

Summary
Plant infection by poleroviruses is restricted to phloem tissues, preventing any classical leaf rub inoculation with viral RNA or virions. Efficient virus inoculation to plants is achieved by viruliferous aphids that acquire the virus by feeding on infected plants. The use of promoter‐driven infectious cDNA is an alternative means to infect plants and allows reverse genetic studies to be performed. Using Beet mild yellowing virus isolate 2ITB (BMYV‐2ITB), we produced a full‐length infectious cDNA clone of the virus (named BMYV‐EK) placed under the control of the T7 RNA polymerase and the Cauliflower mosaic virus 35S promoters. Infectivity of the engineered BMYV‐EK virus was assayed in different plant species and compared with that of the original virus. We showed that in vitro‐ or in planta‐derived transcripts were infectious in protoplasts and in whole plants. Importantly, the natural aphid vector Myzus persicae efficiently transmitted the viral progeny produced in infected plants. By comparing agroinoculation and aphid infection in a host range assay, we showed that the engineered BMYV‐EK virus displayed a similar host range to BMYV‐2ITB, except for Nicotiana benthamiana, which proved to be resistant to systemic infection with BMYV‐EK. Finally, both the BMYV‐EK P0 and the full‐length clone were able to strongly interfere with post‐transcriptional gene silencing.
Introduction
Positive‐stranded RNA phytoviruses are usually directly infectious following rub inoculation of infected plant sap, purified viral particles or in vitro transcribed genomic RNAs (gRNAs), as exemplified by Tobacco mosaic virus (Fraenkel‐Conrat, 1957, 1962) or Beet necrotic yellow vein virus (Quillet et al., 1989). However, members of the Luteoviridae family are restricted to phloem cells, preventing any rub inoculation to produce an infection, except when co‐inoculated with either umbravirus Pea enation mosaic virus‐2 or Groundnut rosette virus (Mayo et al., 2000; Ryabov et al., 2001). In nature, Luteoviridae members are delivered directly into phloem cells by aphids during feeding (Ziegler‐Graff and Brault, 2008). Studies on the biology of members of the Polerovirus genus (Luteoviridae) require virus acquisition by aphids on naturally infected plants or via aphid membrane feeding on purified viral particles in sucrose solution (Brault et al., 1995).
The production of full‐length infectious clones of viral genomes has proven to be essential for reverse genetic studies. For poleroviruses, such clones can be used to infect plants without aphids via Agrobacterium tumefaciens‐mediated gene transfer in a process called ‘agroinfection’. In vivo transcribed viral RNA from the Agrobacterium‐delivered DNA initiates the expression of nonstructural viral proteins involved in replication and RNA silencing suppression, allowing the initiation of a viral cycle of replication and subsequent systemic spread of the virus throughout the plant (Leiser et al., 1992; Prufer et al., 1995).
Beet mild yellowing virus (BMYV) is a polerovirus, and represents the major cause of the yellowing disease of sugar beet (Stevens et al., 2005). BMYV is transmitted efficiently by the aphid Myzus persicae in a circulative and nonpropagative manner (Gray and Gildow, 2003). BMYV forms nonenveloped icosahedral particles (T = 3) containing a 5.7‐kb linear positive‐stranded RNA encoding six proteins and linked at the 5′ end to a viral genome‐linked protein (VPg) (Guilley et al., 1995). To date, an infectious cDNA clone of a German isolate (BMYV‐IPP) has been reported (Stephan and Maiss, 2006). An additional green fluorescent protein (GFP)‐expressing clone of BMYV‐BB (Broom's Barn) was also constructed, but its infectivity was assayed only in protoplasts (Stevens and Vigano, 2007).
To initiate reverse genetic studies on BMYV‐2ITB collected from infected sugar beet (Guilley et al., 1995; Lemaire et al., 1995) and maintained on Beta vulgaris, and to avoid genetic drift and selection or adaptation as a result of successive plant passages, we introduced full‐length cDNA clones under the T7 or Cauliflower mosaic virus (CaMV) 35S promoter to produce in vitro and in vivo transcripts, respectively. Both in vitro and in vivo transcripts were fully infectious in protoplasts and in several hosts, including sugar beet. In addition, the new replicating virus, referred to as BMYV‐EK, was fully aphid transmissible. By comparing the host range of BMYV‐EK with that of the original BMYV‐2ITB isolate, we identified Nicotiana benthamiana as being resistant to systemic infection to BMYV‐EK, in contrast with the parental isolate which was infectious on this host. Previous studies have demonstrated that the P0 protein of some poleroviruses acts as a viral suppressor of RNA silencing (VSR) (Kozlowska‐Makulska et al., 2010; Pfeffer et al., 2002). We therefore investigated the RNA silencing suppressor activity of the constructed BMYV‐EK clone and its encoded P0 protein, and showed that the P0 protein expressed ectopically or from the full‐length infectious clone displayed a silencing suppressor activity. Moreover, we demonstrated that the P0 silencing suppressor activity of BMYV‐EK was not responsible for the lack of infection of N. benthamiana.
Results
Construction of full‐length cDNA clones under the control of the T7 and CaMV 35S promoters
Viral RNA extracted from virions prepared from BMYV‐2ITB‐infected plants was polyadenylated using polyA polymerase. The polyadenylated viral RNA served as a template for cDNA synthesis using oligo‐dT and primer 2 (Table 1) to produce three cDNA fragments by polymerase chain reaction (PCR) (Fig. 1). Oligonucleotides (Fig. 1 and Table 1) were selected based on the sequence of BMYV‐2ITB and on the naturally occurring NsiI, SalI and BamHI restriction sites in the cDNA copy of the gRNA (NC_003491). Amplicons were assembled to obtain full‐length cDNA clones under the control of the T7 or 35S promoter (Fig. 1). The newly engineered virus was named BMYV‐EK (GenBank KC121026). When compared with the parent BMYV‐2ITB (GenBank X83110) genome sequence (Guilley et al., 1995), an additional T residue in the intergenic region after nucleotide 3519 in the 2ITB sequence was found in the BMYV‐EK genome, leading to a 5723‐nucleotide genome, one base longer than BMYV‐2ITB (Table 2). Moreover, base transitions affected 12 pyrimidines and nine purines, whereas only one transversion was observed (Table 2). Such nucleotide variations were either silent or induced amino acid changes within the encoded proteins (Table 2). Overall, the sequence identity between BMYV‐EK and BMYV‐2ITB was 99.6%.
Table 1.
Sequences of the oligonucleotides used to construct the full‐length cDNA clone of Beet mild yellowing virus (BMYV). The number by which each primer is referred is indicated in the first column. Restriction enzyme sites within sequences are in bold. BMYV sequences are underlined. Promoter sequences are italicized
| Sequence position in | Sequence 5′–3′ | ||
|---|---|---|---|
| BMYV | 35S promoter | ||
| 1 | 1–28 | TTTGGTACC TAATACGACTCACTATAGG ACAAAAGAAACCAGCGAGGATCTAGCAG | |
| 2 | 2379–2351 | GTGC GTCGAC CGTAAGCAACATACGGGAC | |
| 3 | 2366–2392 | TACG GTCGAC GCACGCACAGAGGCTGG | |
| 4 | 4131–4104 | TGTAGA GGATCC TGAATTGGTCCTTGGC | |
| 5 | 4114–4141 | AATTCA GGATCC TCTACAAAGGCAATGG | |
| 6 | 5697–5722 | TTCTGCAGTTACTAGTTTTTTTTTTTTTACACCGAAGTGCCGTAGGGAGTTATC | |
| 7 | 1–10 | GCTCGGTACC CCCCTACTCC | |
| 8 | 11–1 | 401–385 | GTTTCTTTTGT CCTCTCCAAATGAAATG |
| 9 | 1–18 | 396–401 | GAGAGG ACAAAAGAAACCAGCGAG |
| 10 | 1040–1021 | GCTACTAGTTTATGCATCAACAACTGATCCC | |
Figure 1.
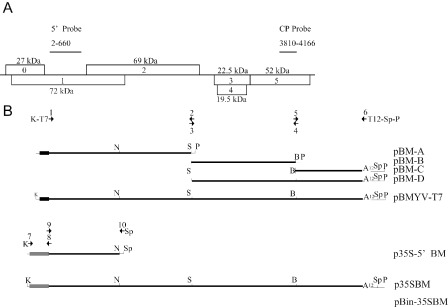
Beet mild yellowing virus (BMYV) genome organization, and cloning strategies used to produce BMYV‐T7 and BMYV‐EK clones. (A) Genome organization of BMYV. Open reading frames (ORFs) are boxed and numbered with the molecular weight of the encoded protein indicated. (B) Cloning strategies used to construct the full‐length cDNA clone of BMYV under the control of the T7 RNA polymerase promoter (small dark box) or the Cauliflower mosaic virus 35S promoter (large grey box). Numbering of arrows depicts primers corresponding to Table 1. The names of the resulting plasmids are indicated on the right. B, BamHI; K, KpnI; N, NsiI; P, PstI; Sp, SpeI. Thick lines correspond to the viral sequences and thin lines represent vector polylinker sequences. A12 represents a 12‐adenosine stretch added during cloning.
Table 2.
Nucleotide changes observed between Beet mild yellowing virus (BMYV)‐2ITB and BMYV‐EK genomic sequences. Nucleotide positions refer to the 2ITB (single and top) and EK (single and bottom) sequences. The first line of letters corresponds to the 2ITB sequence and the second line to the EK sequence. The mutation incidence is indicated for each coding region, except for the P2 open reading frame (ORF). Mutations within P2 (2131–3310) are silent and therefore are not detailed. The proteins affected by the mutation are shown above the changes. The first letter refers to the 2ITB amino acid, the number to its position in the protein sequence and the second letter to the EK amino acid. –, silent
| 229 | 236 | 752 | 1286 | 1457 | 1594 | 1747 | 2131 | 2401 | 2431 | 3310 | 3519 | 4065 | 4073 | 4086 | 4199 | 4547 | 5450 | 5516 | 5520 | 5547 | 5605 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3520 | 4066 | 4074 | 4087 | 4200 | 4548 | 5451 | 5517 | 5521 | 5548 | 5606 | |||||||||||
| C | A | G | T | T | C | T | C | T | C | C | – | G | T | G | G | C | A | G | A | A | C |
| T | G | A | C | C | T | C | T | C | T | T | +T | A | C | A | C | T | G | A | G | G | T |
| P0 | P3 | P5 | |||||||||||||||||||
| – | T69A | A157T | – | A164T | – | – | – | – | K642E | T651A | S671F | ||||||||||
| P1 | P4 | ||||||||||||||||||||
| S25L | – | – | – | – | A480V | L531S | – | L149P | – |
Infectivity of full‐length transcript
Five or ten micrograms of in vitro transcripts derived from the pBMYV‐T7 KpnI–SpeI double‐digested template were used to transfect Chenopodium quinoa protoplasts in the presence or absence of 25 μg of yeast carrier RNA. Protoplast aliquots were harvested at 0, 24, 48 and 72 h post‐inoculation (hpi). Total RNA was extracted and analysed by Northern blot using a BMYV‐coat protein (CP) specific probe (Figs 1 and 2). No signal was observed in the noninoculated sample (Fig. 2, mock) and the input RNA was not detected in the samples harvested immediately after infection (Fig. 2, 0 hpi). Signals corresponding to both the gRNA and subgenomic RNA (sgRNA) were detected with increasing intensities in protoplast samples collected at 24 and 48 hpi. An increase in viral RNA accumulation after 48 hpi was not observed reproducibly, however. In addition, in two independent experiments, the infection of protoplasts was more efficient when yeast total RNA was omitted (Fig. 2, compare lanes at 48 hpi). These data show the infectivity of the engineered BMYV‐EK cDNA clone constructed under the control of the T7 promoter.
Figure 2.
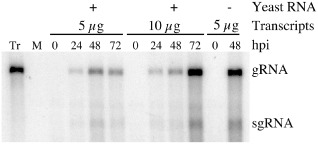
Chenopodium quinoa protoplast infection time course with Beet mild yellowing virus (BMYV)‐T7 in vitro transcripts. Protoplasts were transfected with transcripts in the presence (+) or absence (–) of yeast carrier RNA. Viral RNAs were detected using a BMYV‐CP‐specific probe. Positions of the genomic (gRNA) and subgenomic (sgRNA) RNAs are indicated on the right; 2.5 ng of transcripts (Tr) were loaded as a migration control. M, mock infection; hpi, hours post‐inoculation.
35S promoter‐driven BMYV infection
The BMYV‐EK cDNA sequence was placed under the control of the CaMV 35S promoter to produce the binary vector pBin‐35SBM. This construct was delivered via Agrobacterium tumefaciens to both Arabidopsis thaliana and Montia perfoliata by leaf infiltration. Three weeks post‐inoculation, total RNA from noninoculated leaves was purified and subjected to Northern blot analysis using the 5′ probe (Fig. 1) to avoid the background generated by the CP probe on infected plant total RNAs (data not shown). BMYV gRNA was reproducibly detected in the systemic leaves of infiltrated A. thaliana (Fig. 3A) and M. perfoliata (Fig. 3B), although the level of viral RNA accumulation varied between the plants. These results indicate that these two plants are hosts for BMYV‐EK. Based on existing sequence similarities between Turnip yellows virus (TuYV)‐ and BMYV‐CPs, and the cross‐reactivity of TuYV‐directed antibodies towards BMYV (Duffus and Russell, 1975; Herrbach et al., 1991), we used TuYV antisera to immunodetect BMYV‐CP in infected plants. A band of about 22 kDa, corresponding to the BMYV‐CP molecular weight, was visualized by Western blot in infected M. perfoliata plants (Fig. 3C). The signals corresponding to BMYV‐CP varied among plants, but were in perfect correlation with the level of RNA accumulation observed by Northern blot (Fig. 3B).
Figure 3.
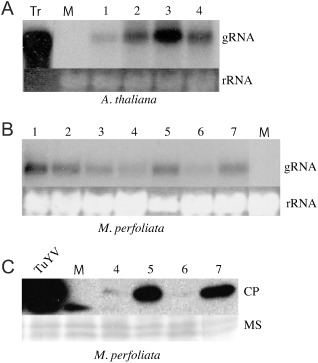
Plant agroinfection using the pBin‐35SBM construct. (A) Detection of the genomic Beet mild yellowing virus (BMYV)‐EK RNA in four independent Arabidopsis thaliana upper noninoculated leaves. Five nanograms of in vitro transcripts (Tr) were used as a control for positioning the genomic RNA (gRNA). (B) Detection of the genomic BMYV‐EK RNA in seven independent Montia perfoliata noninoculated leaves. (C) Detection of the BMYV‐EK coat protein (CP) in M. perfoliata noninoculated leaves of plants exhibiting different levels of RNA accumulation using an antiserum raised against Turnip yellows virus (TuYV)‐CP. Equal loadings of total RNA or proteins were visualized by ethidium bromide staining (rRNA) and membrane staining (MS). Numbers correspond to the individual plants tested. Viral RNAs were detected using the 5′ probe (A and B). Plants 4–7 from (B) were analysed in (C). M, mock infection; TuYV, TuYV‐infected plant extract.
BMYV‐EK viral particle formation and transmission
The production of fully functional viral particles from the BMYV‐EK full‐length clone can be addressed by performing virus transmission tests using the aphid vector M. persicae and infected plants, or purified particles, as virus source (Brault et al., 2000). As polerovirus acquisition by the vector requires appropriate viral particle formation (Brault et al., 2003, 2007; Reinbold et al., 2001), this assay provides insights into both virion assembly and transmission efficiency of the full‐length clone‐derived virus. BMYV‐EK‐agroinfected A. thaliana plants were used as virus source in the transmission assay. After a 48‐h feeding period on infected plants, the aphids were transferred to 18 M. perfoliata test plants that were evaluated by enzyme‐linked immunosorbent assay (ELISA) 3 weeks later. Transmission was 100% efficient (data not shown), demonstrating that the infectious BMYV‐EK clone produced fully functional aphid‐transmissible virions.
BMYV‐EK host range and comparison of the inoculation methods
A comparative analysis of the host range of BMYV‐EK and BMYV‐2ITB was conducted using two different means of virus inoculation (agroinfiltration and inoculation by aphids). Similar numbers of plants, known or predicted hosts for BMYV, were inoculated with either Agrobacterium tumefaciens carrying pBin‐35SBM or viruliferous aphids carrying the BMYV‐2ITB virus. The presence of the virus in the upper noninoculated leaves was assayed by ELISA 3 weeks later (Table 3). The Brassicaceae A. thaliana infection efficiency and viral titre were identical between the two viruses (Table 3, Mann–Whitney test, P = 0.45). Similarly, for Beta macrocarpa and Spinacia oleraceae Géant d'hiver, in the Chenopodiaceae family, no difference in virus accumulation was observed between the two viruses (Table 3). Conversely, virus accumulation in the infected B. vulgaris was statistically lower for BMYV‐EK than for BMYV‐2ITB (Table 3, Mann–Whitney test, P = 0.002). In the same manner, S. oleraceae Monstrueux de Viroflay was a poorly susceptible host for BMYV‐EK; both the infection rate and virus titer were strongly reduced for BMYV‐EK when compared with BMYV‐2ITB (Table 3, chi‐squared and Mann–Whitney tests, P ≤ 0.05). Both viruses were also poorly infectious in the solanaceous host N. benthamiana (Table 3); agroinfection with BMYV‐EK was unsuccessful and the aphid‐mediated transmission rate of BMYV‐2ITB was only 50% efficient (Table 3, chi‐squared test, P = 0.026). BMYV‐2ITB accumulation in N. benthamiana was also strongly reduced compared with virus accumulation in the other plant species tested.
Table 3.
Infectivity of Beet mild yellowing virus (BMYV)‐EK and BMYV‐2ITB in different plants using agroinoculation and virus transmission by aphids, respectively
| BMYV‐EK | BMYV‐2ITB | χ 2 | Mann–Whitney | |||
|---|---|---|---|---|---|---|
| Infected/totala | A 405 ± SDb | Infected/total | A 405 ± SD | P c | P d | |
| Brassicaceae | ||||||
| Arabidopsis thaliana | 5/5 | 0.48 ± 0.110 | 5/5 | 0.50 ± 0.070 | Na | 0.45 |
| Mock | 0.11 ± 0.006 | |||||
| Chenopodiaceae | ||||||
| Beta macrocarpa | 8/8 | 0.53 ± 0.250 | 8/8 | 0.47 ± 0.090 | Na | 0.88 |
| Mock | 0.11 ± 0.006 | |||||
| B. vulgaris | 6/7 | 0.56 ± 0.180 | 7/7 | 1.00 ± 0.200 | 0.29 | 0.002 |
| Mock | 0.11 ± 0 | |||||
| Spinacia oleraceae | ||||||
| Géant d'hiver | 9/10 | 0.65 ± 0.400 | 10/10 | 0.46 ± 0.220 | 0.30 | 0.41 |
| Mock | 0.10 ± 0 | |||||
| S. oleraceae | ||||||
| Monstrueux de Viroflay | 3/10 | 0.33 ± 0.050 | 10/10 | 0.72 ± 0.110 | 0.001 | 0.007 |
| Mock | 0.10 ± 0.006 | |||||
| Solanaceae | ||||||
| Nicotiana benthamiana | 0/7 | 0.11 ± 0.008 | 5/10 | 0.22 ± 0.020 | 0.026 | Na |
| Mock | 0.10 ± 0.006 | |||||
Number of infected plants versus inoculated plants.
The mean enzyme‐linked immunosorbent assay (ELISA) values correspond to infected plants only and mock‐inoculated plants, or, in the case of N. benthamiana challenged with BMYV‐EK, noninfected plants.
χ 2 test was used to compare the inoculation method.
Mann–Whitney test was applied to compare the infectivity potential.
P, P > 0.05 refers to a nonsignificant difference; Na, not applicable.
Comparison of BMYV‐EK and BMYV‐2ITB aphid transmissibility
To address the ability of both viruses to be transmitted by aphids, we performed aphid transmission assays using purified virus prepared from BMYV‐EK‐ or BMYV‐2ITB‐infected M. perfoliata plants as viral source. After a 24‐h acquisition period, aphids were transferred to five different plant species belonging to the Brassicaceae, Chenopodiaceae and Solanaceae families. Three weeks after aphid removal, viral accumulation was assayed in the noninoculated leaves by ELISA. As observed previously, A. thaliana, B. macrocarpa, B. vulgaris and S. oleraceae Géant d'hiver sustained the virus accumulation of both viruses (Table 4). In contrast with the former experiment, the infection of N. benthamiana plants with BMYV‐EK was achieved in 50% of plants loaded with viruliferous aphids, whereas agroinoculation with the infectious clone was unsuccessful (Table 3). Virus accumulation of BMYV‐EK in infected N. benthamiana plants was statistically lower when compared with plants infected with BMYV‐2ITB (Table 4). Taken together, these results showed that BMYV‐EK behaves like BMYV‐2ITB on Brassicaceae and Chenopodiaceae plants whatever the inoculation mode, but is less infectious than BMYV‐2ITB on N. benthamiana plants.
Table 4.
Aphid transmission test using viruses purified from Beet mild yellowing virus (BMYV)‐EK‐agroinfected or BMYV‐2ITB‐aphid‐infected plants
| BMYV‐EK | BMYV‐2ITB | ||||
|---|---|---|---|---|---|
| Infected/totala | A 405 ± SDb | Infected/total | A 405 ± SD | P c | |
| Brassicaceae | |||||
| Arabidopsis thaliana | 10/10 | 1.58 ± 0.590 | 10/10 | 1.36 ± 0.450 | 0.36 |
| Mock | 0.10 ± 0.007 | ||||
| Chenopodiaceae | |||||
| Beta macrocarpa | 5/5 | 1.13 ± 0.250 | 5/5 | 0.91 ± 0.280 | 0.55 |
| Mock | 0.10 ± 0 | ||||
| B. vulgaris | 12/12 | 1.26 ± 0.200 | 12/12 | 1.26 ± 0.470 | 0.81 |
| Mock | 0.11 ± 0.006 | ||||
| Spinacia oleraceae | |||||
| Géant d'hiver | 12/12 | 1.23 ± 0.320 | 12/12 | 1.02 ± 0.360 | 0.11 |
| Mock | 0.10 ± 0.006 | ||||
| Solanaceae | |||||
| Nicotiana benthamiana | 6/12 | 0.30 ± 0.150 | 11/12 | 0.49 ± 0.110 | 0.01 |
| Mock | 0.10 ± 0.006 | ||||
Number of plants infected [determined by double antibody sandwich‐enzyme‐linked immunosorbent assay (DAS‐ELISA)]/number of plants tested.
The ELISA values correspond to infected plants (top row) and mock‐inoculated plants (bottom row).
P, P value from Mann–Whitney statistical analysis; P > 0.05 refers to a nonsignificant difference.
Silencing suppression activity driven by BMYV‐EK
We used the patch‐test method (Voinnet et al., 1998) to evaluate the VSR activity of the P0 protein encoded by BMYV‐EK. As a control, we infiltrated TuYV‐P0 (Pfeffer et al., 2002), which exhibits a strong inhibition of gene silencing activity (Fig. 4). As agroinoculation did not lead to systemic infection of N. benthamiana, we also tested the VSR activity of P0 when expressed in its viral context. Therefore, the full‐length cDNA clone was used in the patch‐test assay and its VSR activity was compared with that of the TuYV full‐length cDNA clone. Four days post‐infiltration, a faint fluorescent patch was observed on N. benthamiana 16C leaves infiltrated with Agrobacterium tumefaciens carrying the GFP‐RNA silencing trigger construct and bacteria expressing the empty binary vector (Fig. 4A, lane 2). However, when pBin‐GFP was co‐expressed with pBin‐P0‐TuYV, pBin‐P0‐BMYV‐EK, pBin‐35STuYV or pBin‐35SBM, patches were brightly fluorescent, indicating over‐expression of the GFP protein and a strong suppression activity in the infiltrated areas (Fig. 4A, lanes 3–6). When compared with mock‐infiltrated 16C plants (Fig. 4B, Mock), Northern blot analyses revealed a slight increase in the GFP mRNA in pBin‐Ø patches. Silencing of GFP was, however, correlated with the high production of GFP siRNAs (Fig. 4B, pBin‐Ø). Conversely, when P0‐BMYV‐EK was expressed, the over‐accumulation of GFP mRNA was accompanied by a drastic reduction in GFP siRNA levels, demonstrating the silencing suppression activity of P0 encoded by the engineered BMYV‐EK virus. This silencing suppression activity was similar to that observed with P0 encoded by TuYV (Fig. 4, lanes 3 and 5). Similarly, when both full‐length infectious clones were used (Fig. 4B, lanes 4 and 6), GFP siRNA levels were also strongly reduced, showing that the silencing suppression activity was effective with both full‐length clones, BMYV‐EK and TuYV. The presence of both viruses in the patch was confirmed by the detection of both gRNA and sgRNA by Northern blot (Fig. 4B) and by immunodetection of the major CP (Fig. 4C), strengthening the fact that the initial transcripts could efficiently be replicated in the infiltrated leaves. Although the sequences of the BMYV and TuYV probes were 81% identical, cross‐hybridization led to weaker signals than the genuine hybridization (Fig. 4B). Taken together, these results suggest that the initial steps of polerovirus infection in planta lead to the expression of P0 VSR, even though this protein has never been successfully detected by Western blotting in virus‐infected plants (Pfeffer et al., 2002).
Figure 4.
Suppression of RNA silencing by the P0 protein and the full‐length clones of Beet mild yellowing virus (BMYV)‐EK and Turnip yellows virus (TuYV). Leaves of Nicotiana benthamiana 16C were infiltrated with buffer (Mock) or mixtures of agrobacteria containing the binary vectors encoding the green fluorescent protein (GFP) and no viral suppressor of RNA silencing (VSR) (Ø, lane 2), BMYV‐P0 (lane 3), TuYV‐P0 (lane 5) or full‐length infectious clones (35SBM and 35STuYV, lanes 4 and 6, respectively). (A) GFP fluorescence was visualized 4 days post‐infiltration under UV light and photographed using a Schott Green VG5 filter (Schott Glass Technologies, Inc., Elmsford, NY, USA). (B) Total RNA was extracted from the agroinfiltrated patches and Northern blotted to detect the presence of the GFP mRNA and GFP siRNA using a GFP cDNA probe. Viral genomic (gRNA) and subgenomic (sgRNA) RNAs were detected using a TuYV DNA probe (TuYV 3′ probe) and after membrane stripping with a BMYV DNA probe (BMYV 3′ probe). Each probe hybridized on both BMYV and TuYV 3′ conserved RNA sequences (81% identity). RNA load represents ethidium bromide staining of the membranes. (C) BMYV and TuYV coat proteins (CPs) were immunodetected 4 days post‐infiltration using antibodies raised against TuYV. Protein loading was visualized by membrane staining (MS).
Discussion
BMYV‐EK clone produces a systemic infection and the virions are efficiently transmitted by aphids
The full‐length cDNA sequence of the BMYV‐2ITB isolate was obtained and placed under the control of the T7 RNA polymerase or the CaMV 35S RNA polymerase II promoter to produce the virus referred to as BMYV‐EK. Amino acid changes were found in the P0, P1, P3–P5 and P4 sequences of BMYV‐EK when compared with the corresponding BMYV‐2ITB proteins, whereas all the nucleotide changes observed within the P2 open reading frame (ORF) of BMYV‐EK were silent (Table 2). T7‐driven in vitro transcripts produced from the BMYV‐EK full‐length cDNA clone replicated efficiently in protoplasts and produced the expected genomic and subgenomic viral RNAs (Fig. 2), despite the presence of an additional nucleotide at position 3520 and two extra G moieties at the 5′ extremity of the BMYV‐EK RNA.
When tested by agroinoculation, the BMYV‐EK full‐length cDNA clone placed under the control of the CaMV 35S promoter was able to infect systemically A. thaliana and M. perfoliata plants, as shown by the detection of both viral gRNA and CP in noninoculated leaves. The CP detection reflected the efficient sgRNA production ensured by viral replication, and allowed the use of ELISA to evaluate the infection potential of the BMYV‐EK full‐length cDNA clone. Finally, the BMYV‐EK virus was efficiently transmitted, indicating that the residue changes within both the CP and the P3–P5 fusion protein, also referred to as the read‐through protein (Table 2, P3–P5), did not affect the virus transmission efficiency by aphids. In addition, by being aphid transmitted, the BMYV‐EK gRNA was shown to be encapsidated. Taken together, these experiments prove that the BMYV‐EK virus is able to fulfil an entire viral cycle.
By comparing agroinoculation and aphid transmission to deliver both BMYV isolates to plants, we observed differences in the ability to infect some hosts between the natural and cloned isolates. All the plant species tested in this study have been reported to be hosts for BMYV (Hauser et al., 2002; Stephan and Maiss, 2006). The BMYV‐2ITB isolate and the BMYV‐EK clone infect similarly A. thaliana, B. macrocarpa and S. oleraceae Géant d'hiver (Table 3, P > 0.05), whatever the inoculation method (agroinfiltration or aphid inoculation). Conversely, although the rate of infection of B. vulgaris was comparable between the two viruses (Table 3, P = 0.29), virus accumulation of BMYV‐EK was reduced when infection was initiated via agrobacteria. In B. vulgaris and S. oleraceae Monstrueux de Viroflay, BMYV‐2ITB accumulated at a higher viral titre when compared with BMYV‐EK.
To exclude any host range effect of Agrobacterium tumefaciens for the delivery of BMYV‐EK, we compared the infectivity of both viruses after aphid transmission of purified virus (Table 4). When similar virus concentrations were used as a source, we confirmed that A. thaliana and all the Chenopodiaceae hosts were infected similarly with the two viruses (Table 4, Mann–Whitney test, P > 0.05). However, in N. benthamiana plants, the infection rate of BMYV‐2ITB was significantly higher than that of BMYV‐EK (P = 0.01), suggesting that certain steps of the infection process are impaired in this host. The natural BMYV‐2ITB isolate consists of quasispecies, which can adapt to fit to the host (Holmes, 2010; Lancaster and Pfeiffer, 2012). By contrast, the BMYV‐EK clone represents one variant originating from the BMYV‐2ITB isolate and may not initiate a compatible interaction with N. benthamiana plants. Moreover, when compared with the other host plants tested, N. benthamiana sustains less efficient multiplication of the BMYV‐2ITB isolate, even when it is delivered by aphids. Interestingly, the full‐length infectious clone of the German isolate BMYV‐IPP (DQ132996), which was adapted to N. benthamiana (Stephan and Maiss, 2006), displayed 3.9% nucleotide divergence from BMYV‐EK (data not shown). Nucleotide substitutions were unequally distributed along the genome. One‐ and two‐nucleotide changes were found in the 5′ and 3′ untranslated region (UTR), respectively. The other substitutions were either silent or affected P0 (44 nucleotides modified, 19 residue changes including nine similar residues out of 239 amino acids), P1 (63 nucleotides modified, 28 residue changes including six similar residues out of 656 amino acids, with changes affecting mostly the N‐terminus of P1), P2 (53 nucleotides modified, eight residue changes including one similar residue out of 625 amino acids), P3–P5 (82 nucleotides modified, 19 residue changes including 10 similar residues out of 669 amino acids) and P4 (14 nucleotides modified, 12 residue changes including three similar residues out of 175 amino acids). Some of these nucleotide and amino acid residue variations may account for the difference in host adaptation observed between these two viruses.
One of the initial steps that could be affected by the nucleotide divergence observed in BMYV‐EK could be the inhibition of RNA silencing by the P0 VSR. Therefore, we tested whether the silencing suppression was functional for both the P0 expressed ectopically or from the viral full‐length clone. We found that the BMYV‐EK‐encoded P0 VSR protein displayed silencing activity comparable with the well‐characterized TuYV‐P0 VSR. Moreover, when using the agroinoculation of full‐length cDNA clones, we showed that the BMYV‐EK virus provided effective silencing suppression activity, which was comparable with that displayed by TuYV. This observation infers that the VSR activity was not the cause of the low accumulation rate of BMYV‐EK in N. benthamiana. Although BMYV‐EK was never detected in the noninoculated leaves of N. benthamiana‐infiltrated plants, sgRNA and CP were observed in the infiltrated leaf, suggesting that replication occurred, but that movement of BMYV‐EK was impaired and could not be attributed to a nonfunctional VSR. Further investigations are required to identify the viral products involved in N. benthamiana host restriction. To this aim, the construction of viral chimeras between BMYV‐IPP and BMYV‐EK may provide some clues to the molecular basis leading to host compatibility or incompatibility.
Experimental Procedures
Molecular cloning
The purification of viral particles from plants infected with the BMYV‐2ITB isolate was performed as described for TuYV (Brault et al., 1995). Viral RNA was obtained after phenol–chloroform extraction performed on viral particles, followed by ethanol precipitation. Viral RNA (2.5 μg) was incubated for 10 min at 70 °C before a polyA tailing using polyA polymerase (Invitrogen™, Life Technologies SAS, Saint Aubin, France) for 15 min at 37 °C, following the manufacturer's recommendations. Polyadenylated viral RNA was purified and reverse transcribed using oligo‐dT primer and primer 2 (Table 1; Fig. 1). Complementary DNAs served as template for the following PCRs. Amplicon A containing the T7 promoter and the first 2379 nucleotides of the 5′ region was produced with primers 1 and 2 (Table 1). After digestion with KpnI and SalI, the fragment was cloned into the KpnI–SalI sites of the pBluescript® vector using the same restriction sites to yield pBM‐A. Amplicons B (1765 bp) and C (1608 bp) were produced using primers 3 and 4, and primers 5 and 6, respectively. Both amplicons were cloned into pGEM‐T. Nonviral PstI and SpeI sites were incorporated within primer 6 for further cloning steps. The insert sequences of pBM‐A, pBM‐B and pBM‐C were verified by sequence analysis. pBM‐C was digested with BamHI and PstI, and the released fragment was inserted between the BamHI and PstI sites of pBM‐B to produce pBM‐D (nucleotides 2351–5722). Finally, the SalI–PstI insert from pBM‐D was subcloned into the SalI–PstI sites of pBM‐A to obtain a full‐length copy of BMYV cDNA fused to the T7 promoter. The 5′ sequence of BMYV cDNA was placed under the control of the CaMV 35S promoter by a PCR megaprimer strategy. Briefly, the 35S promoter amplicon tagged with the first 11 nucleotides of the viral cDNA sequence was produced using primers 7 and 8 (Table 1). Similarly, an amplicon tagged with a 35S sequence was obtained using primers 9 and 10. Both amplicons were mixed after gel purification and amplified using primers 7 and 10. The resulting PCR fragment was purified, KpnI and SpeI were digested and inserted between the KpnI and SpeI sites of the pKS vector to obtain p35S‐5′BM. This sequence was then used to substitute for the KpnI–NsiI fragment containing the T7 promoter of pBMYV‐T7 to create p35SBM. Escherichia coli XL1blue strain was used for all the above cloning steps. The KpnI–SpeI sequence of p35SBM was cloned into the pBin19 binary vector to produce pBin‐35SBM (Fig. 1) in E. coli MC1022 strain. This plasmid was further introduced into Agrobacterium tumefaciens GV3101. pBin‐Ø, pBin‐GFP, pBin‐P0‐TuYV and pBin‐35STuYV have been described elsewhere (Pfeffer et al., 2002). pBin‐P0‐BMYV‐EK was obtained by PCR cloning using XbaI–BamHI‐tagged primers, similar to that described for TuYV‐P0 cloning (Pfeffer et al., 2002). All the full‐length infectious clones were verified by sequencing the plasmid inserts.
Hybridization procedures
PCR primers were used in standard annealing conditions according to the manufacturer's recommendations. The Expand High FidelityPLUS PCR system (Roche Diagnostics, Meylan, France) was used in all amplification reactions. 32P‐radiolabelled probes for Northern blot hybridization were produced with a Prime‐a‐Gene® labelling system (Promega, Madison, WI, USA), using the manufacturer's recommendations, on purified amplicons corresponding to nucleotides 2–660 and 3810–4166 for 5′ and 3′ probes, respectively.
Inoculation procedures
The pBMYV‐T7 plasmid digested with both KpnI and SpeI enzymes served as a template for the production of run‐off in vitro transcripts. In vitro transcription was performed in the presence of cap analogue (New England Biolabs, Evry, France) as described previously (Quillet et al., 1989). The preparation of C. quinoa protoplasts was performed as described for TuYV (Veidt et al., 1992). Briefly, 250 000 protoplasts were inoculated by electroporation with in vitro transcripts. Plant agroinoculations, aphid transmission tests and ELISA were performed as described elsewhere (Brault et al., 1995) and adapted with the following conditions. Three weeks after agroinoculation, aphids were reared on infected plants for 48 h. Ten aphids were then transferred to each target plant for 96 h before insecticide treatment. Viral RNAs, siRNAs and protein analyses were performed as described previously (Kozlowska‐Makulska et al., 2010).
Statistical analysis
Statistical Analysis Software (version 9.2; SAS Institute Inc., Cary, NC, USA) was used to analyse the data.
Acknowledgements
The authors are grateful to Professor M. Welten for statistical suggestions, Dr W. A. Miller for critical reading of the manuscript, Dr O. Lemaire for providing BMYV‐2ITB virus, Malek Alioua for DNA sequencing, Louis Wiss for aphid breeding, and Daniele Scheidecker for protoplast isolation and technical assistance.
References
- Brault, V. , van den Heuvel, J.F. , Verbeek, M. , Ziegler‐Graff, V. , Reutenauer, A. , Herrbach, E. , Garaud, J.C. , Guilley, H. , Richards, K. and Jonard, G. (1995) Aphid transmission of beet western yellows luteovirus requires the minor capsid read‐through protein P74. EMBO J. 14, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault, V. , Mutterer, J. , Scheidecker, D. , Simonis, M.T. , Herrbach, E. , Richards, K. and Ziegler‐Graff, V. (2000) Effects of point mutations in the readthrough domain of the beet western yellows virus minor capsid protein on virus accumulation in planta and on transmission by aphids. J. Virol. 74, 1140–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault, V. , Bergdoll, M. , Mutterer, J. , Prasad, V. , Pfeffer, S. , Erdinger, M. , Richards, K.E. and Ziegler‐Graff, V. (2003) Effects of point mutations in the major capsid protein of beet western yellows virus on capsid formation, virus accumulation, and aphid transmission. J. Virol. 77, 3247–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault, V. , Herrbach, E. and Reinbold, C. (2007) Electron microscopy studies on luteovirid transmission by aphids. Micron, 38, 302–312. [DOI] [PubMed] [Google Scholar]
- Duffus, J.E. and Russell, G.E. (1975) Serological relationship between beet western yellows and beet mild yellowing viruses. Phytopathology, 68, 811–815. [Google Scholar]
- Fraenkel‐Conrat, H. (1957) Structure and infectivity of tobacco mosaic virus. Harvey Lect. 53, 56–68. [PubMed] [Google Scholar]
- Fraenkel‐Conrat, H. (1962) Infectious ribonucleic acid. Surv. Biol. Prog. 4, 59–92. [PubMed] [Google Scholar]
- Gray, S. and Gildow, F.E. (2003) Luteovirus–aphid interactions. Annu. Rev. Phytopathol. 41, 539–566. [DOI] [PubMed] [Google Scholar]
- Guilley, H. , Richards, K.E. and Jonard, G. (1995) Nucleotide sequence of beet mild yellowing virus RNA. Arch. Virol. 140, 1109–1118. [DOI] [PubMed] [Google Scholar]
- Hauser, S. , Stevens, M. , Beuve, M. and Lemaire, O. (2002) Biological properties and molecular characterization of beet chlorosis virus (BChV). Arch. Virol. 147, 745–762. [DOI] [PubMed] [Google Scholar]
- Herrbach, E. , Lemaire, O. , Ziegler‐Graff, V. , Lot, H. , Rabenstein, F. and Bouchery, Y. (1991) Detection of BMYV and BWYV‐isolates using monoclonal antibodies and radioactive RNA probes, and relationships among luteoviruses. Ann. Appl. Biol. 118, 127–138. [Google Scholar]
- Holmes, E.C. (2010) The RNA virus quasispecies: fact or fiction? J. Mol. Biol. 400, 271–273. [DOI] [PubMed] [Google Scholar]
- Kozlowska‐Makulska, A. , Guilley, H. , Szyndel, M.S. , Beuve, M. , Lemaire, O. , Herrbach, E. and Bouzoubaa, S. (2010) P0 proteins of European beet‐infecting poleroviruses display variable RNA silencing suppression activity. J. Gen. Virol. 91, 1082–1091. [DOI] [PubMed] [Google Scholar]
- Lancaster, K.Z. and Pfeiffer, J.K. (2012) Viral population dynamics and virulence thresholds. Curr. Opin. Microbiol. 15, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser, R.M. , Ziegler‐Graff, V. , Reutenauer, A. , Herrbach, E. , Lemaire, O. , Guilley, H. , Richards, K. and Jonard, G. (1992) Agroinfection as an alternative to insects for infecting plants with beet western yellows luteovirus. Proc. Natl. Acad. Sci. USA, 89, 9136–9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire, O. , Herrbach, E. , Stevens, M. , Bouchery, Y. and Smith, H.G. (1995) Detection of sugarbeet infecting beet mild yellowing luteovirus isolates with specific RNA probe. Phytopathology, 85, 1513–1518. [Google Scholar]
- Mayo, M. , Ryabov, E. , Fraser, G. and Taliansky, M. (2000) Mechanical transmission of Potato leafroll virus. J. Gen. Virol. 81, 2791–2795. [DOI] [PubMed] [Google Scholar]
- Pfeffer, S. , Dunoyer, P. , Heim, F. , Richards, K.E. , Jonard, G. and Ziegler‐Graff, V. (2002) P0 of Beet western yellows virus is a suppressor of posttranscriptional gene silencing. J. Virol. 76, 6815–6824. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Prufer, D. , Wipf‐Scheibel, C. , Richards, K. , Guilley, H. , Lecoq, H. and Jonard, G. (1995) Synthesis of a full‐length infectious cDNA clone of cucurbit aphid‐borne yellows virus and its use in gene exchange experiments with structural proteins from other luteoviruses. Virology, 214, 150–158. [DOI] [PubMed] [Google Scholar]
- Quillet, L. , Guilley, H. , Jonard, G. and Richards, K. (1989) In vitro synthesis of biologically active beet necrotic yellow vein virus RNA. Virology, 172, 293–301. [DOI] [PubMed] [Google Scholar]
- Reinbold, C. , Gildow, F.E. , Herrbach, E. , Ziegler‐Graff, V. , Goncalves, M.C. , van Den Heuvel, J.F. and Brault, V. (2001) Studies on the role of the minor capsid protein in transport of Beet western yellows virus through Myzus persicae . J. Gen. Virol. 82, 1995–2007. [DOI] [PubMed] [Google Scholar]
- Ryabov, E.V. , Fraser, G. , Mayo, M.A. , Barker, H. and Taliansky, M. (2001) Umbravirus gene expression helps potato leafroll virus to invade mesophyll tissues and to be transmitted mechanically between plants. Virology, 286, 363–372. [DOI] [PubMed] [Google Scholar]
- Stephan, D. and Maiss, E. (2006) Biological properties of Beet mild yellowing virus derived from a full‐length cDNA clone. J. Gen. Virol. 87, 445–449. [DOI] [PubMed] [Google Scholar]
- Stevens, M. and Vigano, F. (2007) Production of a full‐length infectious GFP‐tagged cDNA clone of Beet mild yellowing virus for the study of plant–polerovirus interactions. Virus Genes, 34, 215–221. [DOI] [PubMed] [Google Scholar]
- Stevens, M. , Freeman, B. , Liu, H.Y. , Herrbach, E. and Lemaire, O. (2005) Beet poleroviruses: close friends or distant relatives? Mol. Plant Pathol. 6, 1–9. [DOI] [PubMed] [Google Scholar]
- Veidt, I. , Bouzoubaa, S.E. , Leiser, R.M. , Ziegler‐Graff, V. , Guilley, H. , Richards, K. and Jonard, G. (1992) Synthesis of full‐length transcripts of beet western yellows virus RNA: messenger properties and biological activity in protoplasts. Virology, 186, 192–200. [DOI] [PubMed] [Google Scholar]
- Voinnet, O. , Vain, P. , Angell, S. and Baulcombe, D.C. (1998) Systemic spread of sequence‐specific transgene RNA degradation in plants is initiated by localized introduction of ectopic promoterless DNA. Cell, 95, 177–187. [DOI] [PubMed] [Google Scholar]
- Ziegler‐Graff, V. and Brault, V. (2008) Role of vector‐transmission proteins. Methods Mol. Biol. 451, 81–96. [DOI] [PubMed] [Google Scholar]