

Summary
Several endogenous viral elements (EVEs) have been identified in plant genomes, including endogenous pararetroviruses (EPRVs). Here, we report the first characterization of EPRV sequences in the genome of African yam of the Dioscorea cayenensis‐rotundata complex. We propose that these sequences should be termed ‘endogenous Dioscorea bacilliform viruses' (eDBVs). Molecular characterization of eDBVs shows that they constitute sequences originating from various parts of badnavirus genomes, resulting in a mosaic structure that is typical of most EPRVs characterized to date. Using complementary molecular approaches, we show that eDBVs belong to at least four distinct Badnavirus species, indicating multiple, independent, endogenization events. Phylogenetic analyses of eDBVs support and enrich the current taxonomy of yam badnaviruses and lead to the characterization of a new Badnavirus species in yam. The impact of eDBVs on diagnosis, yam germplasm conservation and movement, and breeding is discussed.
Keywords: badnavirus, Dioscorea cayenensis‐rotundata complex, endogenous pararetrovirus, phylogeny, yam
Introduction
A wealth of endogenous viral elements (EVEs) has been discovered during the sequencing of genomes within the last 10 years, showing that the integration of viral sequences in eukaryotic genomes is a general phenomenon (Feschotte and Gilbert, 2012). In plants, the existence of EVEs has been documented for two decades, and recent advances in genome sequencing have shown that plant genomes host a wide range of EVEs originating from DNA and RNA viruses (Chiba et al., 2011; Teycheney and Geering, 2011). In contrast with animal retroviruses, the integration of viral sequences in plant genomes is not an obligatory step during virus replication. It is considered to result from horizontal gene transfer (HGT) following illegitimate recombination during the repair of double‐stranded DNA breakages (Staginnus and Richert‐Pöggeler, 2006). The first characterized plant EVEs belong to two virus families with DNA genomes: the Geminiviridae and Caulimoviridae. Geminivirus‐related DNA (GRD) was found only in the genome of Nicotiana spp. (Ashby et al., 1997; Bejarano et al., 1996). In contrast, a wide range of EVEs belonging to five genera of the Caulimoviridae have been characterized in several plant species (Teycheney and Geering, 2011). They are termed endogenous pararetroviruses (EPRVs).
The genomic organization and distribution of EPRVs are diverse, ranging from short, dispersed, repetitive viral sequences to longer stretches of near full‐length viral genomes. Most EPRVs are fragmented, rearranged and have inactivating mutations. They are therefore replication defective, and hence noninfectious. For example, a large array of EPRVs showing a high diversity is dispersed in the genome of banana (Musa spp.; D'Hont et al., 2012; Gayral and Iskra‐Caruana, 2009; Geering et al., 2005). However, a few EPRVs are infectious, making them the only infectious EVEs known in the plant kingdom. Infectious EPRVs exist in the genomes of Musa balbisiana (Chabannes et al., 2013; Gayral et al., 2008; Ndowora et al., 1999), petunia (Richert‐Pöggeler et al., 2003) and Nicotiana edwardsonii (Lockhart et al., 2000), and contain complete but mostly rearranged viral genomes. The mechanisms leading to functional infectious genomes from rearranged endogenous copies remain elusive, although the transcription of EPRVs and recombination may play a role (Chabannes and Iskra‐Caruana, 2013; Richert‐Pöggeler et al., 2003). The importance of both biotic and abiotic stresses in the activation of infectious EPRVs is well established (Côte et al., 2010; Dallot et al., 2001; Lockhart et al., 2000; Richert‐Pöggeler et al., 2003). Such activation occurs in natural and artificial interspecific hybrids and is suspected to be responsible for Banana streak virus (BSV) outbreaks worldwide within the last 20 years, raising concern that infectious EPRVs could significantly affect plant germplasm movement and breeding.
Yam (Dioscorea spp.) is an important staple food worldwide, particularly in West Africa and the South Pacific. Dioscorea cayenensis and D. rotundata are the predominant yam species in Africa and are considered to be related (Chaïr et al., 2005). Yam plants are generally propagated vegetatively through their tubers and this has resulted in the accumulation of viruses in yam germplasm. Many viruses have been reported in yams, including the two badnavirus species Dioscorea bacilliform alata virus (DBALV; Briddon et al., 1999) and Dioscorea bacilliform sansibarensis virus (DBSNV; Seal and Muller, 2007). DBALV can induce leaf distortion with veinal chlorosis symptoms, but infected plants can also be asymptomatic (Kenyon et al., 2008). It is naturally transmitted from D. alata to other Dioscorea species by mealybugs (e.g. Planococcus citri), but can also be transmitted mechanically (Phillips et al., 1999). In addition to the complete sequence of the genomes of DBALV and DBSNV, many partial badnaviral sequences have been generated using badnavirus degenerate primers from yam germplasm of uncertain virus infection status (Bousalem et al., 2009; Eni et al., 2008; Kenyon et al., 2008). These sequences encode the RT‐RNaseH domain, which is commonly used for phylogenetic studies of Badnavirus species (King et al., 2012). Comparison of this region led to Kenyon et al. (2008) proposing that there are at least 11 yam badnavirus species in the South Pacific region alone, highlighting the important genetic variability among yam badnaviruses. Subsequently, Bousalem et al. (2009) carried out a global classification also based on a similar, but slightly shorter, region. In this study, we have chosen to use predominantly the ‘Group 1–11’ nomenclature of Kenyon et al. (2008), as this use the exact region recommended by the International Committee on Taxonomy of Viruses (ICTV; King et al., 2012).
The presence of badnavirus EPRVs in the genome of yam species from the D. cayenensis‐rotundata complex has long been suspected following observations that a high proportion of plants tested positive when indexed by polymerase chain reaction (PCR) for the presence of badnaviruses (Bousalem et al., 2009), and from a comparison of serological and nucleic acid studies on West African material (Seal et al., 2014). The insertion of badnavirus EPRVs in yam genomes could interfere with molecular indexing tests for these viruses and affect yam germplasm conservation and distribution, as well as breeding, because of the risks associated with infectious EPRVs. Therefore, the identification and characterization of badnavirus EPRVs in yam germplasm are essential to implement reliable diagnostic methods and to investigate the potential release of viral particles from EPRVs. In this article, we provide the first proof of badnavirus EPRVs interspersed in the genomes of African yam of the D. cayenensis‐rotundata complex. The characterization of the EPRVs also shows that these sequences in yam (Dioscorea spp.) cluster to at least four distinct Badnavirus species. Our data support and complete the taxonomy of yam badnaviruses proposed by Kenyon et al. (2008), with the creation of an additional putative species, termed ‘Group 12’. Impacts on the diagnosis of badnaviruses in yam and the safe conservation and movement of yam germplasm are also discussed.
Results
Characterization of two rearranged badnavirus sequences from D. rotundata: G1Dr and S3G1Dr
The partial genome sequence of an uncharacterized yam badnavirus was amplified from symptomatic D. rotundata sample GN155 (Seal et al., 2014). In order to obtain the full‐length genome of this putative new virus, an outward facing primer pair, 63‐F2/49‐R2 (Table 1), was designed and used in a long PCR amplification. Surprisingly, one of the cloned amplification products, called G1Dr (accession number KF830002), was a highly rearranged badnavirus sequence of 5253 bp encompassing 12 fragments of open reading frame 3 (ORF3), the end of ORF1 and the beginning of ORF2, and four fragments of the intergenic region (Fig. 1, Table 2). Four of the ORF3 fragments (4, 7, 17 and 19) contained the RT‐RNaseH domain which is commonly used for badnavirus diversity studies. All four RT‐RNaseH sequences shared 95%–99% nucleotide identity with the previously reported sequence BN4Dr (registered under accession number AM944586; Eni et al., 2008), which belongs to putative species ‘Group 9’ of yam badnaviruses defined by Kenyon et al. (2008). ORF3 fragments 7, 17 and 19 and intergenic region fragments 6 and 15 resulted from the duplication of identical or near‐identical sequences (Fig. 2A). Other observed rearrangements include duplications–reversions in some ORF3 fragments with 99%–100% homology to each other (fragments 4 and 19, fragments 5 and 16, and fragments 11 and 13, respectively) and a frameshift between ORF3 fragments 1 and 2 (Fig. 2A). Interestingly, several parts of G1Dr had no significant homology to sequences from GenBank (fragment 10 and parts of fragments 6 and 15; Fig. 1A), and are likely to be yam host plant sequences for which little sequence data are publicly available.
Table 1.
Sequences of the oligonucleotide primers used in this study
| Primer name | Primer sequence | Position on Fig. 1, C |
|---|---|---|
| 49‐R2 | 5′‐TTTGAAACAGTTGTCCATCTTTCTTTG‐3′ | |
| 63‐F2 | 5′‐CGGAGGACTTCATCGCAGTCTATATTG‐3′ | |
| verifG1F | 5′‐GCCTTTGCAGGTAGTGGACT‐3′ | 2 |
| verifG1R | 5′‐ATCTTGCCCGGGTACCAAAG‐3′ | 4 |
| purifG1F | 5′‐ATGGCATGACCAGCCATTCA‐3′ | 1 |
| purifG1R | 5′‐GCAACAAACAGGGGCAATGT‐3′ | 3 |
| B389‐1F | 5′‐TAGTTCGGAAGGTCAAGAAG‐3′ | |
| B389‐2F | 5′‐TGATCCCACCAGAAGAATGC‐3′ | 5 |
| B389‐3R | 5′‐GCATGCTCCTCTTGTGACC‐3′ | 7 |
| B389‐4R | 5′‐CAGTGCCACGGAAGCAGTT‐3′ | 6 |
Figure 1.
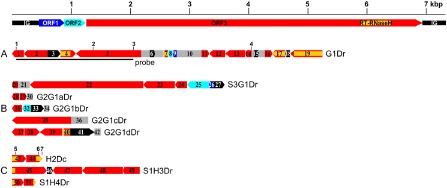
Schematic representation of badnavirus endogenous pararetroviruses (EPRVs) of Dioscorea cayenensis‐rotundata. A scaled linear view of the genome organization of Dioscorea bacilliform alata virus (DBALV) is shown in the top panel. The intergenic region (IG) and open reading frames (ORFs) appear with the following colour codes: IG, black; ORF1, dark blue; ORF2, light blue; ORF3, red. Rearranged badnavirus EPRVs of the D. cayenensis‐rotundata complex are shown in (A–C) using the same colour code. Rearranged fragments are numbered individually and the direction of the arrows indicates sequence orientation. Yellow horizontal lines and grey boxes indicate RT‐RNaseH domains of ORF3 sequences and uncharacterized sequences, respectively. Vertical numbered bars show the locations of the primers used for polymerase chain reaction (PCR) amplifications: 1, purifG1F; 2, verifG1F; 3, purifG1R; 4, verifG1R; 5, B389‐2F; 6, B389‐4R; 7, B389‐3R (see Table 1). (A) Dioscorea rotundata G1Dr sequence amplified from the GN155 plant. The part of the G1Dr sequence that was used as a probe in Southern blot experiments is shown by a horizontal bar. (B) Dioscorea rotundata sequences generated using the verifG1F/R primer pair. S3G1Dr was amplified from the Sd3 seedling and the four other sequences from the GN290 plant. (C) H2Dc sequence amplified from D. cayenensis HT1 plant, and S1H3Dr and S1H4Dr sequences amplified from D. rotundata Sd1 seedling using B389‐2F/3R and B389‐2F/4R primers, respectively.
Table 2.
Endogenous badnavirus‐related sequences obtained in this study
| Sequence name | Accession number | Amplification method | Primers | Size (bp) | Plant of origin (yam species) | Also present in: (yam species) | Related RT‐RNaseH partb | eDBV groupc |
|---|---|---|---|---|---|---|---|---|
| G1Dr | KF830002 | Outward facing primers PCR |
49‐R2 63‐F2 |
5253 | GN155 (D. rotundata) | NDa | BN4Dr (95%–99%) | eDBV9a |
| S3G1Dr | KF830010 | Direct PCR |
verifG1F verifG1R |
3595 | Sd3 (D. rotundata) | Sd2 (D. rotundata) | NDa | |
| G2G1aDr | KF830003 | Direct PCR |
verifG1F verifG1R |
348 | GN290 (D. rotundata) | GP425, CRB146, CRB152 (D. cayenensis) | NDa | |
| G2G1bDr | KF830004 | Direct PCR |
verifG1F verifG1R |
624 | GN290 (D. rotundata) | GP425, CRB146, CRB152 (D. cayenensis) | NDa | |
| G2G1cDr | KF830005 | Direct PCR |
verifG1F verifG1R |
1285 | GN290 (D. rotundata) | GP425, CRB146, CRB152 (D. cayenensis) | NDa | |
| G2G1dDr | KF830006 | Direct PCR |
verifG1F verifG1R |
1499 | GN290 (D. rotundata) | GP425, CRB146, CRB152 (D. cayenensis) | BN4Dr (100%) | eDBV9a |
| H2Dc | KF830007 | Outward facing primers PCR |
B389‐1F B389‐3R |
514 | HT1 (D. cayenensis) | GP425, CRB146, CRB152 (D. cayenensis) | FJ60a_Dr (100%) | eDBV9b |
| S1H3Dr | KF830008 | Direct PCR |
B389‐2F B389‐3R |
2160 | Sd1 (D. rotundata) | GN290 (D. rotundata) | FJ60a_Dr (100%) | eDBV9b |
| S1H4Dr | KF830009 | Direct PCR |
B389‐2F B389‐4R |
405 | Sd1 (D. rotundata) | GP425, CRB146, CRB152 (D. cayenensis); GN290 (D. rotundata) | FJ60a_Dr (100%) | eDBV9b |
eDBV, endogenous Dioscorea bacilliform viruses.
Not determined.
Most closely related badnavirus RT‐RNaseH sequence available in GenBank and its percentage of nucleotide identity to the sequence from this study.
Association of eDBV group and subgroup is based on the phylogenetic analysis shown in Fig. 5.
Figure 2.
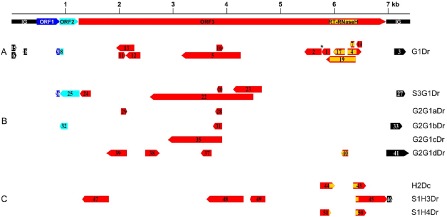
Map indicating the position of the rearranged fragments of Dioscorea cayenensis‐rotundata badnavirus endogenous pararetroviruses (EPRVs). A scaled linear view of Dioscorea bacilliform alata virus (DBALV) genome organization is shown in the top panel as a reference with the same colour code and fragment numbers as in Fig. 1. The direction of the arrows indicates sequence orientation. (A) G1Dr sequence amplified from D. rotundata GN155 plant. *Frameshift between fragments 1 and 2. (B) Sequences amplified from D. rotundata using the verifG1F/R primer pair. (C) H2Dc sequence amplified from D. cayenensis HT1 plant, and S1H3Dr and S1H4Dr sequences amplified from D. rotundata Sd1 seedling using B389‐2F/3R and B389‐2F/4R primers, respectively. Overlapping boxes indicate duplicated sequences within given EPRVs.
Similarly rearranged sequences were searched for in virus‐free D. rotundata plants by designing a primer pair (verifG1F/R; Table 1) from the G1Dr nonrepetitive ORF3 fragments 5 and 14 (see Fig. 1A). This primer pair was used in PCR experiments performed on total DNA from three seedlings, Sd1, Sd2 and Sd3, arising from seeds of a D. rotundata natural cross which were germinated and grown in vitro to prevent the possibility of virus infection. Two distinct amplification patterns were obtained: no amplification was detected from Sd1 (lane 1; Fig. 3A), whereas a 3595‐bp amplification product was raised from Sd2 and Sd3, which exceeded the expected size (lanes 2 and 3; Fig. 3A). Both amplification products were cloned, resulting in sequences S2G1Dr and S3G1Dr, respectively, and sequenced, showing 99% homology to each other. Therefore, sequence S3G1Dr (accession number KF830010) was used for further analyses (Table 2). It differs structurally from the G1Dr region from which primer pair verifG1F/R was designed, and harbours parts of all three badnavirus ORFs and intergenic region (Fig. 1B). Its 3′‐end includes a nonrearranged sequence encompassing the end of ORF1, the complete sequence of ORF2 and the beginning of ORF3 (fragments 26, 25 and 24, respectively; Fig. 2B).
Figure 3.
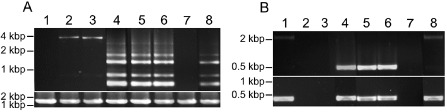
Polymerase chain reaction (PCR) amplification of badnavirus endogenous pararetroviruses (EPRVs) from total DNA of Dioscorea rotundata and D. cayenensis. PCR amplifications were performed on total genomic DNA extracted from D. rotundata seedlings and D. rotundata and D. cayenensis plants using the primer pairs verifG1F/R (A, top panel), atpB1/B2 (A, bottom panel), B389‐2F/3R (B, top panel) and B389‐2F/4R (B, bottom panel). Lane 1, D. rotundata seedling Sd1; lane 2, D. rotundata seedling Sd2; lane 3, D. rotundata seedling Sd3; lane 4, D. cayenensis GP425; lane 5, D. cayenensis CRB146; lane 6, D. cayenensis CRB152; lane 7, D. rotundata CRB439; lane 8, D. rotundata GN290.
To demonstrate the integration of G1Dr‐related sequences in D. rotundata, a Southern blot was performed on total DNA extracted from seedlings Sd1, Sd2 and Sd3 using the 5′‐end of G1Dr as a probe (see Fig. 1A). Undigested and XbaI‐digested DNAs showed strong hybridization to the probe with different patterns from those observed for the infected control (lanes 1–8; Fig. 4A). In particular, hybridization patterns of XbaI‐digested DNA from all three seedlings showed two bands whose respective sizes of 12.5 and 9.5 kbp exceeded that of linearized badnavirus genomic DNA. The hybridization patterns obtained for seedlings Sd1, Sd2 and Sd3 shared three bands in common with all seedlings, with respective sizes of 12.5, 5.5 and 2.5 kbp, whereas an intense band of 9.5 kbp was specific to Sd2 and Sd3. Seedling Sd1 displayed a different pattern with additional faint bands which may result from incomplete restriction digestion (lane 4; Fig. 4A). These results definitively demonstrate that G1Dr‐related sequences are integrated into the genome of D. rotundata in a genotype‐dependent fashion.
Figure 4.
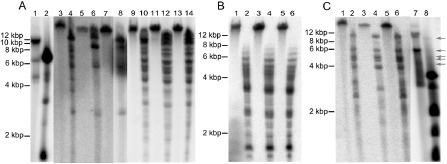
Southern blot hybridizations of Dioscorea rotundata and D. cayenensis total DNA with three different sequence probes. Undigested (odd lanes) and digested (even lanes) total DNAs from D. rotundata and D. cayenensis were hybridized with radiolabelled probes corresponding to the 5′‐end of sequence G1Dr (A), sequence S1H3Dr (B) and sequence ‘1.13’ (C). Total plant DNA was digested with XbaI (A) or HindIII (B, C). (A) Lanes 1–2, badnavirus‐infected D. trifida CRB577; lanes 3–4, D. rotundata seedling Sd1; lanes 5–6, D. rotundata seedling Sd2; lanes 7–8, D. rotundata seedling Sd3; lanes 9–10, D. cayenensis GP425; lanes 11–12, D. cayenensis CRB146; lanes 13–14, D. cayenensis CRB152. (B) Lanes 1–2, D. cayenensis GP425; lanes 3–4, D. cayenensis CRB146; lanes 5–6, D. cayenensis CRB152. (C) Lanes 1–2, D. rotundata seedling Sd1; lanes 3–4, D. rotundata seedling Sd2; lanes 5–6, D. rotundata seedling Sd3, lanes 7–8: badnavirus‐infected D. rotundata GN290. Arrows show the position of restriction fragments originating from plant genomic DNA.
The genomes of symptomless D. rotundata and D. cayenensis host integrated rearranged sequences related to G1Dr
The presence of G1Dr‐related sequences within yam genomes was investigated by PCR performed on total DNA extracted from three symptomless D. cayenensis plants (GP425, CRB146 and CRB152) and two additional symptomless D. rotundata plants (CRB439 and GN290) using the verifG1F/R primer pair. PCR amplification again resulted in two distinct patterns; although no amplification could be raised from sample CRB439 (lane 7; Fig. 3A), samples GP425, CRB146, CRB152 and GN290 displayed a similar amplification pattern with multiple bands (lanes 4, 5, 6 and 8, respectively; Fig. 3A). The four main products amplified from each plant were cloned and sequenced. They share 99% identity to each other; therefore, only G2G1aDr (348 bp; accession number KF830003), G2G1bDr (624 bp; KF830004), G2G1cDr (1285 bp; KF830005) and G2G1dDr (1499 bp; KF830006) sequences from the GN290 plant were used for further analyses (Table 2). All four sequences included mostly partial sequences of badnavirus ORF3 and putative yam sequences at their 3′‐end (fragments 30, 34, 36 and 42, respectively; Fig. 1B). Only G2G1dDr included a 100‐nucleotide sequence of the badnavirus RT‐RNaseH domain (fragment 40; Fig. 1B), which displayed 100% homology with BN4Dr and G1Dr sequences (fragment 19; Fig. 1A). Moreover, fragments 28 from G2G1aDr, 31 from G2G1bDr, 35 from G2G1cDr and 37 from G2G1dDr were 100% homologous to fragment 5 from G1Dr, confirming that D. cayenensis and D. rotundata host similar endogenous badnavirus sequences.
A Southern blot was also performed on total DNA extracted from three D. cayenensis plants, using the same probe as above (lanes 10–14; Fig. 4A). Similar hybridization patterns were observed for all plants. Numerous bands were also observed here for XbaI‐digested plant DNA, including three (13, 11 and 10 kbp) that were larger than badnavirus genome size bands. These results confirm that endogenous badnavirus sequences related to the G1Dr sequence are also present in the genome of D. cayenensis.
A badnavirus sequence from D. cayenensis is integrated into the genomes of D. cayenensis and D. rotundata
The partial sequence of badnavirus Group 9 (FJ60a_Dr; accession number AM072658) was reported in D. rotundata (Bousalem et al., 2009; Kenyon et al., 2008) and also amplified from D. cayenensis plant HT1 (D. Filloux, unpublished data). Based on this partial sequence, outward facing primers, B389‐1F/3R (Table 1), were designed and used for PCR amplification, in order to obtain the full‐length genome of this putative new virus. A 514‐bp amplification product was raised, cloned and sequenced. This rearranged sequence, called H2Dc (accession number KF830007), contained two partial badnavirus ORF3 fragments (Fig. 1, Table 2), including parts of the RT‐RNaseH domain. As expected, sequence comparisons showed that sequence H2Dc is most closely related to sequence FJ60a_Dr.
The presence of sequence H2Dc in D. rotundata was investigated by PCR screening of seedlings Sd1, Sd2 and Sd3 using two sets of primer pairs designed on sequence H2Dc: B389‐2F/3R and B389‐2F/4R (Fig 1C, Table 1). No amplification product could be raised from seedlings Sd2 and Sd3, regardless of the primer pair (lanes 2 and 3; Fig. 3B). By contrast, amplification products were raised from seedling Sd1 using either primer pair (lane 1; Fig. 3B), and were subsequently cloned and sequenced. Sequence S1H4Dr (accession number KF830009), raised using the B389‐2F/4R primer pair, had a similar size and organization as sequence H2Dc (Figs 1, 2C). Sequence S1H3Dr (accession number KF830008), raised using the B389‐2F/3R primer pair, was very different in size (2160 bp) and structure (Fig. 1C, Table 2). Its sequence was composed of rearranged fragments of badnaviral ORF3 sequence, except for fragment 46, which corresponds to a badnavirus intergenic region. The corresponding part of fragment 45 of sequence S1H3Dr was identical to fragment 43 of sequence H2Dc (Fig. 2C).
H2Dc‐related sequences were searched for by PCR using both B389‐2F/3R and B389‐2F/4R primer pairs in additional D. rotundata (CRB439 and GN290) and D. cayenensis (GP425, CRB146 and CRB152) plants. Three different amplification patterns were obtained (lanes 4–8; Fig. 3B). Sequences of the expected size were amplified from D. cayenensis plants GP425, CRB146 and CRB152 with both sets of primers, and these sequences showed 99% identity with sequence H2Dc. By contrast, no amplification product could be raised from D. rotundata plant CRB439 using either primer set (lane 7; Fig. 3B). Amplification products of 2160 and 405 bp were obtained from D. rotundata plant GN290 using B389‐2F/3R and B389‐2F/4R primer pairs, respectively (lane 8; Fig. 3B), cloned and sequenced. They displayed 100% homology with sequences S1H3Dr and S1H4Dr that were amplified from D. rotundata seedling Sd1 (Table 2).
The presence of H2Dc‐related sequences in the genomes of D. cayenensis was confirmed by Southern blot performed on genomic DNA extracted from the plants used for PCR screening and using S1H3Dr as a probe (Fig. 4B). Hybridization of the probe could be observed for high‐molecular‐weight DNA in lanes containing undigested plant genomic DNA and for restriction fragments larger than the probe in lanes containing HindIII‐digested plant genomic DNA. Overall, these results show that badnaviral sequences of similar origin and related to yam badnavirus Group 9 are present in the genomes of both D. cayenensis and D. rotundata.
Diversity of badnavirus EPRVs in D. rotundata seedlings
Our results show that the genome of D. rotundata hosts several endogenous badnavirus sequences. The genetic diversity of these sequences was investigated. To this aim, sequences of the RT‐RNaseH domain were amplified from total DNA extracted from D. rotundata seedlings Sd1 and Sd2 using the degenerate primer pair Badna‐FP/RP (Yang et al., 2003). A total of 47 sequences was generated (accession numbers KF829953–KF829999) and compared with fragment 19 of G1Dr, which carries a sufficiently long RT‐RNaseH sequence to perform phylogenetic analyses (Fig. 2A), and with yam badnavirus sequences previously reported by Kenyon et al. (2008), Eni et al. (2008) and Bousalem et al. (2009). Phylogenetic analyses showed that all sequences generated in this study belong to the genus Badnavirus and can be classified into four distinct groups (Fig. 5). A first group of 22 sequences belongs to Group 8 as defined by Kenyon et al. (2008), which includes DBALV and is similar to DBV‐A(A) described by Bousalem et al. (2009). These sequences display homology levels of 83%–100% between each other and can be separated into three subgroups (8a, 8b and 8c) with 83%–86% identity between subgroups. A second group of nine highly similar sequences sharing 95%–100% homology does not fit into any of the groups previously defined by Kenyon et al. (2008). An additional group was created to accommodate these sequences, and numbered ‘Group 12’. It corresponds to the DBV‐A(B) group defined by Bousalem et al. (2009). A third group of 12 sequences sharing 83%–100% identity fits into Group 9 defined by Kenyon et al. (2008), and can be separated into two subgroups (9a and 9b) with 83%–87% identity between subgroups. As expected, this group also contains fragment 19 of G1Dr, and all EPRV sequences generated in this study that contain RT‐RNaseH parts are related to it (Table 2). A fourth and last group of four sequences sharing 89%–100% identity fits into Group 5 defined by Kenyon et al. (2008), and is similar to DBV‐C of Bousalem et al. (2009).
Figure 5.
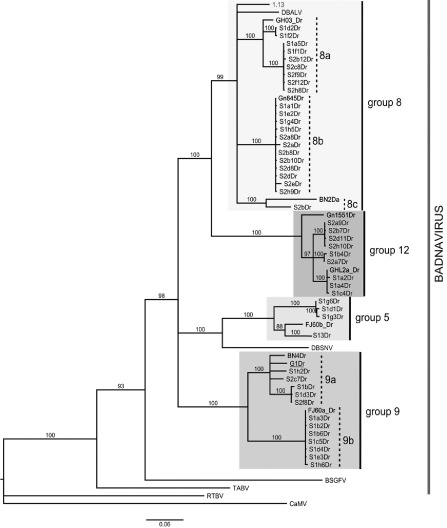
Phylogenetic neighbour‐joining tree built from the nucleotide sequences of badnavirus RT‐RNaseH domain amplified from Dioscorea rotundata seedlings Sd1 and Sd2. Primer sequences were removed. Bootstrap values of 1000 replicates are given above the nodes when above 80% (Tamura et al., 2004) and the evolutionary distances were computed using the HKY model. The first letter and number of the sequences refer to the seedling names, with S1 referring to Sd1 and S2 to Sd2. G1Dr (underlined), sequence of the RT‐RNaseH domain of the rearranged sequence G1Dr (fragment 19; see Fig. 1A) and sequence ‘1.13’ from a strain of DBALV, which was used as a probe in Southern blot experiments, are included. Additional sequences of the badnavirus RT/RNaseH domain were used: Dioscorea bacilliform alata virus (DBALV) and Dioscorea bacilliform sansibarensis virus (DBSNV) sequences and previously reported sequences from yam badnaviruses. Banana streak Goldfinger virus (BSGFV) and Taro bacilliform virus (TABV), used as members of the genus Badnavirus, and Cauliflower mosaic virus (CaMV) and Rice tungro bacilliform virus (RTBV), used as outgroups, are also shown. The scale bar shows the number of substitutions per base.
DBALV‐related sequences are present in the genome of D. rotundata
Most of the RT‐RNaseH badnaviral sequences generated from D. rotundata seedlings belong to yam badnavirus Group 8, which includes DBALV, one of the two yam badnaviruses whose genome has been entirely sequenced (Fig. 5). DBALV was originally isolated from D. alata, but infects many other yam species, including D. rotundata (Kenyon et al., 2008). Group 8 also includes the RT‐RNaseH domain of sequence ‘1.13’, which was generated by rolling circle amplification (RCA) performed on total DNA extracted from a D. trifida plant showing typical symptoms of DBALV infection. This 6‐kbp nonrearranged sequence covers a near‐complete badnavirus genome, missing ORF3 nucleotide positions 90–1480 (accession number KF829952). The RT‐RNaseH domain of sequence ‘1.13’ is 89% homologous to that of DBALV, and ‘1.13’ can therefore be considered as a partial sequence of a DBALV strain. This sequence was used as a probe in a Southern blot experiment performed on genomic DNA extracted from seedlings Sd1, Sd2 and Sd3, in order to search for badnavirus EPRVs related to DBALV in D. rotundata.
Hybridization patterns of HindIII‐digested DNA differed between seedling Sd2 and seedlings Sd1 and Sd3 (lanes 2, 4 and 6; Fig. 4C). All three seedlings shared several hybridized restriction fragments with respective sizes of 10, 6.5, 5, 4.5, 4, 2.5 and 2 kbp. However, two additional fragments of 3 and 2.5 kbp were specific to Sd1 and Sd3. No restriction fragment matching those of the episomal form of the DBALV‐related genome of the infected GN290 plant (lanes 7 and 8; Fig. 4C) could be detected in any of the three D. rotundata seedlings. Furthermore, the presence in the digested DNA from all three seedlings of a 10.5‐kbp band whose size exceeds that of linearized badnavirus genomic DNA confirms that the genome of D. rotundata hosts endogenous sequences related to DBALV.
Discussion
This article reports the first characterization of badnavirus EPRVs in the genome of African yams of the D. cayenensis‐rotundata complex. Using complementary molecular approaches, such as PCR and Southern blot, it provides the first evidence that the genome of D. cayenensis‐rotundata spp. hosts EPRVs from four distinct badnavirus species. The use of seedlings grown in vitro from seeds ensured that virus‐free plant material was employed in this study and that viral sequences that were amplified or hybridized were of endogenous origin, as badnaviruses are not seed transmitted. Although the EPRVs reported in this work are very diverse in size and molecular organization (Fig. 1), they share a highly rearranged structure and are composed of sequences originating from various parts of badnavirus genomes (Fig. 2). Such a mosaic structure is characteristic of most plant EVEs, especially EPRVs (Teycheney and Geering, 2011), including infectious endogenous BSVs (Gayral et al., 2008). We propose that these yam badnavirus EPRVs should be termed ‘endogenous Dioscorea bacilliform viruses’ (eDBVs) according to the nomenclature proposed by Geering et al. (2010).
Phylogeny of eDBVs
Sequence analyses performed on the RT‐RNaseH domain of the eDBVs reported in this work support the diversity and classification of yam badnaviruses proposed by Kenyon et al. (2008), and subsequently by Bousalem et al. (2009). According to the current ICTV Badnavirus species delineation criteria (King et al., 2012), these eDBVs belong to four distinct badnavirus species, including a new one that we propose to create in order to accommodate nucleotide sequences forming a homogeneous cluster that differs by more than 20% from sequences of any other group (Fig. 5). Kenyon et al. (2008) originally defined 13 groups of yam badnaviruses, but further phylogenetic analyses showed that sequences of Group 12 and Group 13 in their analysis do not belong to the genus Badnavirus (Bousalem et al., 2009). Therefore, we propose that these groups are removed and that the new group proposed here is considered as Badnavirus Group 12. Based on our data, we hypothesize that several independent events of endogenization have occurred in the D. rotundata genome, resulting in the presence of eDBVs from at least four distinct badnavirus species.
Most of the sequences of the eDBV RT/RNaseH domain generated by PCR using the Badna‐FP/RP primer pair belong to Group 8 and Group 9 (Fig. 5 and Table 2). Group 9 can be divided into subgroups 9a and 9b. Sequences from subgroup 9b share 99%–100% identity with each other, and probably originate from a single integration locus in the same seedling (Sd1). Similarly, sequences from subgroup 8b share 98%–100% homology with each other, but nevertheless originate from distinct seedlings (Sd1 and Sd2). Phylogenetic analyses performed on rearranged sequences from D. cayenensis‐rotundata samples, whose endogenous nature was confirmed by Southern blot, show that they belong to Group 9 (Table 2). Our work shows that these sequences are closely related to other partial sequences previously reported by Kenyon et al. (2008) and Bousalem et al. (2009), which are likely to be endogenous. The remaining eDBVs reported in this work belong to Group 5 and the new Group 12 (Fig. 5), which display a limited diversity and need to be investigated further.
Previous phylogenetic studies of yam badnaviruses unveiled a high level of diversity among episomal and putative endogenous sequences (Bousalem et al., 2009; Kenyon et al., 2008). This situation is similar to that encountered for other badnaviruses (Borah et al., 2013), especially banana and sugarcane badnaviruses, which are both polyphyletic (Harper et al., 2005; Iskra‐Caruana et al., 2014; Muller et al., 2011). In banana, both episomal and endogenous badnavirus sequences exist and are classified into three separate clades of the phylogeny proposed by Iskra‐Caruana et al. (2014). Sequences from Clade I are both episomal and endogenous, and include infectious endogenous BSVs, whereas sequences from Clade II are exclusively endogenous and sequences from Clade III are most probably episomal (Gayral and Iskra‐Caruana, 2009; Iskra‐Caruana et al., 2010; James et al., 2011a; M. Chabannes et al., CIRAD, Montpellier, unpublished data). Likewise, badnaviruses of sugarcane, which are exclusively episomal, are classified into Clade I and Clade III, and display an important molecular diversity (Muller et al., 2011). All badnavirus partial sequences amplified to date from yams belong to Badnavirus Clade II and correspond to both episomal (DBALV and DBSNV) and endogenous (this study) sequences (Muller et al., 2011). Overall, our data confirm that it is currently impossible to correlate the episomal or endogenous nature of a badnavirus sequence with its phylogenetic position, as endogenous sequences do not form a well‐defined phylogenetic group, but are dispersed over different clades or groups instead.
Structure and age of eDBVs
Dispersed repetitive elements, such as EPRVs, have a significant impact on the complexity and evolution of plant genomes (Jakowitsch et al., 1999), and can be used to analyse plant phylogenies and to date polyploidization events (Mette et al., 2002). Considering that yam genomes host diverse eDBVs and that distinct yam genomes host similar eDBVs, these sequences could be useful for unravelling the evolution of yam genomes, similar to the studies achieved for banana (Gayral et al., 2010).
It has been shown that endogenous forms of Banana streak OL virus (eBSOLV) and Banana streak GF virus (eBSGFV) are integrated into the genome of M. balbisiana as allelic forms (Chabannes et al., 2013). Infectious and noninfectious eBSOLV and eBSGFV alleles can be differentiated by Southern blot hybridization. Although preliminary, our data suggest that eDBV could also exist as allelic forms. For example, D. rotundata seedlings Sd1, Sd2 and Sd3 display distinct PCR amplification and Southern blot patterns for an eDBV that falls into Group 9 of Kenyon et al. (2008) (termed eDBV9), as shown in Figs 3A, B and 4A. Likewise, Sd1 and Sd3 display additional bands in Southern blot hybridization using a DBALV‐like probe, when compared with Sd2 (Fig. 4C), suggesting that allelic forms of eDBV8 may also exist in D. rotundata.
Access to full genome sequences has greatly facilitated the molecular characterization of EPRVs in several crops, including banana, rice and grapevine. Similar resources should soon be available for yam genomes, making it possible to better characterize eDBVs and their allelic structure. Identical eDBV9 sequences were identified in D. rotundata and D. cayenensis (Table 2), but it is not yet possible to demonstrate that these sequences relate to orthologous loci, or to investigate whether endogenization events have occurred in a common ancestor prior to the speciation of D. rotundata and D. cayenensis, which may belong to the same species (Chaïr et al., 2005). Therefore, the dating of insertion events of eDBV in the genome of D. rotundata and D. cayenensis might not be possible until several complete yam genome sequences are available.
Impact of eDBVs on diagnosis, yam germplasm movement and breeding
The characterization of eDBVs was primarily undertaken in order to evaluate the suitability of existing tools for the detection of yam badnaviruses. Access to disease‐free planting material is critical to food security, particularly in countries in which yam is a major staple crop, and yam smallholder farmers need to increase yam tuber yields. Considering the high prevalence of badnaviruses in yam germplasm (Bousalem et al., 2009), reliable detection methods are needed for the safe conservation, multiplication and distribution of yam planting material. The presence and diversity of eDBVs in yam genomes reported in this work complicates the molecular detection of episomal yam badnaviruses, as existing PCR‐based diagnostic tools will generate amplification products from eDBVs. The infection status of a yam plant will therefore not be known, as demonstrated by Seal et al. (2014). The situation in yam in this regard is similar to that of M. balbisiana, for which BSV indexing had to be optimized in order to avoid ‘false’ positives resulting from the presence of endogenous BSVs (James et al., 2011a, b; Le Provost et al., 2006). Similar to BSV, RCA could be used for the specific detection of episomal forms of yam badnaviruses. However, the analysis of RCA products requires a digestion step by single cutting restriction enzymes in viral genomes. To date, only two full‐length sequences of yam badnaviruses are available, and hence the correct selection of restriction enzyme is difficult and complicates the use of RCA as a diagnostic method. A more polyvalent alternative would be to optimize direct binding‐PCR (DB‐PCR) or immunocapture‐PCR (IC‐PCR) with mono‐ or polyclonal antibody (Kenyon et al., 2008). However, current antisera for yam badnaviruses do not detect all strains (Seal et al., 2014); therefore, a promising solution would be to develop a multiplex DB‐PCR, including an additional set of primers targeting yam genomic DNA to detect plant DNA contaminations and a DNaseI treatment step prior to PCR, as deployed successfully for the detection of episomal Pineapple bacilliform virus sequences (Gambley, 2008).
It has been hypothesized that some EVEs may be involved in silencing‐based antiviral defence mechanisms targeting cognate viruses (Bertsch et al., 2009; Mette et al., 2002). However, our Southern hybridization studies show that badnavirus‐infected D. rotundata plant GN290 possesses plant high‐molecular‐weight restriction fragments with high homology to a DBALV‐like probe (see arrows in lane 8; Fig. 4C). Hence, this D. rotundata plant hosts eDBV8 and is nevertheless infected by a strain closely related to DBALV that also falls into Group 8 of Kenyon et al. (2008). This suggests that endogenization of some badnaviral sequences in yams may not provide protection against cognate viruses.
The eDBVs characterized for the first time in this study are highly fragmented and our data provide no support for their infectious nature. Nevertheless, highly rearranged EPRVs have been shown to be infectious in M. balbisiana (Chabannes and Iskra‐Caruana, 2013; Chabannes et al., 2013; Gayral et al., 2008; Iskra‐Caruana et al., 2010), and to have a negative impact on the genetic improvement of banana. If infectious eDBVs are also present in yam genomes, a similar challenge will be posed for yam improvement. It is hoped that access to complete annotated sequences of yam genomes will soon assist in elucidating whether some eDBVs are infectious and might pose a threat to yam breeding and multiplication programmes globally.
Experimental Procedures
Origin of plant material
Two D. rotundata (GN155 and GN290) plants and one D. cayenensis (HT1) plant were collected in 2002 and 2012 from Guinea and Haiti, respectively. Seeds that generated seedlings Sd1, Sd2 and Sd3 were collected from the same progeny of a natural cross of D. rotundata in 2002 from Benin. The seeds were rescued in an in vitro culture laboratory on classical medium and therefore did not have the opportunity to come into contact with badnaviruses. GN290 and GP425, a D. cayenensis plant collected from Guadeloupe in 2009, were grown in the yam quarantine facility of Montpellier (France). CRB146, CRB152 (D. cayenensis), CRB439 (D. rotundata), CRB574 and CRB577 (D. trifida) plants were provided by the Guadeloupe Biological Resource's Center for Tropical Plants (CRB‐PT).
Total DNA extractions from yam leaves
Five hundred milligrams of yam leaves were frozen in liquid nitrogen and ground to a fine powder, which was then mixed with 5 mL of extraction buffer [100 mm tris(hydroxymethyl)aminomethane (Tris)‐HCl, pH 8, 1.4 m NaCl, 20 mm ethylenediaminetetraacetic acid (EDTA), 2% w/v mixed alkyltrimethylammonium bromide, 1% w/v PEG6000 and 0.5% w/v Na2SO3 added freshly]. Samples were incubated at 74 °C for 30 min with 2 mg/mL RNase (Qiagen, Courtaboeuf, France), extracted twice by 5 mL of chloroform–isoamyl alcohol (24:1) and precipitated with 5 mL of isopropanol and 300 mm sodium acetate. Extracted DNAs were purified through Tip100 columns (Qiagen), according to the manufacturer's instructions, and resuspended in 200 μL of sterile distilled deionized water. After quantification, DNA quality was assessed by PCR using the atpB1/B2 primer pair (Soltis et al., 1999), according to the authors' protocol.
Production of rearranged sequences using outward facing primers
Long amplifications were performed on DNA from GN155 and HT1 plants, using outward facing primers located within the Gn1551Dr sequence (AM503380, 63‐F2 and 49‐R2 primers) and FJ60a_Dr sequence (AM072658, B389‐1F and B389‐3R primers), respectively (Table 1). PCRs were performed using Expand+ High Fidelity thermostable DNA polymerase (Roche, Meylan, France). The PCR conditions were 2 min at 92 °C; 30 cycles of 10 s at 92 °C, 30 s at 57 °C and 8 min at 68 °C; and a final elongation step of 7 min at 68 °C. Amplification products were cloned in TOPO‐XL vector (Invitrogen, St Aubin, France). Selected clones were sequenced by primer walking (Beckman Coulter, Takeley, Essex, UK) and named G1Dr (KF830002) and H2Dc (KF830007) for the sequences obtained from GN155 and HT1 plants, respectively.
Screening D. cayenensis‐rotundata plants
Specific primers were designed from G1Dr and H2Dc sequences (Fig. 1A, C). Primers verifG1F and verifG1R were designed from the G1Dr sequence so as to avoid amplification of episomal badnavirus genomes (Table 1) and to raise a 2713‐bp product (see Fig. 1A). The two primers sets B389‐2F/3R and B389‐2F/4R (Table 1) were designed from the H2Dc sequence so as to raise 469‐ and 405‐bp amplification products, respectively (see Fig. 1C). DNAs extracted from D. cayenensis‐rotundata (50 ng) were used with these three sets of primers with GoTaq HotStart enzyme (Promega, Charbonnières, France) employing similar amplification conditions (5 min at 95 °C; 25 cycles of 30 s at 95 °C, 30 s at 60 °C, 10 min at 72 °C; and a final elongation step of 10 min at 72 °C). PCR products were cloned into pGEM®‐T vector (Promega) and sequenced (Beckman Coulter).
Production of RT‐RNaseH sequences from seedlings
Amplification was performed on 50 ng of DNA extracted from virus‐free D. rotundata Sd1 and Sd2 seedlings, using the generic badnavirus primer pair Badna‐FP/RP (Yang et al., 2003) and GoTaq HotStart (Promega). PCR conditions were 5 min at 95 °C; 25 cycles of 30 s at 95 °C, 30 s at 55 °C, 1 min at 72 °C; and a final elongation step of 10 min at 72 °C. PCR products were cloned into pGEM®‐T vector (Promega). Recombinant clones were fingerprinted according to Geering et al. (2005), and selected clones were sequenced (Beckman Coulter).
Phylogenetic analyses
Phylogenetic analyses were performed on 529‐bp RT‐RNaseH domains of badnavirus ORF3 from sequences generated in this work (KF829953–KF829999), fragment 19 of G1Dr and badnavirus sequences, including that of DBALV (X94582 and X94575), DBSNV (DQ822073), BSGFV (Banana streak Goldfinger virus; AY493509), TABV (Taro bacilliform virus; AF357836), Gn845Dr (AM503397), GH03_Dr (AM072664), BN2Da (AM944584), Gn1551Dr (AM503380), GHL2a_Dr (AM072665), BN4Dr (AM944586), FJ60a_Dr (AM072658) and FJ60b_Dr (AM072659). Sequences from a tungrovirus, RTBV (Rice tungro bacilliform virus; X57924), and a caulimovirus, CaMV (Cauliflower mosaic virus; V00141), were used as outgroups from other genera within the Caulimoviridae family. Alignments were performed using clustalw (Larkin et al., 2007), and the phylogenetic tree was built with mega5 using the neighbour‐joining method based on the HKY model (Hasegawa et al., 1985; Tamura et al., 2011). Only sequences displaying more than one base substitution to each other were retained for the construction of the phylogenetic tree in order to avoid redundancy.
Amplification of ‘1.13’ sequence using RCA
DNA from D. trifida CRB574 plant leaves was extracted using a DNeasy Plant Mini Kit (Qiagen). One microlitre of total DNA was employed to amplify circular badnavirus sequences using the TempliPhi™ kit (GE Healthcare, Vélizy‐Villacoublay, France) with the addition of a badnavirus primer cocktail to improve the reaction efficiency (A. D. W. Geering, QAAFI, Brisbane, unpublished data). Amplification products were digested by ApaI and ligated into ApaI‐digested pBlueScript vector (Stratagene, Les Ulis, France). Selected clone ‘1.13’ (KF829952) was sequenced by primer walking (Beckman Coulter).
Southern blot hybridization
Total yam DNA (20–25 μg) was digested overnight by 50 U of HindIII or XbaI, which do not cut in sequence G1Dr and are single cutters in sequence ‘1.13’. Purified digested DNA was electrophoresed in a 1% w/v agarose gel for 16 h at 40 V, and transferred to positively charged nylon membranes (Hybond N+, GE Healthcare), according to the manufacturer's instructions. Membranes were hybridized overnight at 64 °C with radiolabelled probes prepared using the Prime‐a‐Gene® kit (Promega), according to the manufacturer's instructions, under stringency conditions preventing hybridization when target sequences display less than 80% homology to the probe. Autoradiography was performed using a phosphorimager (Typhoon FLA 9000, GE Healthcare) following a 24‐h exposure time.
Acknowledgements
The authors wish to thank Franciane Gamiette, Suzia Gelabale and David Lange for providing yam plants from the Biological Resource's Center for Tropical Plants (CRB‐PT) collections, and Manon Rousselet Mayras for technical help. This work was supported by the European Regional Development Fund.
References
- Ashby, M.K. , Warry, A. , Bejarano, E.R. , Kashoggi, A. , Burrell, M. and Lichtenstein, C.P. (1997) Analysis of multiple copies of geminiviral DNA in the genome of four closely related Nicotiana species suggests a unique integration event. Plant Mol. Biol. 35, 313–321. [DOI] [PubMed] [Google Scholar]
- Bejarano, E.R. , Khashoggi, A. , Witty, M. and Lichtenstein, C. (1996) Integration of multiple repeats of geminiviral DNA into the nuclear genome of tobacco during evolution. Proc. Natl. Acad. Sci. USA, 93, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertsch, C. , Beuve, M. , Dolja, V.V. , Wirth, M. , Pelsy, F. , Herrbach, E. and Lemaire, O. (2009) Retention of the virus‐derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol. Direct. 4, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah, B.K. , Sharma, S. , Kant, R. , Johnson, A.M.A. , Saigopal, D.V.R. and Dasgupta, I. (2013) Bacilliform DNA‐containing plant viruses in the tropics: commonalities within a genetically diverse group. Mol. Plant Pathol. 14, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousalem, M. , Durand, O. , Scarcelli, N. , Lebas, B.S.M. , Kenyon, L. , Marchand, J.‐L. , Lefort, F. and Seal, S.E. (2009) Dilemmas caused by endogenous pararetroviruses regarding the taxonomy and diagnosis of yam (Dioscorea spp.) badnaviruses: analyses to support safe germplasm movement. Arch. Virol. 154, 297–314. [DOI] [PubMed] [Google Scholar]
- Briddon, R.W. , Phillips, S. , Brunt, A. and Hull, R. (1999) Analysis of the sequence of Dioscorea alata Bacilliform Virus; comparison to other members of the badnavirus group. Virus Genes, 18, 277–283. [DOI] [PubMed] [Google Scholar]
- Chabannes, M. and Iskra‐Caruana, M.‐L. (2013) Endogenous pararetroviruses—a reservoir of virus infection in plants. Curr. Opin. Virol. 3, 615–620. [DOI] [PubMed] [Google Scholar]
- Chabannes, M. , Baurens, F.‐C. , Duroy, P.‐O. , Bocs, S. , Vernerey, M.‐S. , Rodier‐Goud, M. , Barbe, V. , Gayral, P. and Iskra‐Caruana, M.‐L. (2013) Three infectious viral species lying in wait in the banana genome. J. Virol. 87, 8624–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaïr, H. , Perrier, X. , Agbangla, C. , Marchand, J.‐L. , Dainou, O. and Noyer, J. (2005) Use of cpSSRs for the characterisation of yam phylogeny in Benin. Genome, 48, 674–684. [DOI] [PubMed] [Google Scholar]
- Chiba, S. , Kondo, H. , Tani, A. , Saisho, D. , Sakamoto, W. , Kanematsu, S. and Suzuki, N. (2011) Widespread endogenization of genome sequences of non‐retroviral RNA viruses into plant genomes. PLoS Pathog. 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côte, F.X. , Galzi, S. , Follioti, M. , Lamagnère, Y. , Teycheney, P.‐Y. and Iskra‐Caruana, M.‐L. (2010) Micropropagation by tissue culture triggers differential expression of infectious endogenous Banana streak virus sequences (eBSV) present in the B genome of natural and synthetic interspecific banana plantains. Mol. Plant Pathol. 11, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallot, S. , Acuña, P. , Rivera, C. , Ramírez, P. , Côte, F. , Lockhart, B.E.L. and Caruana, M.‐L. (2001) Evidence that the proliferation stage of micropropagation procedure is determinant in the expression of Banana streak virus integrated into the genome of the FHIA 21 hybrid (Musa AAAB). Arch. Virol. 146, 2179–2190. [DOI] [PubMed] [Google Scholar]
- D′Hont, A. , Denoeud, F. , Aury, J.‐M. , Baurens, F.‐C. , Carreel, F. , Garsmeur, O. , Noel, B. , Bocs, S. , Droc, G. , Rouard, M. , Da Silva, C. , Jabbari, K. , Cardi, C. , Poulain, J. , Souquet, M. , Labadie, K. , Jourda, C. , Lengellé, J. , Rodier‐Goud, M. , Alberti, A. , Bernard, M. , Correa, M. , Ayyampalayam, S. , McKain, M.R. , Leebens‐Mack, J. , Burgess, D. , Freeling, M. , Mbéguié‐A‐Mbéguié, D. , Chabannes, M. , Wicker, T. , Panaud, O. , Barbosa, J. , Hribova, E. , Heslop‐Harrison, P. , Habas, R. , Rivallan, R. , Francois, P. , Poiron, C. , Kilian, A. , Burthia, D. , Jenny, C. , Bakry, F. , Brown, S. , Guignon, V. , Kema, G. , Dita, M. , Waalwijk, C. , Joseph, S. , Dievart, A. , Jaillon, O. , Leclercq, J. , Argout, X. , Lyons, E. , Almeida, A. , Jeridi, M. , Dolezel, J. , Roux, N. , Risterucci, A.‐M. , Weissenbach, J. , Ruiz, M. , Glaszmann, J.‐C. , Quétier, F. , Yahiaoui, N. and Wincker, P. (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature, 488, 213–217. [DOI] [PubMed] [Google Scholar]
- Eni, A.O. , Hugues, J. d'A. , Asiedu, R. and Rey, M.E.C. (2008) Sequence diversity among badnavirus isolates infecting yam (Dioscorea spp.) in Ghana, Togo, Benin and Nigeria. Arch. Virol. 153, 2263–2272. [DOI] [PubMed] [Google Scholar]
- Feschotte, C. and Gilbert, C. (2012) Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13, 283–296. [DOI] [PubMed] [Google Scholar]
- Gambley, C. (2008) The aetiology of pineapple mealybug wilt disease: the role of viruses. PhD Thesis, University of Queensland, St Lucia, Qld.
- Gayral, P. and Iskra‐Caruana, M.‐L. (2009) Phylogeny of Banana streak virus reveals recent and repetitive endogenization in the genome of its banana host (Musa sp.). J. Mol. Evol. 69, 65–80. [DOI] [PubMed] [Google Scholar]
- Gayral, P. , Noa‐Carrazana, J.‐C. , Lescot, M. , Lheureux, F. , Lockhart, B.E.L. , Matsumoto, T. , Piffanelli, P. and Iskra‐Caruana, M.‐L. (2008) A single banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J. Virol. 82, 6697–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayral, P. , Blondin, L. , Guidolin, O. , Carreel, F. , Hippolyte, I. , Perrier, X. and Iskra‐Caruana, M.‐L. (2010) Evolution of endogenous sequences of banana streak virus: what can we learn from banana (Musa sp.) evolution? J. Virol. 84, 7346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering, A.D.W. , Olszewski, N.E. , Harper, G. , Lockhart, B.E.L. , Hull, R. and Thomas, J.E. (2005) Banana contains a diverse array of endogenous badnaviruses. J. Gen. Virol. 86, 511–520. [DOI] [PubMed] [Google Scholar]
- Geering, A.D.W. , Scharaschkin, T. and Teycheney, P.‐Y. (2010) The classification and nomenclature of endogenous viruses of the family Caulimoviridae . Arch. Virol. 155, 123–131. [DOI] [PubMed] [Google Scholar]
- Harper, G. , Hart, D. , Moult, S. , Hull, R. , Geering, A.D.W. and Thomas, J. (2005) The diversity of banana streak virus isolates in Uganda. Arch. Virol. 150, 2407–2420. [DOI] [PubMed] [Google Scholar]
- Hasegawa, M. , Kishino, H. and Yano, T. (1985) Dating the human–ape split by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174. [DOI] [PubMed] [Google Scholar]
- Iskra‐Caruana, M.‐L. , Baurens, F.‐C. , Gayral, P. and Chabannes, M. (2010) A four‐partner plant–virus interaction: enemies can also come from within. Mol. Plant–Microbe Interact. 23, 1394–1402. [DOI] [PubMed] [Google Scholar]
- Iskra‐Caruana, M.‐L. , Duroy, P.‐O. , Chabannes, M. and Muller, E. (2014) The common evolutionary history of badnaviruses and banana. Infect. Genet. Evol. 21, 83–89. [DOI] [PubMed] [Google Scholar]
- Jakowitsch, J. , Mette, M.F. , van der Winden, J. , Matzke, M.A. and Matzke, A.J.M. (1999) Integrated pararetroviral sequences define a unique class of dispersed repetitive DNA in plants. Proc. Natl. Acad. Sci. USA, 96, 13 241–13 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, A.P. , Geijskes, R.J. , Dale, J.L. and Harding, R.M. (2011a) Molecular characterization of six badnavirus species associated with leaf streak disease of banana in East Africa. Ann. Appl. Biol. 158, 346–353. [Google Scholar]
- James, A.P. , Geijskes, R.J. , Dale, J.L. and Harding, R.M. (2011b) Development of a novel Rolling‐Circle Amplification technique to detect Banana streak virus that also discriminates between integrated and episomal virus sequences. Plant Dis. 95, 57–62. [DOI] [PubMed] [Google Scholar]
- Kenyon, L. , Lebas, B.S.M. and Seal, S.E. (2008) Yams (Dioscorea spp.) from the South Pacific Islands contain many novel badnaviruses: implications for international movement of yam germplasm. Arch. Virol. 153, 877–889. [DOI] [PubMed] [Google Scholar]
- King, A.M.Q. , Adams, M.J. , Carstens, E.B. and Lefkowitz, E.J. (2012) Virus Taxonomy: IXth Report of the International Committee on Taxonomy of Viruses, Vol. 9 London: Elsevier/Academic Press. [Google Scholar]
- Larkin, M.A. , Blackshields, G. , Brown, N.P. , Chenna, R. , McGettigan, P.A. , McWilliam, H. , Valentin, F. , Wallace, I.M. , Wilm, A. , Lopez, R. , Thompson, J.D. , Gibson, T.J. and Higgins, D.G. (2007) ClustalW and ClustalX version 2.0. Bioinformatics, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Le Provost, G. , Iskra‐Caruana, M.‐L. , Acina, I. and Teycheney, P.‐Y. (2006) Improved detection of episomal banana streak viruses by multiplex immunocapture PCR. J. Virol. Methods, 137, 7–13. [DOI] [PubMed] [Google Scholar]
- Lockhart, B.E.L. , Menke, J. , Dahal, G. and Olszewski, N.E. (2000) Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol. 81, 1579–1585. [DOI] [PubMed] [Google Scholar]
- Mette, M.F. , Kanno, T. , Aufsatz, W. , Jakowitsch, J. , van der Winden, J. , Matzke, M.A. and Matzke, A.J.M. (2002) Endogenous viral sequences and their potential contribution to heritable virus resistance in plants. EMBO J. 21, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, E. , Dupuy, V. , Blondin, L. , Bauffe, F. , Daugrois, J.‐H. , Laboureau, N. and Iskra‐Caruana, M.‐L. (2011) High molecular variability of sugarcane bacilliform viruses in Guadeloupe implying the existence of at least three new species. Virus Res. 160, 414–419. [DOI] [PubMed] [Google Scholar]
- Ndowora, T. , Dahal, G. , LaFleur, D. , Harper, G. , Hull, R. , Olszewski, N.E. and Lockhart, B.E.L. (1999) Evidence that badnavirus infection in Musa can originate from integrated pararetroviral sequences. Virology, 255, 214–220. [DOI] [PubMed] [Google Scholar]
- Phillips, S. , Briddon, R.W. , Brunt, A.A. and Hull, R. (1999) The partial characterization of badnavirus infecting the Greater asiatic or water yam (Dioscorea alata). J. Phytopathol. 147, 265–269. [Google Scholar]
- Richert‐Pöggeler, K.R. , Noreen, F. , Schwarzacher, T. , Harper, G. and Hohn, T. (2003) Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J. 22, 4836–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal, S. , Turaki, A. , Muller, E. , Kumar, P.L. , Kenyon, L. , Filloux, D. , Galzi, S. , Lopez‐Montes, A. and Iskra‐Caruana, M.‐L. (2014) The prevalence of badnaviruses in West African yams (Dioscorea cayenensis‐rotundata) and evidence of endogenous pararetrovirus sequences in their genomes. Virus Res. doi: 10.1016/j.virusres.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Seal, S.E. and Muller, E. (2007) Molecular analysis of a full‐length sequence of a new yam badnavirus from Dioscorea sansibarensis . Arch. Virol. 152, 819–825. [DOI] [PubMed] [Google Scholar]
- Soltis, P.S. , Soltis, D.E. and Chase, M.W. (1999) Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature, 402, 402–404. [DOI] [PubMed] [Google Scholar]
- Staginnus, C. and Richert‐Pöggeler, K.R. (2006) Endogenous pararetroviruses: two faceted travelers in the plant genome. Trends Plant Sci. 11, 485–491. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Nei, M. and Kumar, S. (2004) Prospects for inferring very large phylogenies by using the neighbor‐joining method. Proc. Natl. Acad. Sci. USA, 101, 11 030–11 035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. and Kumar, S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teycheney, P.‐Y. and Geering, A.D.W. (2011) Endogenous viral sequences in plant genomes In: Recent Advances in Plant Virology (Caranta C., Aranda M.A., Tepfer M. and Lopez‐Moya J.J., eds), pp. 343–362. Norfolk: Caister Academic Press. [Google Scholar]
- Yang, I.C. , Hafner, G.J. , Revill, P.A. , Dale, J.L. and Harding, R.M. (2003) Sequence diversity of South Pacific isolates of Taro bacilliform virus and the development of a PCR‐based diagnostic test. Arch. Virol. 148, 1957–1968. [DOI] [PubMed] [Google Scholar]