

Summary
Haustoria of biotrophic rust fungi are responsible for the uptake of nutrients from their hosts and for the production of secreted proteins, known as effectors, which modulate the host immune system. The identification of the transcriptome of haustoria and an understanding of the functions of expressed genes therefore hold essential keys for the elucidation of fungus–plant interactions and the development of novel fungal control strategies. Here, we purified haustoria from infected leaves and used 454 sequencing to examine the haustorial transcriptomes of Phakopsora pachyrhizi and Uromyces appendiculatus, the causal agents of soybean rust and common bean rust, respectively. These pathogens cause extensive yield losses in their respective legume crop hosts. A series of analyses were used to annotate expressed sequences, including transposable elements and viruses, to predict secreted proteins from the assembled sequences and to identify families of candidate effectors. This work provides a foundation for the comparative analysis of haustorial gene expression with further insights into physiology and effector evolution.
Introduction
Rust fungi are obligate biotrophic pathogens dependent on living host tissue for propagation (Voegele et al., 2009). The haustorium is a hallmark structure of rusts and other biotrophic fungi through which they interact with host cells (Voegele and Mendgen, 2003). Haustoria are conduits for nutrient acquisition, metabolic and catabolic processes, and the synthesis and delivery of proteins, known as effectors, which modulate the host immune system (Catanzariti et al., 2006; Dodds et al., 2009; Voegele et al., 2009; Voegele and Mendgen, 2011). Therefore, effectors play key roles in establishing and maintaining biotrophic interactions (Oliva et al., 2010). The identification of the sets of genes that are expressed in haustoria will thus provide critical information needed to understand the fungal and host processes that underlie the outcomes of their interactions.
Genome and transcriptome sequencing, accompanied by bioinformatic analyses, provides opportunities to identify the genes expressed in the haustoria of rust fungus pathogens (De Wit et al., 2009; Duplessis et al., 2012). These approaches are facilitated by the ability to obtain RNA directly from haustoria. The isolation of intact haustoria from infected tissue can generate sufficient RNA for cDNA library construction, sequencing and expression profiling (Hahn and Mendgen, 1997; Link and Voegele, 2008). These materials, coupled with next‐generation sequencing techniques, enable the assembly of haustorial transcriptomes with high coverage (Garnica et al., 2013; Weßling et al., 2012).
We are interested in the interactions between rust fungi and legume crop plants. The rust pathogens Phakopsora pachyrhizi and Uromyces appendiculatus are globally distributed and cause the diseases soybean rust and common bean rust, respectively (Araya et al., 2004; Goellner et al., 2010; Li et al., 2010; Liebenberg and Pretorius, 2010; Schneider et al., 2005). Uromyces appendiculatus has also served as an important model organism for cytological and physiological studies of rust fungi (Zhou et al., 1991). Despite the importance of these pathogens, their genome sequences are not yet available. There are, however, collections of sequences expressed at various life cycle stages, including a small number of haustorial sequences (Posada‐Buitrago and Frederick, 2005; Puthoff et al., 2008; Tremblay et al., 2009, 2012). This general lack of sequence information hinders our ability to understand the physiology and interactions of these rust fungi with their crop hosts. Two examples of the power of such information come from the recent sequencing of the Puccinia graminis f. sp. tritici and Melampsora larici‐populina genomes, combined with expression profiling data, which have provided an excellent resource for the identification of candidate effectors from rust fungi (Duplessis et al., 2011).
As a starting point to gain deeper insight into the physiology of haustoria and the effectors deployed in legume–rust interactions, we sequenced the haustorial transcriptomes of U. appendiculatus and P. pachyrhizi. Bioinformatic approaches were used to annotate the resulting sequences, to identify gene families and to predict the secreted proteins. We show that the gene functions associated with the core physiology of these organisms overlap, but that there is less conservation in the suites of candidate effector proteins produced by each. Interestingly, sequences related to transposable elements (TEs) and a nonoverlapping set of viruses are abundant in the haustoria of both fungi.
Results
Haustorial transcriptome assemblies and gene ontology (GO) annotation
Total RNA was extracted from haustoria prepared from infected leaves collected at 10 days post‐inoculation (dpi) (U. appendiculatus) and 12 dpi (P. pachyrhizi) (Fig. 1). Copy DNA was synthesized from these two RNA pools, as well as from polyA+ mRNA prepared from U. appendiculatus total RNA. Sequencing of P. pachyrhizi cDNA produced 1 051 753 reads, and 11 872 contigs longer than 100 bp were assembled (Fig. S1A, see Supporting Information). The average size of the 2668 contigs longer than 500 bp was 832 bp, with the longest spanning 4079 bp. A total of 4483 unique P. pachyrhizi contigs were identified following application of a series of bioinformatic filters (Fig. 2, File S1, see Supporting Information). For U. appendiculatus, the 894 873 reads produced from total RNA and polyA+ mRNA were combined and assembled into 14 581 contigs longer than 100 bp (Fig. S1B). The average size of the 6818 contigs longer than 500 bp was 1114 bp, with the longest spanning 9639 bp. A total of 7582 unique U. appendiculatus contigs remained after application of a series of bioinformatic filters (Fig. 2, File S2, see Supporting Information).
Figure 1.
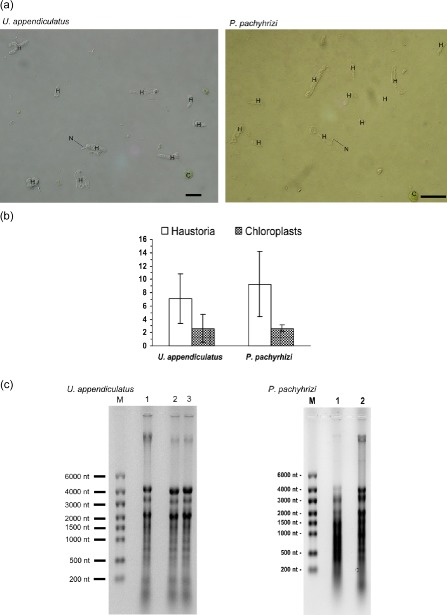
Purification of haustoria and haustorial RNA. (a) Micrographs of haustoria preparations. Haustorial suspensions after elution from the concanavalin A (Con‐A) column were examined microscopically. Haustoria (H) and chloroplasts (C) are indicated, as well as two representative haustorial necks (N). Bar, 10 μm. (b) Counts of haustoria and chloroplasts in preparations. The graphs show means of 23 haustoria preparations each. (c) Agarose gel electrophoresis of total RNA prepared from haustoria. Visible bands represent rRNA from haustoria and chloroplasts, and degraded RNAs can also be seen. nt, nucleotide.
Figure 2.
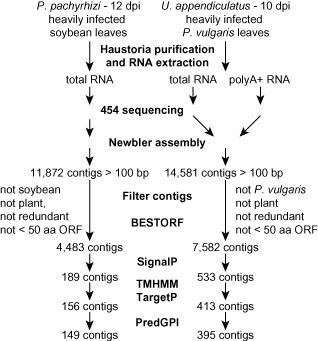
Work flow for the identification of putative secreted proteins expressed in haustoria of Phakopsora pachyrhizi and Uromyces appendiculatus. The BESTORF program was used to predict the open reading frames present in high‐confidence fungal contigs. Amino acid sequences of proteins predicted by BESTORF were used in the subsequent analyses with SignalP to predict secreted proteins. The prediction of proteins most likely to be secreted outside of fungal cells was further refined using the TMHMM, TargetP and PredGPI programs.
The P. pachyrhizi and U. appendiculatus contigs were compared with the GenBank nonredundant (nr) protein database (22 December 2010) using blastx (Altschul et al., 1997), and alignments that met the significance threshold of E ≤ 10−3 were obtained for 2585 and 6229 contigs, respectively. This output was used with the blast2go suite (Conesa et al., 2005; Götz et al., 2008) to generate annotation for 1568 (35%) P. pachyrhizi contigs (Fig. 3a, Table S1, see Supporting Information) and 3650 (48%) U. appendiculatus contigs (Fig. 3c, Table S2, see Supporting Information). Comparison of GO categories did not reveal significant differences between the categories of genes expressed in the haustoria of the two fungi.
Figure 3.
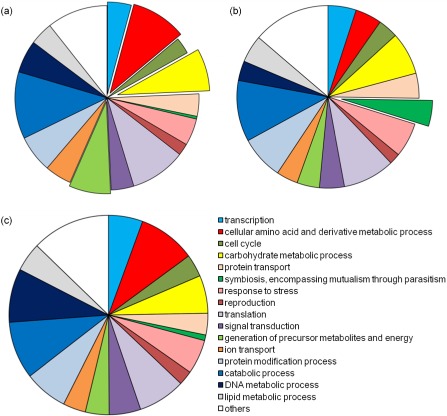
Annotation of the Phakopsora pachyrhizi and Uromyces appendiculatus transcriptomes: (a) P. pachyrhizi haustoria; (b) P. pachyrhizi urediospores and germ tubes; (c) U. appendiculatus haustoria. For the two P. pachyrhizi stages, the over‐represented gene ontology (GO) categories are emphasized by offset slices.
Comparison of the P. pachyrhizi haustorial transcriptome with expressed sequence tags (ESTs) from resting urediospores and germ tubes
We next compared these haustorial sequences with sequences from resting or water‐germinated urediospores to identify GO categories of transcripts that are putatively over‐represented or specific to haustoria. This was possible because of the 34 329 P. pachyrhizi ESTs available from GenBank (Posada‐Buitrago and Frederick, 2005). A similar dataset including ESTs from urediospores and germ tubes was not available for U. appendiculatus. These ESTs were assembled into 3365 contigs and 3383 singletons, which were labelled PpGI (P. pachyrhizi Gene Index). Of these, 2881 contigs were unique to water‐germinated spores, 289 were unique to resting spores and 213 were unique to infected leaves. We removed 138 sequences of plant or bacterial origin and, because our primary interest was a comparison with sequences from resting or water‐germinated urediospores, we removed 426 PpGI contigs and singletons unique to infected leaves, which probably included haustorial expressed sequences. Of the resulting 4955 unique PpGI sequences, 1863 could be assigned GO annotation as described above (Fig. 3b, Table S3, see Supporting Information).
To gain an insight into the physiology of the biotrophic interaction, we compared the GO categories of the P. pachyrhizi haustorial transcriptome with the PpGI ESTs from resting urediospores and water‐germinated spores (Table 1). All strongly over‐represented GO categories for resting urediospores or water‐germinated spores are mainly a result of one contig (PpGI_contig3375) which comprises 5073 reads. Three contigs with high similarity to PpGI_contig3375 were also found in the haustorial sequences (Pp_contig03605, Pp_contig06191 and Pp_contig05246), but all three combined only accounted for 25 reads (0.04%). In the opposite direction, the higher ratio of reads from haustoria compared with reads from resting or water‐germinated urediospores for the GO categories ‘generation of precursor metabolites and energy’, ‘cellular amino acid and derivative metabolic process’, ‘carbohydrate metabolic process’ and ‘transcription’ is consistent with the role of haustoria in the uptake of sugars and amino acids and more active gene expression (Wirsel et al., 2001).
Table 1.
Gene ontology (GO) categories with the most pronounced differing expression between pre‐haustorial cell types and haustoria
| GO level | GO category | Description | Pre‐haustorial reads* | Haustorial reads | Ratio h/pa | q b, c |
|---|---|---|---|---|---|---|
| 1 | GO:0008150 | Biological processd | 11120 | 60369 | 1 | 1 |
| 2 | GO:0032502 | Developmental process | 5471 | 2318 | 0.0780 | 0 |
| 3 | GO:0044419 | Interspecies interaction between organisms | 5300 | 65 | 0.0023 | 0 |
| 4 | GO:0006091 | Generation of precursor metabolites and energy | 255 | 9743 | 7.0379 | 0 |
| 4 | GO:0006519 | Cellular amino acid and derivative metabolic process | 276 | 4702 | 3.1381 | 0 |
| 4 | GO:0005975 | Carbohydrate metabolic process | 643 | 1204 | 4.0690 | 0 |
| 4 | GO:0044403 | Symbiosis, encompassing mutualism through parasitism | 5300 | 65 | 0.0023 | 0 |
| 6 | GO:0006350 | Transcription | 423 | 9869 | 4.2976 | 0 |
*Reads from resting urediospores and water‐germinated spores.
Ratio of haustorial (h) to pre‐haustorial (p) reads normalized by the ‘biological process’ totals.
q value near 0 (<10−500), ratio >4; apparent redundancies and artificially increased categories were removed.
Probability values from Fisher's test with correction for multiple testing.
Biological process was added to the table as reference to the totals.
Prediction of haustorial secreted proteins
Our bioinformatics pipeline identified 156 P. pachyrhizi contigs containing open reading frames (ORFs) encoding putative secretion signals and unlikely to be targeted to a membrane or mitochondria (Fig. 2, File S3, Table S5, see Supporting Information). Seven of these proteins also contained a glycosylphosphatidylinositol (GPI) anchor predicted by PredGPI. These proteins represent 3.5% of the haustorial transcriptome, and 40 (26%) of these had GO annotations. These putative secreted proteins include homologues to haustoria‐expressed secreted proteins (HESP379, HESP767, HESP‐C49; Catanzariti et al., 2006) from flax rust, and to rust transferred proteins (RTPs) in Uromyces species (Pretsch et al., 2013).
For U. appendiculatus, 413 contigs encode predicted secreted proteins (File S4, Table S5, see Supporting Information), 18 of which also contain a predicted GPI anchor. These predicted secreted proteins represent 5.4% of the transcriptome, and 117 (28%) had GO annotations among which the terms ‘carbohydrate metabolic process’ (including plant cell wall‐degrading enzymes and fungal cell wall‐modifying enzymes) and ‘response to biotic stimulus’ were significantly enriched. The secreted proteins include expansin homologues, one protein with a small secreted protein (SSP) annotation and three RTPs. Two have been identified previously, Ua‐RTP1p and Ua‐RTP2p (Puthoff et al., 2008), but the third is more distantly related and can be classified as RTP9p‐like (Pretsch et al., 2013). The 15 reads for Ua_contig00666 suggest that Ua‐RTP9 is weakly expressed relative to Ua‐RTP1 (Ua_contig06414, 355 reads) and Ua‐RTP2 (Ua_contig06302, 296 reads).
Families of putative secreted proteins
We performed single linkage clustering (Graham et al., 2004) to identify 134 families that contained at least two putative secreted proteins (Table S5). This analysis included the haustorial transcriptomes, PpGI, U. appendiculatus hyphal and haustorial sequences (Puthoff et al., 2008), and haustorial secreted proteins identified by the yeast signal sequence trap for U. appendiculatus (Ua‐HSPs; T. I. Link, unpublished results). We also included EST (Jakupovic et al., 2006) and secretome (Link and Voegele, 2008) sequences from U. fabae. To gain further insight into the function of the members in each family, we searched them for conserved sequences and used this information to include sequences from other fungi that shared this conservation. Features of the largest families are summarized in Fig. 4 and in the following text. Because the sequences are not necessarily complete at the N‐terminus and the N‐terminus is necessary for signal peptide prediction, we expect that putative secreted proteins are likely to be under‐represented among the gene families.
Figure 4.

Summary of properties of the 16 largest clusters of proteins with predicted secretion motifs. Single linkage clustering was used to group the contigs, and the cluster ID, corresponding to Table S5, is provided to the left of the graph. The graph shows the numbers of genes (unique contigs) per cluster, which is subdivided into those predicted to be soluble secreted proteins (black) and all others (white). Roman numbers indicate the specificities of the clusters: I, specific to rust fungi; II, specific to Pucciniaceae; III, specific to Uromyces. The columns on the right give additional information. (H), Hacquard et al. (2012); (S), Saunders et al. (2012).
The 29 members of cluster 112 that are predicted to be secreted represent the highest number of any cluster (Fig. 4). Although the longest member of the cluster was 967 amino acids in length (PGTG_14242), most were short, with an average length of 162 amino acids. Cluster 112 members possess a 94‐amino‐acid region containing 10 conserved cysteine residues (Fig. 5a). A [YFW]xC motif (Godfrey et al., 2010) was found at cysteine eight, and a similar [AFY]xC motif was found at cysteine two. At least one member of this gene family was identified in all five rust species analysed, but no member was found in any other fungal species, indicating that the family is rust specific. Phylogenetic analysis of the family suggests that it was present in an ancestral rust species and that there was further expansion of the family after the species separated (Fig. 5b). Interestingly, some closely related genes cluster in the P. graminis f. sp. tritici genome, and one gene (PGTG_12466) is among the 100 most highly up‐regulated genes following wheat infection compared with resting urediospores (Duplessis et al., 2011).
Figure 5.
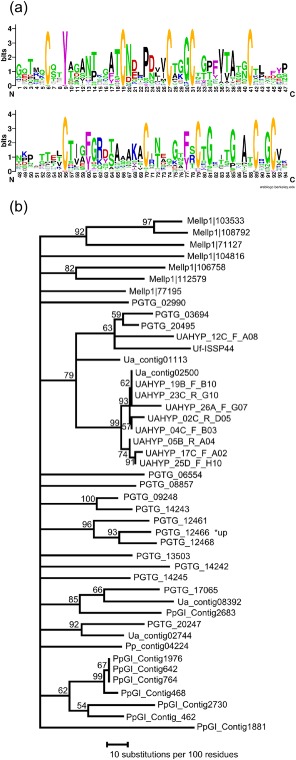
Gene family cluster 112. (a) Conserved profile of the central 94 amino acids (WebLogo). (b) Phylogeny: neighbour‐joining analysis of protein alignment; consensus tree of 100 bootstraps; only bootstrap values greater than 50 are shown (PAUP*). *up, among the 100 most strongly up‐regulated genes in infected leaves versus urediospores (Duplessis et al., 2011). Mellp, Melampsora larcici‐populina; PGTG, Puccinia graminis f. sp. tritici; Pp, Phakopsora pachyrhizi; Ua, Uromyces appendiculatus.
Cluster 2456 contains 27 members that are up to 234 amino acids in length. The mature proteins have a completely conserved CxC motif near their centres, and are rich in serine and glutamine, which occur in stretches as well as in conserved patterns. There is a conserved Rx[FLIM] motif very near the predicted signal peptide. The proteins in this cluster occur in Uromyces species and P. graminis f. sp. tritici, but not in P. pachyrhizi and M. larici‐populina, suggesting that they are restricted to the family Pucciniaceae. Phylogenetic analysis (Fig. S2, see Supporting Information) shows that P. graminis f. sp. tritici proteins cluster together, as do the proteins from U. fabae and U. appendiculatus. This indicates that the family has proliferated and diversified after the split between Puccinia and Uromyces. Strikingly, Ua‐ISSP24 and Uf‐ISSP41 (infection structure‐secreted proteins) only differ by one amino acid. The genes in this family may function during different stages. For example, Uf‐ISSP41 was only observed in cell types occurring before haustoria formation (Fig. S3, see Supporting Information), but PGTG_11318 is among the 100 most strongly up‐regulated genes in infected wheat (Duplessis et al., 2011).
Cluster 398 (12 members) is represented in all five rust species analysed. Its members contain 12 conserved cysteines and are up to 219 amino acids long, except for Mellp1|85995, which encodes a predicted 492‐amino‐acid protein. The family has a [YFW]xC motif that is part of a larger [YFW]xCxGxC motif (Fig. 6a). The phylogeny indicates that the proliferation of the family took place before and after the split between the different species (Fig. 6b). The family contains some of the most highly expressed genes encoding putative secreted proteins. Uf‐HSP42 is the most abundant Uf‐HSP and is not expressed in pre‐haustorial cell types (Link and Voegele, 2008), and Ua_contig14788 (807 reads) and Ua_contig08771 (686 reads) are among the most abundant haustorial transcripts. The same is true for Pp_contig03139 (229 reads) and Pp_contig08880 (195 reads). Consistent with this, three PGTG proteins are among the most highly up‐regulated proteins in infected wheat leaves (Duplessis et al., 2011).
Figure 6.
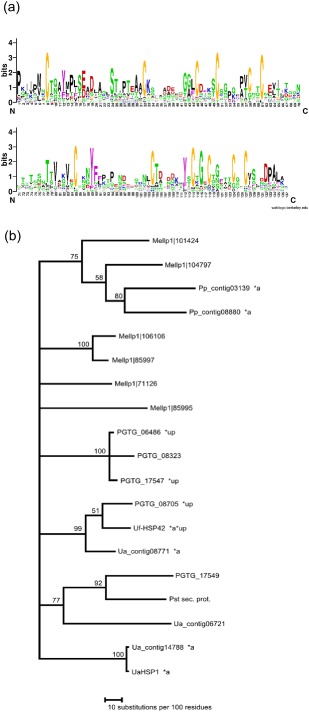
Gene family cluster 398. (a) Conserved profile of 137 central amino acids (WebLogo). (b) Phylogeny: neighbour‐joining analysis of protein alignment (PAUP*); consensus tree of 100 bootstraps; only bootstrap values greater than 50 are shown. *a, abundant transcripts; *up, among 100 most strongly up‐regulated genes in infected leaves versus urediospores; *a*up, abundant and up‐regulated in haustorium against pre‐haustorial cell types (Duplessis et al., 2011; Link and Voegele, 2008). Mellp, Melampsora larcici‐populina; PGTG, Puccinia graminis f. sp. tritici; Pp, Phakopsora pachyrhizi; Ua, Uromyces appendiculatus; Uf, Uromyces fabae. Pst sec. prot., secreted protein from Puccinia striiformis f. sp. tritici.
Cluster 1_2 (38 members) is a large family of cysteine‐rich proteins represented in the five rust species analysed, and, in general, proteins from individual species cluster together in the phylogeny. The predicted proteins range in size from 200 to 250 amino acids, and have eight conserved cysteines. The most strongly conserved region in this family forms a CLWxG motif (Fig. S4a, see Supporting Information). Members of this family also contain an RxY motif near their N termini. At 969 amino acids in length, PGTT_20487 is an outlier that encodes eight transmembrane domains and is annotated as a chloride channel. It possesses the conserved cysteines as well as the CLWxG and RxY motifs. The family is specific to rust fungi—only PGTT_20487 has homologues in other species. Family members from both P. graminis f. sp. tritici and M. larici‐populina are highly up‐regulated during infection (Fig. S4b), as is Uf‐HSP44. By contrast, Uf‐HSP39 is only weakly expressed in haustoria (Duplessis et al., 2011; Link and Voegele, 2008), suggesting that members of the family are differentially regulated.
The five members of cluster 20 have six conserved cysteines, the second of which is included in a YGxC motif. Family members were also present in P. graminis f. sp. tritici and M. larici‐populina, and, surprisingly, also in the necrotrophic ascomycete plant pathogens Aspergillus, Penicillium, Pyrenophora and Phaeosphaeria spp., and Glomerella graminicola and Botryotinia fuckeliana. The most distant species with homologues is Phytophthora infestans, an oomycete. We did not find homologues in any Basidiomycetes apart from the Pucciniales. The phylogeny (Fig. S5, see Supporting Information) implies that rust fungi acquired the gene once and that it subsequently diversified. The presence of the protein family in these diverse pathogens, and its absence in nonpathogens, strongly indicates a role in pathogenesis. Four of the P. graminis f. sp. tritici homologues are strongly up‐regulated following infection of wheat (Duplessis et al., 2011).
Not all of the families have members in all five of the rust species as in the previous examples. Cluster 1_0_152 (22 members) is an example of a family that is specific to U. appendiculatus. The family consists of small proteins ranging from 59 to 107 amino acids in length. There are two conserved cysteines in the predicted signal peptides, but none in the mature proteins. The proteins contain three [RHK]x[LMIFYW] sites (Fig. S6, see Supporting Information).
Transposon‐ and virus‐like sequences
In P. pachyrhizi and U. appendiculatus, 221 and 328 contigs, respectively, were classified as transposon, retrotransposon or polyprotein (Table 2). For P. pachyrhizi, transposons and retrotransposons accounted for 4.4% and polyproteins for 4.9% of all reads comprising contigs ≥100 bp. Similarly for U. appendiculatus, transposons and retrotransposons accounted for 4.5% and polyproteins 2.2% of all reads comprising contigs ≥100 bp. Interestingly, only five contigs corresponding to 0.3% of the reads were annotated as TEs in P. pachyrhizi sequences derived from resting urediospores and water‐germinated spores.
Table 2.
Transposon‐related sequence descriptions for Phakopsora pachyrhizi and U romyces appendiculatus
| Category | Number of contigs | ||
|---|---|---|---|
| P. pachyrhizi | U. appendiculatus | ||
| Pre‐haustorial | Haustoria | Haustoria | |
| Transposons | 1 | 3 | |
| Retrotransposons | 1 | 29 | 54 |
| Transposable elements | 3 | ||
| Retrotransposable elements | 25 | 163 | |
| Transposases | 1 | 14 | 1 |
| Polyproteins | 2 | 138 | 71 |
| Mutator‐like elements | 1 | 2 | 9 |
| Others | 15 | 29 | |
| Sum | 5 | 221 | 328 |
We identified 10 P. pachyrhizi and four U. appendiculatus contigs of probable mycoviral origin (viruses of fungi; Table 3). The virus‐like sequences from each fungus were dissimilar, and thus appear to be organism specific. In P. pachyrhizi, 9315 reads (2.3%), and, in U. appendiculatus, 10 777 reads (1.1%), were assembled into contigs related to mycoviral origin. Amino acid sequences predicted from ORFs longer than 300 bp were used in blastp searches against GenBank nr, and the top hits are provided in Table 3.
Table 3.
Sequence features of virus‐like contigs found in P hakopsora pachyrhizi and U romyces appendiculatus.
| Contig | Reads* | Lengtha | ORFb | RFc | AAd | Top blast hit, E value** | E |
|---|---|---|---|---|---|---|---|
| P. pachyrhizi | |||||||
| 03854 | 3194 | 3518 | 109–3483 | +1 | 1124 | RdRp [Tobacco mild green mosaic virus] | 3 × 10−27 |
| 02383 | 2862 | 5179 | 49–2529 | −2 | 826 | RdRp [Xanthophyllomyces dendrorhous virus L1A] | 7 × 10−103 |
| 2526–4976 | −3 | 816 | Major CP [Saccharomyces cerevisiae virus L‐A (L1)] | 3 × 10−5 | |||
| 03662 | 2510 | 3728 | 84–3626 | −1 | 1180 | RdRp [Blackberry yellow vein‐associated virus] | 6 × 10−22 |
| 02716 | 474 | 2309 | 791–1030 | −2 | 79 | RdRp, putative [Ophiostoma mitovirus 5] | 1 × 10−22 |
| 00799 | 80 | 2197 | 1–2046 | −2 | 682 | RdRp [Xanthophyllomyces dendrorhous virus L2] | 2 × 10−116 |
| 04598 | 67 | 2296 | 347–1987 | +2 | 546 | RdRp [Xanthophyllomyces dendrorhous virus L2] | 1 × 10−136 |
| 1944–2237 | +3 | 294 | No blast with E < 10−4 | ||||
| 00012 | 55 | 1017 | 154–1016 | +1 | 287 | RdRp [Xanthophyllomyces dendrorhous virus L1b] | 1 × 10−36 |
| 02568 | 47 | 1827 | 400–1002 | −1 | 200 | RNA polymerase [Tuber aestivum virus 1] | 8 × 10−45 |
| 1–382 | −3 | 127 | RNA polymerase, putative [unidentified blackcurrant Totiviridae] | 1 × 10−06 | |||
| 1208–1546 | −3 | 112 | RdRp [Xanthophyllomyces dendrorhous virus L1b] | 6 × 10−13 | |||
| 00672 | 20 | 1305 | 230–1304 | +2 | 358 | RdRp [Xanthophyllomyces dendrorhous virus L2] | 8 × 10−94 |
| 05068 | 6 | 449 | 3–446 | −1 | 148 | RdRp [Xanthophyllomyces dendrorhous virus L1b] | 2 × 10−31 |
| 67–447 | +1 | 127 | No blast with E < 10−4 | ||||
| U. appendiculatus | |||||||
| 13226 | 5113 | 9639 | 44–3321 | +2 | 1055 | 1a protein [Cassia yellow blotch virus] | 1 × 10−26 |
| 3502–5388 | +1 | 628 | 129‐kDa replicase [Frangipani mosaic virus] | 2 × 10−33 | |||
| 5755–6957 | +1 | 400 | 59‐kDa readthrough protein [Sorghum chlorotic spot virus] | 4 × 10−38 | |||
| 6981–9551 | +3 | 856 | CI protein [Onion yellow dwarf virus] | 2 × 10−11 | |||
| 06136 | 3142 | 7466 | 165–7325 | +3 | 2386 | Polyprotein [Beet mosaic virus] | 6 × 10−56 |
| 00006 | 1425 | 3575 | 46–3501 | −3 | 1151 | RNA replicase [Kyuri green mottle mosaic virus] | 2 × 10−23 |
| 00002 | 1097 | 3445 | 102–3395 | +3 | 1097 | 136‐kDa protein [Pepper ringspot virus] | 2 × 10−32 |
CP, capsid protein; RdRp, RNA‐dependent RNA polymerase.
*Number of reads in the contig. Contigs are sorted by this column for each species.
Nucleotide length of the contig.
Starting and ending nucleotide of the open reading frame (ORF). Only ORFs encoding more than 100 amino acids are listed with the exception of P. pachyrhizi contig02716 for which the longest predicted ORF encodes a 79‐amino‐acid polypeptide.
RF, reading frame of the ORF relative to the contig sequence; note that orientation was not preserved during sequencing.
AA, number of amino acids encoded by the ORF.
**A blastp search was performed against the GenBank nonredundant (nr) database using the predicted amino acid sequence from each ORF.
Seven contigs from P. pachyrhizi had high similarity to the RNA‐dependent RNA polymerases (RdRps) of totiviruses (Totiviridae; double‐stranded RNA viruses of fungi; Nuss, 2005). Pp_contig02383, 5179 bases in length, also encodes a capsid protein with a frame‐shifted, overlapping ORF upstream of the RdRp, as is found in the 4580 bases of the type member totivirus Saccharomyces cerevisiae virus L‐A (SCV; Icho and Wickner, 1989). Interestingly, Pp_contig02383 begins with GAAAATTTCT, whereas SCV begins with GAAAAATTTTT, and so we may have captured the correct 5′ end. An extended 3′ end was not found. Nevertheless, Pp_contig02383 plus Pp_contig04598 may represent a nearly complete viral genome. Therefore, based on the sequence similarities, it is likely that these contigs represent a novel totivirus in P. pachyrhizi, although there may be variants. Another contig was similar to the RdRp of a novel mitovirus (Narnaviridae; nonencapsidated RNA viruses of fungi often found in mitochondria; Nuss, 2005). The remaining two virus‐like sequences from P. pachyrhizi were similar to the RdRps of the plant viruses Tobacco mild green mosaic virus (Tobamovirus) and Blackberry yellow vein‐associated virus (Closteroviridae). Interestingly, Sclerotinia sclerotiorum RNA virus L, a mycovirus, has conserved motifs in its RNA replicase that make it related to plant clostero‐ and tobamoviruses (Ghabrial and Suzuki, 2009). Thus, it is possible that the other two contigs may also describe a novel mycovirus.
The four virus‐like sequences in U. appendiculatus also have the highest similarity to plant virus polyproteins. Strikingly, Pp_contig06136 encodes a 2386‐amino‐acid polypeptide that has conserved sequence and genome organization with Beet mosaic virus, a plant potyvirus. Interestingly, mycoviruses of the Hypoviridae (hypoviruses) are phylogenetically related to potyviruses, having linear single‐stranded RNA genomes of 9–13 kb in length encoding polyproteins and with polyA+ tails (Ghabrial and Suzuki, 2009). Cryphonectria hypovirus 1‐EP713, the type member of Hypoviridae, encodes a suppressor of silencing, p29, at the amino terminus of the polyprotein, as do plant potyviruses (Nuss, 2005). Therefore, it is possible that these contigs with similarity to potyviruses represent hypoviruses in U. appendiculatus, and not plant viruses. Similar virus‐like sequences observed by Jakupovic et al. (2006) were not encoded by the fungal genome.
Discussion
Number of transcript assemblies, quality of data and coverage
The rust genomes sequenced to date contain approximately 16 000–18 000 genes (Duplessis et al., 2011), and so we estimate that transcripts corresponding to approximately 25% of the P. pachyrhizi genome and 42% of the U. appendiculatus genome were identified in our study. The estimates suggest that coverage of the U. appendiculatus haustorial transcriptome was better than that for P. pachyrhizi. This difference was probably a result of the greater challenge of preparing RNA from P. pachyrhizi haustoria, which were lower in yield and smaller in size. Consequently, we could not sequence cDNA generated from polyA+ RNA for P. pachyrhizi, as we did for U. appendiculatus, and this possibly led to discrepancies in coverage. Nevertheless, similar average contig lengths suggest that RNA quality for P. pachyrhizi and U. appendiculatus haustoria was sufficient for a project of this kind. The numbers of unique transcripts expressed in haustoria of either species were somewhat fewer than the 12 846 contigs identified in P. striiformis f. sp. tritici (wheat stripe rust) haustoria (Garnica et al., 2013) and the 7077 distinct contigs identified in Golovinomyces orontii (powdery mildew) haustoria (Weßling et al., 2012). 454 sequencing was also used recently by Fernandez et al. (2012) to identify Hemileia vastatrix transcripts expressed during the infection of Coffea arabica leaves. From 6763 H. vastatrix contigs, they identified 382 predicted secreted proteins, which is similar to the numbers of transcripts and predicted secreted proteins found here. These studies utilizing next‐generation sequencing methods have provided much greater coverage of haustorial transcriptomes than previous efforts utilizing Sanger sequencing of cDNAs (Catanzariti et al., 2006; Jakupovic et al., 2006; Joly et al., 2010; Puthoff et al., 2008).
A couple lines of evidence helped to validate our approach of enriching haustoria to identify the haustorial transcriptomes of P. pachyrhizi and U. appendiculatus. The first is that we were able to identify homologues of several previously identified haustorial secreted proteins, such as RTPs and HESPs. This is similar to the case with the P. striiformis haustorial transcriptome in which RTP and HESP homologues were also present (Garnica et al., 2013). The second was that PpGI_contig3375, making up nearly 20% of the reads and 50% of the reads with GO biological process annotation in resting urediospore and germ tube stages of P. pachyrhizi, had a very low expression in haustoria. The massive numbers of reads contributing to this contig skewed the results of GO annotation for over‐represented GO categories in the resting urediospore and germ tube stages. This high expression in urediospores and germ tubes hints at a function in early infection. PpGI_3375 has similarity to the GAS1/GAS2 gEgh16 gene family from the powdery mildew fungus Blumeria graminis, and GAS1 and GAS2 in Magnaporthe grisea. Consistent with a potential early function, GAS1 and GAS2 were only expressed in appressoria of M. grisea and, when these genes were deleted, appressorial penetration was reduced (Xue et al., 2002; Zhang et al., 2008). These observations show that our haustoria transcriptome included the expected genes and excluded highly abundant transcripts expressed at earlier stages.
Haustoria as power plants or refineries
The haustorium is the organ for the uptake of nutrients, sugars and amino acids by rust fungi (Struck et al., 2002, 2004; Voegele et al., 2001). Enzymes that are highly expressed and localized in haustoria convert nutrient sugars to storage compounds (Link et al., 2005; Voegele et al., 2005). Based on these findings, haustoria were named the ‘power plants’ of rust fungi (Voegele and Mendgen, 2003). Some of the earliest experiments on expression profiling for haustoria identified highly induced genes involved in thiamine and amino acid biosynthesis, indicating that haustoria have metabolic roles in addition to nutrient uptake (Hahn and Mendgen, 1997; Jakupovic et al., 2006; Sohn et al., 2000). For P. pachyrhizi, GO categorization suggests that many genes in haustoria are involved in primary metabolism, with emphasis on carbohydrates and amino acids, and affiliated with nutrient uptake. These findings support the idea that haustoria are involved in the conversion of nutrients to form more useful factors for fungal growth and energy production, and confirm the function of haustoria as power plants or, perhaps more aptly, refineries for rust fungi (Voegele and Mendgen, 2003). Our conclusions based on the analysis of GO annotation from the two haustorial transcriptomes are consistent with those of Garnica et al. (2013), who used the differential expression of genes in the haustoria versus germinated spores of P. striiformis f. sp. tritici. They showed that haustoria are equipped to assimilate sugars, amino acids, nitrogen and other nutrients, and are precursors of the reductant, NADPH (reduced form of nicotinamide adenine dinucleotide phosphate). These molecules can readily be used for ATP production and synthesis of macromolecules in haustoria through pathways distinct from earlier life stages.
Families of putative candidate effectors
Initially, effectors were identified as avirulence proteins that are recognized by cognate resistance proteins and trigger hypersensitive disease resistance (De Wit et al., 2009). However, it is now clear that the primary roles of effectors are to alter host cellular functions in favour of pathogens, such as suppressing defences in various ways or altering the host's metabolism (Song et al., 2009). The study of effectors can provide valuable insights into pathogenesis, virulence and host defence responses (Duplessis et al., 2012). Effectors are expected to be excellent targets for the control of pathogens, but, unlike effectors from some other plant pathogens, relatively little is known about rust effectors.
Effectors may be specific to their lineage or their species and belong to gene families that arose during the development of pathogenicity, and may be preferentially expressed during infection (Ellis et al., 2009). The families of genes highlighted here possess hallmarks consistent with their potential roles as candidate effectors. They are lineage or species specific or more broadly conserved among plant pathogens. Based on the members present in the sequenced genomes of P. graminis and M. larici‐populina, the families appear to have expanded during rust evolution, and some are clustered in fungal genomes, suggesting that tandem duplication has contributed to their evolution and expansion. Many genes have been shown previously to be strongly expressed during infection in P. graminis and M. larici‐populina, which is consistent with their identification in haustoria of P. pachyrhizi and U. appendiculatus. We were able to identify families that were unique to P. pachyrhizi or U. appendiculatus, but families with members in both P. pachyrhizi and Uromyces ssp. also had members in at least one other rust species (Table S5). Thus, we were not able to identify putative effectors specific to legume rusts.
Many characterized effectors in fungal pathogens are SSPs and are often rich in cysteine (Stergiopoulos and de Wit, 2009). Many of the gene families we found encode short, cysteine‐rich proteins that could be grouped by their distinct cysteine patterns. Apart from rust fungi (Duplessis et al., 2011), similar genes have been found in several species of plant pathogenic fungi (Rep, 2005) and oomycetes (Kamoun, 2006), and have been implicated in avirulence and effector functions (Stergiopoulos and de Wit, 2009). Cysteines are important for secreted proteins to maintain their structure, but distinct cysteine patterns have been associated with specific functions. For example, the plant defensins have antimicrobial properties (Graham et al., 2008). Other short, cysteine‐rich proteins are implicated in plant signalling (Marshall et al., 2011). Kessler et al. (2010) found that receptors for small, secreted cysteine‐rich proteins can play a role in pollen tube reception and fungal invasion, and it seems probable that cysteine‐rich effector proteins could hijack host signalling pathways.
Recent studies have defined and categorized families of secreted proteins or SSPs in M. larici‐populina, P. graminis and P. striiformis, and have predicted the families most likely to contain effector proteins (Cantu et al., 2011; Hacquard et al., 2012; Saunders et al., 2012). When we compared our most interesting gene families with those of Hacquard et al. (2012), there was little overlap. Our cluster 112 corresponds to their Class III, and our cluster 398 corresponds to their Class VI with their characteristic cysteine motifs. The overlap between our clusters and the tribes identified in the comprehensive study by Saunders et al. (2012) was more extensive. We found that the following clusters and tribes correspond to one another: cluster 112–tribe74, cluster 2456–tribe213, cluster 389–tribe110, cluster 1_2–tribe38 and tribe75, and cluster 20–tribe76 (Fig. 4). Saunders et al. (2012) used an automated computational pipeline, whereas our analyses involved a more manual curation. These approaches produced similar conclusions, as exemplified by cluster 389–tribe110, which was identified as highly interesting by us and has a high probability score to contain effectors in Saunders et al. (2012). The case of cluster 20–tribe76 is an example of a family that we found to be highly interesting, but was overlooked by Saunders et al. (2012).
Because there is a paucity of sequence information available for rust fungi in general, it is noteworthy that increasing sequence information for just a few additional species enhances the knowledge on their gene families. For example, tribe213 is simply defined as lineage specific by Saunders et al. (2012), but we can now more closely define lineage specificity to the family level. The same holds true for tribe63, considered to be highly interesting by Saunders et al. (2012) in part because of its lineage specificity. Tribe63 corresponds to our cluster 1_63, with homologues from both U. appendiculatus and P. pachyrhizi, demonstrating that it is not lineage specific. With the genome sequences of P. graminis f. sp. tritici and M. larici‐populina now published, we can be more confident that the families found to be lineage specific are specific to clades below family level.
Garnica et al. (2013) did not present an analysis of families of haustorial secreted proteins from P. striiformis f. sp. tritici. They did provide an analysis of the annotated proteins, and showed that 15 were annotated as different types of glycoside hydrolases related to chitinase as well as plant cell wall‐degrading enzymes. We also observed predicted proteins with glycoside hydrolase activities, including chitinase (Table S1 and S2, Fig. 4). Garnica et al. (2013) also indicated that cysteine‐rich proteins were abundant, with 191 of 437 predicted haustorial secreted proteins possessing 4–28 cysteines. Their experimental design also allowed them to determine that 85% of haustorial secreted proteins were differentially expressed when compared with germinated spores. They found that 295 were overexpressed in haustoria and 76 were overexpressed in germinated spores. These data are consistent with a majority of candidate effectors being strongly produced in haustoria where they can mediate biotrophic interactions. However, they did not exclude the likely possibility that important effectors are synthesized by other cell types at this and other life stages.
We did not identify a novel sequence motif in the full set of predicted secreted proteins that might indicate a signal used for recognition and transfer of haustorial expressed proteins into the plant cell. Nevertheless, in distinct gene families, we did find the [YFW]xC motif (Godfrey et al., 2010) and conserved sites corresponding to the [RHK]x[LMIFYW] motif (Kale et al., 2010). There has been controversy about the functionality of the latter motif in rust fungi (Gan et al., 2010). Here, the motif occurs primarily in families lacking conserved cysteines. Unlike RXLR in oomycetes, which is typically in close proximity to the signal peptide (Rehmany et al., 2005), the motifs found here were more dispersed. Garnica et al. (2013) also identified similar motifs, and used a statistical analysis indicating that the motifs occurred within the haustorial secreted proteins at frequencies expected on the basis of random chance.
Are TEs or viruses involved in the evolution or regulation of effectors?
In Phytophthora species, accelerated evolution of effectors is connected to the localization of effector loci in gene‐poor and repetitive element‐rich regions of the genome (Raffaele et al., 2010). Similarly, Ustilago maydis effector loci were found in clusters in regions of low sequence conservation, which suggests rapid evolution of these genes (Kämper et al., 2006; Schirawski et al., 2010). For rust fungi, however, no mechanism for accelerated evolution has been demonstrated to date. Nevertheless, Duplessis et al. (2011) found that both M. larici‐populina and P. graminis f. sp. tritici have very high numbers (45%) of TEs in their genomes. Our results indicate that P. pachyrhizi and U. appendiculatus also encode TEs in their genomes, and that these TEs are very active in haustoria because of their high expression levels. For P. pachyrhizi, we propose that transposon activity could contribute to the adaptability and large number of strains observed in the absence of a sexual stage (Bromfield, 1984).
These findings corroborate a previous observation that TE expression is greater in haustoria than in hyphae of U. appendiculatus (Puthoff et al., 2008). Increased TE expression in fungi has been linked to the loss of Dicer‐like and Argonaute genes responsible for gene silencing (Drinnenberg et al., 2009). In U. appendiculatus, we observed contigs with similarity to Argonaute and Dicer‐like genes (contigs 09363, 10579, 00757, 03383, 10462). In P. pachyrhizi, we found Argonaute‐like sequences (contigs 06318, 04581 and 00400), but not Dicer‐like. Dicer‐like genes may not be present because of random chance, low expression levels or absence from the P. pachyrhizi genome. If the silencing system is intact in these two fungi, the data imply that silencing suppression occurs in the haustoria, as proposed by Puthoff et al. (2008). Alternatively, haustoria could inherently lack gene silencing capabilities, or silencing could be suppressed by the host. The presence of hypoviruses that may encode silencing suppressors could also explain the increase in TEs. The activities of TEs and viruses in haustoria of P. pachyrhizi and U. appendiculatus suggest potential modes of biocontrol, with either killer viruses or hypovirulence (Drinnenberg et al., 2011; Nuss, 2005). It may also be possible to develop TE‐ or viral‐based vectors for transient or stable transformation. Despite our evidence for the detection of viral RNAs, we have not discounted the possibility that the RNAs for all of these potential viruses instead originated from the fungal genome (Taylor and Bruenn, 2009). Further examination of these mycoviral species and TEs is warranted.
Conclusions and Further Prospects
By isolating haustoria from infected leaves and assembling transcripts from shotgun pyrosequencing, we have identified genes from P. pachyrhizi and U. appendiculatus that are likely to be important to haustorium biology, biotrophy and pathogenicity of these rust fungi. We found several families of putative secreted proteins that fit the profile of candidate effectors from fungal pathogens based on their lineage specificity, small size, cysteine‐rich nature, high expression during infection and lack of similarity to proteins of known function (Ellis et al., 2009). Transformation of rust fungi to knock down or knock out the expression of candidate effectors may be feasible (Lawrence et al., 2010), but such experiments remain a difficult proposition. Other approaches can be used to establish which genes encode bonafide effectors. For example, heterologous expression in other plant pathogens (Sohn et al., 2007), cell death suppression assays (Bos et al., 2009), identification of interaction partners from the host (Chen et al., 2010), in planta localization (Kemen et al., 2005) and host‐induced gene silencing (Nowara et al., 2010; Nunes and Dean, 2012) can be used to identify and characterize effectors. These studies, combined with the sequencing of additional isolates as well as close relatives, such as U. fabae for U. appendiculatus or P. meibomiae for P. pachyrhizi, will provide more information on species‐specific evolution of pathogenicity in legume‐infecting rusts.
Experimental Procedures
Plant and fungal material; growth and inoculation
Uromyces appendiculatus isolate SWBR1 (laboratory collection Universität Konstanz, Konstanz, Germany) was sprayed on all leaves of susceptible, 21‐day‐old, Phaseolus vulgaris var. nanus cv. Primel using a watery suspension containing 500 mg of spores and 500 mg of milk powder per litre. Plants were kept in the dark at 100% humidity for 24 h and then transferred to the glasshouse (day/night, 14 h/10 h–16 h/8 h; temperatures, 17–22 °C).
Phakopsora pachyrhizi isolate Thai1 (laboratory collection Universität Konstanz) was sprayed on all leaves of susceptible, 21‐day‐old, Glycine max cv. Erin. Inoculation, incubation and growth conditions were the same as for U. appendiculatus.
Preparation of haustorial cDNA
Haustoria were prepared as described by Hahn and Mendgen (1992) with minor modifications. To maximize the yield of haustoria, heavily infected leaves were collected at 10 dpi for U. appendiculatus and 12 dpi for P. pachyrhizi. Leaves were washed under running water whilst rubbing with a gloved hand to remove urediospores, and then dried with paper towels. After homogenization of the leaves, the homogenate was filtered through a 20‐μm nylon mesh (PA‐20/14 Nybolt, Franz Eckert GmbH, Waldkirch, Germany). Because P. pachyrhizi haustoria are smaller, its homogenates were subsequently filtered through a 15‐μm nylon mesh (PA‐15/10 Nybolt). Haustoria were enriched by concanavalin A (Con‐A) binding, and microscopic examination of the suspensions determined that they almost exclusively contained haustoria and chloroplasts (Fig. 1). The haustoria to chloroplast ratios were about 3.5 for P. pachyrhizi and 2.5 for U. appendiculatus. Preparations with ratios >2 were used for RNA isolation. Haustoria were either spun down (P. pachyrhizi) or suspended in RNAlater ® (Applied Biosystems, Foster City, CA, USA) (U. appendiculatus) and stored at −80 °C. Total RNA was extracted using citric acid buffers (Purescript®, Gentra‐Systems Inc., Minneapolis, MN, USA), and its integrity was confirmed by agarose gel electrophoresis (Fig. 1). For the enrichment of mRNA, the Oligotex®‐Mini kit (Qiagen, Hilden, Germany) was used. cDNA preparation and size fractionation were performed with the SMART™ cDNA Library Construction Kit (Clontech, Mountain View, CA, USA) using either 1 μg of total RNA or 0.5 μg of mRNA, and running the long distance polymerase chain reaction (LD‐PCR) with 20 cycles.
Haustoria transcriptome shotgun sequencing, assembly and filtering
The three cDNA preparations (U. appendiculatus haustorial mRNA, U. appendiculatus haustorial total RNA and P. pachyrhizi haustorial total RNA) were lyophilized for storage and reconstituted in 10 mm Tris‐HCl (pH 7.5) and 1 mm ethylenediaminetetraacetic acid (EDTA) at a final concentration of approximately 25 ng/μL each. Epicentre (Madison, WI, USA) Nextera preparation reactions (Roche Titanium‐compatible kit, part number NT09115) were used as specified in the manufacturer's protocol. Nextera Adapter 1, without barcoding, was used for each library. Library quality control steps, emulsion PCR and pyrosequencing were performed as outlined in the Roche‐454 Life Sciences (Branford, CT, USA) GS FLX Titanium methods manual (October 2008) using a GS Titanium LV emPCR (Lib‐L) Kit V2 for the two‐region 70 × 75 PicoTiterPlate and a GS Titanium Sequencing Kit XLR70 (200 cycle). The sequencing reads were assembled into contigs using the 454 Newbler2.3 assembly tool.
For P. pachyrhizi, the 11 872 contigs from the Newbler assembly were filtered to remove those that matched the soybean genome (Schmutz et al., 2010), aligned well with plant proteins but not fungal proteins using blastx searches against the GenBank nr database, were redundant or had ORFs shorter than 50 amino acids and did not yield a hit with GenBank nr using blastx (E ≥ 10−3). The P. pachyrhizi Transcriptome Shotgun Assembly (TSA) project has been deposited at DDBJ/EMBL/GenBank under the accession number GACM00000000. The version described in this article is the first version, GACM01000000. Because our original assembly included all sequences 100 bp or longer and GenBank TSA only allows for submission of sequences of 200 bp or longer, the assembly is also available here as File S1.
For U. appendiculatus, the 14 581 contigs were filtered to remove those with matches to P. vulgaris sequences (P. vulgaris v 0.9, http://www.phytozome.com), the soybean genome (Schmutz et al., 2010) and Arabidopsis plastid and mitochondrial genomes (Sato et al., 1999; Unseld et al., 1997) using blastn (E ≤ 10−14 and %ID ≥ 95%). The U. appendiculatus TSA project has been deposited at DDBJ/EMBL/GenBank under the accession number GACI00000000. The version described in this article is the first version, GACI01000000. For the same reasons as for P. pachyrhizi, the assembly is also available here as File S2.
Assembly of P. pachyrhizi ESTs available from GenBank
Phakopsora pachyrhizi ESTs (34 329) were downloaded from the National Center for Biotechnology Information (NCBI) EST database. ESTs originated from either water‐germinated urediospore (31 433 ESTs), infected soybean leaf (2064 ESTs) or resting urediospore (832 ESTs) libraries. Sequence quality information in each record was used to trim low‐quality sequence. If no quality information was provided, sequences were trimmed by 10% from the 3′ end. blastn (Altschul et al., 1997) against pSport1 (Gibco/BRL Life Technology, Carlsbad, CA, USA) was used to identify and remove vector sequence. ESTs were assembled using CAP3 (Huang and Madan, 1999) using a minimum overlap of 50 bases and a minimum alignment score of 100. Contigs were viewed using contigimage (Center for Computational Genomics and Bioinformatics, University of Minnesota, Minneapolis, MN, USA) and corrected using Consed (Gordon et al., 1998). Following assembly, blastn (E < 10−30) against the soybean EST gene index (Lee et al., 2005) and GenBank was used to remove contaminant sequences. The remaining contigs and singletons comprise the PpGI (File S5).
Clustering of sequences and analysis of families
To identify homologous genes and gene families, we used blastx‐based (Altschul et al., 1997) single linkage clustering (Graham et al., 2004) using E value cut‐offs of 10−7, 10−14 and 10−21 to identify clusters of related sequences. Clustering results were linked to predictions of secretion signals and blastx results of searches against eight basidiomycete genomes (Table S6, see Supporting Information). clustal W (Thompson et al., 1994) was used to align predicted protein sequences within each cluster. HMMbuild (Durbin et al., 1998) was used to create hidden Markov models (HMMs) for conserved domain(s) from trimmed alignments. HMMsearch (Durbin et al., 1998) was used to screen the HMMs against our own data, Uniref100 (Apweiler et al., 2004) and the eight basidiomycete genomes (Table S6). Additional sequences were added to the alignment if they met the following criteria: E < 10−7, presence of a predicted signal peptide (see below) and no additional transmembrane domains. Phylogenies of the families with bootstrap values were built using PAUP* software (PAUP*, portable version 4.0.0d55 for Unix) (Swofford, 2003), and the resulting trees were displayed with TREEVIEW (Page, 1996). Clustering results are summarized in Table S5.
GO annotation and analyses
Annotation was performed using blast2go (Conesa et al., 2005; Götz et al., 2008). Default parameters were used for blastx (Altschul et al., 1997) against GenBank nr [E < 10−3, HSP > 33 bp and annotation with the blast2go rule (E‐value‐Hit‐Filter E < 10−6; annotation cut‐off, 55; GO weight, 5)]. The annotation step was repeated for all sequences with mapped GO terms, but without annotation, using a lower annotation cut‐off of 45. GOslim was run, using either the generic or the yeast setting. Figures related to the annotation were created with blast2go and edited using Inkscape (inkscape.org).
The total numbers of reads of all contigs and singletons in a GO category were summed and TEs and virus‐like sequences were subtracted. Each GO category was normalized to the total reads for all the GO annotated reads to derive a ratio for the expression of a particular GO category in the haustoria relative to the expression in resting urediospores or germ tubes (Table 1). GO categories that were different between resting urediospores or germ tubes and haustoria were determined on the basis of Fisher's exact test, with correction for multiple testing (Bonferroni, 1935; Fisher, 1966) and a two‐fold difference as a cut‐off (Tables 1 and S4, see Supporting Information).
Prediction of secreted proteins
BestORF (SoftBerry, Inc., Mount Kisco, NY, USA) was used to predict ORFs within contigs with organism settings for Puccinia. Signal peptides were predicted using SignalP3 (Bendtsen et al., 2004) with default cut‐offs. Only proteins with a signal peptide predicted by both the neural network (NN) and HMM algorithms were considered for further testing. TMHMM (Sonnhammer et al., 1998) was used to predict putative transmembrane domains. Proteins with putative transmembrane domains and the predicted signal peptide were presumed to be membrane proteins. TargetP (Emanuelsson et al., 2007) was used to predict and exclude potential mitochondrial proteins. The default ‘winner takes all’ setting was used, but only proteins predicted as mitochondrial and falling into reliability classes 1, 2 or 3 (of 5) were considered to be mitochondrial. Protein sequences with a predicted signal peptide that were not mitochondrial and did not include an additional transmembrane domain were considered to be secreted proteins and retained as putative effectors. Finally GPI anchors were predicted using PredGPI (http://gpcr.biocomp.unibo.it/predgpi/pred.htm) (Pierleoni et al., 2008). Only the proteins predicted as ‘highly probable’ were considered to be GPI anchored.
Supporting information
Fig. S1 Sequence length distribution of contigs greater than 100 bp before application of bioinformatics filters: (A) Phakopsora pachyrhizi; (B) Uromyces appendiculatus.
Fig. S2 Phylogeny for gene family cluster 2456.
Fig. S3 Uf‐ISSP41 is expressed in pre‐haustorial in vitro infection structures but not in haustoria.
Fig. S4 Gene family cluster 1_2.
Fig. S5 Phylogeny for gene family cluster 20.
Fig. S6 Conserved amino acid motif in gene family cluster 1_0_152 (WebLogo).
File S1 FASTA file of the Phakopsora pachyrhizi haustorial transcriptome assembly.
File S2 FASTA file of the Uromyces appendiculatus haustorial transcriptome assembly.
File S3 List of Phakopsora pachyrhizi contigs from the haustorial transcriptome assembly encoding proteins with predicted secretion signals.
File S4 List of Uromyces appendiculatus contigs from the haustorial transcriptome assembly encoding proteins with predicted secretion signals.
File S5 FASTA file of the Phakopsora pachyrhizi Gene Index (PpGI) contigs assembled from P. pachyrhizi expressed sequence tag (EST) sequences available in GenBank.
Table S1 Phakopsora pachyrhizi haustorial transcriptome.
Table S2 Uromyces appendiculatus haustorial transcriptome.
Table S3 Phakopsora pachyrhizi pre‐haustorial PpGI expressed sequence tag (EST) contigs.
Table S4 Comparison of transcription between gene ontology (GO) categories in Phakopsora pachyrhizi resting urediospores and water‐germinated spores and P. pachyrhizi haustoria.
Table S5 Single linkage clustering results.
Table S6 Basidiomycete species screened for homologies.
Acknowledgements
We thank Dan Nettleton for assistance with statistical analyses, and Sarah Pierce, Yeunsook Lee and Michael Ernst for technical assistance. We thank Francismar Guimaraes, Andres Cernadas and Tarek Hewezi for helpful discussions. This work was supported by the Deutsche Forschungsgemeinschaft (VO595/3‐1 and VO595/3‐2 to RTV), the Iowa Soybean Association, the Iowa State University Plant Sciences Institute and Hatch Act and State of Iowa Funds. This is a journal paper of the Iowa Agriculture and Home Economics Experiment Station, Ames, IA, USA, project number 3708.
References
- Altschul, S.F. , Madden, T.L. , Schäffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler, R. , Bairoch, A. , Wu, C.H. , Barker, W.C. , Boeckmann, B. , Ferro, S. , Gasteiger, E. , Huang, H. , Lopez, R. , Magrane, M. , Martin, M.J. , Natale, D.A. , O'Donovan, C. , Redaschi, N. and Yeh, L.S. (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya, C.M. , Alleyne, A.T. , Steadman, J.R. , Eskridgne, K.M. and Coyne, D.P. (2004) Phenotypic and genotypic characterization of Uromyces appendiculatus from Phaseolus vulgaris in the Americas. Plant Dis. 88, 830–836. [DOI] [PubMed] [Google Scholar]
- Bendtsen, J.D. , Nielsen, H. , von Heijne, G. and Brunak, S. (2004) Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. [DOI] [PubMed] [Google Scholar]
- Bonferroni, C.E. (1935) Ill calcolo delle assicurazioni su gruppi di teste. Studi in Onore del Professore Salvatore Ortu Carboni 13–60. Rome, Italy. [Google Scholar]
- Bos, J.I. , Chaparro‐Garcia, A. , Quesada‐Ocampo, L.M. , McSpadden Gardener, B.B. and Kamoun, S. (2009) Distinct amino acids of the Phytophthora infestans effector AVR3a condition activation of R3a hypersensitivity and suppression of cell death. Mol. Plant–Microbe Interact. 22, 269–281. [DOI] [PubMed] [Google Scholar]
- Bromfield, K.R. (1984) Soybean Rust. St. Paul, MN: The American Phytopathological Society. [Google Scholar]
- Cantu, D. , Govindarajulu, M. , Kozik, A. , Wang, M. , Chen, X. , Kojima, K.K. , Jurka, J. , Michelmore, R.W. and Dubcovsky, J. (2011) Next generation sequencing provides rapid access to the genome of Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. PLoS ONE, 6, e24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanzariti, A.M. , Dodds, P.N. , Lawrence, G.J. , Ayliffe, M.A. and Ellis, J.G. (2006) Haustorially expressed secreted proteins from flax rust are highly enriched for avirulence elicitors. Plant Cell, 18, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R. , Chang, C. , Tucker, M.L. and Cooper, B. (2010) Affinity purification and mass spectrometry: an attractive choice to investigate protein–protein interactions in plant immunity. Curr. Proteom. 7, 258–264. [Google Scholar]
- Conesa, A. , Götz, S. , Garcia‐Gomez, J.M. , Terol, J. , Talon, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- De Wit, P.J. , Mehrabi, R. , Van den Burg, H.A. and Stergiopoulos, I. (2009) Fungal effector proteins: past, present and future. Mol. Plant Pathol. 10, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds, P.N. , Rafiqi, M. , Gan, P.H. , Hardham, A.R. , Jones, D.A. and Ellis, J.G. (2009) Effectors of biotrophic fungi and oomycetes: pathogenicity factors and triggers of host resistance. New Phytol. 183, 993–1000. [DOI] [PubMed] [Google Scholar]
- Drinnenberg, I.A. , Weinberg, D.E. , Xie, K.T. , Mower, J.P. , Wolfe, K.H. , Fink, G.R. and Bartel, D.P. (2009) RNAi in budding yeast. Science, 326, 544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg, I.A. , Fink, G.R. and Bartel, D.P. (2011) Compatibility with killer explains the rise of RNAi‐deficient fungi. Science, 333, 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplessis, S. , Cuomo, C.A. , Lin, Y.C. , Aerts, A. , Tisserant, E. , Veneault‐Fourrey, C. , Joly, D.L. , Hacquard, S. , Amselem, J. , Cantarel, B.L. , Chiu, R. , Coutinho, P.M. , Feau, N. , Field, M. , Frey, P. , Gelhaye, E. , Goldberg, J. , Grabherr, M.G. , Kodira, C.D. , Kohler, A. , Kues, U. , Lindquist, E.A. , Lucas, S.M. , Mago, R. , Mauceli, E. , Morin, E. , Murat, C. , Pangilinan, J.L. , Park, R. , Pearson, M. , Quesneville, H. , Rouhier, N. , Sakthikumar, S. , Salamov, A.A. , Schmutz, J. , Selles, B. , Shapiro, H. , Tanguay, P. , Tuskan, G.A. , Henrissat, B. , Van de Peer, Y. , Rouze, P. , Ellis, J.G. , Dodds, P.N. , Schein, J.E. , Zhong, S. , Hamelin, R.C. , Grigoriev, I.V. , Szabo, L.J. and Martin, F. (2011) Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA, 108, 9166–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplessis, S. , Joly, D.L. and Dodds, P.N. (2012) Rust effectors In: Effectors in Plant–Microbe Interactions (Martin F. and Kamoun S., eds), pp. 155–193. West Sussex: John Wiley & Sons, Inc.. [Google Scholar]
- Durbin, R. , Eddy, S.R. , Krogh, A. and Mitchison, G. (1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge: Cambridge University Press. [Google Scholar]
- Ellis, J.G. , Rafiqi, M. , Gan, P. , Chakrabarti, A. and Dodds, P.N. (2009) Recent progress in discovery and functional analysis of effector proteins of fungal and oomycete plant pathogens. Curr. Opin. Plant Biol. 12, 399–405. [DOI] [PubMed] [Google Scholar]
- Emanuelsson, O. , Brunak, S. , von Heijne, G. and Nielsen, H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971. [DOI] [PubMed] [Google Scholar]
- Fernandez, D. , Tisserant, E. , Talhinhas, P. , Azinheira, H. , Vieira, A. , Petitot, A.S. , Loureiro, A. , Poulain, J. , Da Silva, C. , Silva Mdo, C. and Duplessis, S. (2012) 454‐pyrosequencing of Coffea arabica leaves infected by the rust fungus Hemileia vastatrix reveals in planta‐expressed pathogen‐secreted proteins and plant functions in a late compatible plant–rust interaction. Mol. Plant Pathol. 13, 17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R.A. (1966) The Design of Experiments. Edinburgh: Oliver and Boyd. [Google Scholar]
- Gan, P.H. , Rafiqi, M. , Ellis, J.G. , Jones, D.A. , Hardham, A.R. and Dodds, P.N. (2010) Lipid binding activities of flax rust AvrM and AvrL567 effectors. Plant Signal. Behav. 5, 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnica, D.P. , Upadhyaya, N.M. , Dodds, P.N. and Rathjen, J.P. (2013) Strategies for wheat stripe rust pathogenicity identified by transcriptome sequencing. PLoS ONE, 8, e67150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabrial, S.A. and Suzuki, N. (2009) Viruses of plant pathogenic fungi. Annu. Rev. Phytopathol. 47, 353–384. [DOI] [PubMed] [Google Scholar]
- Godfrey, D. , Bohlenius, H. , Pedersen, C. , Zhang, Z. , Emmersen, J. and Thordal‐Christensen, H. (2010) Powdery mildew fungal effector candidates share N‐terminal Y/F/WxC‐motif. BMC Genomics, 11, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goellner, K. , Loehrer, M. , Langenbach, C. , Conrath, U. , Koch, E. and Schaffrath, U. (2010) Phakopsora pachyrhizi, the causal agent of Asian soybean rust. Mol. Plant Pathol. 11, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, D. , Abajian, C. and Green, P. (1998) Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202. [DOI] [PubMed] [Google Scholar]
- Götz, S. , Garcia‐Gomez, J.M. , Terol, J. , Williams, T.D. , Nagaraj, S.H. , Nueda, M.J. , Robles, M. , Talon, M. , Dopazo, J. and Conesa, A. (2008) High‐throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, M.A. , Silverstein, K.A. , Cannon, S.B. and VandenBosch, K.A. (2004) Computational identification and characterization of novel genes from legumes. Plant Physiol. 135, 1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, M.A. , Silverstein, K.A.T. and VandenBosch, K.A. (2008) Defensin‐like genes: genomic perspectives on a diverse superfamily in plants. Crop Sci. 48, S3–S11. [Google Scholar]
- Hacquard, S. , Joly, D.L. , Lin, Y.C. , Tisserant, E. , Feau, N. , Delaruelle, C. , Legue, V. , Kohler, A. , Tanguay, P. , Petre, B. , Frey, P. , Van de Peer, Y. , Rouze, P. , Martin, F. , Hamelin, R.C. and Duplessis, S. (2012) A comprehensive analysis of genes encoding small secreted proteins identifies candidate effectors in Melampsora larici‐populina (poplar leaf rust). Mol. Plant–Microbe Interact. 25, 279–293. [DOI] [PubMed] [Google Scholar]
- Hahn, M. and Mendgen, K. (1992) Isolation by ConA binding of haustoria from different rust fungi and comparison of their surface qualities. Protoplasma, 170, 95–103. [Google Scholar]
- Hahn, M. and Mendgen, K. (1997) Characterization of in planta‐induced rust genes isolated from a haustorium‐specific cDNA library. Mol. Plant–Microbe Interact. 10, 427–437. [DOI] [PubMed] [Google Scholar]
- Huang, X. and Madan, A. (1999) CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icho, T. and Wickner, R.B. (1989) The double‐stranded RNA genome of yeast virus L‐A encodes its own putative RNA polymerase by fusing two open reading frames. J. Biol. Chem. 264, 6716–6723. [PubMed] [Google Scholar]
- Jakupovic, M. , Heintz, M. , Reichmann, P. , Mendgen, K. and Hahn, M. (2006) Microarray analysis of expressed sequence tags from haustoria of the rust fungus Uromyces fabae . Fungal Genet. Biol. 43, 8–19. [DOI] [PubMed] [Google Scholar]
- Joly, D.L. , Feau, N. , Tanguay, P. and Hamelin, R.C. (2010) Comparative analysis of secreted protein evolution using expressed sequence tags from four poplar leaf rusts (Melampsora spp). BMC Genomics, 11, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale, S.D. , Gu, B. , Capelluto, D.G. , Dou, D. , Feldman, E. , Rumore, A. , Arredondo, F.D. , Hanlon, R. , Fudal, I. , Rouxel, T. , Lawrence, C.B. , Shan, W. and Tyler, B. (2010) External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell, 142, 284–295. [DOI] [PubMed] [Google Scholar]
- Kamoun, S. (2006) A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 44, 41–60. [DOI] [PubMed] [Google Scholar]
- Kämper, J. , Kahmann, R. , Bölker, M. , Ma, L.J. , Brefort, T. , Saville, B.J. , Banuett, F. , Kronstad, J.W. , Gold, S.E. , Müller, O. , Perlin, M.H. , Wösten, H.A. , de Vries, R. , Ruiz‐Herrera, J. , Reynaga‐Peña, C.G. , Snetselaar, K. , McCann, M. , Pérez‐Martín, J. , Feldbrügge, M. , Basse, C.W. , Steinberg, G. , Ibeas, J.I. , Holloman, W. , Guzman, P. , Farman, M. , Stajich, J.E. , Sentandreu, R. , González‐Prieto, J.M. , Kennell, J.C. , Molina, L. , Schirawski, J. , Mendoza‐Mendoza, A. , Greilinger, D. , Münch, K. , Rössel, N. , Scherer, M. , Vraneš, M. , Ladendorf, O. , Vincon, V. , Fuchs, U. , Sandrock, B. , Meng, S. , Ho, E.C. , Cahill, M.J. , Boyce, K.J. , Klose, J. , Klosterman, S.J. , Deelstra, H.J. , Ortiz‐Castellanos, L. , Li, W. , Sanchez‐Alonso, P. , Schreier, P.H. , Häuser‐Hahn, I. , Vaupel, M. , Koopmann, E. , Friedrich, G. , Voss, H. , Schlüter, T. , Margolis, J. , Platt, D. , Swimmer, C. , Gnirke, A. , Chen, F. , Vysotskaia, V. , Mannhaupt, G. , Güldener, U. , Münsterkötter, M. , Haase, D. , Oesterheld, M. , Mewes, H.W. , Mauceli, E.W. , DeCaprio, D. , Wade, C.M. , Butler, J. , Young, S. , Jaffe, D.B. , Calvo, S. , Nusbaum, C. , Galagan, J. and Birren, B. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature, 444, 97–101. [DOI] [PubMed] [Google Scholar]
- Kemen, E. , Kemen, A.C. , Rafiqi, M. , Hempel, U. , Mendgen, K. , Hahn, M. and Voegele, R.T. (2005) Identification of a protein from rust fungi transferred from haustoria into infected plant cells. Mol. Plant–Microbe Interact. 18, 1130–1139. [DOI] [PubMed] [Google Scholar]
- Kessler, S.A. , Shimosato‐Asano, H. , Keinath, N.F. , Wuest, S.E. , Ingram, G. , Panstruga, R. and Grossniklaus, U. (2010) Conserved molecular components for pollen tube reception and fungal invasion. Science, 330, 968–971. [DOI] [PubMed] [Google Scholar]
- Lawrence, G.J. , Dodds, P.N. and Ellis, J.G. (2010) Transformation of the flax rust fungus, Melampsora lini: selection via silencing of an avirulence gene. Plant J. 61, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. , Tsai, J. , Sunkara, S. , Karamycheva, S. , Pertea, G. , Sultana, R. , Antonescu, V. , Chan, A. , Cheung, F. and Quackenbush, J. (2005) The TIGR Gene Indices: clustering and assembling EST and known genes and integration with eukaryotic genomes. Nucleic Acids Res. 33, D71–D74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Esker, P.D. , Pan, Z. , Dias, A.P. , Xue, L. and Yang, X.B. (2010) The uniqueness of the soybean rust pathosystem – an improved understanding of the risk in different regions of the world. Plant Dis. 94, 796–806. [DOI] [PubMed] [Google Scholar]
- Liebenberg, M.M. and Pretorius, Z.A. (2010) Common bean rust: pathology and control In: Horticultural Reviews (Janick J., ed.), pp. 1–99. Hoboken, NJ: Wiley‐Blackwell. [Google Scholar]
- Link, T. , Lohaus, G. , Heiser, I. , Mendgen, K. , Hahn, M. and Voegele, R.T. (2005) Characterization of a novel NADP+‐dependent d‐arabitol dehydrogenase from the plant pathogen Uromyces fabae . Biochem. J. 389, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link, T.I. and Voegele, R.T. (2008) Secreted proteins of Uromyces fabae: similarities and stage specificity. Mol. Plant Pathol. 9, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, E. , Costa, L.M. and Gutierrez‐Marcos, J. (2011) Cysteine‐rich peptides (CRPs) mediate diverse aspects of cell–cell communication in plant reproduction and development. J. Exp. Bot. 62, 1677–1686. [DOI] [PubMed] [Google Scholar]
- Nowara, D. , Gay, A. , Lacomme, C. , Shaw, J. , Ridout, C. , Douchkov, D. , Hensel, G. , Kumlehn, J. and Schweizer, P. (2010) HIGS: host‐induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis . Plant Cell, 22, 3130–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes, C.C. and Dean, R.A. (2012) Host‐induced gene silencing: a tool for understanding fungal host interaction and for developing novel disease control strategies. Mol. Plant Pathol. 13, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss, D.L. (2005) Hypovirulence: mycoviruses at the fungal–plant interface. Nat. Rev. Microbiol. 3, 632–642. [DOI] [PubMed] [Google Scholar]
- Oliva, R. , Win, J. , Raffaele, S. , Boutemy, L. , Bozkurt, T.O. , Chaparro‐Garcia, A. , Segretin, M.E. , Stam, R. , Schornack, S. , Cano, L.M. , van Damme, M. , Huitema, E. , Thines, M. , Banfield, M.J. and Kamoun, S. (2010) Recent developments in effector biology of filamentous plant pathogens. Cell. Microbiol. 12, 705–715. [DOI] [PubMed] [Google Scholar]
- Page, R.D. (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12, 357–358. [DOI] [PubMed] [Google Scholar]
- Pierleoni, A. , Martelli, P.L. and Casadio, R. (2008) PredGPI: a GPI‐anchor predictor. BMC Bioinformatics, 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada‐Buitrago, M.L. and Frederick, R.D. (2005) Expressed sequence tag analysis of the soybean rust pathogen Phakopsora pachyrhizi . Fungal Genet. Biol. 42, 949–962. [DOI] [PubMed] [Google Scholar]
- Pretsch, K. , Kemen, A.C. , Kemen, E. , Geiger, M. , Mendgen, K. and Voegele, R.T. (2013) The rust transferred proteins – a new family of effector proteins exhibiting protease inhibitor function. Mol. Plant Pathol. 14, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthoff, D.P. , Neelam, A. , Ehrenfried, M.L. , Scheffler, B.E. , Ballard, L. , Song, Q. , Campbell, K.B. , Cooper, B. and Tucker, M.L. (2008) Analysis of expressed sequence tags from Uromyces appendiculatus hyphae and haustoria and their comparison to sequences from other rust fungi. Phytopathology, 98, 1126–1135. [DOI] [PubMed] [Google Scholar]
- Raffaele, S. , Farrer, R.A. , Cano, L.M. , Studholme, D.J. , MacLean, D. , Thines, M. , Jiang, R.H. , Zody, M.C. , Kunjeti, S.G. , Donofrio, N.M. , Meyers, B.C. , Nusbaum, C. and Kamoun, S. (2010) Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science, 330, 1540–1543. [DOI] [PubMed] [Google Scholar]
- Rehmany, A.P. , Gordon, A. , Rose, L.E. , Allen, R.L. , Armstrong, M.R. , Whisson, S.C. , Kamoun, S. , Tyler, B.M. , Birch, P.R.J. and Beynon, J.L. (2005) Differential recognition of highly divergent downy mildew avirulence gene alleles by RPP1 resistance genes from two Arabidopsis lines. Plant Cell, 17, 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rep, M. (2005) Small proteins of plant‐pathogenic fungi secreted during host colonization. FEMS Microbiol. Lett. 253, 19–27. [DOI] [PubMed] [Google Scholar]
- Sato, S. , Nakamura, Y. , Kaneko, T. , Asamizu, E. and Tabata, S. (1999) Complete structure of the chloroplast genome of Arabidopsis thaliana . DNA Res. 6, 283–290. [DOI] [PubMed] [Google Scholar]
- Saunders, D.G. , Win, J. , Cano, L.M. , Szabo, L.J. , Kamoun, S. and Raffaele, S. (2012) Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PLoS ONE, 7, e29847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirawski, J. , Mannhaupt, G. , Munch, K. , Brefort, T. , Schipper, K. , Doehlemann, G. , Di Stasio, M. , Rossel, N. , Mendoza‐Mendoza, A. , Pester, D. , Muller, O. , Winterberg, B. , Meyer, E. , Ghareeb, H. , Wollenberg, T. , Munsterkotter, M. , Wong, P. , Walter, M. , Stukenbrock, E. , Guldener, U. and Kahmann, R. (2010) Pathogenicity determinants in smut fungi revealed by genome comparison. Science, 330, 1546–1548. [DOI] [PubMed] [Google Scholar]
- Schmutz, J. , Cannon, S.B. , Schlueter, J. , Ma, J. , Mitros, T. , Nelson, W. , Hyten, D.L. , Song, Q. , Thelen, J.J. , Cheng, J. , Xu, D. , Hellsten, U. , May, G.D. , Yu, Y. , Sakurai, T. , Umezawa, T. , Bhattacharyya, M.K. , Sandhu, D. , Valliyodan, B. , Lindquist, E. , Peto, M. , Grant, D. , Shu, S. , Goodstein, D. , Barry, K. , Futrell‐Griggs, M. , Abernathy, B. , Du, J. , Tian, Z. , Zhu, L. , Gill, N. , Joshi, T. , Libault, M. , Sethuraman, A. , Zhang, X.C. , Shinozaki, K. , Nguyen, H.T. , Wing, R.A. , Cregan, P. , Specht, J. , Grimwood, J. , Rokhsar, D. , Stacey, G. , Shoemaker, R.C. and Jackson, S.A. (2010) Genome sequence of the palaeopolyploid soybean. Nature, 463, 178–183. [DOI] [PubMed] [Google Scholar]
- Schneider, R.W. , Hollier, C.A. , Whitam, H.K. , Palm, M.E. , McKemy, J.M. , Hernández, J.R. , Levy, L. and DeVries‐Paterson, R. (2005) First report of soybean rust caused by Phakopsora pachyrhizi in the continental United States. Plant Dis. 89, 774. [DOI] [PubMed] [Google Scholar]
- Sohn, J. , Voegele, R.T. , Mendgen, K. and Hahn, M. (2000) High level activation of vitamin B1 biosynthesis genes in haustoria of the rust fungus Uromyces fabae . Mol. Plant–Microbe Interact. 13, 629–636. [DOI] [PubMed] [Google Scholar]
- Sohn, K.H. , Lei, R. , Nemri, A. and Jones, J.D. (2007) The downy mildew effector proteins ATR1 and ATR13 promote disease susceptibility in Arabidopsis thaliana . Plant Cell, 19, 4077–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Win, J. , Tian, M. , Schornack, S. , Kaschani, F. , Ilyas, M. , van der Hoorn, R.A. and Kamoun, S. (2009) Apoplastic effectors secreted by two unrelated eukaryotic plant pathogens target the tomato defense protease Rcr3. Proc. Natl. Acad. Sci. USA, 106, 1654–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer, E.L. , von Heijne, G. and Krogh, A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182. [PubMed] [Google Scholar]
- Stergiopoulos, I. and de Wit, P.J.G.M. (2009) Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263. [DOI] [PubMed] [Google Scholar]
- Struck, C. , Ernst, M. and Hahn, M. (2002) Characterization of a developmentally regulated amino acid transporter (AAT1p) of the rust fungus Uromyces fabae . Mol. Plant Pathol. 3, 23–30. [DOI] [PubMed] [Google Scholar]
- Struck, C. , Mueller, E. , Martin, H. and Lohaus, G. (2004) The Uromyces fabae UfAAT3 gene encodes a general amino acid permease that prefers uptake of in planta scarce amino acids. Mol. Plant Pathol. 5, 183–189. [DOI] [PubMed] [Google Scholar]
- Swofford, D.L. (2003) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Taylor, D.J. and Bruenn, J. (2009) The evolution of novel fungal genes from non‐retroviral RNA viruses. BMC Biol. 7, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D. , Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, A. , Li, S. , Scheffler, B. and Matthews, B.F. (2009) Laser capture microdissection and expressed sequence tag analysis of uredinia formed by Phakopsora pachyrhizi, the causal agent of Asian soybean rust. Physiol. Mol. Plant Pathol. 73, 163–174. [Google Scholar]
- Tremblay, A. , Hosseini, P. , Alkharouf, N.W. and Matthews, B.F. (2012) Identification of genes expressed by Phakopsora pachyrhizi, the pathogen causing soybean rust, at a late stage of infection of susceptible soybean leaves. Plant Pathol. 61, 773–786. [Google Scholar]
- Unseld, M. , Marienfeld, J.R. , Brandt, P. and Brennicke, A. (1997) The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 15, 57–61. [DOI] [PubMed] [Google Scholar]
- Voegele, R.T. and Mendgen, K. (2003) Rust haustoria: nutrient uptake and beyond. New Phytol. 159, 93–100. [DOI] [PubMed] [Google Scholar]
- Voegele, R.T. and Mendgen, K.W. (2011) Nutrient uptake in rust fungi: how sweet is parasitic life? Euphytica, 179, 41–55. [Google Scholar]
- Voegele, R.T. , Struck, C. , Hahn, M. and Mendgen, K. (2001) The role of haustoria in sugar supply during infection of broad bean by the rust fungus Uromyces fabae . Proc. Natl. Acad. Sci. USA, 98, 8133–8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegele, R.T. , Hahn, M. , Lohaus, G. , Link, T. , Heiser, I. and Mendgen, K. (2005) Possible roles for mannitol and mannitol dehydrogenase in the biotrophic plant pathogen Uromyces fabae . Plant Physiol. 137, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegele, R.T. , Hahn, M. and Mendgen, K. (2009) The Uredinales: cytology, biochemistry, and molecular biology In: The Mycota V. Plant Relationships (Deising H., ed.), pp. 69–98. Berlin, Heidelberg: Springer. [Google Scholar]
- Weßling, R. , Schmidt, S.M. , Micali, C.O. , Knaust, F. , Reinhardt, R. , Neumann, U. , Ver Loren van Themaat, E. and Panstruga, R. (2012) Transcriptome analysis of enriched Golovinomyces orontii haustoria by deep 454 pyrosequencing. Fungal Genet. Biol. 49, 470–482. [DOI] [PubMed] [Google Scholar]
- Wirsel, S.G. , Voegele, R.T. and Mendgen, K.W. (2001) Differential regulation of gene expression in the obligate biotrophic interaction of Uromyces fabae with its host Vicia faba . Mol. Plant–Microbe Interact. 14, 1319–1326. [DOI] [PubMed] [Google Scholar]
- Xue, C. , Park, G. , Choi, W. , Zheng, L. , Dean, R.A. and Xu, J.R. (2002) Two novel fungal virulence genes specifically expressed in appressoria of the rice blast fungus. Plant Cell, 14, 2107–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Qu, Z. , Zheng, W. , Liu, B. , Wang, X. , Xue, X. , Xu, L. , Huang, L. , Han, Q. , Zhao, J. and Kang, Z. (2008) Stage‐specific gene expression during urediniospore germination in Puccinia striiformis f. sp tritici . BMC Genomics, 9, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X.L. , Stumpf, M.A. , Hoch, H.C. and Kung, C. (1991) A mechanosensitive channel in whole cells and in membrane patches of the fungus Uromyces . Science, 253, 1415–1417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Sequence length distribution of contigs greater than 100 bp before application of bioinformatics filters: (A) Phakopsora pachyrhizi; (B) Uromyces appendiculatus.
Fig. S2 Phylogeny for gene family cluster 2456.
Fig. S3 Uf‐ISSP41 is expressed in pre‐haustorial in vitro infection structures but not in haustoria.
Fig. S4 Gene family cluster 1_2.
Fig. S5 Phylogeny for gene family cluster 20.
Fig. S6 Conserved amino acid motif in gene family cluster 1_0_152 (WebLogo).
File S1 FASTA file of the Phakopsora pachyrhizi haustorial transcriptome assembly.
File S2 FASTA file of the Uromyces appendiculatus haustorial transcriptome assembly.
File S3 List of Phakopsora pachyrhizi contigs from the haustorial transcriptome assembly encoding proteins with predicted secretion signals.
File S4 List of Uromyces appendiculatus contigs from the haustorial transcriptome assembly encoding proteins with predicted secretion signals.
File S5 FASTA file of the Phakopsora pachyrhizi Gene Index (PpGI) contigs assembled from P. pachyrhizi expressed sequence tag (EST) sequences available in GenBank.
Table S1 Phakopsora pachyrhizi haustorial transcriptome.
Table S2 Uromyces appendiculatus haustorial transcriptome.
Table S3 Phakopsora pachyrhizi pre‐haustorial PpGI expressed sequence tag (EST) contigs.
Table S4 Comparison of transcription between gene ontology (GO) categories in Phakopsora pachyrhizi resting urediospores and water‐germinated spores and P. pachyrhizi haustoria.
Table S5 Single linkage clustering results.
Table S6 Basidiomycete species screened for homologies.