

Summary
The necrotrophic fungal pathogen Pyrenophora tritici‐repentis causes tan spot, a major disease of wheat, throughout the world. The proteinaceous effector ToxA is responsible for foliar necrosis on ToxA‐sensitive wheat genotypes. The single copy ToxA gene was deleted from a wild‐type race 1 P. tritici‐repentis isolate via homologous recombination of a knockout construct. Expression of the ToxA transcript was found to be absent in transformants (toxa), as was ToxA protein production in fungal culture filtrates. Plant bioassays were conducted to test transformant pathogenicity. The toxa strains were unable to induce necrosis on ToxA‐sensitive wheat genotypes. To our knowledge, this is the first demonstration of a targeted gene knockout in P. tritici‐repentis. The ability to undertake gene deletions will facilitate the characterization of other pathogenicity effectors of this economically significant necrotroph.
Keywords: homologous recombination, transformation, yellow leaf spot, yellow spot, necrotrophic effector, toxin, Triticum aestivum
Introduction
The necrotrophic fungus Pyrenophora tritici‐repentis (Died.) Drechs. [anamorph: Drechslera tritici‐repentis (Died.) Shoem.] is an economically significant pathogen and is the causal agent of tan (or yellow) spot, a devastating foliar disease of wheat. This leaf spotting disease affects the major wheat (Triticum aestivum L.) and durum wheat (T. turgidum L.) growing areas of the world, and causes severe yield losses by reducing the leaf photosynthetic area (De Wolf et al., 1998). It is the most economically damaging disease of wheat in Australia (Murray and Brennan, 2009).
Pyrenophora tritici‐repentis produces at least three effectors (host‐selective toxins), referred to as ToxA, ToxB and ToxC (Lamari et al., 2003). These effectors interact in a highly specific manner with the host plant (Tan et al., 2010), leading to the development of two distinct foliar symptoms: tan necrosis and/or extensive chlorosis. The type of symptom depends on both the effectors produced by a particular isolate and the susceptibility genes present in the infected wheat host. So far, eight races of P. tritici‐repentis have been defined, based on their ability to produce the three effectors alone or in combination (Lamari et al., 2003). ToxA causes the most severe damage by inducing necrosis on the leaves of ToxA‐sensitive wheat genotypes (possessing the Tsn1 susceptibility gene) (Faris et al., 2010), whereas ToxB and ToxC both induce chlorosis, although on different wheat genotypes harbouring the Tsc2 and Tsc1 loci, respectively (Effertz et al., 2002; Friesen and Faris, 2004). ToxA is the predominant effector in the tan spot–wheat pathosystem, and is present in the majority of isolates worldwide (Friesen et al., 2006). A single copy gene (ToxA) encodes the small (13.2 kDa) secreted ToxA protein (Ciuffetti et al., 1997; Tuori et al., 1995). The ToxA gene is sufficient for the pathogenicity of P. tritici‐repentis, as transformation of a non‐pathogenic isolate with ToxA was sufficient to render that isolate pathogenic on ToxA‐sensitive wheat lines (Ciuffetti et al., 1997).
ToxA was first identified over 20 years ago as a necrosis toxin (Ballance et al., 1989), and isolation of the ToxA gene followed 8 years later (Ciuffetti et al., 1997). However, there have been no reports of ToxA gene knockout mutants, and moreover, to our knowledge, there have been no reports of any gene deletions of P. tritici‐repentis. Recent work has been successful in the generation of partial knockdown mutants of P. tritici‐repentis using a sense‐ and antisense‐mediated RNA‐silencing mechanism to reduce the expression of ToxB and an exo‐1,3‐β‐glucanase gene (Aboukhaddour et al., 2012; Fu et al., 2013). Here, we report the successful generation of a ToxA knockout strain, and investigate mutant pathogenicity on a set of differential wheat genotypes. To our knowledge, this is the first study in which a gene has been successfully deleted in P. tritici‐repentis.
Results
ToxA gene disruption
Using a polymerase chain reaction (PCR) fusion strategy, a phleomycin resistance cassette (PhleoR) was integrated into the ToxA target site by means of flanking sequences homologous to the targeted ToxA locus. Protoplasts of an Australian P. tritici‐repentis isolate (M4) were transformed with the knockout construct, and putative transformants were screened via PCR for the absence of the ToxA gene. Four transformants found to have undergone homologous recombination were selected for further study, and were designated as toxa‐1, toxa‐2, toxa‐3 and toxa‐4. Absence of ToxA gene expression was confirmed in all four transformants based on reverse transcription‐polymerase chain reaction (RT‐PCR) analysis (Fig. 1a).
Figure 1.
Confirmation of four independent ToxA knockout strains. (a) Reverse transcription‐polymerase chain reaction (RT‐PCR) detection of ToxA transcript (PCR product size of 393 bp) in the four knockout strains and wild‐type (WT), as visualized by agarose gel electrophoresis. Act1 was included as a positive control (PCR product size of 150 bp). (b) Correct integration of the ToxA gene deletion cassette at 5′ and 3′ ends in four replicates of the knockout strains. PCR product sizes for the 5′ and 3′ amplicons are 1.7 kb and 1.6 kb, respectively. (c) Phleomycin resistance cassette (PhleoR) copy number as detected by quantitative PCR (qPCR). Error bars depict standard deviation. (d) Sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) of culture filtrate proteins. Open arrows indicate ToxA (13.2 kDa). Two independent wild‐type (WT) culture filtrate samples were analysed.
To exclude the possibility that the ToxA gene deletion construct had integrated ectopically elsewhere in the genome, mutants were screened via PCR to ensure that the construct had integrated at the intended homologous site (Fig. 1b). Correct targeted integration was confirmed for all four transformants.
The copy number of the phleomycin cassette was determined for each of the four mutants via quantitative polymerase chain reaction (qPCR) (Fig. 1c). As expected, the phleomycin resistance gene was not detected in the wild‐type isolate. The PhleoR copy numbers revealed for each of the transformants were closely correlated with the known single copy of the Act1 actin gene in the P. tritici‐repentis genome sequence, thus confirming single integration of the ToxA knockout construct.
ToxA protein production was evaluated by sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) of crude culture filtrates of wild‐type, mutants and the race 5 isolate DW5, which does not produce ToxA (but instead produces ToxB) (Fig. 1d). The presence of notable bands with a mass of approximately 13.2 kDa, corresponding to the expected size of the ToxA protein (Tuori et al., 1995), was detected in the wild‐type isolate, but not in the four transformants or DW5, although faint bands of a similar size were visualized in the knockouts and race 5 isolate. Therefore, to confirm the absence of ToxA, these gel bands were excised from wild‐type, toxa‐1 and DW5, and subjected to peptide analysis by electrospray ionization mass spectrometry. As expected, ToxA was only identified within the wild‐type isolate, and was absent from knockout and DW5 culture filtrates, which both contained other low‐molecular‐weight proteins (Fig. S1, see Supporting Information).
Functional analysis of ToxA knockout mutants
In order to assess mutants for pathogenicity and to functionally confirm the deletion of ToxA, a set of five tan spot differential wheat genotypes, which differ in their effector sensitivities, were inoculated (Table 1) (Ciuffetti et al., 1997; Lamari et al., 1998, 2003). Also included was the ToxA‐sensitive line BG261 (a Parastagonospora nodorum ToxA differential line) (Friesen et al., 2006). Plants were inoculated with conidial suspensions of the wild‐type isolate and two of the independent knockout mutants (toxa‐1 and toxa‐2). As expected, the spreading necrosis typical of ToxA was observed on the ToxA‐sensitive lines (cv. Glenlea, BG261 and cv. Katepwa) following infection with the wild‐type isolate (Fig. 2). However, this spreading necrosis was absent on these lines following inoculation with the two mutants, thus functionally confirming toxa‐1 and toxa‐2 as lacking ToxA. The average disease score of the wild‐type isolate was determined to be significantly higher than that of the mutants on these wheat lines (Fig. 3).
Table 1.
Differential wheat genotypes and their corresponding effector sensitivities (adapted from Lamari et al., 1998). N and C denote necrosis and chlorosis, respectively, and a dash indicates insensitivity
| Effector | Differential wheat line | ||||
|---|---|---|---|---|---|
| Glenlea | 6B662 | Katepwa | 6B365 | Auburn | |
| ToxA | N | – | N | – | – |
| ToxB | – | C | C | – | – |
| ToxC | – | – | – | C | – |
Figure 2.

Reactions of a differential set of wheat lines to inoculation with wild‐type (WT), toxa mutants and DW5. Inoculation with 0.25% gelatin (Ctrl) is included as a negative control. Images were taken at 7 days post‐inoculation and show representative second leaf symptoms.
Figure 3.
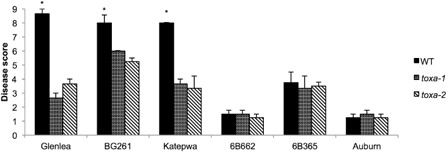
Pathogenicity assays of the ToxA knockout strains. Average disease scores of individual plants from one round of infection. Leaf symptoms were assessed for disease severity at 7 days post‐inoculation using a nine point scale, where 1 represents absence of infection and 9 denotes total necrosis. Analysis of variance (ANOVA) revealed a significant effect of fungal strain on disease score (P ≤ 0.05). Average disease scores with asterisks are significantly different as determined by Fisher's least‐significant difference (LSD) post‐hoc analysis (P ≤ 0.01).
All three strains were unable to induce chlorosis on the ToxB‐sensitive wheat line 6B662, although the wild‐type isolate and both mutants induced chlorosis on the ToxC‐sensitive wheat line 6B365 (Figs 2 and 3). No strain was able to successfully infect cv. Auburn, which is resistant to all races and insensitive to all known effectors produced by P. tritici‐repentis (Ciuffetti et al., 1997). Katepwa has been reported to be resistant to race 3 (ToxC‐producing) isolates (Lamari et al., 1998), and so the observation of mild chlorosis following inoculation with the ToxA knockouts is purportedly the result of an as yet unknown effector.
To further functionally validate the ToxA deletion, the differential wheat lines were infiltrated with crude culture filtrates of wild‐type, toxa‐1 and toxa‐2 mutant strains and purified ToxA protein (Fig. 4). The knockout mutants were unable to cause necrosis on the three ToxA‐sensitive lines (cv. Glenlea, BG261 and cv. Katepwa).
Figure 4.
Sensitivity of differential wheat lines infiltrated with culture filtrates of wild‐type (WT) and toxa mutants. Infiltrations with purified ToxA protein (ToxA) and Fries medium (Ctrl) are included as positive and negative controls, respectively. Images were taken at 10 days post‐infiltration and show representative second leaf symptoms.
The wild‐type isolate (M4) used in this study was collected from Australia, where, so far, all isolates tested (including M4) have been found to possess ToxA and to lack ToxB (Antoni et al., 2010). Inoculation with M4 conidia and culture filtrate infiltration induced chlorosis on the ToxC differential wheat line 6B365, thus confirming that this isolate also produces ToxC and hence belongs to race 1 (ToxA+, ToxB–, ToxC+). Both of the toxa mutants tested also retained this chlorosis‐inducing ability (Figs 2 and 4).
No observable effect on colony morphology, growth rate, sporulation or spore germination was detected in the knockouts compared with the wild‐type (data not shown).
Discussion
Deletion of ToxA from P. tritici‐repentis significantly reduced the extent of disease caused by this pathogen on the ToxA‐sensitive (Tsn1) wheat genotypes tested, and thus emphasizes the role of ToxA as a major necrotrophic effector. Although non‐ToxA‐containing isolates are found in certain parts of the world (e.g. North America, the Middle East and North Africa) (Friesen et al., 2005; Lamari et al., 2005; Strelkov et al., 2002), the majority of P. tritici‐repentis isolates worldwide produce ToxA (Friesen et al., 2006; Lamari and Strelkov, 2010). In recent years, an increase in the prevalence of tan spot disease, particularly in Europe, has been reported (Crop Monitor, 2008/2009, Jorgensen and Olsen, 2007); however, the proportion of isolates that produce ToxA has yet to be determined, and the distribution of ToxA within P. tritici‐repentis isolates worldwide requires further research.
The ToxA–Tsn1 interaction is the best characterized host–effector interaction identified in wheat. The dominant Tsn1 gene was identified on the long arm of chromosome 5B (Faris et al., 1996), and has been demonstrated to confer sensitivity to purified ToxA using Tsn1‐disrupted mutants (Liu et al., 2006). Cloning of Tsn1 revealed disease resistance gene features, such as serine/threonine protein kinase (S/TPK) and nucleotide‐binding site‐leucine‐rich repeat (NBS‐LRR) domains. All three domains are required for ToxA sensitivity, as demonstrated via induced mutagenesis (Faris et al., 2010). Although Tsn1 is required to mediate ToxA recognition, it is unlikely to be the actual ToxA receptor. The Tsn1 protein does not contain any apparent transmembrane domains, and thus is probably located within the cell cytoplasm, and yeast two‐hybrid assays suggest that the protein does not interact directly with ToxA (Faris et al., 2010). However, ToxA has been reported to interact with a chloroplast‐localized protein known as ToxABP1 (Manning et al., 2007). Viral‐induced gene silencing of ToxABP1 in wheat has been reported to reduce the extent of ToxA‐induced necrosis, although it is likely that other ToxA–host protein interactions are required for full necrosis (Manning et al., 2010).
In order to fully understand the P. tritici‐repentis–wheat pathosystem, the role of ToxA in disease needs to be determined for all ToxA‐producing races (races 1, 2, 7 and 8). An early report found that ToxA insensitivity was associated with resistance to race 2 (ToxA+ ToxB– ToxC–) (Lamari and Bernier, 1991). However, in another study, inoculation of this same race 2 isolate on different ToxA‐insensitive wheat genotypes resulted in necrotic lesions, although they developed more slowly compared with those on Tsn1 genotypes (Friesen et al., 2003). Therefore, although the production of ToxA is sufficient for pathogenicity (Ciuffetti et al., 1997), it does not appear to be necessary for disease on all host genotypes. Previous work to determine whether ToxA insensitivity equates to race 1 (ToxA+ ToxB– ToxC+) resistance has shown that wheat lines with mutations for ToxA insensitivity are susceptible following inoculation with two race 1 isolates (Friesen et al., 2002). This earlier work demonstrated that host sensitivity to ToxA is not necessarily equivalent to resistance to race 1, and one or more other effectors are in play, including ToxC. Indeed, our results agree with this, as we observed symptoms on BG261. The determination of the precise role of ToxA in tan spot disease will require further characterization of the ToxA knockout described herein, against a range of wheat genotypes, as well as the generation of toxa mutant strains of other races.
The ToxA–Tsn1 interaction has also been evaluated using segregating wheat lines. For example, a population of recombinant inbred lines (derived from Salamouni and Katepwa) has been evaluated for reaction to race 1 and race 2 isolates (Faris et al., 2012). Unsurprisingly, the Tsn1 locus was significantly associated with disease for all isolates tested. However, the amount of variation explained by Tsn1 varied considerably (ranging between 5% and 30%), which is suggestive of possible ToxA gene regulation variation among P. tritici‐repentis isolates. This has been demonstrated for ToxA‐producing isolates of another necrotrophic fungal pathogen of wheat, Parastagonospora nodorum (syn. Stagonospora nodorum; teleomorph: Phaeosphaeria nodorum), which expresses ToxA at different levels, such that expression of ToxA at higher levels causes more disease on Tsn1 wheat (Faris et al., 2011). It is also conceivable that broad‐spectrum or race non‐specific resistance mechanisms may impede ToxA–Tsn1 interactions (Faris and Friesen, 2005), or that the consequences of host–effector interactions are reduced or masked as a result of epistatic effects with other host–effector interactions (Friesen et al., 2008).
The ToxA gene has been successfully deleted in P. nodorum and, similar to the approach presented herein, culture filtrates of these mutants were infiltrated on the P. nodorum differential line BG261 and were unable to induce necrosis, demonstrating that ToxA is necessary for complete virulence on Tsn1 wheat lines (Friesen et al., 2006). The authors also performed infection assays on a wheat population segregating for Tsn1 (BR34 × Grandin), and found that the Tsn1 quantitative trait locus (QTL) associated with the disease phenotype was eliminated following inoculation with ToxA‐disrupted P. nodorum mutants. In the same study, expression of either ToxA from P. tritici‐repentis or from P. nodorum in an avirulent (ToxA‐lacking) P. nodorum strain was sufficient to confer virulence that co‐segregated with Tsn1. Despite predicted amino acid differences, this demonstrates that P. tritici‐repentis ToxA and P. nodorum ToxA are functionally identical in their interaction with Tsn1, and there is evidence that the ToxA gene was horizontally transferred from P. nodorum to P. tritici‐repentis (Friesen et al., 2006). The full suite of native effectors will differ between the two pathogens, and so it is important to dissect separately the role of ToxA within each pathosystem.
The work presented herein opens up several new vistas for future research. First, removal of ToxA, the predominant effector of P. tritici‐repentis, will facilitate the identification of other, as yet uncharacterized, effectors, whose effects have so far been masked by extensive ToxA‐induced necrosis. This is in agreement with previous work, whereby inoculation of Tsn1‐disrupted mutants still resulted in disease (Friesen et al., 2003), and sensitivity to ToxA was a non‐significant factor in disease development following inoculation of the BR34 × Grandin population with race 1 and 2 isolates (Faris and Friesen, 2005).
Second, the generation of toxa mutants will enable the screening of Tsn1 wheat cultivars for sensitivity to other novel effectors. In particular, this will permit the screening of ToxA‐sensitive wheat mapping populations for novel effector sensitivity and disease resistance QTLs, and thus facilitate the identification of molecular markers for wheat breeders and potential targets for crossing. Thus far, the identification of novel QTLs has been limited to just a handful of mapping populations with ToxA‐insensitive parents.
Third, the development of a targeted gene knockout method for such an economically significant and global wheat pathogen is noteworthy. Over the last 2 years, an RNA‐silencing method has been successfully developed to reduce gene expression in P. tritici‐repentis (Aboukhaddour et al., 2012; Fu et al., 2013). This technique was used to create ToxB‐silenced transformants, with ToxB production ranging from 15% to 81% of Western blot band intensity relative to the wild‐type (Aboukhaddour et al., 2012). In the case of ToxB, such an approach was well justified as the ToxB gene is found in multiple copies, the number of which varies among isolates (Martinez et al., 2001; Strelkov et al., 2005). Thus, to knock out all individual copies of ToxB would be inherently more challenging than RNA silencing. However, the RNA‐silencing strategy is less suitable for single copy effector gene discovery, as it results in partial gene knockdowns with variable expression levels, and thus will not eliminate gene expression entirely. Here, we show, as proof‐of‐concept, that P. tritici‐repentis possesses the necessary machinery to bring about the precise integration of exogenous sequences through homologous recombination, and paves the way for the creation of knockouts of other potential genes of interest, and the introduction of specific mutations into a target gene.
The results of this study reiterate the need to increase the area sown to ToxA‐insensitive wheat varieties and, ultimately, to phase out Tsn1 cultivars, particularly in wheat‐growing regions with a high proportion of ToxA‐expressing isolates. A recent wheat cultivar trial found that there was no yield penalty associated with growing ToxA‐insensitive varieties and, moreover, in the presence of disease, ToxA‐insensitive lines substantially outperformed the sensitive lines (Oliver et al., 2014).
In Australia, effector‐assisted breeding has been adopted in response to the significant combined losses caused by ToxA‐producing pathogens ($212 million caused by tan spot and $108 million caused by Septoria nodorum blotch) (Murray and Brennan, 2009). Semi‐purified ToxA has been delivered to wheat breeders since 2009 as a selection tool towards the development of disease‐resistant germplasm, with current delivery of 30 000 doses per annum (Vleeshouwers and Oliver, 2014). As a result, there has been a considerable decrease in the area sown to ToxA‐sensitive wheat varieties, a major step to reduce the huge scale of losses caused by tan spot. However, in accordance with this study and earlier findings, screening germplasm with ToxA should not be used in place of fungal inoculation by breeding programmes, as ToxA sensitivity is not always required for susceptibility to race 1. This is probably because of the presence of ToxC and other effectors not yet identified, and there are currently no Australian commercial wheat varieties rated as resistant to tan spot (DAFWA, 2014). A thorough understanding of the role of ToxA and other effectors in tan spot disease is required, and knockout capability can be expected to expedite strategies targeting the release of resistant lines.
Experimental Procedures
Fungal material and growth conditions
The pathogenic P. tritici‐repentis race 1 isolate M4 was collected from Meckering, Western Australia in 2009. PCR amplification of M4 gDNA confirmed the presence of the ToxA gene and the absence of ToxB using the primers ToxAscreeningF/R and TB10f/TB12r, respectively (Antoni et al., 2010). Fungi were grown on V8‐PDA plates (Campbell's V8 juice, 150 mL/L; potato dextrose agar, 10 g/L; CaCO3, 3 g/L; agar, 15 g/L) and incubated at 22 °C under 12‐h cycles of light. Sporulation was induced by flooding the plates with ultrapure water and flattening colonies using an L‐shaped glass rod. Plates were placed under near‐UV and fluorescent lights for 24 h, followed by incubation at 15 °C in darkness for 24 h. Liquid cultures were started with crushed mycelia in Fries medium (Liu et al., 2004), and grown at 27 °C and 100 rpm in darkness. For culture filtrates, liquid cultures were shaken for 3 days, followed by 2.5 weeks of stationary growth. The filtrate was harvested by filtration through sterile gauze, MiraCloth (Merck Millipore, Billerica, MA, USA), and passed through a 0.2‐μm syringe filter unit (Pall Life Sciences, Port Washington, NY, USA). For spore germination assays, 100 conidia suspended in water were germinated per strain on 1.5% agarose at 4 °C. After 17 h, the number of germ tubes was counted per conidium. Three independent replicates were performed per fungal strain.
Development of the ToxA knockout construct
A fusion PCR approach was undertaken for the inactivation of ToxA, whereby two homologous flanking regions and the phleomycin resistance cassette were amplified separately and then fused in a single PCR. Flanking regions of the ToxA gene (PTRG_04889) were amplified from genomic M4 DNA. A 1639‐bp upstream flanking region was amplified using PtrToxA5′f and PtrToxA5′r primers, whereas PtrToxA3′f and PtrToxA3′r primers were used to amplify a 1561‐bp downstream sequence. A phleomycin cassette (PhleoR) was amplified from pAN8‐1 using the primers pAN8f and pAN8r, as described previously (Solomon et al., 2006). Incorporated into the 5′ regions of the PtrToxA3′f and PtrToxA5′r primers were 25 bp and 23 bp of sequence homologous to the 3′ and 5′ ends of the phleomycin fragment. Fragments were gel extracted using a QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany), and equimolar amounts were combined as template for a single fusion PCR using primers PtrToxA5′f and PtrToxA3′r at a final concentration of 50 μm. The fusion PCR was performed using iProof High‐Fidelity Master Mix (Bio‐Rad, Hercules, CA, USA) with the following cycling conditions: 98 °C/30 s; (98 °C/5 s, 67 °C/30 s, 72 °C/5 min) × 35; 72 °C/5 min. The fusion PCR product (5624 bp) was gel extracted and resuspended in sterile water. A further amplification with nested primers (PtrToxA5′Nf and PtrToxA3′Nr) was performed to generate the final gene deletion construct. All primer sequences are detailed in Table 2.
Table 2.
Primers used throughout this study. Bold text refers to sequence complementary to the phleomycin resistance cassette primers (pAN8f and pAN8r)
| Primer name | Sequence |
|---|---|
| PtrToxA5′f | TCCGCTCTCGATTACCGGCTCA |
| PtrToxA5′r | TGTGACTTTTGGTTACGCCGTCTTGTCAATGTCGACTTGGCCGATG |
| PtrToxA3′f | TCTCCTATGAGTCGTTTACCCAGAATCAATGGGAATAAGTCTCCCCACCA |
| PtrToxA3′r | GCGCTCTCGGTACGCTCCTC |
| pAN8f | AGACGGCGTAACCAAAAGTCACA |
| pAN8r | TTCTGGGTAAACGACTCATAGGAGA |
| PtrToxA5′Nf | TGTTCGAGCCTGGTTCAGAT |
| PtrToxA3′Nr | CCTATCTTAAGGGCGGCTTC |
| PtrToxAF2 | ACCGGCAGGACTAATCGCCTCA |
| PtrToxAR2 | CCAACACGTGCCGTTCCGGT |
| Phleo5 | CTCCGTCTTCCGTAGCCGTG |
| Phleo3 | CCAATACGCCGGCCGAAAC |
| Act1F4 | CGAGACCTTCAACGCTCCCGC |
| Act1R4 | GCGTGGGGAAGAGCGAAACCC |
| PhleoF4 | GACCGAGATCGGCGAGCAGC |
| PhleoR4 | TCAAGCTCCTGGGACCCGTGG |
Fungal transformation
A polyethylene glycol (PEG)‐mediated protoplast transformation method was used (Aboukhaddour et al., 2012). Transformations were performed using 5 μg of DNA per 1 × 107 protoplasts. Protoplasts were overlaid with RM agar (Aboukhaddour et al., 2012) amended to a final concentration of 10 μg/mL phleomycin. Resistant colonies were transferred to V8‐PDA agar containing 15 μg/mL phleomycin for a second round of screening. Putative transformants were screened via PCR in order to verify the absence of the ToxA gene (primers PtrToxAF2 and PtrToxAR2) and correct genomic integration of the gene deletion construct (primer combinations PtrToxA5′f/Phleo5 and Phleo3/PtrToxA3′r which amplify the 5′ and 3′ flanking regions from positive transformants, respectively). The thermal cycling conditions were as follows: 94 °C/3 min; (94 °C/30 s, 58 °C/30 s, 72 °C/2 min) × 35; 72 °C/5 min. Single spore re‐isolation was performed for all true transformants to ensure mutant purity.
RNA extraction and transcript expression
Total RNA was isolated from 1‐week‐old fungal liquid cultures using TRIzol Reagent, according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). RNA was reversed transcribed using iScript reverse transcriptase (Bio‐Rad), according to the manufacturer's protocol. The resulting cDNA was quantified using a NanoDrop 2000 UV‐Vis spectrophotometer (ThermoScientific, Waltham, MA, USA). Primers PtrToxAF2 and PtrToxAR2 were used to test for ToxA expression in wild‐type and transformants. As a control, expression of the housekeeping actin gene (Act1) was tested using Act1F4 and Act1R4 primers. PCR products were visualized by agarose gel electrophoresis on a 1.5% agarose gel using SYBR Safe DNA Gel Stain (Life Technologies, Carlsbad, CA, USA).
Determination of copy number
qPCR was performed to confirm single integration of the ToxA knockout construct into the M4 genome, as has been described previously to determine the copy number of introduced gene cassettes in fungal transformants (Solomon et al., 2008). Genomic DNA was extracted from wild‐type and transformants using the Biosprint 15 DNA kit (Qiagen) according to the manufacturer's instructions. For detection of the ToxA knockout construct copy number, a 150‐bp region of the phleomycin resistance cassette (PhleoR) was amplified using the primers PhleoF4 and PhleoR4. As an endogenous control, primers Act1F4 and Act1R4 were used to amplify a 150‐bp fragment from the single copy actin gene (Act1) (Ellwood et al., 2012). Thermal cycling conditions were 95 °C/15 min, (94 °C/15 s, 63 °C/30 s, 72 °C/30 s) × 35, and were performed in a CFX96 Real‐Time PCR Detection System (Bio‐Rad). Each 20‐μL qPCR consisted of 50 ng of DNA, 10 μL of QuantiTect SYBR Green PCR mix (Qiagen) and 300 nm of the appropriate primers. PCR efficiencies of the target (PhleoR) and reference (Act1) qPCR amplifications were tested to be approximately equal. The PhleoR copy number was normalized to the Act1 copy number using the ΔΔC t method. Samples were analysed in triplicate with two technical replicates.
ToxA protein production
Fungal culture filtrates were analysed by SDS‐PAGE performed on a Mini‐PROTEAN 3 vertical gel apparatus (Bio‐Rad). Culture filtrates were passed through PD‐10 desalting columns to remove salts and low‐molecular‐weight impurities, according to the manufacturer's instructions (GE Healthcare, Fairfield, CT, USA). Proteins were resolved via a 16.5% polyacrylamide separating gel using the Tris–tricine buffer separation system (Schagger and Vonjagow, 1987). Approximately 40 μg of each total protein sample was loaded per lane and the Precision Plus Protein Standard (Bio‐Rad) was used as protein molecular weight standard. Gels were fixed and visualized via Coomassie G250 colloidal staining (Neuhoff et al., 1988). Bands of the expected ToxA size were excised individually from the gel for M4, DW5 and toxa‐1, trypsin digested and peptides were extracted according to standard techniques (Bringans et al., 2008). Peptides were analysed by electrospray ionization mass spectrometry using the Shimadzu Prominence nano‐high‐performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan) coupled to a 5600 triple time‐of‐flight mass spectrometer (AB Sciex, Framingham, MA, USA). Tryptic peptides were loaded onto an Agilent Zorbax 300SB‐C18, 3.5 μm (Agilent Technologies, Santa Clara, USA), and separated with a linear gradient of water–acetonitrile–0.1% formic acid (v/v/v). Spectra were analysed to identify proteins of interest using Mascot sequence matching software (Matrix Science, London, UK) with the Ludwig NR database.
Plant material and pathogenicity assays
Wheat seeds (Triticum aestivum L.) were obtained from the Australian Winter Cereals Collection (AWCC), Tamworth, NSW, Australia. For culture filtrate assays, seeds were sown in Grade 2 vermiculite (The Perlite and Vermiculite Factory, Jandakot, WA, Australia) in seed trays, and grown at 20 °C under a 12‐h day/night cycle in a controlled growth chamber. Fully extended leaves of 2‐week‐old wheat plants were infiltrated with crude culture filtrate from the wild‐type or toxa mutants. A needleless 1‐mL syringe was used to infiltrate the adaxial surface of second leaves, and the infiltration boundaries were marked with a permanent marker pen. Leaves were evaluated at 10 days post‐infiltration. All infiltration experiments were repeated twice with consistent results, using a minimum of four plants per line each time. The ToxA protein was purified as described previously (Tan et al., 2012) and infiltrated at 50 μg/mL.
For the infections, pots (10 cm in diameter) containing P500 perlite (The Perlite and Vermiculite Factory) and vermiculite were sown with four seeds and grown at 21 °C under a 12‐h day/night cycle. Inoculum was prepared consisting of approximately 2000 conidia/mL in 0.25% gelatin. Infection assays were performed by evenly spraying 2‐week‐old plants (at the two‐ to three‐leaf stage) with inoculum using a spray bottle until run‐off. The plants were incubated in a misting chamber for 2 days (relative humidity ≥ 95%) with continuous moisture supplied by a humidifier. At 7 days post‐inoculation, the second leaves were harvested and photographed, and plants were visually assessed for disease severity based on the Australian wheat disease resistance ratings scale (DAFWA, 2014), whereby varieties are rated between 1 and 9 (1, absence of infection (resistant); 9, total necrosis (very susceptible)). All infection experiments were independently repeated twice with consistent results, using a minimum of four replicates (pots) per treatment and performed as blind experiments.
Disease scores were analysed by analysis of variance (ANOVA) to determine any significant differences between isolate virulence (P ≤ 0.05), followed by Fisher's least significant difference (LSD) post‐hoc analysis to identify which mean values were significantly different (P ≤ 0.01). Prior to ANOVA, raw data were checked to ensure homogeneity of variance.
Supporting information
Fig. S1 Pyrenophora tritici‐repentis proteins identified from sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) excised bands of culture filtrates. (a) SDS‐PAGE of culture filtrate proteins (as shown in Fig. 1a) with arrows indicating excised bands. Two bands (upper and lower) were excised from three strains: wild‐type (WT), DW5 and toxa‐1. (b) The peptide fragmentation data from liquid chromatography‐tandem mass spectrometry (LC/MS/MS) were searched against the non‐redundant Ludwig NR database using the MASCOT sequence matching software. Only significant hits (score >35) with a significance threshold of P < 0.01 and a minimum of two peptide matches are shown.
Acknowledgements
This work was supported by grants from the Australian Grains Research and Development Corporation (GRDC). We thank T. Friesen (US Department of Agriculture) for the provision of isolate DW5 and wheat lines 6B662 and 6B365. We also thank the Australian Winter Cereals Collection (AWCC) for the provision of seeds.
References
- Aboukhaddour, R. , Kim, Y.M. and Strelkov, S.E. (2012) RNA‐mediated gene silencing of ToxB in Pyrenophora tritici‐repentis . Mol. Plant Pathol. 13, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoni, E.A. , Rybak, K. , Tucker, M.P. , Hane, J.K. , Solomon, P.S. , Drenth, A. , Shankar, M. and Oliver, R.P. (2010) Ubiquity of ToxA and absence of ToxB in Australian populations of Pyrenophora tritici‐repentis . Australas. Plant Pathol. 39, 63–68. [Google Scholar]
- Ballance, G.M. , Lamari, L. and Bernier, C.C. (1989) Purification and characterization of a host‐selective necrosis toxin from Pyrenophora tritici‐repentis . Physiol. Mol. Plant Pathol. 35, 203–213. [Google Scholar]
- Bringans, S. , Eriksen, S. , Kendrick, T. , Gopalakrishnakone, P. , Livk, A. , Lock, R. and Lipscombe, R. (2008) Proteomic analysis of the venom of Heterometrus longimanus (Asian black scorpion). Proteomics, 8, 1081–1096. [DOI] [PubMed] [Google Scholar]
- Ciuffetti, L.M. , Tuori, R.P. and Gaventa, J.M. (1997) A single gene encodes a selective toxin causal to the development of tan spot of wheat. Plant Cell, 9, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crop Monitor (2008/2009) Winter wheat survey, UK. Available at http://www.cropmonitor.co.uk/wwheat/surveys/highlight2009.cfm [accessed on Feb 3, 2014].
- DAFWA (2014) Wheat variety guide for WA 2014. Available at http://grdc.com.au/Resources/Publications/2014/02/Wheat‐Variety‐Guide‐for‐WA‐2014 [accessed on Feb 3, 2014].
- De Wolf, E.D. , Effertz, R.J. , Ali, S. and Francl, L.J. (1998) Vistas of tan spot research. Can. J. Plant Pathol. 20, 349–370. [Google Scholar]
- Effertz, R.J. , Meinhardt, S.W. , Anderson, J.A. , Jordahl, J.G. and Francl, L.J. (2002) Identification of a chlorosis‐inducing toxin from Pyrenophora tritici‐repentis and the chromosomal location of an insensitivity locus in wheat. Phytopathology, 92, 527–533. [DOI] [PubMed] [Google Scholar]
- Ellwood, S.R. , Syme, R.A. , Moffat, C.S. and Oliver, R.P. (2012) Evolution of three Pyrenophora cereal pathogens: recent divergence, speciation and evolution of non‐coding DNA. Fungal Genet. Biol. 49, 825–829. [DOI] [PubMed] [Google Scholar]
- Faris, J.D. and Friesen, T.L. (2005) Identification of quantitative trait loci for race‐nonspecific resistance to tan spot in wheat. Theor. Appl. Genet. 111, 386–392. [DOI] [PubMed] [Google Scholar]
- Faris, J.D. , Anderson, J.A. , Francl, L.J. and Jordahl, J.G. (1996) Chromosomal location of a gene conditioning insensitivity in wheat to a necrosis‐inducing culture filtrate from Pyrenophora tritici‐repentis . Phytopathology, 86, 459–463. [Google Scholar]
- Faris, J.D. , Zhang, Z.C. , Lu, H.J. , Lu, S.W. , Reddy, L. , Cloutier, S. , Fellers, J.P. , Meinhardt, S.W. , Rasmussen, J.B. , Xu, S.S. , Oliver, R.P. , Simons, K.J. and Friesen, T.L. (2010) A unique wheat disease resistance‐like gene governs effector‐triggered susceptibility to necrotrophic pathogens. Proc. Natl. Acad. Sci. USA, 107, 13 544–13 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faris, J.D. , Zhang, Z.C. , Rasmussen, J.B. and Friesen, T.L. (2011) Variable expression of the Stagonospora nodorum effector SnToxA among isolates is correlated with levels of disease in wheat. Mol. Plant–Microbe Interact. 24, 1419–1426. [DOI] [PubMed] [Google Scholar]
- Faris, J.D. , Abeysekara, N.S. , McClean, P.E. , Xu, S.S. and Friesen, T.L. (2012) Tan spot susceptibility governed by the Tsn1 locus and race‐nonspecific resistance quantitative trait loci in a population derived from the wheat lines Salamouni and Katepwa. Mol. Breed. 30, 1669–1678. [Google Scholar]
- Friesen, T.L. and Faris, J.D. (2004) Molecular mapping of resistance to Pyrenophora tritici‐repentis race 5 and sensitivity to Ptr ToxB in wheat. Theor. Appl. Genet. 109, 464–471. [DOI] [PubMed] [Google Scholar]
- Friesen, T.L. , Rasmussen, J.B. , Kwon, C.Y. , Francl, L.J. and Meinhardt, S.W. (2002) Reaction of Ptr ToxA‐insensitive wheat mutants to Pyrenophora tritici‐repentis race 1. Phytopathology, 92, 38–42. [DOI] [PubMed] [Google Scholar]
- Friesen, T.L. , Ali, S. , Kianian, S. , Francl, L.J. and Rasmussen, J.B. (2003) Role of host sensitivity to Ptr ToxA in development of tan spot of wheat. Phytopathology, 93, 397–401. [DOI] [PubMed] [Google Scholar]
- Friesen, T.L. , Ali, S. , Klein, K.K. and Rasmussen, J.B. (2005) Population genetic analysis of a global collection of Pyrenophora tritici‐repentis, causal agent of tan spot of wheat. Phytopathology, 95, 1144–1150. [DOI] [PubMed] [Google Scholar]
- Friesen, T.L. , Stukenbrock, E.H. , Liu, Z.H. , Meinhardt, S. , Ling, H. , Faris, J.D. , Rasmussen, J.B. , Solomon, P.S. , McDonald, B.A. and Oliver, R.P. (2006) Emergence of a new disease as a result of interspecific virulence gene transfer. Nat. Genet. 38, 953–956. [DOI] [PubMed] [Google Scholar]
- Friesen, T.L. , Zhang, Z.C. , Solomon, P.S. , Oliver, R.P. and Faris, J.D. (2008) Characterization of the interaction of a novel Stagonospora nodorum host‐selective toxin with a wheat susceptibility gene. Plant Physiol. 146, 682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H. , Feng, J. , Aboukhaddour, R. , Cao, T. , Hwang, S.F. and Strelkov, S.E. (2013) An exo‐1,3‐beta‐glucanase GLU1 contributes to the virulence of the wheat tan spot pathogen Pyrenophora tritici‐repentis . Fungal Biol. 117, 673–681. [DOI] [PubMed] [Google Scholar]
- Jorgensen, L.N. and Olsen, L.V. (2007) Control of tan spot (Drechslera tritici‐repentis) using cultivar resistance, tillage methods and fungicides. Crop Prot. 26, 1606–1616. [Google Scholar]
- Lamari, L. and Bernier, C.C. (1991) Genetics of tan necrosis and extensive chlorosis in tan spot of wheat caused by Pyrenophora‐tritici‐repentis . Phytopathology, 81, 1092–1095. [Google Scholar]
- Lamari, L. and Strelkov, S.E. (2010) The wheat/Pyrenophora tritici‐repentis interaction: progress towards an understanding of tan spot disease. Can. J. Plant Pathol. 32, 4–10. [Google Scholar]
- Lamari, L. , Gilbert, J. and Tekauz, A. (1998) Race differentiation in Pyrenophora tritici‐repentis and survey of physiologic variation in western Canada. Can. J. Plant Pathol. 20, 396–400. [Google Scholar]
- Lamari, L. , Strelkov, S.E. , Yahyaoui, A. , Orabi, J. and Smith, R.B. (2003) The identification of two new races of Pyrenophora tritici‐repentis from the host center of diversity confirms a one‐to‐one relationship in tan spot of wheat. Phytopathology, 93, 391–396. [DOI] [PubMed] [Google Scholar]
- Lamari, L. , Strelkov, S.E. , Yahyaoui, A. , Amedov, M. , Saidov, M. , Djunusova, M. and Koichibayev, M. (2005) Virulence of Pyrenophora tritici‐repentis in the countries of the Silk Road. Can. J. Plant Pathol. 27, 383–388. [Google Scholar]
- Liu, Z.H. , Faris, J.D. , Meinhardt, S.W. , Ali, S. , Rasmussen, J.B. and Friesen, T.L. (2004) Genetic and physical mapping of a gene conditioning sensitivity in wheat to a partially purified host‐selective toxin produced by Stagonospora nodorum . Phytopathology, 94, 1056–1060. [DOI] [PubMed] [Google Scholar]
- Liu, Z.H. , Friesen, T.L. , Ling, H. , Meinhardt, S.W. , Oliver, R.P. , Rasmussen, J.B. and Faris, J.D. (2006) The Tsn1–ToxA interaction in the wheat–Stagonospora nodorum pathosystem parallels that of the wheat–tan spot system. Genome, 49, 1265–1273. [DOI] [PubMed] [Google Scholar]
- Manning, V.A. , Hardison, L.K. and Ciuffetti, L.M. (2007) Ptr ToxA interacts with a chloroplast‐localized protein. Mol. Plant–Microbe Interact. 20, 168–177. [DOI] [PubMed] [Google Scholar]
- Manning, V.A. , Chu, A.L. , Scofield, S.R. and Ciuffetti, L.M. (2010) Intracellular expression of a host‐selective toxin, ToxA, in diverse plants phenocopies silencing of a ToxA‐interacting protein, ToxABP1. New Phytol. 187, 1034–1047. [DOI] [PubMed] [Google Scholar]
- Martinez, J.P. , Ottum, S.A. , Ali, S. , Franci, L.J. and Ciuffetti, L.M. (2001) Characterization of the ToxB gene from Pyrenophora tritici‐repentis . Mol. Plant–Microbe Interact. 14, 675–677. [DOI] [PubMed] [Google Scholar]
- Murray, G.M. and Brennan, J.P. (2009) Estimating disease losses to the Australian wheat industry. Australas. Plant Pathol. 38, 558–570. [Google Scholar]
- Neuhoff, V. , Arold, N. , Taube, D. and Ehrhardt, W. (1988) Improved staining of proteins in polyacrylamide gels including isoelectric‐focusing gels with clear background at nanogram sensitivity using Coomassie brilliant blue G‐250 and R‐250. Electrophoresis, 9, 255–262. [DOI] [PubMed] [Google Scholar]
- Oliver, R. , Lichtenzveig, J. , Tan, K.C. , Waters, O. , Rybak, K. , Lawrence, J. , Friesen, T. and Burgess, P. (2014) Absence of detectable yield penalty associated with insensitivity to Pleosporales necrotrophic effectors in wheat grown in the West Australian wheat belt. Plant Pathol. DOI: 10.1111/ppa.12191. [DOI] [Google Scholar]
- Schagger, H. and Vonjagow, G. (1987) Tricine sodium dodecyl‐sulfate polyacrylamide‐gel electrophoresis for the separation of proteins in the range from 1‐kDa to 100‐kDa. Anal. Biochem. 166, 368–379. [DOI] [PubMed] [Google Scholar]
- Solomon, P.S. , Rybak, K. , Trengove, R.D. and Oliver, R.P. (2006) Investigating the role of calcium/calmodulin‐dependent protein kinases in Stagonospora nodorum . Mol. Microbiol. 62, 367–381. [DOI] [PubMed] [Google Scholar]
- Solomon, P.S. , Ipcho, S.V.S. , Hane, J.K. , Tan, K.C. and Oliver, R.P. (2008) A quantitative PCR approach to determine gene copy number. Fungal Genet. Rep. 55, 5–8. [Google Scholar]
- Strelkov, S.E. , Lamar, L. , Sayoud, R. and Smith, R.B. (2002) Comparative virulence of chlorosis‐inducing races of Pyrenophora tritici‐repentis . Can. J. Plant Pathol. 24, 29–35. [Google Scholar]
- Strelkov, S.E. , Kowatsch, R.F. , Ballance, G.M. and Lamari, L. (2005) Characterization of the ToxB gene from North African and Canadian isolates of Pyrenophora tritici‐repentis . Physiol. Mol. Plant Pathol. 67, 164–170. [Google Scholar]
- Tan, K.C. , Oliver, R.P. , Solomon, P.S. and Moffat, C.S. (2010) Proteinaceous necrotrophic effectors in fungal virulence. Funct. Plant Biol. 37, 907–912. [Google Scholar]
- Tan, K.C. , Ferguson‐Hunt, M. , Rybak, K. , Waters, O.D.C. , Stanley, W.A. , Bond, C.S. , Stukenbrock, E.H. , Friesen, T.L. , Faris, J.D. , McDonald, B.A. and Oliver, R.P. (2012) Quantitative variation in effector activity of ToxA isoforms from Stagonospora nodorum and Pyrenophora tritici‐repentis . Mol. Plant–Microbe Interact. 25, 515–522. [DOI] [PubMed] [Google Scholar]
- Tuori, R.P. , Wolpert, T.J. and Ciuffetti, L.M. (1995) Purification and immunological characterization of toxic components from cultures of Pyrenophora‐tritici‐repentis . Mol. Plant–Microbe Interact. 8, 41–48. [DOI] [PubMed] [Google Scholar]
- Vleeshouwers, V.G.A.A. and Oliver, R.P. (2014) Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol. Plant–Microbe Interact. 27, 196–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Pyrenophora tritici‐repentis proteins identified from sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) excised bands of culture filtrates. (a) SDS‐PAGE of culture filtrate proteins (as shown in Fig. 1a) with arrows indicating excised bands. Two bands (upper and lower) were excised from three strains: wild‐type (WT), DW5 and toxa‐1. (b) The peptide fragmentation data from liquid chromatography‐tandem mass spectrometry (LC/MS/MS) were searched against the non‐redundant Ludwig NR database using the MASCOT sequence matching software. Only significant hits (score >35) with a significance threshold of P < 0.01 and a minimum of two peptide matches are shown.