 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, Imielinski M, Hu X, Ling S, Akbani R, Rosenberg M, Cibulskis C, Ramachandran A, Collisson EA, Kwiatkowski DJ, Lawrence MS, Weinstein JN, Verhaak RG, Wu CJ, Hammerman PS, Cherniack AD, Getz G, Cancer N Genome Atlas Research, Artyomov MN, Schreiber R, Govindan R, Meyerson M, Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas, Nat Genet, 48 (2016) 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Vavala T, Monica V, Lo Iacono M, Mele T, Busso S, Righi L, Papotti M, Scagliotti GV, Novello S, Precision medicine in age-specific non-small-cell-lung-cancer patients: Integrating biomolecular results into clinical practice-A new approach to improve personalized translational research, Lung Cancer, 107 (2017) 84–90. [DOI] [PubMed] [Google Scholar]
- [67].Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansen S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Janne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M, Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing, Cell, 150 (2012) 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yin S, Yang J, Lin B, Deng W, Zhang Y, Yi X, Shi Y, Tao Y, Cai J, Wu CI, Zhao G, Hurst LD, Zhang J, Hu L, Kong X, Exome sequencing identifies frequent mutation of MLL2 in non-small cell lung carcinoma from Chinese patients, Sci Rep, 4 (2014) 6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Choi M, Kadara H, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J, Chow C, Kim K, Kalhor N, Moran C, Rimm D, Swisher S, Gibbons DL, Heymach J, Kaftan E, Townsend JP, Lynch TJ, Schlessinger J, Lee J, Lifton RP, Herbst RS, Wistuba II, Mutation profiles in early-stage lung squamous cell carcinoma with clinical follow-up and correlation with markers of immune function, Ann Oncol, 28 (2017) 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Xiong W, Deng Z, Tang Y, Deng Z, Li M, Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer, Biochem Biophys Res Commun, 504 (2018) 129–136. [DOI] [PubMed] [Google Scholar]
- [71].Abudureheman A, Ainiwaer J, Hou Z, Niyaz M, Turghun A, Hasim A, Zhang H, Lu X, Sheyhidin I, High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma, J Cancer Res Clin Oncol, 144 (2018) 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lv S, Ji L, Chen B, Liu S, Lei C, Liu X, Qi X, Wang Y, Lai-Han Leung E, Wang H, Zhang L, Yu X, Liu Z, Wei Q, Lu L, Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4, Oncogene, 37 (2018) 1354–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cho SJ, Yoon C, Lee JH, Chang KK, Lin JX, Kim YH, Kook MC, Aksoy BA, Park DJ, Ashktorab H, Smoot DT, Schultz N, Yoon SS, KMT2C Mutations in Diffuse-Type Gastric Adenocarcinoma Promote Epithelial-to-Mesenchymal Transition, Clin Cancer Res, 24 (2018) 6556–6569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [74].Rahnamoun H, Hong J, Sun Z, Lee J, Lu H, Lauberth SM, Mutant p53 regulates enhancer-associated H3K4 monomethylation through interactions with the methyltransferase MLL4, J Biol Chem, 293 (2018) 13234–13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Roy S, Tomaszowski KH, Luzwick JW, Park S, Li J, Murphy M, Schlacher K, p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLtheta pathways, Elife, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kim DH, Kim J, Lee JW, Requirement for MLL3 in p53 regulation of hepatic expression of small heterodimer partner and bile acid homeostasis, Mol Endocrinol, 25 (2011) 2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, Lee JW, A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4, Proc Natl Acad Sci U S A, 106 (2009) 8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, Huang J, Hua X, Arrowsmith CH, Berger SL, Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth, Nature, 525 (2015) 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Guo C, Chang CC, Wortham M, Chen LH, Kernagis DN, Qin X, Cho YW, Chi JT, Grant GA, McLendon RE, Yan H, Ge K, Papadopoulos N, Bigner DD, He Y, Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways, Proc Natl Acad Sci U S A, 109 (2012) 17603–17608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].MacFawn I, Wilson H, Selth LA, Leighton I, Serebriiskii I, Bleackley RC, Elzamzamy O, Farris J, Pifer PM, Richer J, Frisch SM, Grainyhead-like-2 confers NK-sensitivity through interactions with epigenetic modifiers, Mol Immunol, 105 (2018) 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, Ge K, H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation, Elife, 2 (2013) e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mohan M, Herz HM, Shilatifard A, SnapShot: Histone lysine methylase complexes, Cell, 149 (2012) 498–498 e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Toska E, Castel P, Chhangawala S, Arruabarrena-Aristorena A, Chan C, Hristidis VC, Cocco E, Sallaku M, Xu G, Park J, Minuesa G, Shifman SG, Socci ND, Koche R, Leslie CS, Scaltriti M, Baselga J, PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression, Cell Rep, 27 (2019) 294–306 e295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, Baselga J, PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D, Science, 355 (2017) 1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, Minuesa G, Morse N, Rodon J, Ibrahim Y, Cortes J, Perez-Garcia J, Galvan P, Grueso J, Guzman M, Katzenellenbogen JA, Kharas M, Lewis JS, Dickler M, Serra V, Rosen N, Chandarlapaty S, Scaltriti M, Baselga J, PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer, Sci Transl Med, 7 (2015) 283ra251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ, Altered Hox expression and segmental identity in Mll-mutant mice, Nature, 378 (1995) 505–508. [DOI] [PubMed] [Google Scholar]
- [87].Ayton P, Sneddon SF, Palmer DB, Rosewell IR, Owen MJ, Young B, Presley R, Subramanian V, Truncation of the MLL gene in exon 5 by gene targeting leads to early preimplantation lethality of homozygous embryos, 30 (2001) 201–212. [DOI] [PubMed] [Google Scholar]
- [88].Glaser S, Schaft J, Lubitz S, Vintersten K, van der Hoeven F, Tufteland KR, Aasland R, Anastassiadis K, Ang SL, Stewart AF, Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development, Development, 133 (2006) 1423–1432. [DOI] [PubMed] [Google Scholar]
- [89].Aoshima K, Inoue E, Sawa H, Okada Y, Paternal H3K4 methylation is required for minor zygotic gene activation and early mouse embryonic development, EMBO Rep, 16 (2015) 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Strom SP, Lozano R, Lee H, Dorrani N, Mann J, O’Lague PF, Mans N, Deignan JL, Vilain E, Nelson SF, Grody WW, Quintero-Rivera F, De Novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing, BMC Med Genet, 15 (2014) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, Irving M, Saggar AK, Smithson S, Trembath RC, Deshpande C, Simpson MA, De novo mutations in MLL cause Wiedemann-Steiner syndrome, Am J Hum Genet, 91 (2012) 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhang H, Xiang B, Chen H, Chen X, Cai T, A novel deletion mutation in KMT2A identified in a child with ID/DD and blood eosinophilia, BMC Med Genet, 20 (2019) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zech M, Boesch S, Maier EM, Borggraefe I, Vill K, Laccone F, Pilshofer V, Ceballos-Baumann A, Alhaddad B, Berutti R, Poewe W, Haack TB, Haslinger B, Strom TM, Winkelmann J, Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia, Am J Hum Genet, 99 (2016) 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gorman KM, Meyer E, Kurian MA, Review of the phenotype of early-onset generalised progressive dystonia due to mutations in KMT2B, Eur J Paediatr Neurol, 22 (2018) 245–256. [DOI] [PubMed] [Google Scholar]
- [95].Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LE, Wissink-Lindhout W, Fenckova M, van den Akker WM, Kasri NN, Nillesen WM, Prescott T, Clark RD, Devriendt K, van Reeuwijk J, de Brouwer AP, Gilissen C, Zhou H, Brunner HG, Veltman JA, Schenck A, van Bokhoven H, Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability, American journal of human genetics, 91 (2012) 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Koemans TS, Kleefstra T, Chubak MC, Stone MH, Reijnders MRF, de Munnik S, Willemsen MH, Fenckova M, Stumpel C, Bok LA, Sifuentes Saenz M, Byerly KA, Baughn LB, Stegmann APA, Pfundt R, Zhou H, van Bokhoven H, Schenck A, Kramer JM, Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder, PLoS Genet, 13 (2017) e1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lintas C, Persico AM, Unraveling molecular pathways shared by Kabuki and Kabuki-like syndromes, Clin Genet, 94 (2018) 283–295. [DOI] [PubMed] [Google Scholar]
- [98].Teranishi H, Koga Y, Nakashima K, Morihana E, Ishii K, Sakai Y, Taguchi T, Oda Y, Miyake N, Matsumoto N, Ohga S, Cancer Management in Kabuki Syndrome: The First Case of Wilms Tumor and a Literature Review, J Pediatr Hematol Oncol, 40 (2018) 391–394. [DOI] [PubMed] [Google Scholar]
- [99].Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, Flores ER, Northcott PA, Rajaram V, Li W, Shilatifard A, Sillitoe RV, Chen K, Lee MG, MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes, Mol Cell, 70 (2018) 825–841 e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Feinberg AP, Ohlsson R, Henikoff S, The epigenetic progenitor origin of human cancer, Nat Rev Genet, 7 (2006) 21–33. [DOI] [PubMed] [Google Scholar]
- [101].Feinberg AP, Koldobskiy MA, Gondor A, Epigenetic modulators, modifiers and mediators in cancer aetiology and progression, Nat Rev Genet, 17 (2016) 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Rampias T, Karagiannis D, Avgeris M, Polyzos A, Kokkalis A, Kanaki Z, Kousidou E, Tzetis M, Kanavakis E, Stravodimos K, Manola KN, Pantelias GE, Scorilas A, Klinakis A, The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer, EMBO Rep, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, Dong A, Nguyen KT, Smil D, Bolshan Y, Hajian T, He H, Seitova A, Chau I, Li F, Poda G, Couture JF, Brown PJ, Al-Awar R, Schapira M, Arrowsmith CH, Vedadi M, Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5, Biochem J, 449 (2013) 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tsai IC, McKnight K, McKinstry SU, Maynard AT, Tan PL, Golzio C, White CT, Price DJ, Davis EE, Amrine-Madsen H, Katsanis N, Small molecule inhibition of RAS/MAPK signaling ameliorates developmental pathologies of Kabuki Syndrome, Sci Rep, 8 (2018) 10779. [DOI] [PMC free article] [PubMed] [Google Scholar]