

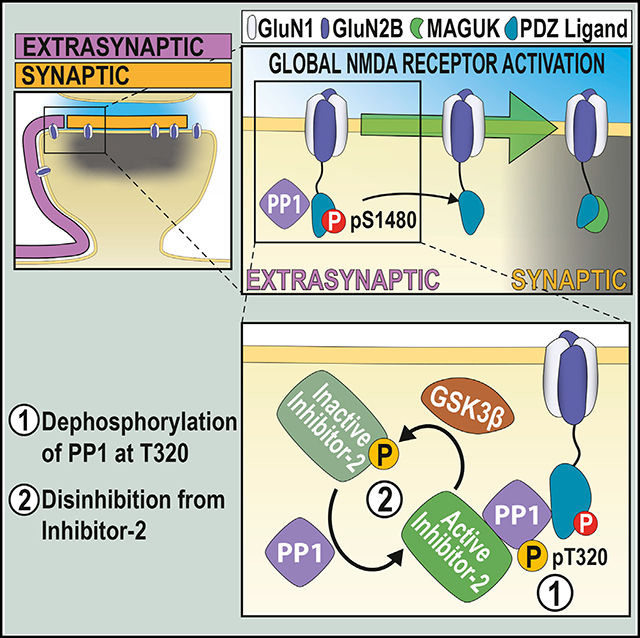
SUMMARY
In mature neurons, postsynaptic N-methyl-D-aspartate receptors (NMDARs) are segregated into two populations, synaptic and extrasynaptic, which differ in localization, function, and associated intracellular cascades. These two pools are connected via lateral diffusion, and receptor exchange between them modulates synaptic NMDAR content. Here, we identify the phosphorylation of the PDZ-ligand of the GluN2B subunit of NMDARs (at S1480) as a critical determinant in dynamically controlling NMDAR synaptic content. We find that phosphorylation of GluN2B at S1480 maintains NMDARs at extrasynaptic membranes as part of a protein complex containing protein phosphatase 1 (PP1). Global activation of NMDARs leads to the activation of PP1, which mediates dephosphorylation of GluN2B at S1480 to promote an increase in synaptic NMDAR content. Thus, PP1-mediated dephosphorylation of the GluN2B PDZ-ligand modulates the synaptic expression of NMDARs in mature neurons in an activity-dependent manner, a process with profound consequences for synaptic and structural plasticity, metaplasticity, and synaptic neurotransmission.
In Brief
The dynamic regulation of synaptically expressed NMDA receptors (NMDARs) is essential for synaptic function. Here, Chiu et al. describe a mechanism controlling this process in mature neurons by showing that increases in NMDAR synaptic content are driven by PP1-mediated dephosphorylation of extrasynaptic NMDARs within their GluN2B PDZ-ligands.
Graphical Abstract
INTRODUCTION
N-methyl-D-aspartate receptors (NMDARs) are ionotropic glutamate receptors essential for excitatory neurotransmission in the brain (Paoletti et al., 2013). In mature neurons, functional postsynaptic NMDARs are segregated in two different locations: synaptic NMDARs, which are encapsulated within the postsynaptic density (PSD), and extrasynaptic NMDARs, which are distributed between perisynaptic sites and dendritic shafts (Papouin and Oliet, 2014). These two populations have distinct functions and trigger different, sometimes opposing, intracellular cascades. The NMDAR content in both populations is plastic and, even in mature neurons, can be dynamically modified in response to different stimuli (Hunt and Castillo, 2012). Bidirectional plasticity of NMDARs has been hypothesized to affect important synaptic functions, such as metaplasticity and homeostatic plasticity, and to modify the integrative properties of the neuron (Dore et al., 2017; Hunt and Castillo, 2012). Mechanistically, both an increase in surface-expressed NMDARs and the synaptic recruitment of extrasynaptic NMDARs contributes to an increase in NMDAR synaptic content (Harney et al., 2008; Tovar and Westbrook, 2002). However, the molecular determinants controlling these processes are not fully understood.
We have previously identified a molecular mechanism controlling the clearance of GluN2B-containing NMDARs from synaptic sites by showing that the phosphorylation of the GluN2B PDZ-ligand (at S1480; GluN2B-pS1480) promotes the lateral diffusion of synaptic NMDARs to extrasynaptic sites (Chen et al., 2012; Sanz-Clemente et al., 2010, 2013). We report here that GluN2B-pS1480-containing NMDARs are expressed at extrasynaptic membranes and must be dephosphorylated in order to enter synaptic sites. We have demonstrated that extrasynaptic NMDARs form a stable protein complex containing phosphatase protein 1 (PP1). Global activation of NMDARs activates PP1 to dephosphorylate GluN2B-pS1480 and promotes the insertion of extrasynaptic GluN2B receptors into the PSD. This mechanism, therefore, controls activity-dependent NMDAR plasticity in mature neurons.
RESULTS
We have previously identified a molecular mechanism promoting the clearance of GluN2B-containing NMDARs from PSDs: the synaptic activity-dependent phosphorylation of the PDZ-ligand of the GluN2B subunit (at S1480; GluN2B-pS1480) mediated by casein kinase 2 (CK2). GluN2B-pS1480 disrupts the interaction of NMDARs with synaptic scaffolding proteins (e.g., PSD-95), which facilitates lateral diffusion to extrasynaptic sites, where phosphorylated receptors are internalized in a clathrin-dependent manner (Chen et al., 2012; Lussier et al., 2015; Sanz-Clemente et al., 2010, 2013). Because GluN2B-containing NMDARs are fated for recycling (Lavezzari et al., 2004) and GluN2B-pS1480 seems to be permissive for lateral diffusion between synaptic and extrasynaptic populations, we initially hypothesized that internalized NMDARs would be recycled and reinserted into the PSD.
We first tested this hypothesis by using an antibody feeding approach in primary hippocampal neurons overexpressing GFP-GluN2B wild-type (WT) or the phospho-mimetic mutant GluN2B S1480E. Despite comparable total expression to GluN2B WT, GluN2B S1480E displays impaired surface expression due to increased internalization (Sanz-Clemente et al., 2010). Consistent with our hypothesis, we found that the phospho-mimetic mutant was readily recycled to the plasma membrane at a level comparable to GluN2B WT (Figure 1A). Next, we evaluated NMDAR surface and synaptic expression by conducting genetic molecular replacement experiments in floxed GluN2A/2B mice as previously described (Chen et al., 2012). Briefly, we used organotypic hippocampal slices co-transfected with either Cre alone, to remove endogenous GluN2A and GluN2B, or Cre with exogenous GluN2B S1480E. Then, we measured both evoked NMDAR-mediated synaptic currents (NMDAR-excitatory postsynaptic currents [EPSCs]) and whole-cell responses to puffed-on NMDA. We found that neurons expressing only the phospho-mimetic mutant GluN2B S1480E largely lacked NMDAR-EPSCs (Figure 1B; Chen et al., 2012), but whole-cell responses to puffed-on NMDA were present in those neurons (Figure 1C), demonstrating surface expression of GluN2B S1480E.
Figure 1. Phosphorylation of the PDZ-Ligand of the GluN2B Subunit (GluN2B-pS1480) of NMDARs Maintains NMDARs Segregated at Extrasynaptic Membranes.

All graphs represent mean ± SEM; ns, non-significant; ***p < 0.001 In a Mann-Whitney U test.
(A) Hippocampal neurons were transfected at DIV7 with GFP-GluN2B WT or the phospho-mimetic mutant S1480E. Recycling ratios were analyzed at DIV13 or DIV14. Scale bar represents 10 μm. n (WT and S1480E) = 22 and 20 in 3 independent experiments. p = 0.54.
(B and C) Organotypic hippocampal slices were prepared from P7 Grin2afl/flGrin2bfl/fl mice, biolistically transfected with either Cre or Cre + GluN2B-S1480E at DIV2-DIV4, and paired whole-cell recordings were obtained from Cre-expressing and neighboring control CA1 pyramidal neurons at DIV18-DIV24. Scatterplots represent peak amplitudes of NMDAR-EPSCs (B) or whole-cell responses to 100 μM NMDA puffed onto the same cells (C) from single pairs (open circles) of transfected and control cells. Filled circles represent mean ± SEM. Dashed lines represent linear regression and 95% confidence interval. For sample traces, control cells are represented black and transfected cells are represented in green or red. Scale bars represent 100 ms and 100 pA (B) or 5 s and 400 pA (C). p = 0.54 (B).
These data indicate that, although a population of phosphorylated receptors can be trafficked to the surface, these receptors are not stabilized at PSDs and are exclusively present at extrasynaptic sites. Based on these data, we formulated our central hypothesis: surface-expressed GluN2B-containing NMDARs must be dephosphorylated within the GluN2B PDZ-ligand prior to integration into the PSD.
Global Activation of NMDARs Promotes PP1-Mediated Dephosphorylation of S1480 on Surface-Expressed GluN2B
If GluN2B needs to be dephosphorylated to enter PSDs, then this reaction should occur on surface-expressed GluN2B-containing NMDARs. To test this hypothesis, we first induced GluN2B dephosphorylation in primary cortical cultures by globally activating NMDARs (bath application of 50 μM NMDA for 10 min; Chung et al., 2004). As expected, acute NMDA treatment did not affect the total expression of GluN2B (not shown), but it promoted a robust dephosphorylation of GluN2B-pS1480 in whole-cell lysate (Figure 2). Next, we isolated surface-expressed NMDARs using two independent methods: biotinylation (Figure 2A) and biochemical isolation of the synaptic plasma membranes (SPMs) (Figure 2B) and quantified the levels of GluN2B-pS1480 by immunoblotting using a custom-generated antibody against GluN2B-pS1480 (Figure S1). Importantly, we observed a similar reduction in GluN2B phosphorylation in total lysate and in surface-expressed proteins, indicating that global NMDAR activation dephosphorylates the NMDAR population with the potential to be integrated into the PSD through lateral insertion.
Figure 2. Co-activation of Synaptic and Extrasynaptic NMDARs Promotes PP1-Mediated Dephosphorylation of S1480 on Surface-Expressed GluN2B.

Levels of normalized GluN2B-pS1480 were analyzed by immunoblotting from cortical primary neurons after the indicated manipulations. Graphs represent mean ± SEM. *p < 0.05; **p < 0.01; ****p < 0.0001 using a Mann-Whitney U test (A and B) or Kruskal-Wallis H test (C-F).
(A) Biotinylation experiment in DIV24-DIV28 neurons after induction of GluN2B-pS1480 dephosphorylation by NMDA incubation (50 μM for 10 min). Tubulin is shown as a control. M denotes marker. n = 5.
(B) Isolation of the synaptic plasma membrane (SPM) fraction from DIV14-DIV21 neurons following NMDA treatment as before. Transferrin receptor (Tfr) is shown as a control. n = 5.
(C) Synaptic, extrasynaptic, or global activation of NMDARs in DIV21-DIV28 neurons. pCreb and Creb are shown as controls. n = 3.
(D) Incubation of DIV14-DIV17 neurons with phosphatase inhibitors for 45 min: fostriecin for PP2A/4 (Fos; 1 μM); FK506 for PP2B (FK; 1 μM); okadaic acid for PP2A at 20 nM and PP2A/PP1 at 50 nM (OA; 20 or 50 nM); and calyculin A for PP1/PP2A (Cal A; 100 nM). n = 10.
(E) Pre-incubation of DIV14-DIV21 neurons with 100 nM Cal A for 45 min before NMDA treatment as before. p = 0.66 (Cal A versus Cal A + NMDA). n = 14.
(F) Lentiviral transduction of the dominant negative forms of GFP-PP1 α and γ1 (PP1α/γ1 D95N) and GFP (as a control) in DIV14-DIV17 neurons for 7–10 days. GluN2B-pS1480 dephosphorylation was induced with NMDA as before. p > 0.99 (PP1α) and p = 0.78 (PP1γ1). n = 6.
See also Figures S1 and S2.
Because previous studies have shown that synaptic NMDAR activation induces GluN2B S1480 phosphorylation and global NMDAR activation promotes dephosphorylation (Chung et al., 2004; Sanz-Clemente et al., 2010; Figures 2A and 2B), we wondered whether selective activation of extrasynaptic NMDARs would be sufficient for GluN2B-pS1480 dephosphorylation. Thus, we utilized an established protocol for the preferential stimulation of extrasynaptic NMDARs in primary cortical cultures and quantified phosphorylated CREB levels as a control (Hardingham et al., 2002). This protocol was unable to maximally de-phosphorylate GluN2B-pS1480 (Figure 2C), suggesting that both synaptic and extrasynaptic NMDAR populations synergize to dephosphorylate GluN2B. Because a major consequence of GluN2B-pS1480 is the decrease in GluN2B surface expression (Sanz-Clemente et al., 2010), we next investigated whether the pharmacological manipulations that control GluN2B-pS1480 also affect GluN2B surface expression. Consistent with our biochemical data (Figure 2C), global activation of NMDARs resulted in a robust increase in surface-expressed GluN2B, whereas preferential activation of extrasynaptic NMDARs failed to significantly modify GluN2B surface expression (Figure S2).
We finally sought to identify the phosphatase directly responsible for GluN2B-pS1480 dephosphorylation. We first used a pharmacologic approach to inhibit the phosphoprotein phosphatases (PPs) in neuronal cultures and probed for changes in baseline GluN2B-pS1480. We observed a robust increase in GluN2B-pS1480 in neurons treated with okadaic acid (OA) (50 nM) and calyculin A (Cal A) (100 nM; Figure 2D). At the concentrations used, both these inhibitors block PP1 and PP2A. Because treatment with fostriecin, a PP2A and PP4 inhibitor, was unable to cause changes in GluN2B S1480 phosphorylation, we hypothesized that PP1 is the phosphatase responsible for GluN2B-pS1480 dephosphorylation. Supporting this idea, low concentrations of OA (under 50 nM), which preferentially inhibit PP2A (Ishihara et al., 1989), did not modulate GluN2B-pS1480 (Figure 2D). Importantly, pre-incubation with Cal A (Figure 2E) or 50 nM OA (not shown) blocked the NMDA-triggered dephosphorylation of GluN2B-pS1480, demonstrating that PP1 also controls the agonist-driven reaction. We confirmed the involvement of PP1 in GluN2B-pS1480 dephosphorylation by using a genetic approach. Specifically, we transduced cultured neurons with lentiviruses expressing the dominant-negative form of the two PP1 isoforms present in dendrites (PP1α and PP1γ1 [D95N]; Strack et al., 1999; Zhang et al., 1996) and tested the levels of GluN2B-pS1480 by immunoblotting. Overexpression of nonfunctional PP1 prevented NMDA-driven dephosphorylation of GluN2B S1480, supporting that PP1 is the phosphatase responsible for GluN2B-pS1480 dephosphorylation (Figure 2F).
Together, our data demonstrate that global NMDAR activation triggers the PP1-mediated dephosphorylation of GluN2B-pS1480 on NMDARs expressed at the cell surface.
Regulation of the PP1-Dependent Dephosphorylation of GluN2B S1480
PP1 is a constitutively active phosphatase whose activity is regulated by several molecular mechanisms (Figure 3A; Cohen, 2002). These include (1) direct inhibitory phosphorylation of the PP1 C terminus, (2) complexing with inhibitor proteins to block accessibility to the active site, and (3) complexing of active PP1 with target specifying proteins to confer specificity and restrict PP1 to a specific subcellular compartment. We next sought to examine which of these modes of PP1 regulation participate in GluN2B-pS1480 dephosphorylation.
Figure 3. Regulation of the PP1-Dependent Dephosphorylation of GluN2B S1480.

All graphs represent mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 in a Kruskal-Wallis H test (B, C, and F) or a Mann-Whitney U test (D, E, H, and J).
(A) PP1 is regulated by (1) inhibitory CDK5 phosphorylation, (2) complexing with endogenous inhibitor proteins, or (3) targeting to a correct substrate.
(B) Immunoblot analysis of GluN2B-pS1480 levels from DIV14 cortical primary neurons after pharmacological blockade of GSK3 to prevent I-2 inactivation (CHIR99021; 10 μM for 2 h) prior to NMDA-triggered dephosphorylation of GluN2B-pS1480. n = 8.
(C) GluN2B-pS1480 levels were assessed by immunoblotting from DIV14-DIV17 cortical primary neurons after lentiviral transduction of constitutively active GFP-I-2 (I-2 T73A) or GFP and GFP-I-2 WT(as controls) in cortical neurons for 7–10 days. GluN2B-pS1480 dephosphorylation was induced with NMDA as before. p = 0.63 (I-2 T73A). n = 6.
(D) NMDA-induced translocation to the plasma membrane of GFP-PP1 and GFP-CaMKII (as a control) was evaluated in DIV14 hippocampal cultures by TIRF microscopy. Baseline fluorescence was recorded for 5 min prior to the addition of 50 μM NMDA. Graph represents changes in TIRF signal over time. Scale bar represents 10 μm. n = 11 in 4 independent experiments. See also Videos S1 and S2.
(E) The SPM fraction of DIV14-DIV21 cortical neurons was isolated and the relative expression of the indicated proteins was evaluated by immunoblotting. Protein levels were normalized to transferrin receptor (Tfr) in each condition and then normalized to control surface expression levels. n = 4.
(F) Levels of PP1 present in the SPM fraction of mice of the indicated age were assessed by immunoblotting. n = 8.
(G) Levels of GluN2B-pS1480 present in the SPM fraction of mice of the indicated age were assessed by immunoblotting.
(H) Immunoblot analysis of GluN2B-pS1480 from cortical primary neurons of different DIVs after induction of GluN2B-pS1480 dephosphorylation. p = 0.68 (DIV7). n = 5.
(I) The extrasynaptic fraction from rat cortex was isolated and the association of GluN2B, PP1, and I-2 was analyzed by immunoblotting following co-immunoprecipitation with an anti-GluN2B antibody. Graph represents fold of enrichment normalized to input. n = 3.
(J) cLTD (20 μM NMDA for 3 min and 30 min recovery) was initiated in acute hippocampal slices from P72-P84 mice. GluN2B-pS1480 and GluA1-pS845 levels were assessed by immunoblotting. n = 6.
See also Figure S3.
As previously reported (Hou et al., 2013), we found that NMDA treatment causes a global reduction in PP1-pT320, a CDK5-mediated phosphorylation that inhibits phosphatase activity by blocking the access of PP1 to its substrates (Figure S3A).
Neurons express several inhibitor proteins that complex with PP1 to block access to the active site, including inhibitor 1 (I-1), inhibitor-2 (I-2), and dopamine- and cyclic AMP (cAMP)-regulated phosphoprotein-32 (DARPP-32). In turn, these inhibitors are regulated by kinase and phosphatase activity to allow for signal integration (Eto and Brautigan, 2012). Pharmacological manipulations to modulate I-1 and DARPP-32 activation failed to control GluN2B-pS1480 dephosphorylation, excluding them as molecular players in this process (Figures S2B–S2D). We next tested the involvement of I-2, which functions as an active inhibitor in its dephosphorylated state (Siddoway et al., 2013), by inhibiting GSK3β, the kinase responsible for inactivating I-2. GSK3 blockade prevented NMDA-mediated GluN2B-pS1480 dephosphorylation, suggesting a role for I-2 in this process (Figure 3B). To further confirm I-2 involvement, we generated lentiviruses expressing a phospho-deficient (active) form of I-2 (I-2 T73A) to constitutively inhibit PP1. As expected, overexpression of this mutant prevented NMDA-triggered GluN2B-pS1480 dephosphorylation (Figure 3C).
The last key piece of PP1 regulation is correct targeting and complexing of PP1 with its substrate (Hendrickx et al., 2009). Because PP1 is a cytosolic protein that is not associated to the plasma membrane, but dephosphorylation occurs on NMDARs expressed at the cell surface (Figures 2A and 2B), we hypothesized that PP1 might translocate to a plasma membrane-associated population in response to NMDAR activation. To test this possibility, we first utilized total internal reflection fluorescence (TIRF) microscopy, a technique that enables examination of dynamic processes that occur within 100 nm of the plasma membrane. We overexpressed GFP-PP1 in hippocampal neurons and tracked its targeting to the surface in response to NMDA treatment overtime. As a positive control, we used GFP-CaMKII (Merrill et al., 2005). As expected, CaMKII showed robust surface trafficking in response to NMDA treatment, but surprisingly, PP1 showed no change in surface levels (Figure 3D; Videos S1 and S2). Because overexpression of GFP-PP1 may saturate the system, we next isolated the SPM fraction following NMDA treatment in primary cortical neurons and quantified the levels of endogenous PP1 associated with SPMs by immunoblotting (Figure 3E). Again, we observed a strong enrichment of CaMKII in the SPM fraction after NMDA treatment, with no increase in SPM-associated PP1. These data indicate that the PP1 population responsible for GluN2B S1480 dephosphorylation is present at the plasma membrane likely already associated with NMDAR complexes.
GluN2B-pS1480 Dephosphorylation Is Enhanced by Neuronal Maturation
Our finding that GluN2B-pS1480 dephosphorylation is dependent on PP1 associated with SPMs resembles the regulation of the complementary process, phosphorylation of GluN2B at S1480, which is strongly regulated by the level of SPM-associated CK2 (Sanz-Clemente et al., 2013). The association of CK2 with SPMs is developmentally regulated and peaks around the second postnatal week in rodents, coincident with the highest level of GluN2B-pS1480 (Sanz-Clemente et al., 2010). Therefore, we next investigated whether the association of PP1 with SPMs also has a developmental component and found low amounts of PP1 in the SPM fraction isolated from immature mouse brain (postnatal day 7 [P7]), whereas the association of PP1 with SPMs dramatically increased throughout brain maturation. This is not a mere consequence of changes in PP1 total expression, as PP1 is strongly expressed since early development (Figure 3F). These data suggest that the developmentally controlled association to SPMs of both the kinase (CK2) and the phosphatase (PP1) synergize to reduce the levels of GluN2B-pS1480 in adulthood (Figure 3G; Sanz-Clemente et al., 2010).
Our data suggest that NMDAR activity-driven GluN2B-pS1480 dephosphorylation might also be differentially controlled at different stages of synaptic maturation. We tested this possibility by inducing NMDA-dependent GluN2B-pS1480 dephosphorylation in primary cortical neurons at different stages of maturation (from day in vitro 7 [DIV7] to DIV28 or DIV29). As expected, we found a strong influence of maturation in the dephosphorylation process: in immature neurons (DIV7), NMDA treatment promoted a small increase in GluN2B-pS1480 rather than the expected decrease observed beginning at DIV14, which became increasingly enhanced throughout neuronal maturation (Figure 3H).
GluN2B-pS1480 Dephosphorylation Is Mediated by a Population of PP1 Not Activated during LTD
Because GluN2B-pS1480 receptors are exclusively expressed at extrasynaptic membranes (Figure 1B) and there is no translocation of PP1 to the surface upon NMDA treatment (Figures 3D and 3E), we hypothesized that the PP1 population mediating this reaction is also extrasynaptically located. This idea is consistent with the need for extrasynaptic NMDAR activation to induce dephosphorylation (Figure 2C) and would explain the differential outcome of NMDA treatment in immature cultures (Figure 3H), as the compartmentalization between synaptic and extrasynaptic sites has not yet occurred. We tested this possibility by isolating GluN2B-containing protein complexes from adult rat brain extrasynaptic membranes by performing co-immunoprecipitation with an anti-GluN2B antibody. As shown in Figure 3I, both PP1 and I-2 co-immunoprecipitated with GluN2B. Whereas GluN2B was enriched ~2-fold in our co-immunoprecipitation (co-IP) experiments, the enrichment of PP1 was higher (~4-fold; Figure 3I). This indicates the existence of multiple copies of PP1 associated with a single GluN2B, likely indirectly, through the binding of several target specifiers that link PP1 with GluN2B, such as spinophilin (Baucum et al., 2013), and yotiao (Lin et al., 1998). Together, our results indicate that extrasynaptic GluN2B-containing NMDARs interact with PP1 and I-2.
The existence of an extrasynaptic pool of PP1 (Figure 3I), in combination with our data showing the lack of PP1 recruitment to synapses after NMDA treatment (Figures 3D and 3E) is in sharp contrast with the reported synaptic recruitment of PP1 after the induction of long-term depression (LTD) (Morishita et al., 2001). This suggests the existence of distinct PP1 populations that can be differentially controlled. To test this possibility, we activated synaptic PP1 (Hu et al., 2007) by inducing chemical LTD (cLTD) in ex vivo hippocampal slices from adult (>P70) mice (Babiec et al., 2014). As a control, we quantified the levels of GluA1 phosphorylation at S845 (GluA1-pS845), because synaptic PP1 mediates GluA1-pS845 dephosphorylation (Diering and Huganir, 2018). As shown in Figure 3J, we found a drastic difference between the regulation of the PP1-mediated GluN2B and GluA1 dephosphorylation after activation of synaptic PP1: whereas levels of GluA1-p845 decreased as expected, the levels of GluN2B-pS1480 increased after cLTD, presumably due to synaptic NMDARs activation.
Together with our developmental data, these findings suggest that the GluN2B-pS1480 dephosphorylation reaction is mediated by strict spatiotemporal trafficking of PP1 to extrasynaptic membranes.
PP1 Activity Modulates NMDAR Synaptic Localization
If our hypothesis that GluN2B-pS1480 maintains NMDARs at extrasynaptic membranes is correct, then modulation of PP1 activity against GluN2B S1480 should alter both synaptic signaling and synaptic GluN2B receptor content (Figure 4A). To test this idea, we first examined changes in ERK phosphorylation (pERK), because activation of synaptic NMDARs leads to an increase in pERK, whereas extrasynaptic NMDAR activation decreases pERK levels (Hardingham and Bading, 2010). Specifically, we modulated GluN2B-pS1480 in cultured neurons with NMDA (50 μM for 10 min) or OA (50 nM for 45 min) to induce the potential redistribution of NMDARs, and after drug withdrawal, we increased synaptic activity using a standard pharmacological approach (20 μM bicuculline and 100 μM 4-AP incubation [Bic/4-AP]). As expected, cells treated with Bic/4-AP showed a robust increase in ERK phosphorylation that, consistent with our hypothesis, was potentiated in neurons pre-treated with NMDA. Conversely, cells pre-treated with OA to block PP1 activity displayed a reduced increase in ERK phosphorylation after Bic/4-AP treatment in comparison with control cells (please note the expected elevated basal ERK phosphorylation in OA pre-treated neurons in comparison to controls; Figure 4B).
Figure 4. PP1 Activity Modulates NMDAR Synaptic Localization.

All graphs represent mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 in a Mann-Whitney U test.
(A) Global synaptic and extrasynaptic NMDAR activation triggers PP1 activity to promote dephosphorylation of GluN2B, which modulates its synaptic content.
(B) DIV18-DIV21 cortical neurons were pre-treated with OA (50 nM for 45 min) or NMDA (50 μM for 10 min), and synaptic NMDAR stimulation was performed by incubating with 20 μM bicuculline and 100 μM 4-AP for 30 min. Levels of pERK and ERK were evaluated by immunoblotting. n = 7.
(C) Super-resolution SIM micrographs were acquired from GluN2B WT transfected neurons (Ci). Total numbers of PSD-95 (Cii) and surface-expressed GluN2B (Ciii) puncta were quantified. To analyze the synaptic expression of exogenous receptors, Nikon NIS-Elements software identified colocalized puncta (synaptic GluN2B, white) (containing surface GFP-GluN2B [red] and endogenous PSD-95 [blue]; Civ). Values are presented as synaptic GluN2Bversus total GluN2B puncta after pharmacological manipulations (Cv). Samples from each processing step are presented in (Civ). Scale bars represent 10 μm (Ci) and 1 μm (Civ). n (Con, OA, and NMDA) = 55, 18, and 29 in 4 independent experiments.
(D) PSD fractions were isolated from acute hippocampal slices from P70-P96 mice treated with NMDA (50 μM for 15 min) or Cal A (100 nM for 30 to 60 min) to promote or inhibit GluN2B-pS1480 dephosphorylation. Levels of GluN2B-pS1480 and synaptic GluN2B were analyzed by immunoblotting and normalized to the synaptic marker Homer1. n (Cal A and NMDA) = 9 and 7.
(E) Model to predict synaptic GluN2B content based on GluN2B-pS1480 levels. Blue circles represent data from acute slices treated with NMDA (50 μM for 15 min), and red circles represent slices treated with Cal A (100 nM for 30 to 60 min). Data points were fitted to an exponential regression. Trendline follows the equation 1.4478e−.37118(GluN2B−pS1480/GluN2B) (p < 0.001).
We next employed super-resolution microscopy in primary hippocampal cultures after transfection of GFP-GluN2B to investigate its synaptic expression after pharmacological treatment. Specifically, we labeled overexpressed receptors located at the surface and endogenous PSD-95 (as a synaptic marker) and used structured illumination microscopy (SIM) to acquire ~120-nm-resolution images (Figure 4Ci). We first quantified the number of PSD-95 puncta and found that the pharmacological manipulations do not affect the global number of synaptic sites (Figure 4Cii). Because NMDA treatment increases the surface expression of GluN2B (surface versus intracellular; Figure S2), we also quantified the net number of GluN2B puncta expressed at the surface of analyzed neurons to ensure that they displayed comparable levels of surface-expressed GluN2B, and therefore, we could utilize consistent analytical parameters in all conditions (Figure 4Ciii). Finally, we generated a mask containing the overlapped areas between GluN2B and PSD-95 puncta to identify synaptic NMDARs (Figure 4Civ). Quantification of the overlapped GluN2B and PSD- 95 areas revealed that incubation with NMDA to promote GluN2B-pS1480 dephosphorylation increased the synaptic expression of GFP-GluN2B, whereas OA treatment, which enhances GluN2B-pS1480, decreased GluN2B synaptic content (Figure 4Cv).
To confirm these data in a more intact preparation, we employed ex vivo PP1 manipulation in acute hippocampal slices. NMDA treatment to induce dephosphorylation of GluN2B S1480 resulted in a robust increase in the normalized synaptic content of endogenous GluN2B. Conversely, slices treated with calyculin A, the most effective PP1 inhibitor in acute slices, displayed an increase in GluN2B-pS1480 and a concomitant decrease in GluN2B synaptic content (Figure 4D). To unbiasedly assess the importance of GluN2B-pS1480 in determining GluN2B synaptic localization, we first tested the strength of correlation between normalized GluN2B-pS1480 levels and normalized GluN2B synaptic content using the data obtained from pharmacologically treated brain slices (Figure 4D). Specifically, we calculated the Spearman’s rank correlation and found a strong association between these parameters (p < 0.01). We, therefore, fit the data to an exponential model with the following equation: Synaptic GluN2B = 1.4478e−.37118(GluN2B−pS1480/GluN2B) (p < 0.001), which predicts synaptic NMDAR content based on levels of GluN2B-pS1480 (Figure 4E). Together, these data demonstrate that PP1 activity against the GluN2B PDZ-ligand is a crucial regulator of GluN2B-containing NMDAR trafficking and controls NMDAR synaptic content in mature neurons.
DISCUSSION
In this study, we identify PPI-mediated dephosphorylation of the PDZ-ligand of GluN2B (at S1480) as a critical determinant for enhancing NMDAR synaptic content in mature neurons. Our findings expand our previous work showing that CK2 phosphorylates GluN2B S1480 to disrupt the interaction of NMDARs with MAGUK proteins and promote NMDAR synaptic clearance and internalization (Chung et al., 2004; Sanz-Clemente et al., 2010, 2013). Therefore, the dynamic regulation of the phosphorylation state of the GluN2B PDZ-ligand is a key modulator of synaptic NMDAR content.
Given the essential functional and regulatory roles displayed by synaptic NMDARs, modification of synaptic NMDAR density has emerged as a key mechanism for controlling neuronal function. The most evident consequence of this process is the modulation of metaplasticity, which modifies the induction threshold of AMPAR plasticity. However, other neuronal properties, such as the integration of synaptic inputs or the control of homeostatic plasticity may also be affected (Dore et al., 2017; Hunt and Castillo, 2012). The precise molecular mechanisms underlying NMDAR synaptic content modulation remain unclear, although the involvement of the co-activation of NMDARs and GPCRs (i.e., mGluR1/5 or mAChRs) has been reported (Dore et al., 2017). Here, we identify a molecular mechanism that controls the synaptic recruitment of extrasynaptic NMDARs to synaptic sites, a process previously identified to contribute to NMDAR synaptic content increases (Dupuis et al., 2014; Harney et al., 2008). Regulation of synaptic-extrasynaptic NMDAR exchange is important because elevated extrasynaptic NMDAR signaling triggers excitotoxicity and alterations in NMDAR balance have been associated with several neurological disorders, including Huntington’s disease, traumatic brain injury, and epilepsy (Parsons and Raymond, 2014). Therefore, impairments in the PP1-mediated regulation of NMDAR trafficking may contribute to disease pathophysiology.
PP1 is a ubiquitously expressed serine-threonine phosphatase, although in neurons, it is enriched in dendritic spines (Strack et al., 1999). Our finding that activation of PP1 increases NMDAR synaptic content by dephosphorylating the GluN2B PDZ-ligand is unexpected, as phosphatases, and particularly PP1, have been associated with synaptic activity depression rather than potentiation (Woolfrey and Dell’Acqua, 2015). In fact, compelling evidence supports a model in which PP1 induces AMPAR-LTD (reduction of synaptic AMPAR content) by dephosphorylating the GluA1 subunit of AMPAR (at S845) and CaMKII (at T256; Siddoway et al., 2014). Although they can occur independently, alterations in NMDAR and AMPAR synaptic content are often co-induced (Hunt et al., 2013). How then can PP1 activity drive opposite effects in synaptic NMDAR and AMPAR density (i.e., increasing NMDAR but decreasing AMPAR synaptic content)? We propose that the activation of different pools of PP1 is a critical factor for the different outcomes. The stimuli classically used to induce AMPAR-LTD would activate synaptic-resident PP1, but not the PP1 pool associated with GluN2B at extrasynaptic membranes (as extrasynaptic NMDARs would not be activated). Conversely, strong stimuli required to increase synaptic NMDAR content would cause a robust release of glutamate, enough to spillover and activate both synaptic and extrasynaptic NMDARs. This would result in an increase in both AMPAR (by CaMKII activation and AMPAR phosphorylation) and NMDAR (by PP1 activation and dephosphorylation of GluN2B-pS1480) synaptic content. The existence of different pools of PP1 that can be differentially activated is consistent with the differential regulation of PP1-mediated dephosphorylation of GluN2B-pS1480 and GluA1-pS845 (Figure 3J) and is supported by the fact that the stimulus used to induce AMPAR-LTD in culture recruits PP1 to synaptic sites (Morishita et al., 2001), whereas our biochemical and TIRF data indicate that global NMDAR activation does not (Figures 3D and 3E). In summary, our data reveal a critical role for PP1 in controlling NMDAR synaptic content in mature neurons by mediating the dephosphorylation of GluN2B upon global activation of NMDARs.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Antonio Sanz-Clemente (antonio.sanz-clemente@northwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The care and use of animals were in accordance to the guidelines set by the Northwestern University and UCSF IACUCs.
Primary Neuronal Cultures
Primary neuronal cultures were generated from male and female E18 Sprague-Dawley rats. Dissociated neurons were plated on poly-D-lysine coated plates or coverglasses. Cells were maintained in Neurobasal media (GIBCO) supplemented with B-27 (GIBCO) and glutamine (20 mM) (GIBCO). For biochemical experiments, primary cortical neuronal cultures at a density of ~150,000 cells/cm2 were used due to the ease of generating enough material for analysis. For imaging-based experiments, hippocampal primary neuronal cultures were utilized at a density of ~40,000 cells/cm2.
Animals
C57/B6 male and female mice were cared for by the Northwestern Center for Comparative Medicine. Mice were group housed (up to 5 mice per cage), maintained on a 14/10 light-dark schedule, and had access to food and water ad libitum. Mice of the indicated ages were used to generate either slice cultures or acute slices for ex vivo experiments.
METHOD DETAILS
Lentivirus Generation
Lentiviruses were generated by transfecting Lenti-X cells (Clontech) with psPAX2, pMD2.G, and the pLVX backbone containing the indicated construct. The viral supernatant was harvested 48 and 72 hr post transfection and concentrated using polyethylene glycol for 24 hr. Concentrated virus was resuspended 100× in PBS and stored at −80°C until use.
Immunoflouresence Microscopy
For recycling experiments, hippocampal neurons were transfected at DIV7 with GFP-tagged GluN2B (WT or S1480E), and surface-expressed receptors were labeled with anti-GFP antibody for 15 min at RT 4–7 days post transfection. Following internalization receptors (30 min at 37°C), remaining surface receptors were blocked with Fab (20 μg/ml) for 20 min at RT. After allowing for recycling of receptors back to the surface (45 min at 37°C), cells were washed and fixed with 4% PFA in PBS containing 4% sucrose. Surface receptors were labeled with Alexa 555-conjugated secondary antibody (shown in white for clarity). The intracellular pool of receptors was identified by permeabilizing cells with 0.25% Triton X-100 and labeling anti-GFP tagged receptors with Alexa 633-conjugated secondary antibodies (shown in green). Cells were imaged on a Zeiss LSM 510 confocal microscope. Serial optical sections collected at 0.35 μm intervals were used to create maximum projection images. Quantification was performed by analyzing the fluorescence intensity of 3–4 independent areas per neuron using MetaMorph 6.0 software (Universal Imaging Corp) and is presented as ratio surface/intracellular intensities (mean ± SEM).
For surface expression experiments, cells were transfected on DIV14 and subject to pharmacologic manipulation immediately prior to labeling with an anti-GFP antibody for 15 min at RT surface receptors and subsequent labeling with Alexa 555-conjugated secondary antibody. The intracellular pool of receptors was identified by permeabilizing cells with 0.25% Triton X-100 and labeling anti-GFP tagged receptors with Alexa 647-conjugated secondary antibodies. Images were acquired on a Nikon A1 confocal microscope. Serial optical sections collected at 0.35 μm intervals were used to create maximum projection images. Quantification was performed by analyzing the fluorescence intensity of 5 independent areas per neuron using FIJI and is presented as ratio surface/intracellular intensities (mean ± SEM).
Electrophysiology in Organotypic Slice Cultures
Double-floxed GluN2A/GluN2B (Grin2afl/flGrin2bfl/fl) mice were generated as previously described (Akashi et al., 2009; Gray et al., 2011; Mishina and Sakimura, 2007). Cultured slices were prepared and transfected as previously described (Schnell et al., 2002). Briefly, hippocampi were dissected from P7 Grin2afl/flGrin2bfl/fl mice and biolistically co-transfected after 2–4 days in culture with pFUGW-Cre:mCherry (expressing a nuclear targeted Cre:mCherry fusion protein) and either pCAGGS-GFP or pCAGGS-GluN2B-S1480E-IRES-GFP. Slices were cultured for an additional 14–20 days before recording. Slices were recorded in a submersion chamber on an upright Olympus microscope, perfused in room temperature normal ACSF saturated with 95% O2/5% CO2. Picrotoxin (0.1 mM) and NBQX (10 μM) were added to the ACSF to block GABAa and AMPA receptors respectively. CA1 pyramidal cells were visualized by infrared differential interference contrast microscopy and transfected neurons were identified by epifluorescence microscopy. The intracellular solution contained (in mM): CsMeSO4 135, NaCl 8, HEPES 10, Na-GTP 0.3, Mg-ATP 4, EGTA 0.3, and QX-314 5. Cells were recorded with 3 to 5MΩ borosilicate glass pipettes, following stimulation of Schaffer collaterals with bipolar electrodes placed in stratum radiatum of the CA1 region. Series resistance was monitored and not compensated, and cells in which series resistance varied by 25% during a recording session were discarded. Synaptic responses were collected with a Multiclamp 700B amplifier (Axon Instruments), filtered at 2 kHz, and digitized at 10 Hz. All paired recordings involved simultaneous whole-cell recordings from a transfected neuron and a neighboring untransfected neuron. NMDAR-EPSCs were recorded at +40 mV in the presence of 10 μM NBQX. The stimulus was adjusted to evoke a measurable, monosynaptic EPSC in both cells. Whole cell NMDA responses were evoked with 100 μM NMDA (in ACSF) pressure-ejected (puffed) from a glass pipette by a Picospritzer III (Parker-Hannifin). Slices were aligned such that NMDA was puffed over the soma of the cell pair and perfused along the apical dendrites following the bath flow direction. Paired amplitude data were analyzed with a Wilcoxon signed-rank test and comparison of paired data groups were performed using a Mann-Whitney U test. Linear regressions were obtained using the least-squares method. Data are presented as mean ± SEM.
Generation of Neuronal Membrane Fractions
The crude membrane fraction was generated as previously described (Sanz-Clemente, et. al. 2010). Briefly, cortical neurons were harvested in PBS containing 0.1 mM CaCl2 and 1 mM MgCl2, and pelleted by centrifugation at 20,000 g. Pellets were resuspended in hypotonic buffer (10 mM Tris, 1 mM NaVO4) and sonicated. Membranes were isolated by centrifugation at 20,000 g and resuspended in SDS loading buffer.
Biochemical Subcellular Fractionation
Biochemical subcellular fraction followed established protocols. Briefly, brain tissue or cells were homogenized in homogenization buffer (0.32M HEPES-buffered sucrose) containing protease (Roche) and phosphatase (Sigma) inhibitors and centrifuged for 10 min at 1,000 g to remove nuclei and large debris. The supernatant was centrifuged at 10,000 g for 15 min to generate the synaptosomal fraction (P2). P2 was resuspended in hypotonic buffer, sonicated, and centrifuged at 25,000 g to generate the SPM fraction (P3). To separate PSDs from the extrasynaptic fraction, SPMs were resuspended in PBS containing 1% Triton X-100 and ultracentrifuged at 100,000 g for 1 hour. Pellets containing the PSD were resuspended in PBS.
Preferential Extrasynaptic NMDAR Activation
Selective NMDAR stimulation was carried out as follows. Synaptic NMDARs were activated by treatment with 50 μM bicuculline for 20 min. Extrasynaptic NMDARs were preferentially activated by initially inhibiting synaptic NMDARs by promoting their opening in the presence of the irreversible NMDAR blocker MK-801 (50 μM bicuculline in the presence of 10 μM MK-801 for 10 min) and subsequent bath NMDAR treatment (50 μM NMDA for 10 min). Global NMDAR activation was carried out by treatment with 50 μM NMDA for 10 min. pCreb and Creb were utilized as controls.
Total Internal Reflection Microscopy
For total internal reflection microscopy, primary hippocampal neurons were plated in glass bottom dishes (Matek). On DIV10, cells were transfected with either GFP-PP1α/γ1 or GFP-CaMKII. Cells were imaged on DIV14 on a NIKON X1 spinning disk confocal microscope. During imaging, cells were maintained at 37°C with appropriate humidity and gas using a Tokai Hit incubation chamber. A timelapse series captured images every 30 s to establish at least 5 min of baseline and continued capturing images for 10 min following treatment with 50 μM NMDA. Timelapse series were acquired with NIKON elements software and changes in fluorescence at each time point were analyzed using FIJI. Changes in fluorescence intensity from baseline are reported as mean ± SEM.
Co-immunoprecipitation from extrasynaptic membranes
For co-immoprecipitation, the SPM fraction was obtained from rat brain cortex as explained before, dissolved in PBS 1% Triton X-100 and ultracentrifuged at 100,000 g for 1 hr. The pellet (PSD) was discarded, and the extrasynaptic membranes fraction (SN) was incubated with 2 μg anti-GluN2B antibody (or IgG as a control) for 1 hr at 4°C with rotation. BSA pre-blocked protein A-Sepharose beads were added to the lysate and incubated for 1 additional hr. After 3×10 min washes with cold PBS 1% TX-100, samples were resuspended in SDS loading buffer.
Immunoblotting
Follow preparation, samples were then run on SDS-PAGE and immunoblotted with the indicated antibodies. For phosphorylation-state specific and total immunoblots, the same membrane was used following antibody stripping. Quantification was performed by FIJI and presented as mean ± SEM.
Super-resolution Microscopy
For super-resolution microscopy, hippocampal primary cultured neurons were transfected with GFP-GluN2B at DIV14 and processed at DIV21–23. Surface-expressed receptors were labeled with an anti-GFP antibody and cells were fixed and permeabilized as before. Endogenous PSD-95 was labeled with a specific antibody as a synaptic marker. Super-resolution images were acquired using a Nikon N-SIM super-resolution structured illumination microscope and reconstructed with NIS-Elements software. One secondary dendrite was selected and imaged from each neuron. Reconstruction parameters (0.74;0.83;0.21) were kept consistent throughout the experiment. Imaging and reconstruction parameters were empirically determined with the assistance of the expertise of the Nikon Imaging Center at Northwestern University. Reconstructed images were then opened in FIJI software. After thresholding both channels, a colocalized channel was created containing the overlapped area of GFP-GluN2B and PSD95 channels. Particle analysis was conducted on all three channels to get the total area and number of GFP-GluN2B, PSD95, and colocalized proteins independently. Synaptic GluN2B was determined by the ratio between colocalized puncta area and total GFP-GluN2B puncta area expressed on cell surface and are reported as mean ± SEM.
Pharmacologic Manipulation of Acute Slices
Acute cortical slices were generated from adult (8 to 14 week) C57/B6 mice. Mice were anesthetized with isoflurane prior to decapitation. Coronal slices (300 μm thick) were cut in ice-cold ACSF containing the following composition (in mM): NaCl 130, KCl 3.5, Glucose 10, NaH2PO4 1.25, NaHCO3 24, CaCl2 1, MgCl2 2. The slices were incubated for 30 min at 30–32°C and allowed to rest for at least 1 hr at room temperature in normal ACSF containing (in mM): NaCl 130, KCl 3.5, Glucose 10, NaH2PO4 1.25, NaHCO3 24, CaCl2 2, MgCl2 1. For pharmacologic manipulation, slices were incubated at 32°C and treated with either 50 μM NMDA for 15 min (NMDA), 100 nM Cal A for 30 to 60 min (Cal A), or 20 μM NMDA for 3 min with 30 min of recovery in drug-free ACSF (cLTD). Following pharmacologic manipulation, slices were flash frozen and subject to subsequent fractionation.
Modeling of GluN2B Synaptic content
Data from acute slices treated with either NMDA or Cal A were pooled. A Spearman’s rank correlation was calculated to verify association between normalized GluN2B-pS1480 levels and normalized synaptic GluN2B content. Subsequently, the data were fit to an exponential model following the form y = eax + b. Model was obtained by regressing the natural log of synaptic GluN2B content against GluN2B-pS1480 levels using the Im function in R. F-test was used to calculate statistical significance of the model.
Our antibody against phosphorylation state-specific GluN2B S1480 (Ac-CGHVYEKLSSIE(pS)DV-OH) was generated by New England Peptide. Antibodies against GluN2B and PSD-95 were obtained from Neuromab. transferrin receptor, CaMKII, and PP1γ were obtained from Thermo Fisher. PP1 antibody was obtained from Santa Cruz. All drugs and inhibitors were obtained from Tocris Biosciences.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification methods for individual experiments are described as above in method details. Statistical tests and significance values are reported in figure legends. All data is presented as mean ± SEM. N represents number of experimental repeats. For imaging experiments, n represents the number of neurons sampled. Prism 7 (Graphpad) was used for all statistical tests. R was used to generate the GluN2B mathematical model. Tests utilized include Wilcoxon signed-rank test, Mann-Whitney U Test, Kruskal-Wallis H Test, and F test. For all analysis, ns = non-significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DATA AND CODE AVAILABILITY
The uncropped blots utilized for assembling the figures in this study can be found at: https://data.mendeley.com/datasets/769vrs9v2v/draft?a=7901a11c-4c68-4817-ad94-7b09aab4ab1a.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GluN2B-pS1480 | New England Peptide (NEP) | N/A |
| Anti-GluN2B | Neuromab | Cat#75-101; RRID:AB_2232584 |
| Anti-GFP | Invitrogen | Cat#A11122; RRID:AB_221569 |
| Anti-Tubulin | Sigma-Aldrich | Cat#T2200; RRID:AB_262133 |
| Anti-Transferrin Receptor | Invitrogen | Cat#13-6800 RRID:AB_2533029 |
| Anti-PhosphoCREB | Cell Signaling Technology | Cat# 9198; RRID:AB_2561044 |
| Anti-CREB | Millipore | Cat# MAB5432; RRID:AB_2085900 |
| Anti-CaMKII | Thermo Fisher | Cat# MA1-048 RRID:AB_325403 |
| Anti-PP1 | Santa Cruz Biotechnologies | Cat#sc-7482; RRID:AB_628177 |
| Anti-Inhibitor-2 | R&D Systems | Cat# AF4719; RRID:AB_2168877 |
| Anti-GluA1-pS845 | Cell Signaling Technology | Cat#D10G5 RRID:AB_10860773 |
| Anti-GluA1 | Millipore | Cat#MAB2263; RRID:AB_11212678 |
| Anti-PhophoERK | Cell Signaling Technology | Cat#4370 RRID:AB_2315112 |
| Anti-ERK | Cell Signaling Technology | Cat#4695 RRID:AB_390779 |
| Anti-Homer | Synaptic Systems | Cat#160 003 RRID:AB_887730 |
| Anti-PP1α-pT320 | Cell Signaling Technologies | Cat#2581; RRID:AB_330823 |
| Anti-PSD-95 | Neuromab | Cat#75-028 RRID:AB_2292909 |
| Anti-Rabbit IgG, Alexa 555 conjugated | Life Technologies | Cat#A21429; RRID:AB_2535850 |
| Anti-Rabbit IgG, Alexa 647 conjugated | Life Technologies | Cat#A21245; RRID:AB_2535813 |
| Anti-Mouse IgG, Alexa 647 conjugated | Life Technologies | Cat#A21236; RRID:AB_2535805 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NMDA | Tocris | Cat#0114; CAS: 6384-92-5 |
| (−)-Bicuculline methiodide | Tocris | Cat#2503; CAS: 40709-69-1 |
| (+)-MK 801 maleate | Tocris | Cat#0924; CAS: 77086-22-7 |
| Fostriecin Sodium Salt | Tocris | Cat#1840 CAS: 87860-39-7 |
| FK 506 | Tocris | Cat#3631; CAS: 104987-11-3 |
| Okadaic Acid | Tocris | Cat#1136; CAS: 78111-17-8 |
| Calyculin A | Tocris | Cat#1336 CAS: 101932-71-2 |
| CHIR99021 | Tocris | Cat#4423; CAS: 252917-06-9 |
| 4-AP | Tocris | Cat#0940; CAS: 504-24-5 |
| D-AP5 | Tocris | Cat#0106; CAS: 79055-68-8 |
| TTX | Tocris | Cat#1078; CAS: 4368-28-9 |
| KCl | Sigma | Cat#P9333; CAS: 7447-40-7 |
| Forskolin | Tocris | Cat#1099; CAS: 66575-29-9 |
| H-89 | Tocris | Cat#2910; CAS: 130964-39-5 |
| Glutamate | Tocris | Cat# 0218; CAS: 56-86-0 |
| Antarctic Phosphatase | NEB | Cat#M0289 |
| Deposited Data | ||
| Raw data | This paper | https://data.mendeley.com/datasets/769vrs9v2v/draft?a=7901a11c-4c68-4817-ad94-7b09aab4ab1a |
| Experimental Models: Organisms/Strains | ||
| Rat, Sprague-Dawley | Charles River | Cat# Crl:CD(SD); RRID: RGD_734476 |
| Mouse, C57BL/6J | The Jackson Laboratory | Cat#000664; RRID:IMSR_JAX:000664 |
| Mouse, Grin2afl/flGrin2bfl/fl | Mishina and Sakimura, 2007 | N/A |
| Recombinant DNA | ||
| pcDNA-GluN2B wild-type | Sanz-Clemente et al., 2010 | N/A |
| pcDNA-GluN2B-S1480E | Sanz-Clemente et al., 2010 | N/A |
| pFUGW-Cre:mCherry | Chen et al., 2012 | N/A |
| pCAGGS-GFP | Chen et al., 2012 | N/A |
| pCAGGS-GluN2B-S1480E-IRES-GFP | Chen et al., 2012 | N/A |
| pEGFP-CaMKII | Sanz-Clemente et al., 2013 | N/A |
| pEGFP-PP1α | This paper | N/A |
| pEGF-PP1γ1 | This paper | N/A |
| pLVX-eGFP | This paper | N/A |
| pLVX-PP1α-D95N | This paper | N/A |
| pLVX-PP1γ1-D95N | This paper | N/A |
| pLVX-I-2 | This paper | N/A |
| pLVX-I-2-T73A | This paper | N/A |
| Software and Algorithms | ||
| Prism 7 | Graphpad | RRID:SCR_002798 |
| FIJI | FIJI | RRID:SCR_002285 |
| NIS Elements | Nikon | RRID:SCR_014329 |
| Metamorph 6.0 | Universal Imaging Corp | RRID:SCR_002368 |
| R | R Project | RRID:SCR 001905 |
Highlights.
GluN2B PDZ-ligand phosphorylation maintains NMDARs at extrasynaptic sites
Extrasynaptic NMDARs form a stable protein complex containing PP1
Global NMDAR activation triggers PP1 activation to increase NMDAR synaptic content
A PP1 population not involved in LTD mediates GluN2B PDZligand dephosphorylation
ACKNOWLEDGMENTS
We thank Alec M. Chiu for assistance with statistical analysis and Luca Zangrandi for technical assistance. We also thank the Northwestern Center for Advanced Microscopy for their assistance in planning and analyzing imaging experiments. This research was supported by NIGMS (T32GM008061; A.M.C.), NIMH (K08MH100562; J.A.G.), and NIA (K99AG041225; A.S.-C.) and a NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (no. 24133; A.S.-C.). P.H. was supported by the J.W. Fulbright commission.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.030.
REFERENCES
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M, and Sakimura K (2009). NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J. Neurosci 29, 10869–10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiec WE, Guglietta R, Jami SA, Morishita W, Malenka RC, and O’Dell TJ (2014). Ionotropic NMDA receptor signaling is required for the induction of long-term depression in the mouse hippocampal CA1 region. J. Neurosci 34, 5285–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ 2nd, Brown AM, and Colbran RJ (2013). Differential association of postsynaptic signaling protein complexes in striatum and hippocampus. J. Neurochem 124, 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Gray JA, Sanz-Clemente A, Wei Z, Thomas EV, Nicoll RA, and Roche KW (2012). SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep. 2, 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, and Huganir RL (2004). Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J. Neurosci 24, 10248–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PT (2002). Protein phosphatase 1-targeted in many directions. J. Cell Sci 115, 241–256. [DOI] [PubMed] [Google Scholar]
- Diering GH, and Huganir RL (2018). The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore K, Stein IS, Brock JA, Castillo PE, Zito K, and Sjöström PJ (2017). Unconventional NMDA receptor signaling. J. Neurosci 37, 10800–10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis JP, Ladépêche L, Seth H, Bard L, Varela J, Mikasova L, Bouchet D, Rogemond V, Honnorat J, Hanse E, and Groc L (2014). Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO J. 33, 842–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, and Brautigan DL (2012). Endogenous inhibitor proteins that connect Ser/Thr kinases and phosphatases in cell signaling. IUBMB Life 64, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, and Nicoll RA (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, and Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, and Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci 5, 405–414. [DOI] [PubMed] [Google Scholar]
- Harney SC, Jane DE, and Anwyl R (2008). Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J. Neurosci 28, 11685–11694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickx A, Beullens M, Ceulemans H, Den Abt T, Van Eynde A, Nicolaescu E, Lesage B, and Bollen M (2009). Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol 16, 365–371. [DOI] [PubMed] [Google Scholar]
- Hou H, Sun L, Siddoway BA, Petralia RS, Yang H, Gu H, Nairn AC, and Xia H (2013). Synaptic NMDA receptor stimulation activates PP1 by inhibiting its phosphorylation by Cdk5. J. Cell Biol 203, 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XD, Huang Q, Yang X, and Xia H (2007). Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus. J. Neurosci 27, 4674–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, and Castillo PE (2012). Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr. Opin. Neurobiol 22, 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Puente N, Grandes P, and Castillo PE (2013). Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat. Neurosci 16, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. (1989). Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun 159, 871–877. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, and Roche KW (2004). Subunit-specific regulation of NMDA receptor endocytosis. J. Neurosci 24, 6383–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, and Sheng M (1998). Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J. Neurosci 18, 2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier MP, Sanz-Clemente A, and Roche KW (2015). Dynamic regulation of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J. Biol. Chem 290, 28596–28603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MA, Chen Y, Strack S, and Hell JW (2005). Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol. Sci 26, 645–653. [DOI] [PubMed] [Google Scholar]
- Mishina M, and Sakimura K (2007). Conditional gene targeting on the pure C57BL/6 genetic background. Neurosci. Res 58, 105–112. [DOI] [PubMed] [Google Scholar]
- Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, and Malenka RC (2001). Regulation of synaptic strength by protein phosphatase 1. Neuron 32, 1133–1148. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, and Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Papouin T, and Oliet SH (2014). Organization, control and function of extra-synaptic NMDA receptors. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, and Raymond LA (2014). Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Matta JA, Isaac JT, and Roche KW (2010). Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 67, 984–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA, and Roche KW (2013). Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 3, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, and Nicoll RA (2002). Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc. Natl. Acad. Sci 99, 13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway BA, Altimimi HF, Hou H, Petralia RS, Xu B, Stellwagen D, and Xia H (2013). An essential role for inhibitor-2 regulation of protein phosphatase-1 in synaptic scaling. J. Neurosci 33, 11206–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway B, Hou H, and Xia H (2014). Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology 78, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Kini S, Ebner FF, Wadzinski BE, and Colbran RJ (1999). Differential cellular and subcellular localization of protein phosphatase 1 isoforms in brain. J. Comp. Neurol 413, 373–384. [PubMed] [Google Scholar]
- Tovar KR, and Westbrook GL (2002). Mobile NMDA receptors at hippocampal synapses. Neuron 34, 255–264. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, and Dell’Acqua ML (2015). Coordination of protein phosphorylation and dephosphorylation in synaptic plasticity. J. Biol. Chem 290, 28604–28612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang Z, Brew K, and Lee EY (1996). Mutational analysis of the catalytic subunit of muscle protein phosphatase-1. Biochemistry 35, 6276–6282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The uncropped blots utilized for assembling the figures in this study can be found at: https://data.mendeley.com/datasets/769vrs9v2v/draft?a=7901a11c-4c68-4817-ad94-7b09aab4ab1a.