

Summary
The rice root nematode Hirschmanniella oryzae is the most abundant plant‐parasitic nematode in flooded rice fields and is distributed world‐wide. Although it is economically less important than sedentary nematodes, it can cause severe yield reductions and economic losses in specific environmental conditions. No transcriptome data for this genus were available until now. We have performed 454 sequencing on a mixed life stages population to gain an insight into nematode–plant interactions and nematode survival strategies. The results of two assembly strategies were combined to reduce the redundancy of the data, generating a final dataset of 21 360 contigs. The data were screened for putative plant cell wall‐modifying proteins, which facilitate nematode migration through host roots. A β‐mannanase, previously not reported in nematodes, was detected in the dataset. The data were screened for putative effector proteins that may alter the host defence mechanism. Two enzymes, chorismate mutase and isochorismatase, thought to be involved in the salicyclic acid pathway, were identified. Experimental treatments of H. oryzae with artificial seawater showed that late embryogenesis abundant (LEA) proteins and SXP/RAL‐2 are induced, suggesting that these proteins are involved in the process of anhydrobiosis. The newly generated data can highlight potential differences between sedentary and migratory nematodes, and will be useful in the further study of host–nematode interactions and the developmental biology of this nematode.
Introduction
The rice root nematode Hirschmanniella oryzae is a migratory endoparasite with Oryza sativa as its main host. Hirschmanniella oryzae is the most common nematode in flooded rice ecosystems all over the world (Prot and Rahman, 1994). The nematode is adapted to anaerobic conditions and this makes it the most abundant nematode species in these ecosystems (Babatola, 1981; Maung et al., 2010).
Symptoms of damage caused by H. oryzae are difficult to interpret. In many cases, there are no visible symptoms above ground, although some chlorosis and growth retardation can appear (Babatola and Bridge, 1979; Ichinohe, 1988; Khuong, 1987). Hirschmanniella oryzae prefers to enter the root through lateral roots or root tips, thereby using common invasion sites. It migrates within the aerenchyma of the root, which is well developed in flooded rice, leaving a trace of necrotic tissue along its path. This necrosis, together with the secondary invasion of microorganisms, causes general browning of rice roots (Babatola and Bridge, 1980). The yield loss as a result of nematode invasion varies between geographical regions and population density. Yield losses of up to more than 30% have been reported (Babatola and Bridge, 1979; Ichinohe, 1988; Prot and Rahman, 1994). Nematodes can survive in wet soil for 7 months without a host interaction and can persist at high population densities in several weeds and food crops (Bridge et al., 2005). Hirschmanniella oryzae reproduces sexually and, under favourable conditions, the life cycle can be completed in 33 days, during which the nematode undergoes four moulting stages. It has been reported that all larval and adult life stages can feed on the host (Karakas, 2004).
The majority of nematode sequence data are from sedentary species, but recent advances in this field have provided more data on migratory nematodes (Haegeman et al., 2011; Jacob et al., 2008; Kikuchi et al., 2011; Nicol et al., 2012). Sedentary species have a huge impact on yield losses for many important food crops world‐wide (Sikora and Fernandez, 2005; Wesemael et al., 2011). In the USA, Heterodera glycines alone accounts for almost 30% of the total losses in soybean production (Wrather and Koenning, 2006). However, we must not marginalize the economic importance of migratory species, such as Pratylenchus spp., Hirschmanniella spp. and Radopholus similis, which have a broad host range and can have a large impact on several crops (Moens and Perry, 2009).
The expressed sequence tag (EST) data generated in this project have been used to identify secreted putative cell wall‐degrading enzymes (CWDEs) and putative effector proteins, secreted molecules which manipulate the host for the benefit of the pathogen. The mechanisms behind the successful infection of a host plant were investigated, as were the molecular tools used by the nematode to cope with detrimental environmental circumstances, such as drought and potential bacterial infection. Until now, no sequence data were available for the genus Hirschmanniella, except for some β‐1,4‐endoglucanases (Rybarczyk‐Mydlowska et al., 2012). The generated transcriptome data will provide the scientific community with a new source of information about this genus and migratory nematodes.
Results
Dataset characteristics
RNA extracted from mixed stage populations of H. oryzae was sequenced using a Roche 454 sequencer; 450 171 reads were generated; following quality control, 134 205 and 106 911 assembled sequences longer than 150 bp were produced using Newbler and CLC assembly programs, respectively. The program CLC produced more than twice as many contigs as Newbler (48 347 and 21 706 contigs, respectively) (Table 1). As CLC software is thought to generate a less redundant assembly, these data were used in further analysis (Kumar and Blaxter, 2010). To allow a greater confidence in the predicted sequences, only those CLC‐assembled contigs that showed high similarity with the contigs predicted by Newbler software were retained. This resulted in a batch of 22 321 sequences. To reduce the amount of contaminating sequences, contigs with high similarity to certain plant‐pathogenic or soil bacteria, fungi or rice proteins were removed from the dataset. This led to a final set of 21 360 predicted contigs (Table 1). This final dataset is enriched in contigs with a relatively high number of reads. Roughly 30% of the contigs consisting of two to five reads were kept in the final dataset, whereas about 80% of the contigs containing more than five reads were retained (Table 2).
Table 1.
Dataset characteristics from the two different assemblies using software programs (CLC and Newbler 2.3). The last column contains the characteristics of the final dataset that was used in the different analyses
| CLC | Newbler | Final dataset | |
|---|---|---|---|
| Number of reads | 450171 | 450171 | 204524 |
| Average read length | 385 nt | 385 nt | – |
| Number of contigs | 48347 | 23196 | 21360 |
| Mean contig length | 600 nt | 549 nt | 680 nt |
| Number of singletons | 58564 | 128557 | – |
| Mean singleton length | 374 nt | 349 nt | – |
nt, nucleotide.
Table 2.
Comparison of the original dataset (CLC) with the final dataset, with regard to the number of reads per contig. The last column shows the percentages of sequences from the original dataset that were retained in the final dataset
| Reads/contig | # CLC contigs | # Final contigs | % retained |
|---|---|---|---|
| 2–5 | 34334 | 9650 | 28.11 |
| 6–10 | 8125 | 6563 | 80.78 |
| 11–20 | 3886 | 3457 | 88.96 |
| 21–50 | 1577 | 1363 | 86.43 |
| 51–100 | 301 | 239 | 79.40 |
| 101+ | 122 | 88 | 72.13 |
Comparison with protein databases and annotation
Almost all sequences, except for 22 contigs, were predicted by ORFpredictor to have an open reading frame (ORF), with a mean length of 144 amino acids; 16 656 of these expected protein sequences started with methionine; 1356 sequences were predicted to have a putative signal peptide and no transmembrane domain. Blastx (bitscore > 50) of all sequences against the Swiss‐Prot and Trembl protein databases was used to search for similar sequences, and this resulted in 14 248 significant hits. The resulting identifiers from this search were used as query for a functional annotation according to the Gene Ontology (GO) terminology for cellular component, biological process and molecular function. The most abundant GO terms are summarized in Fig. 1. A total of 153 906 GO terms were assigned to the sequences; 6473 of these terms were unique. Figure 1 shows that most proteins reside in the cytoplasm and the nucleus. Most of the sequences are predicted to have a function in ATP or protein binding. In the ‘biological process’ category, the term ‘oxidation–reduction process’ is over‐represented compared with the other terms in this category.
Figure 1.
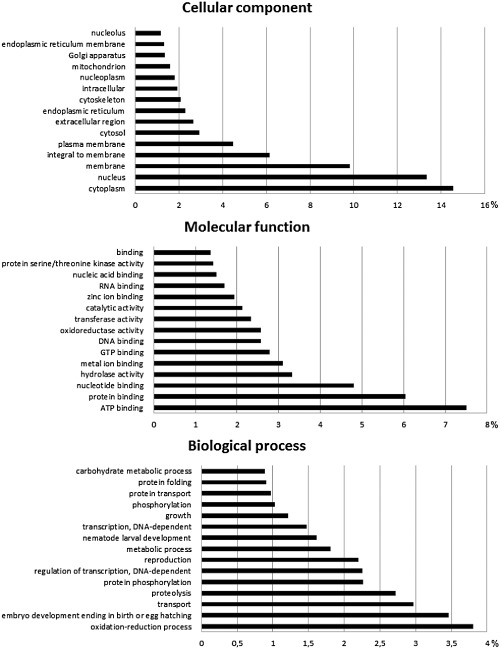
Percentage of Hirschmanniella oryzae contigs assigned to a certain Gene Ontology (GO) term as predicted by QuickGO from EBI.
HMMscan was used to identify possible Pfam domains. Of the 7090 sequences without similarity to Swiss‐Prot or Trembl databases, 173 were suspected to have a Pfam domain. The 10 most abundant Pfam domains are listed in Table 3; 66.7% of the predicted contigs were similar to sequences from Swiss‐Prot and Trembl databases. Predicted protein sequences were scanned for the occurrence of Pfam domains. Most of the Pfam domains mentioned in Table 3 take part in multiple processes. Protein kinases are involved in a multitude of cellular processes, including development, intercellular communication and differentiation (Manning et al., 2002). The WD‐repeat is also implicated in a wide range of functions (Smith et al., 1999), as are the cysteine proteases (Chapman et al., 1997; Grudkowska and Zagdanska, 2004). Other Pfam domains, such as Ras and Arf, are involved in a variety of signalling networks and regulatory pathways (Donaldson and Honda, 2005; Downward, 1998). Cysteine proteases with the Peptidase_C1 domain have very diverse functions; they can even act as manipulators of plant defence if secreted by plant‐pathogenic bacteria (Brix et al., 2008; Shindo and Van der Hoorn, 2008). RNA recognition motifs (RRMs) are one of the most abundant protein domains in eukaryotes and are involved in post‐transcriptional gene expression processes (Dreyfuss et al., 2002). These Pfam domains, or the corresponding InterPro domains, have been reported previously to be among the most abundant InterPro domains in an assembly of nematode transcriptomes (Parkinson et al., 2004).
Table 3.
Ten most common protein families with their accession numbers of the Pfam and InterPro databases. The target names and descriptions are specified in the last two columns. The fourth column shows the share of contigs assigned with that Pfam domain
| Pfam | InterPro | Number | Percentage | Target name | Description of target |
|---|---|---|---|---|---|
| PF00069 | IPR000719 | 178 | 1.67 | Pkinase | Protein kinase domain |
| PF07714 | IPR001245 | 135 | 1.27 | Pkinase_Tyr | Protein tyrosine kinase |
| PF00400 | IPR001680 | 115 | 1.08 | WD40 | WD domain, G‐β repeat |
| PF00071 | IPR013753 | 111 | 1.04 | Ras | Ras family |
| PF00112 | IPR000668 | 104 | 0.98 | Peptidase_C1 | Papain family cysteine protease |
| PF00153 | IPR001993 | 86 | 0.81 | Mito_carr | Mitochondrial carrier protein |
| PF00025 | IPR006689 | 84 | 0.79 | Arf | ADP‐ribosylation factor family |
| PF08477 | IPR013684 | 79 | 0.74 | Miro | Miro‐like protein |
| PF00076 | IPR000504 | 71 | 0.67 | RRM_1 | RNA recognition motif |
| PF14259 | IPR000505 | 64 | 0.60 | RRM_6 | RNA recognition motif |
A total of 47.1% of the sequences showed similarity to the protein sequences of Caenorhabditis elegans, 46.6% with Meloidogyne incognita and 53.3% with Bursaphelenchus xylophilus; 1323 sequences had homologues in M. incognita and/or B. xylophilus, but not in C. elegans. Of these sequences, 78 were predicted to have a putative signal peptide and no transmembrane domain, indicating that these proteins could be involved in plant parasitism. Nine are known to be involved in the infection process: expansin (5), endoglucanase (2), pectate lyase (1) and chorismate mutase (1). Of the contigs without similarity to these three organisms, 773 were predicted to have a putative secretion signal without a transmembrane domain. Most of these did not show similarities to the Swiss‐Prot or Trembl database (585 contigs), or were assigned as an uncharacterized protein (71 contigs).
Most abundant transcripts
To look for transcripts with high expression, the 100 contigs which contained the greatest number of mapped reads were selected. As a result of normalization of the data, it is not possible to quantify expression absolutely, but the number of reads can provide an idea of the abundance of the transcripts. These 100 contigs are built with an average of 163 reads, compared with an average of 10 reads per contig in the full dataset. The GO of these 100 contigs showed a bias towards GO terms involving reproduction and development (GO:0009792; GO:0000003; GO:0002119) in the category of ‘biological process’. These three terms accounted for 22% of the assigned terms in this category, indicating the importance of these processes during the nematode's life cycle. About 60 of the 100 contigs were annotated as housekeeping genes; 35 contigs were not annotated with a specific function, nine of which were predicted to have a signal peptide. The transcript with the highest expression consisted of 923 reads and did not show any similarity to other sequences in public databases. The longest possible ORF for this transcript was only 52 amino acids long. The transcript containing the second greatest number of reads (402 reads) was similar to a galactose‐binding lectin from C. elegans, LEC‐8. It has been suggested that this protein is involved in the defence process against bacterial infection (Ideo et al., 2009). Some other highly expressed transcripts code for expansin, a thaumatin‐like protein (TLP), glycoside hydrolase family (GHF)25 lysozyme and a lysozyme with a destabilase domain. These last three are probably involved in protection against bacterial pathogens (Evans et al., 2008; Zavalova et al., 2006). It has been shown that TLPs are up‐regulated on bacterial infection in C. elegans (Fasseas et al., 2012). Seven TLPs were found in the full dataset, four of which contained a predicted secretion signal. The localization of the expression of one of the TLPs was checked by in situ hybridization, showing expression in the pharyngeal region (Fig. 2A). A protein Blast was performed for the GHF25 lysozyme and the lysozyme with a destabilase domain. They showed highest similarity with LYS‐8 from Caenorhabditis briggsae and ILYS‐3 from C. elegans, respectively, a protist‐type and an invertebrate‐type lysozyme (Schulenburg and Boehnisch, 2008). Both are known to be up‐regulated on bacterial infection (Irazoqui et al., 2010; Mallo et al., 2002).
Figure 2.
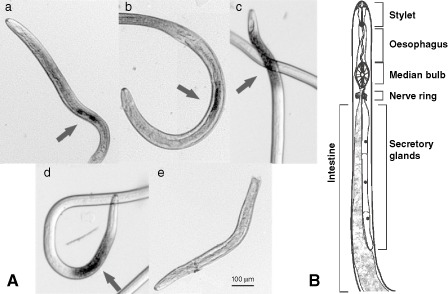
(A) Whole‐mount in situ hybridization of Hirschmanniella oryzae showing the spatial expression pattern of β‐mannanase (a), xylanase (GHF30) (b), thaumatin‐like protein (c) and chorismate mutase (d). The negative control (e) showed no signal. Arrows indicate the stained region of the pharyngeal region (c) or pharyngeal glands (a, b, d). Magnification, 128×. (B) Anatomy of the pharyngeal region of H. oryzae. Contrary to some other plant‐parasitic nematodes (e.g. Heterodera sp.), the pharyngeal glands overlap the intestine.
Survival in dry conditions
Hirschmanniella oryzae can survive in dry soils for several months without a host in a state of anhydrobiosis (Mathur and Prasad, 1973). We used an artificial seawater solution to induce anhydrobiosis and mimic drought stress. Nematodes were able to survive a 24‐h treatment in 40% artificial seawater solution. When higher concentrations were used, the nematode did not revive in distilled water. Several genes have been reported to play a role in this process in nematodes, including hydrophilic late embryogenesis abundant (LEA) proteins (Browne et al., 2002; Goyal et al., 2005). Six different sequences with similarities to LEA proteins were present in the dataset. All six were hydrophilic and had a negative grand average of hydropathicity (GRAVY) index, with a mean index of −0.817. The expression of the LEA protein with the lowest GRAVY index was investigated by quantitative reverse transcription‐polymerase chain reaction (Q‐RT‐PCR) on soaking in artificial seawater solution for 24 h. The expression values of two other genes that could be involved in this process were also considered: glutathione peroxidase and sxp/ral‐2 (Tyson et al., 2012). The results are shown in Fig. 3. The transcripts encoding LEA and SXP/RAL‐2 were highly induced on artificial seawater treatment, with the most significant activation in 30% solution. Glutathione peroxidase was not significantly up‐ or down‐regulated at any concentration. Expression ratios of both reference genes varied around unity according to REST 2009 software, indicating that the expression of these genes was not influenced by the treatments. The overall expression ratios were 1.013 and 1.039 for FMRFamide‐like neuropeptide 14 and elongation factor 1α, respectively.
Figure 3.
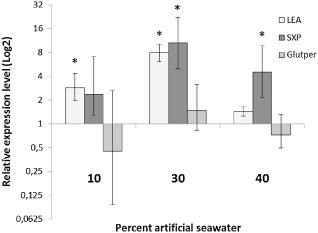
Changes in expression level of a late embryogenesis abundant (LEA) protein, sxp/ral‐2 (SXP) and a glutathione peroxidase (Glutper) from Hirschmanniella oryzae. Nematodes were soaked in 10%, 30% or 40% artificial seawater solution for 24 h after which RNA was extracted and the relative expression levels were estimated relative to a 24‐h soak in water. Each value is the mean of two biological replicates, each with three technical replicates. Asterisks indicate significant differences (P < 0.05). The bars represent the standard error.
Mining the transcriptome for cell wall‐modifying proteins
The Blastx output was used to scan the dataset for putative cell wall‐modifying proteins. Individual protein sequence similarity searches were performed on the translated ESTs to confirm the results from the Blastx output using Swiss‐Prot and Trembl databases. Seven different putative cell wall‐modifying proteins were discovered (Table 4). Expansin is the most abundant sequence in this category, followed by endoglucanase and pectate lyase. The discovered GHF30 xylanase shows similarities to the xylanase from the migratory nematode Radopholus similis (Haegeman et al., 2009). A single β‐mannanase (member of GHF5) was also observed in the transcriptome of H. oryzae, as well as genes encoding polygalacturonases and a poly‐α‐d‐galacturonosidase. Most of these proteins contained a putative signal peptide, indicating secretion. The spatial expression pattern of some of these putative CWDEs was tested by performing an in situ hybridization. The results of the hybridization of β‐mannanase and xylanase (GHF30) are shown in Fig. 2A. Both showed staining in the gland cell area, whereas the negative control showed no staining. The pharyngeal gland cells in Hirschmanniella species usually overlap the intestine and are unequal in length (Fig. 2B).
Table 4.
Summary of the number of sequences with similarity to cell wall‐degrading proteins. The last column indicates whether at least one sequence of a family of enzymes was predicted to have a signal peptide
| Enzyme | Family | # Contigs | # Reads | SP |
|---|---|---|---|---|
| Expansin | Beta | 8 | 487 | Yes |
| β‐1,4‐Endoglucanase | GHF5 | 15 | 136 | Yes |
| Pectate lyase | PL | 6 | 108 | Yes |
| β‐Mannanase | GHF5 | 1 | 58 | Yes |
| Polygalacturonase | GHF28 | 3 | 13 | No |
| Poly‐α‐d‐galacturonosidase | GHF28 | 1 | 4 | No |
| Xylanase | GHF30 | 1 | 4 | Yes |
GHF, glycoside hydrolase family; SP, signal peptide.
Chorismate mutase and isochorismatase
Next to effector proteins that degrade the plant cell wall, nematodes also secrete proteins that can alter the defence mechanism of the plant. One of these proteins is chorismate mutase (CM) (Lambert et al., 1999). This protein contains a signal peptide and its gland cell spatial expression was confirmed by in situ hybridization (Fig. 2A). The protein has a CM type 2 domain (PF01817). A protein sequence similarity search (Blastp) using the catalytic domain sequence revealed high similarity with CMs from several Burkholderia species. When we performed a tBlastn against the nucleotide or EST database, the top hits came from plant‐parasitic nematode species, both from Meloidogyne arenaria. A phylogenetic tree was constructed based on Bayesian inference. CM is in the same clade as other plant‐parasitic nematodes, with its closest relative being a CM from Pratylenchus coffeae (Fig. 4).
Figure 4.
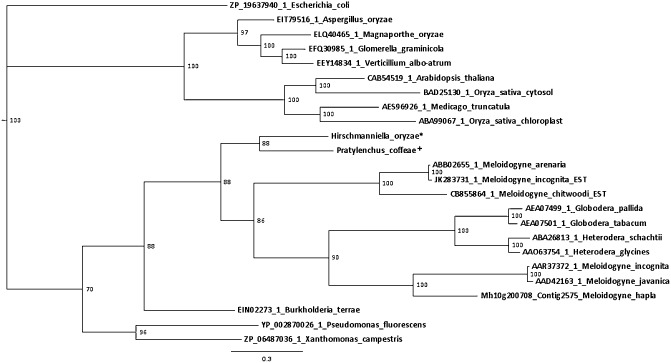
Phylogenetic tree of chorismate mutase (CM) constructed with MrBayes software. Posterior probabilities are shown for each node. The closest homologue of the CM of Hirschmanniella oryzae comes from Pratylenchus coffeae. Nematode sequences cluster together with CM sequences from plant‐pathogenic bacteria. Plant sequences are in the same cluster as plant‐pathogenic fungi. Taxa are indicated as follows: proteinID_species. An asterisk (*) indicates in‐house data. A plus sign (+) indicates data from Haegeman et al. (2011). The suffix ‘EST’ indicates a translated EST sequence instead of a protein sequence.
Next to CM, two contigs with an isochorismatase (ICM) domain (PF 00857) were also present in the dataset (ICM1 and ICM2). These sequences did not contain a predicted signal peptide. Using Blastp with the ICM1 sequence as a query revealed that it is mostly similar to plant‐pathogenic bacteria. A tBlastn against the EST database at the National Center for Biotechnology Information (NCBI) discovered homologues in other plant‐parasitic nematodes exclusively. A search with the protein sequence of ICM2 against the nonredundant protein database only revealed similarity to eukaryotic sequences. Most of these sequences were predicted to have a mitochondrial localization in their annotation. ICM2 was compared with nematode ESTs (tBlastn) and showed similarity to ESTs derived from nematodes with different lifestyles (plant‐parasitic, animal‐parasitic and free‐living). Both ICM sequences were used to construct a phylogenetic tree (Fig. 5). ICM2 is in a cluster together with other ICM sequences originating from nematode species, not limited to plant‐parasitic nematodes. ICM1 homologues in nematodes are restricted to the plant‐parasitic species, which could indicate that it is involved in plant parasitism. ICM1 and ICM2 do not show any similarity with each other. No significant similarities were observed between sequences clustered together with ICM1 and sequences from the same organism clustering together with ICM2. A conserved domain search of both ICM sequences revealed that ICM1 has a cysteine hydrolase domain (cd00431, 1.5e‐40), whereas ICM2 has an YcaC‐related domain (cd01012, 3.59e‐54). Both CM and ICM are involved in the salicylic acid (SA) biosynthesis pathway in plants (Dempsey et al., 2011). It is tempting to speculate that CM and ICM could interfere with SA production, thereby altering the defence mechanism of the host.
Figure 5.
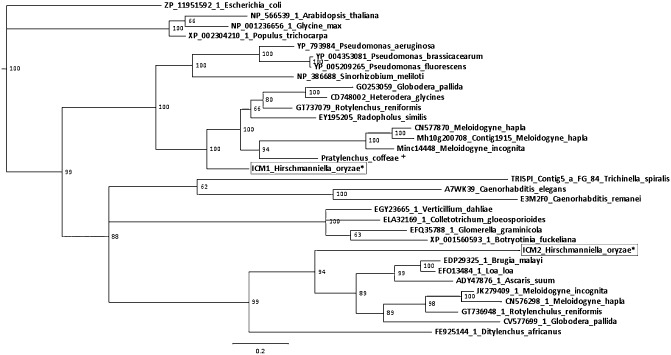
Phylogenetic tree of isochorismatase (ICM) constructed with MrBayes software. Posterior probabilities are shown for each node. Homologues in expressed sequence tag (EST) and protein databases on the National Center for Biotechnology Information (NCBI) server were used to construct this tree. Taxa are indicated as follows: proteinID_species. An asterisk (*) indicates in‐house data. ICM1 and ICM2 cluster in different branches of the tree. A plus sign (+) indicates data from Haegeman et al. (2011).
Discussion
The transcriptome of H. oryzae was studied to gain an insight into the survival strategies of this organism and to investigate the parasitic interaction with its host, O. sativa. The ESTs were generated using the 454 FLX Titanium Platform on a mixed stages population of H. oryzae. 454 sequencing produces longer reads than Illumina sequencing; the longer reads are beneficial for transcriptome assembly (Pop and Salzberg, 2008). A total of 450 171 reads, with an average read length of 385 nucleotides, was assembled by a combination of CLC and Newbler software. As there are several different drawbacks to each assembly strategy, we combined the two strategies to have more confidence in each contig. The disadvantage, however, is that several contigs only predicted by one program were discarded during this combined assembly process. The dataset was further reduced by rejecting the sequences with high similarity (bitscore > 200) to possible contaminating sources. By doing so, potential horizontal gene transfer events (Mitreva et al., 2009) might be overlooked. This led us to a final dataset consisting of 21 360 contigs.
To survive, H. oryzae must be able to adapt to environmental conditions and must be able to successfully infect a host plant to be capable of feeding and reproducing. During the growing season, rice is flooded and deprived of oxygen. In between growing seasons, however, the field can dry out. In addition, the soil is inhabited by bacteria which may be harmful to nematodes (Costa et al., 2012; Hodgkin et al., 2000; Piskiewicz et al., 2007). Hirschmanniella oryzae has been reported to survive in anaerobic conditions (Babatola, 1981). We only detected the presence of a few genes coding for enzymes that are important in anaerobic metabolism pathways, such as fumarate reductase and malate dehydrogenase, but not enough to construct a full pathway (Muller et al., 2012). This is probably caused by the fact that the analysed nematodes were not harvested under anaerobic conditions.
There is a vast bacterial community thriving in the soil and rhizosphere of rice plants (Lu et al., 2006). Most bacteria are harmless to nematodes; however, some species are nematode pathogens, such as certain Pasteuria and Pseudomonas species (Siddiqui and Mahmood, 1999). Several antimicrobial peptides, as well as lysozymes and lectins with antibacterial activity, have been described in C. elegans (Bogaerts et al., 2010; Irazoqui et al., 2010; Mallo et al., 2002). The top 100 most abundant transcripts mirror the need for H. oryzae to protect itself against microbial invaders. Several proteins with (potential) antimicrobial properties were detected. First, two different types of lysozyme were identified: GHF25 lysozyme (lys25) and a lysozyme with a destabilase domain (dest‐lys). lys25 is up‐regulated in the intestinal cells of C. elegans on pathogen infection (Mallo et al., 2002). Dest‐lys is up‐regulated on bacterial infection in C. elegans (Irazoqui et al., 2010), and is also known for its antifungal properties (Yudina et al., 2012; Zavalova et al., 2006). In addition, several lectins are involved in the innate immune system against bacteria. A galactose‐binding lectin with strong similarity to LEC‐8 of C. elegans was the second most abundant transcript in the dataset. Lec‐8 is up‐regulated on bacterial infection and probably functions as a competitive binding protein for glycolipid molecules that are targeted by the bacterial invader (Ideo et al., 2009; Mallo et al., 2002). The dataset contained several TLPs, one of which was present in the list of most abundant transcripts (113 reads). Spatial analysis by in situ hybridization showed this gene to be expressed in the pharyngeal region. In plants, TLPs are classified as pathogenesis‐related proteins family 5 and have antifungal properties. In nematodes, they have been reported to be involved in the innate immune system (Fasseas et al., 2012; Golden and Melov, 2004; O'Rourke et al., 2006). Although most antibacterial proteins involved in the innate immune system are expressed in the intestines of the nematode, some are also expressed in the gland cells or pharyngeal region. For instance, LYS‐8, an antibacterial lysozyme in C. elegans, is up‐regulated on bacterial infection and its expression has been reported in the pharyngeal region (Mallo et al., 2002). Among several other activities, TLPs also show β‐1,3‐glucanase activity, implicating that this nematode could secrete this protein as a protection against fungi or to degrade callose in the plant (Liu et al., 2010).
Next to being challenged by parasitic bacteria and/or fungi, another big challenge is surviving long periods of drought. Hirschmanniella oryzae is used to a life in flooded rice fields (Prot and Rahman, 1994). Although these rice fields are flooded most of the time, they can dry out in between growing seasons. The nematode has to rapidly adapt to this new situation to overcome the dry period. Several nematodes are known to be able to survive long periods of drought (Reardon et al., 2010; Tyson et al., 2007). They are adapted to these extreme situations by the differential expression of several genes. One of these genes is lea, also found in plants in which it accumulates in response to water loss. These LEA proteins have been found in nematodes, where they are strongly induced on anhydrobiosis (Browne et al., 2002). Other proteins also play a role during this process. In Panagrolaimus superbus, an anhydrobiotic nematode, it was found that SXP/RAL‐2 is the most abundant transcript in the transcriptome (Tyson et al., 2012). This led the authors to believe that it is involved in survival during desiccation. Glutathione peroxidases have also been shown to be involved in drought tolerance (Reardon et al., 2010). When a state of anhydrobiosis was induced by soaking H. oryzae in a diluted solution of artificial seawater for 24 h, thereby mimicking desiccation stress, sxp/ral‐2 and lea were both significantly up‐regulated. Although the up‐regulation of certain glutathione peroxidases on dehydration of nematodes has been described previously (Reardon et al., 2010), this was not observed in this experiment. To our knowledge, this is the first time that the up‐regulation of sxp/ral‐2 on anhydrobiosis has been described. This family of proteins is specific to nematodes. It is present in free‐living and animal‐parasitic nematodes, as well as in plant‐parasitic nematodes (Tyson et al., 2012). The role of these proteins has not been elucidated to date. It has been proposed that they are involved in plant parasitism, for instance in feeding cell induction (Tytgat et al., 2005). However, the presence of this family across the phylum may indicate a more general function. The results presented here suggest that SXP/RAL‐2 has a role in protection against desiccation. This apparent contradiction can be explained by the fact that there may be several SXP/RAL‐2 isoforms within a species, as shown previously for Globodera rostochiensis (Jones et al., 2000). These proteins are grouped within the same family, but have a different spatial expression pattern. The expression of two different sxp/ral‐2 genes has been reported in either the amphids or hypodermis of G. rostochiensis, and one sxp/ral‐2 gene was expressed in the subventral pharyngeal glands of M. incognita (Jones et al., 2000; Tytgat et al., 2005). This could be an indication that diverse members of this family exhibit different functions.
To successfully infect the host plant, the nematode must overcome the cell wall as a structural barrier, which mainly consists of cellulose, hemicelluloses, pectins and structural proteins (Vogel, 2008). To overcome this hurdle, nematodes hold an arsenal of CWDEs thought to have been acquired through horizontal gene transfer from bacteria and/or fungi (Jones et al., 2005). Several cell wall‐modifying proteins have already been described in nematodes, such as expansin, β‐1,4‐endoglucanase, pectate lyase, polygalacturonase and xylanase, which were also found in the H. oryzae ESTs (Jaubert et al., 2002; Kikuchi et al., 2006; Mitreva‐Dautova et al., 2006; Qin et al., 2004; Rosso et al., 1999).
A putative new CWDE not previously found in nematodes was identified in the H. oryzae ESTs: β‐mannanase. It is possibly involved in hemicellulose degradation. The hemicellulose fraction is more abundant in cell walls of monocots relative to dicots (Vogel, 2008), which could explain the presence of this enzyme in H. oryzae, whereas it is absent in the genomes of M. incognita, M. hapla and B. xylophilus. β‐Mannanase is important in the degradation of β‐(1,4)‐linked mannans. This enzyme has previously been found in plants, fungi and bacteria. Its presence has been confirmed in several plant‐pathogenic fungi and bacteria, where it has a role in cell wall degradation (Couturier et al., 2012; Kim et al., 2011; Pham et al., 2010). This is the first report of this gene in nematodes. The presence and activity of this type of β‐mannanase have been reported recently in an insect species (Acuna et al., 2012). An in situ hybridization showed a spatial expression pattern in the region of the gland cells, ruling out the possibility that this contig originated from bacterial contamination. It might be possible that H. oryzae acquired this gene by horizontal gene transfer. A search using Blastp against the nonredundant protein database revealed β‐mannanase as top hit. This sequence originated from Opitutus terrae, an obligate anaerobic bacterium which is known to reside in rice paddy soils (Chin et al., 2001). The fact that the β‐mannanases from both organisms have a high similarity (51% identical residues), and that these two organisms share the same ecosystem, could point to horizontal gene transfer. GHF30 xylanases have been found in plant‐parasitic nematodes previously, although there is some discussion about the designation of these enzymes to GHF5 or GHF30 (Haegeman et al., 2009). The presence of all of these enzymes shows that H. oryzae is well equipped to break through the monocot plant cell wall and migrate through the root. As migratory nematodes must continuously pass cell walls during their whole life cycle, it is not surprising that they possess an arsenal of enzymes to degrade hemicellulose.
Sedentary nematodes secrete effector proteins which aid in the establishment of a specialized feeding site and attenuate the host defence mechanism (Hewezi and Baum, 2013). As migratory nematodes do not form specialized feeding cells and do not stay in one place in the host, one could claim that they are not in need of these specific effector proteins. This used to be a generally accepted idea for necrotrophic fungi, but it was later shown that they are able to manipulate the host's defence to their advantage (Oliver and Solomon, 2010). Therefore, migratory nematodes might also secrete effector proteins to suppress the plant immune system. CM was found in our dataset and in the transcriptome of Pratylenchus coffeae (Haegeman et al., 2011). CM is involved in the shikimate pathway in plants, where it catalyses the step from chorismate to prephenate. Secretion of CM was suggested by the presence of a putative secretion signal and expression in the region of the pharyngeal gland cells. The secreted CM is probably involved in the deregulation of the SA pathway in the host plant, making it more vulnerable to pathogen attack. This has been shown recently for a secreted CM from Ustilago maydis, which lowers the total SA content in infected leaves (Djamei et al., 2011).
In addition to CM, another enzyme involved in the SA pathway in plants was also discovered, ICM. ICM converts isochorismate to 2,3‐dihydroxybenzoate and pyruvate, thereby depleting the pool of isochorismate and reducing SA synthesis. ICM has been found in the secretome of phytopathogenic fungi and is thought to have a function in reducing the SA content of the host (El‐Bebany et al., 2010; Soanes et al., 2008). Two sequences with similarity to ICM have been found in the dataset. ICM1 clusters together with other plant‐parasitic nematode sequences in a phylogenetic tree and does not have any homologues in nonparasitic nematodes. This is an indication that it could be involved in plant parasitism. ICM2 has homologues in plant‐parasitic nematodes, as well as free‐living nematodes and animal‐parasitic nematodes, and has a conserved YcaC‐related domain, but does not show similarity to ICM1. A Blastp sequence similarity search revealed that it also has putative orthologues in other eukaryotic species, most of which were predicted to have a mitochondrial localization. YcaC‐related ICM is probably involved indirectly in energy transduction. It has been shown that it can bind to creatine kinase, which catalyses the transfer of a phosphate group from phosphocreatine to ADP, yielding ATP and creatine. This is supported by the fact that YcaC‐related ICM is mainly localized in mitochondria, where ATP production takes place (Jiang et al., 2008). These results indicate that ICM1 could be an interesting effector protein for the nematode, whereas ICM2 is involved in basic cellular functioning.
In conclusion, the transcriptome of H. oryzae provides insights into the proteins needed by this nematode to survive in unfavourable environmental conditions. In addition, it augments our understanding of the strategies used to infect the host plant, from CWDEs to putative effector proteins. As the available data for migratory species are scarce compared with the economically more important cyst and root‐knot nematodes, these new data are a convenient source to perform comparative studies.
Experimental Procedures
RNA/DNA extraction and sequencing
Juvenile and adult stages of H. oryzae obtained from Myanmar were extracted from fresh rice roots using a modified Baermann funnel. RNA was extracted from the nematodes using the RNeasy kit (Qiagen, Hilden, Germany). The RNA was sent to LGC Genomics (Berlin, Germany), where mRNA was isolated and a cDNA library was constructed with the Mint Universal cDNA synthesis kit (Evrogen, Moscow, Russia). The cDNA was normalized using the TRIMMER kit (Evrogen), size selected (>800 bp) and transformed into the vector pDNR‐lib. The library was analysed by 454 FLX Titanium sequencing in a run on one‐half of a picotitre plate. The raw data have been submitted to the Sequence Read Archive (SRA) database of the NCBI under accession number SRA048498.
Clean up and assembly
The reads were processed with Newbler 2.3 and CLC Genomics workbench 4.0.2 software. Contaminating vector and adaptor sequences were trimmed from the reads and an assembly was performed using standard settings. Singletons and sequences shorter than 150 bp were removed from the dataset. Contigs generated by CLC software were Blast searched (Blastn) against contigs predicted by Newbler 2.3 with a cut‐off bitscore of 200.
Blast searches and annotation
The remaining contigs in the dataset were subsequently Blast searched (Blastx, cut‐off bitscore of 200) against protein sequences of several plant‐pathogenic bacteria and fungi, soil bacteria and rice to remove possible contamination. Predicted proteins from the O. sativa genome (version 7.0) were downloaded from the website of the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/). The data for the other organisms were downloaded from the NCBI server (http://www.ncbi.nlm.nih.gov/): Bacillus sp., Pseudomonas fluorescens, Rhizobium sp., Azospirillum sp., Agrobacterium sp., Gluconobacter sp., Flavobacterium sp., Herbaspirillum sp., Thiobacillus sp., Xanthomonas oryzae, Magnaporthe grisea, Magnaporthe salvinii, Rhizoctonia solani, Cochliobolus miyabeanus, Gibberella fujikuroi and Ustilaginoidea virens. The remaining sequences were subjected to a Blastx (cut‐off bitscore of 50) against the Swiss‐Prot and Trembl databases. The output was used for annotation. By using the software application QuickGO from EBI (http://www.ebi.ac.uk/QuickGO/GAnnotation), GOs were downloaded for the protein identifiers of the Blastx top hits. Sequences were compared with (Blastx, cut‐off bitscore of 50) protein data of C. elegans, M. incognita and B. xylophilus downloaded from Wormbase (http://www.wormbase.org, release WS229), http://www7.inra.fr/meloidogyne_incognita and http://www.genedb.org/Homepage/Bxylophilus (version 1.2), respectively.
Protein domain search and sequence analysis
Putative proteins were predicted using the OrfPredictor tool (Min et al., 2005). The protein sequences were scanned for the presence of putative signal peptides and the absence of transmembrane domains using SignalP 4.0 (Petersen et al., 2011). Pfam HMM motifs were downloaded from the Pfam website (release 26.0). HMMscan (HMMER3.0, reporting threshold 30, hmmer.org) was used to look for possible domains in the contigs. The molecular weight of a protein was calculated using the ProtParam tool from the ExPASy website (http://web.expasy.org/protparam/). To calculate the GRAVY index, the Kyte–Doolittle scale was used (Kyte and Doolittle, 1982). Conserved domains from the Conserved Domain Database (CDD) were retrieved through CD‐search in the NCBI server.
Dehydration treatment
Nematodes of mixed stages were soaked in an artificial seawater solution or in distilled water as a control. The seawater solution was adapted from Feng et al. (2006). It contained 1692.00 mm NaCl, 9.00 mm KCl, 9.27 mm CaCl2, 22.94 mm MgCl2.6H2O, 25.50 mm MgSO4.7H2O and 2.14 mm NaHCO3. Approximately 500 nematodes were soaked in six‐well plates for 24 h in water, 10%, 30% and 40% artificial seawater.
RNA extraction and cDNA synthesis
RNA was extracted with the RNeasy kit (Qiagen). To remove all contaminating DNA, 1 μg of the extracted RNA was treated with 1 μL DNaseI (1 U/μL; Fermentas, Waltham, MA, USA), 0.5 μL RNAsin ribonuclease inhibitor (Promega Fitchburg, WI, USA) and 3 μL DNaseI buffer (10×, Fermentas) in a total volume of 27 μL. The mixture was incubated at 37 °C for 30 min, after which 3 μL of 50 mm ethylenediaminetetraacetic acid (EDTA) was added and incubated for 10 min at 65 °C to stop the reaction. First‐strand cDNA synthesis was started by adding 1 μL oligo dT (700 ng/μL) to 30 μL DNAse‐treated RNA and incubating the mixture for 5 min at 70 °C. Afterwards, the following products were added: 10 μL of Goscript 5× reaction buffer, 5 μL of 25 mm MgCl2, 2.5 μL of PCR nucleotide mix, 1 μL of Goscript reverse transcriptase (Promega) and 0.5 μL of water to a total volume of 50 μL. The mixture was incubated for 5 min at 25 °C, 2 h at 42 °C and 15 min at 70 °C to stop the reaction.
Q‐RT‐PCR
The SensiMix SYBR No‐ROX kit (Bioline, London, UK) was used to perform Q‐RT‐PCR. Each reaction contained 10 μL of 2 × SensiMix, 500 nm of each primer and 1 μL of cDNA in a total volume of 20 μL. All reactions were performed in three technical replicates on a Rotor‐Gene 3000 (Corbett Life Science, Hilden, Germany) and analysed with Rotor‐Gene 6000 software version 1.7. The PCR conditions were as follows: 10 min of initial denaturation at 95 °C, followed by 45 cycles of 25 s at 95 °C, 25 s at 58 °C and 20 s at 72 °C. The melting curve was generated by gradually increasing the temperature from 72 °C to 95 °C after the last cycle to test the specificity of the amplicon. The data were analysed by REST 2009 software to determine statistically significant differences (Pfaffl et al., 2002). Two reference genes identified in the contigs were used; an FMRFamide‐like neuropeptide 14 and an elongation factor 1α. Primers are listed in Table S1 (see Supporting Information).
Construction of a phylogenetic tree
To construct a phylogenetic tree, homologues of CM or ICM were downloaded from the NCBI server. EST sequences were first translated into protein sequences. The alignment was performed by Clustal Omega on the EBI server (http://www.ebi.ac.uk/Tools/msa/clustalo/). The output format was converted into the nexus format using Readseq (http://www.ebi.ac.uk/Tools/sfc/readseq/). The nexus file was loaded into MrBayes 3.2.1 to construct a phylogenetic tree using Bayesian inference with a gamma distributed model (Ronquist et al., 2012). At least 50 000 generations were performed to achieve a standard deviation below 0.05.
Whole‐mount in situ hybridization
Genes were cloned in pGEM‐T (Promega), using the standard cloning techniques. The primers used for cloning and probe synthesis are listed in Table S1. Whole‐mount in situ hybridization was performed as described previously by de Boer et al. (1998) with some minor modifications with regard to the fixation of the nematodes and the hybridization temperature. The nematodes were fixed in 2% formaldehyde for 16 h at 4 °C, followed by an additional incubation for 4 h at room temperature. Hybridization was performed at 47 °C. Photographs were taken with a Leica S8AP0 (Wetzlar, Germany) stereomicroscope.
Supporting information
Table S1 Primer sequences used to clone genes, make in situ hybridization (ISH) probes and perform quantitative reverse transcription‐polymerase chain reaction (Q‐RT‐PCR).
Acknowledgements
AH and TK are postdoctoral fellows funded by the Research Foundation Flanders (FWO‐Vlaanderen). The authors wish to thank Zin Thu Zar Maung and Pa Pa Win for sending rice roots infected with H. oryzae. They would also like to thank Dr Peter Thorpe for the careful reading of the manuscript. Financial support for this project was received from Ghent University (BOF 01B00308). The authors declare that there is no conflict of interest.
References
- Acuna, R. , Padilla, B.E. , Florez‐Ramos, C.P. , Rubio, J.D. , Herrera, J.C. , Benavides, P. , Lee, S.J. , Yeats, T.H. , Egan, A.N. , Doyle, J.J. and Rose, J.K. (2012) Adaptive horizontal transfer of a bacterial gene to an invasive insect pest of coffee. Proc. Natl. Acad. Sci. USA, 109, 4197–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babatola, J.O. (1981) Effect of pH, oxygen and temperature on the activity and survival of Hirschmanniella spp. Nematologica, 27, 285–291. [Google Scholar]
- Babatola, J.O. and Bridge, J. (1979) Pathogenicity of Hirschmanniella oryzae, Hirschmanniella spinicaudata and Hirschmanniella imamuri on rice. J. Nematol. 11, 128–132. [PMC free article] [PubMed] [Google Scholar]
- Babatola, J.O. and Bridge, J. (1980) Feeding behavior and histopathology of Hirschmanniella oryzae, H. imamuri and H. spinicaudata on rice. J. Nematol. 12, 48–53. [PMC free article] [PubMed] [Google Scholar]
- de Boer, J.M. , Yan, Y. , Smant, G. , Davis, E.L. and Baum, T.J. (1998) In‐situ hybridization to messenger RNA in Heterodera glycines . J. Nematol. 30, 309–312. [PMC free article] [PubMed] [Google Scholar]
- Bogaerts, A. , Beets, I. , Schoofs, L. and Verleyen, P. (2010) Antimicrobial peptides in Caenorhabditis elegans . Invert. Surviv. J. 7, 45–52. [Google Scholar]
- Bridge, J. , Plowright, R.A. and Peng, D. (2005) Nematode parasites of rice In: Plant Parasitic Nematodes in Subtropical and Tropical Agriculture (Luc M., Sikora R.A. and Bridge J., eds), pp. 87–130. Wallingford, Oxfordshire: CABI Publishing. [Google Scholar]
- Brix, K. , Dunkhorst, A. , Mayer, K. and Jordans, S. (2008) Cysteine cathepsins: cellular roadmap to different functions. Biochimie, 90, 194–207. [DOI] [PubMed] [Google Scholar]
- Browne, J. , Tunnacliffe, A. and Burnell, A. (2002) Anhydrobiosis: plant desiccation gene found in a nematode. Nature, 416, 38. [DOI] [PubMed] [Google Scholar]
- Chapman, H.A. , Riese, R.J. and Shi, G.P. (1997) Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 59, 63–88. [DOI] [PubMed] [Google Scholar]
- Chin, K.J. , Liesack, W. and Janssen, P.H. (2001) Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division ‘Verrucomicrobia’ isolated from rice paddy soil. Int. J. Syst. Evol. Microbiol. 51, 1965–1968. [DOI] [PubMed] [Google Scholar]
- Costa, S.R. , Kerry, B.R. , Bardgett, R.D. and Davies, K.G. (2012) Interactions between nematodes and their microbial enemies in coastal sand dunes. Oecologia, 170, 1053–1066. [DOI] [PubMed] [Google Scholar]
- Couturier, M. , Navarro, D. , Olive, C. , Chevret, D. , Haon, M. , Favel, A. , Lesage‐Meessen, L. , Henrissat, B. , Coutinho, P.M. and Berrin, J.G. (2012) Post‐genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis . BMC Genomics, 13, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey, D.A. , Vlot, A.C. , Wildermuth, M.C. and Klessig, D.F. (2011) Salicylic acid biosynthesis and metabolism. Arabidopsis Book, 9, e0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djamei, A. , Schipper, K. , Rabe, F. , Ghosh, A. , Vincon, V. , Kahnt, J. , Osorio, S. , Tohge, T. , Fernie, A.R. , Feussner, I. , Feussner, K. , Meinicke, P. , Stierhof, Y.D. , Schwarz, H. , Macek, B. , Mann, M. and Kahmann, R. (2011) Metabolic priming by a secreted fungal effector. Nature, 478, 395–398. [DOI] [PubMed] [Google Scholar]
- Donaldson, J.G. and Honda, A. (2005) Localization and function of Arf family GTPases. Biochem. Soc. Trans. 33, 639–642. [DOI] [PubMed] [Google Scholar]
- Downward, J. (1998) Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 8, 49–54. [DOI] [PubMed] [Google Scholar]
- Dreyfuss, G. , Kim, V.N. and Kataoka, N. (2002) Messenger‐RNA‐binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3, 195–205. [DOI] [PubMed] [Google Scholar]
- El‐Bebany, A.F. , Rampitsch, C. and Daayf, F. (2010) Proteomic analysis of the phytopathogenic soilborne fungus Verticillium dahliae reveals differential protein expression in isolates that differ in aggressiveness. Proteomics, 10, 289–303. [DOI] [PubMed] [Google Scholar]
- Evans, E.A. , Kawli, T. and Tan, M.W. (2008) Pseudomonas aeruginosa suppresses host immunity by activating the DAF‐2 insulin‐like signaling pathway in Caenorhabditis elegans . PLoS Pathog. 4, e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasseas, M.K. , Fasseas, C. , Mountzouris, K.C. and Syntichaki, P. (2012) Effects of Lactobacillus salivarius, Lactobacillus reuteri, and Pediococcus acidilactici on the nematode Caenorhabditis elegans include possible antitumor activity. Appl. Microbiol. Biotechnol. 97, 2109–2118. [DOI] [PubMed] [Google Scholar]
- Feng, S.P. , Han, R.C. , Qiu, X.H. , Cao, L. , Chen, J.H. and Wang, G.H. (2006) Storage of osmotically treated entomopathogenic nematode Steinernema carpocapsae . Insect Sci. 13, 263–269. [Google Scholar]
- Golden, T.R. and Melov, S. (2004) Microarray analysis of gene expression with age in individual nematodes. Aging Cell, 3, 111–124. [DOI] [PubMed] [Google Scholar]
- Goyal, K. , Pinelli, C. , Maslen, S.L. , Rastogi, R.K. , Stephens, E. and Tunnacliffe, A. (2005) Dehydration‐regulated processing of late embryogenesis abundant protein in a desiccation‐tolerant nematode. FEBS Lett. 579, 4093–4098. [DOI] [PubMed] [Google Scholar]
- Grudkowska, M. and Zagdanska, B. (2004) Multifunctional role of plant cysteine proteinases. Acta Biochim. Pol. 51, 609–624. [PubMed] [Google Scholar]
- Haegeman, A. , Vanholme, B. and Gheysen, G. (2009) Characterization of a putative endoxylanase in the migratory plant‐parasitic nematode Radopholus similis . Mol. Plant Pathol. 10, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegeman, A. , Joseph, S. and Gheysen, G. (2011) Analysis of the transcriptome of the root lesion nematode Pratylenchus coffeae generated by 454 sequencing technology. Mol. Biochem. Parasitol. 178, 7–14. [DOI] [PubMed] [Google Scholar]
- Hewezi, T. and Baum, T.J. (2013) Manipulation of plant cells by cyst and root‐knot nematode effectors. Mol. Plant–Microbe Interact. 26, 9–16. [DOI] [PubMed] [Google Scholar]
- Hodgkin, J. , Kuwabara, P.E. and Corneliussen, B. (2000) A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans . Curr. Biol. 10, 1615–1618. [DOI] [PubMed] [Google Scholar]
- Ichinohe, M. (1988) Current research on the major nematode problems in Japan. J. Nematol. 20, 184–190. [PMC free article] [PubMed] [Google Scholar]
- Ideo, H. , Fukushima, K. , Gengyo‐Ando, K. , Mitani, S. , Dejima, K. , Nomura, K. and Yamashita, K. (2009) A Caenorhabditis elegans glycolipid‐binding galectin functions in host defense against bacterial infection. J. Biol. Chem. 284, 26 493–26 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irazoqui, J.E. , Troemel, E.R. , Feinbaum, R.L. , Luhachack, L.G. , Cezairliyan, B.O. and Ausubel, F.M. (2010) Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus . PLoS Pathog. 6, e1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob, J. , Mitreva, M. , Vanholme, B. and Gheysen, G. (2008) Exploring the transcriptome of the burrowing nematode Radopholus similis . Mol. Genet. Genomics, 280, 1–17. [DOI] [PubMed] [Google Scholar]
- Jaubert, S. , Laffaire, J.B. , Abad, P. and Rosso, M.N. (2002) A polygalacturonase of animal origin isolated from the root‐knot nematode Meloidogyne incognita . FEBS Lett. 522, 109–112. [DOI] [PubMed] [Google Scholar]
- Jiang, S. , Sun, X. and Zhang, S. (2008) The ycaC‐related protein from the amphioxus Branchiostoma belcheri (BbycaCR) interacts with creatine kinase. FEBS J. 275, 4597–4605. [DOI] [PubMed] [Google Scholar]
- Jones, J.T. , Smant, G. and Blok, V.C. (2000) SXP/RAL‐2 proteins of the potato cyst nematode Globodera rostochiensis: secreted proteins of the hypodermis and amphids. Nematology, 2, 887–893. [Google Scholar]
- Jones, J.T. , Furlanetto, C. and Kikuchi, T. (2005) Horizontal gene transfer from bacteria and fungi as a driving force in the evolution of plant parasitism in nematodes. Nematology, 7, 641–646. [Google Scholar]
- Karakas, M. (2004) Life cycle and mating behavior of Hirschmanniella oryzae (nematoda: Pratylenchidae) on excised Oryzae sativa roots. Fen Bilimleri Dergisi, 25, 1–6. [Google Scholar]
- Khuong, N.B. (1987) Hirschmanniella spp in rice fields of Vietnam. J. Nematol. 19, 82–84. [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, T. , Shibuya, H. , Aikawa, T. and Jones, J.T. (2006) Cloning and characterization of pectate lyases expressed in the esophageal gland of the pine wood nematode Bursaphelenchus xylophilus . Mol. Plant–Microbe Interact. 19, 280–287. [DOI] [PubMed] [Google Scholar]
- Kikuchi, T. , Cotton, J.A. , Dalzell, J.J. , Hasegawa, K. , Kanzaki, N. , McVeigh, P. , Takanashi, T. , Tsai, I.J. , Assefa, S.A. , Cock, P.J. , Otto, T.D. , Hunt, M. , Reid, A.J. , Sanchez‐Flores, A. , Tsuchihara, K. , Yokoi, T. , Larsson, M.C. , Miwa, J. , Maule, A.G. , Sahashi, N. , Jones, J.T. and Berriman, M. (2011) Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus . PLoS Pathog. 7, e1002219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.Y. , Ham, S.J. , Lee, H.J. , Kim, Y.J. , Shin, D.H. , Rhee, Y.H. , Son, K.H. and Park, H.Y. (2011) A highly active endo‐β‐1,4‐mannanase produced by Cellulosimicrobium sp. strain HY‐13, a hemicellulolytic bacterium in the gut of Eisenia fetida . Enzyme Microb. Tech. 48, 365–370. [DOI] [PubMed] [Google Scholar]
- Kumar, S. and Blaxter, M.L. (2010) Comparing de novo assemblers for 454 transcriptome data. BMC Genomics, e, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte, J. and Doolittle, R.F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Lambert, K.N. , Allen, K.D. and Sussex, I.M. (1999) Cloning and characterization of an esophageal‐gland‐specific chorismate mutase from the phytoparasitic nematode Meloidogyne javanica . Mol. Plant–Microbe Interact. 12, 328–336. [DOI] [PubMed] [Google Scholar]
- Liu, J.J. , Sturrock, R. and Ekramoddoullah, A.K. (2010) The superfamily of thaumatin‐like proteins: its origin, evolution, and expression towards biological function. Plant Cell Rep. 29, 419–436. [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Rosencrantz, D. , Liesack, W. and Conrad, R. (2006) Structure and activity of bacterial community inhabiting rice roots and the rhizosphere. Environ. Microbiol. 8, 1351–1360. [DOI] [PubMed] [Google Scholar]
- Mallo, G.V. , Kurz, C.L. , Couillault, C. , Pujol, N. , Granjeaud, S. , Kohara, Y. and Ewbank, J. (2002) Inducible antibacterial defense system in C. elegans . Curr. Biol. 12, 1209–1214. [DOI] [PubMed] [Google Scholar]
- Manning, G. , Plowman, G.D. , Hunter, T. and Sudarsanam, S. (2002) Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520. [DOI] [PubMed] [Google Scholar]
- Mathur, V.K. and Prasad, S.K. (1973) Survival and host range of the rice root nematode, Hirschmanniella oryzae . Indian J. Nematol. 3, 88–93. [Google Scholar]
- Maung, Z.T.Z. , Kyi, P.P. , Myint, Y.Y. and De Waele, D. (2010) Occurrence of the rice root nematode Hirschmanniella oryzae on monsoon rice in Myanmar. Trop. Plant Pathol. 35, 3–10. [Google Scholar]
- Min, X.J. , Butler, G. , Storms, R. and Tsang, A. (2005) OrfPredictor: predicting protein‐coding regions in EST‐derived sequences. Nucleic Acids Res. 33, W677–W680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitreva, M. , Smant, G. and Helder, J. (2009) Role of horizontal gene transfer in the evolution of plant parasitism among nematodes. Methods Mol. Biol. 532, 517– 535. [DOI] [PubMed] [Google Scholar]
- Mitreva‐Dautova, M. , Roze, E. , Overmars, H. , de Graaff, L. , Schots, A. , Helder, J. , Goverse, A. , Bakker, J. and Smant, G. (2006) A symbiont‐independent endo‐1,4‐beta‐xylanase from the plant‐parasitic nematode Meloidogyne incognita . Mol. Plant–Microbe Interact. 19, 521–529. [DOI] [PubMed] [Google Scholar]
- Moens, M. and Perry, R.N. (2009) Migratory plant endoparasitic nematodes: a group rich in contrasts and divergence. Annu. Rev. Phytopathol. 47, 313–332. [DOI] [PubMed] [Google Scholar]
- Muller, M. , Mentel, M. , van Hellemond, J.J. , Henze, K. , Woehle, C. , Gould, S.B. , Yu, R.Y. and van der Giezen M., Tielens, A.G. and Martin, W.F. (2012) Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 76, 444–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol, P. , Gill, R. , Fosu‐Nyarko, J. and Jones, M.G. (2012) De novo analysis and functional classification of the transcriptome of the root lesion nematode, Pratylenchus thornei, after 454 GS FLX sequencing. Int. J. Parasitol. 42, 225– 237. [DOI] [PubMed] [Google Scholar]
- O'Rourke, D. , Baban, D. , Demidova, M. , Mott, R. and Hodgkin, J. (2006) Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum . Genome Res. 16, 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver, R.P. and Solomon, P.S. (2010) New developments in pathogenicity and virulence of necrotrophs. Curr. Opin. Plant Biol. 13, 415–419. [PubMed] [Google Scholar]
- Parkinson, J. , Mitreva, M. , Whitton, C. , Thomson, M. , Daub, J. , Martin, J. , Schmid, R. , Hall, N. , Barrell, B. , Waterston, R.H. , McCarter, J.P. and Blaxter, M.L. (2004) A transcriptomic analysis of the phylum Nematoda. Nat. Genet. 36, 1259–1267. [DOI] [PubMed] [Google Scholar]
- Petersen, T.N. , Brunak, S. and von Heijne G. and Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods, 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Pfaffl, M.W. , Horgan, G.W. and Dempfle, L. (2002) Relative expression software tool (REST) for group‐wise comparison and statistical analysis of relative expression results in real‐time PCR. Nucleic Acids Res. 30, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, T.A. , Berrin, J.G. , Record, E. , To, K.A. and Sigoillot, J.C. (2010) Hydrolysis of softwood by Aspergillus mannanase: role of a carbohydrate‐binding module. J. Biotechnol. 148, 163–170. [DOI] [PubMed] [Google Scholar]
- Piskiewicz, A.M. , Duyts, H. , Berg, M.P. , Costa, S.R. and van der Putten, W.H. (2007) Soil microorganisms control plant ectoparasitic nematodes in natural coastal foredunes. Oecologia, 152, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop, M. and Salzberg, S.L. (2008) Bioinformatics challenges of new sequencing technology. Trends Genet. 24, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prot, J.‐C. and Rahman, M.L. (1994) Nematode ecology, economic importance, and management in rice ecosystems in South and Southeast Asia In: Rice Pest Science and Management (Teng P.S., Heong K.L. and Moody K., eds), pp. 129–144. Manila: IRRI. [Google Scholar]
- Qin, L. , Kudla, U. , Roze, E.H.A. , Goverse, A. , Popeijus, H. , Nieuwland, J. , Overmars, H. , Jones, J.T. , Schots, A. , Smant, G. , Bakker, J. and Helder, J. (2004) Plant degradation: a nematode expansin acting on plants. Nature, 427, 30. [DOI] [PubMed] [Google Scholar]
- Reardon, W. , Chakrabortee, S. , Pereira, T.C. , Tyson, T. , Banton, M.C. , Dolan, K.M. , Culleton, B.A. , Wise, M.J. , Burnell, A.M. and Tunnacliffe, A. (2010) Expression profiling and cross‐species RNA interference (RNAi) of desiccation‐induced transcripts in the anhydrobiotic nematode Aphelenchus avenae . BMC Mol. Biol. 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko, M. and van der Mark P., Ayres, D.L. , Darling, A. , Hohna, S. , Larget, B. , Liu, L. , Suchard, M.A. and Huelsenbeck, J.P. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso, M.N. , Favery, B. , Piotte, C. , Arthaud, L. , de Boer, J.M. , Hussey, R.S. , Bakker, J. , Baum, T.J. and Abad, P. (1999) Isolation of a cDNA encoding a beta‐1,4‐endoglucanase in the root‐knot nematode Meloidogyne incognita and expression analysis during plant parasitism. Mol. Plant–Microbe Interact. 12, 585–591. [DOI] [PubMed] [Google Scholar]
- Rybarczyk‐Mydlowska, O.K. , Maboreke, H.R. and van Megen H., van den Elsen, S. , Mooyman, P. , Smant, G. , Bakker, J. and Helder, J. (2012) Rather than by direct acquisition via lateral gene transfer, GHF5 cellulases were passed on from early Pratylenchidae to root‐knot and cyst nematodes. BMC Evol. Biol. 12, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulenburg, H. and Boehnisch, C. (2008) Diversification and adaptive sequence evolution of Caenorhabditis lysozymes (Nematoda: Rhabditidae). BMC Evol. Biol. 8, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo, T. and Van der Hoorn, R.A. (2008) Papain‐like cysteine proteases: key players at molecular battlefields employed by both plants and their invaders. Mol. Plant Pathol. 9, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui, Z.A. and Mahmood, I. (1999) Role of bacteria in the management of plant parasitic nematodes: a review. Bioresour. Technol. 69, 167–179. [Google Scholar]
- Sikora, R.A. and Fernandez, E. (2005) Nematode parasites of vegetables In: Plant Parasitic Nematodes in Subtropical and Tropical Agriculture (Luc M., Sikora R.A. and Bridge J., eds), pp. 319–392. Wallingford, UK: CABI Publishing. [Google Scholar]
- Smith, T.F. , Gaitatzes, C. , Saxena, K. and Neer, E.J. (1999) The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24, 181–185. [DOI] [PubMed] [Google Scholar]
- Soanes, D.M. , Alam, I. , Cornell, M. , Wong, H.M. , Hedeler, C. , Paton, N.W. , Rattray, M. , Hubbard, S.J. , Oliver, S.G. and Talbot, N.J. (2008) Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. PLoS ONE, 3, e2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson, T. , Reardon, W. , Browne, J.A. and Burnell, A.M. (2007) Gene induction by desiccation stress in the entomopathogenic nematode Steinernema carpocapsae reveals parallels with drought tolerance mechanisms in plants. Int. J. Parasitol. 37, 763–776. [DOI] [PubMed] [Google Scholar]
- Tyson, T. , O'Mahony, Z.G. , Wong, S. , Skelton, M. , Daly, B. , Jones, J.T. , Mulvihill, E.D. , Elsworth, B. , Phillips, M. , Blaxter, M. and Burnell, A.M. (2012) A molecular analysis of desiccation tolerance mechanisms in the anhydrobiotic nematode Panagrolaimus superbus using expressed sequenced tags. BMC Res. Notes, 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytgat, T. , Vercauteren, I. , Vanholme, B. , De, M.J. , Vanhoutte, I. , Gheysen, G. , Borgonie, G. , Coomans, A. and Gheysen, G. (2005) An SXP/RAL‐2 protein produced by the subventral pharyngeal glands in the plant parasitic root‐knot nematode Meloidogyne incognita . Parasitol. Res. 95, 50–54. [DOI] [PubMed] [Google Scholar]
- Vogel, J. (2008) Unique aspects of the grass cell wall. Curr. Opin. Plant Biol. 11, 301–307. [DOI] [PubMed] [Google Scholar]
- Wesemael, W.M.L. , Viaene, N. and Moens, M. (2011) Root‐knot nematodes (Meloidogyne spp.) in Europe. Nematology, 13, 3–16. [Google Scholar]
- Wrather, J.A. and Koenning, S.R. (2006) Estimates of disease effects on soybean yields in the United States 2003 to 2005. J. Nematol. 38, 173–180. [PMC free article] [PubMed] [Google Scholar]
- Yudina, T.G. , Guo, D. , Piskunkova, N.F. , Pavlova, I.B. , Zavalova, L.L. and Baskova, I.P. (2012) Antifungal and antibacterial functions of medicinal leech recombinant destabilase‐lysozyme and its heated‐up derivative. Front. Chem. Sci. Eng. 6, 203–209. [Google Scholar]
- Zavalova, L.L. , Yudina, T.G. , Artamonova, I.I. and Baskova, I.P. (2006) Antibacterial non‐glycosidase activity of invertebrate destabilase‐lysozyme and of its helical amphipathic peptides. Chemotherapy, 52, 158–160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences used to clone genes, make in situ hybridization (ISH) probes and perform quantitative reverse transcription‐polymerase chain reaction (Q‐RT‐PCR).