

SUMMARY
High-accuracy next-generation DNA sequencing promises a paradigm shift in early cancer detection by enabling the identification of mutant cancer molecules in minimally invasive body fluid samples. We demonstrate 80% sensitivity for ovarian cancer detection using ultra-accurate Duplex Sequencing to identify TP53 mutations in uterine lavage. However, in addition to tumor DNA, we also detect low-frequency TP53 mutations in nearly all lavages from women with and without cancer. These mutations increase with age and share the selection traits of clonal TP53 mutations commonly found in human tumors. We show that low-frequency TP53 mutations exist in multiple healthy tissues, from newborn to centenarian, and progressively increase in abundance and pathogenicity with older age across tissue types. Our results illustrate that subclonal cancer evolutionary processes are a ubiquitous part of normal human aging, and great care must be taken to distinguish tumor-derived from age-associated mutations in high-sensitivity clinical cancer diagnostics.
Graphical Abstract
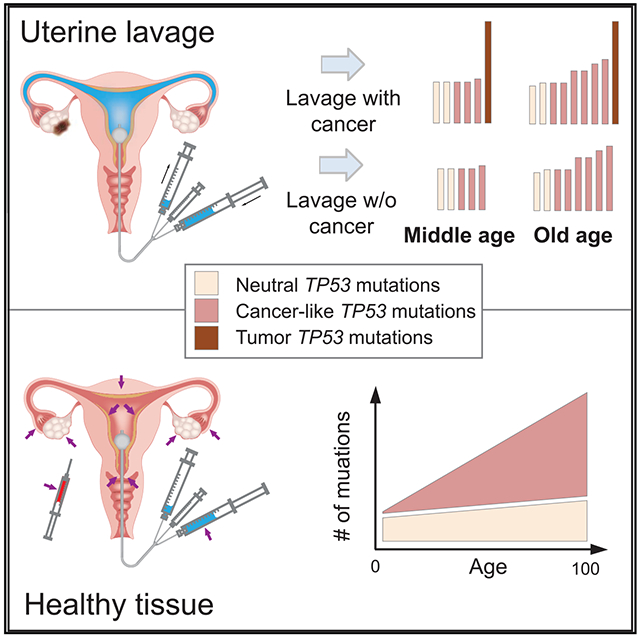
In Brief
Salk et al. demonstrate that ultra-sensitive DNA sequencing to identify TP53 mutations among cells shed into uterine fluid shows potential for minimally invasive ovarian cancer detection. Yet they also reveal ubiquitous age-related accumulations of cancer-like TP53 mutations in the normal tissues of healthy women. This highlights an important challenge of using tumor driver mutations for cancer screening.
INTRODUCTION
Worldwide, >250,000 new cases of ovarian cancer are diagnosed each year, and two-thirds of these women die from the disease (Bray et al., 2018). This high mortality is largely due to the high frequency of metastasis before diagnosis and a lack of effective screening and early detection methods. More than 60% of cases are diagnosed at an advanced stage, when the 5-year survival rate is only 29% (Siegel et al., 2017). In contrast, survival for women with localized disease is 92%, indicating that early ovarian cancer detection could vastly decrease mortality, yet diagnosis at this stage is rare. The most used approach for ovarian cancer screening involves a combination of serum protein CA-125 level and transvaginal ultrasound, but this has not demonstrated survival benefit, and it may result in harm due to false-positives, such as unnecessary surgeries in cancer-free women (Henderson et al., 2018). Thus, the US Preventive Services Task Force recommends against its use (Grossman et al., 2018). Better tools for early ovarian cancer detection remains an urgent and unmet clinical need (Drescher and Anderson, 2018).
In recent years, it has been demonstrated that cancers can be non-invasively detected in “liquid biopsies,” that is, blood or other body fluids in which cancers shed cells or DNA (Diaz and Bardelli, 2014). Proof-of-principle for this approach in ovarian cancer detection was initially accomplished via the identification of tumor-derived mutations in DNA extracted from routine Papanicolaou (Pap) tests (Kinde et al., 2013). Although the sensitivity for ovarian cancer detection was only 41%, these findings supported the exciting possibility that ovarian cancer could be detected by the genetic identification of cancer cells disseminated into the gynecological tract. A follow-up study recently reported that improved sensitivity, up to 63%, could be obtained by combining mutation detection in Pap tests and plasma. In addition, sampling with an intrauterine brush also improved sensitivity, probably due to increased tumor cell recovery by more proximal collection to the anatomical site of tumors (Wang et al., 2018).
An alternate means for tumor cell collection, developed by members of our team, consists of trans-cervical lavage of the uterine cavity (Figure 1A; Maritschnegg et al., 2015). This method improves the efficiency of collection by rinsing the uterus and fallopian tubes, the latter being the site of origin of most serous ovarian cancers (Labidi-Galy et al., 2017). This lavage technique demonstrated 80% sensitivity for ovarian cancer detection (Maritschnegg et al., 2015). The challenge, however, was that cancer-derived mutations, particularly those from early-stage tumors, were often present in a very small fraction of the total lavage DNA. To detect these mutations, digital droplet PCR (ddPCR) was required, which is a sensitive method but not practical for prospective screening because the tumor mutation needs to be known a priori.
Figure 1. Detection of Ovarian Cancer Using UL and DS.

(A) UL is carried out by passing a small catheter through the cervix, followed by concurrent flushing and aspiration with 10 mL of saline.
(B) After cell isolation by centrifugation, DNA is extracted, fragmented, and ligated with DS adapters, which include degenerate molecular tags (α and β). Following amplification, hybrid capture, and sequencing, reads sharing the same tags are grouped and mutations are scored only if present in both strands of each original DNA molecule.
(C and D) Each spot on the 2D surface represents one of the 1,179 coding positions in TP53. The y axis indicates mutant allele frequency (MAF). By standard NGS, all positions show false mutations (C). DS of the same sample (case 6 in E) eliminates errors and reveals only true mutations (D).
(E and F) TP53 mutations identified by DS in UL from women with ovarian cancer (E) and women who are cancer-free (controls; F). Fuchsia bars, matching tumor mutation; blue bars, biological background mutations. Mutations are sorted by ascending MAF within each patient and patients are sorted by age.
Next-generation DNA sequencing (NGS) is a widely used, variant-agnostic form of mutation detection, but it has a background error rate of up to ~1%, which precludes confident identification of lower-frequency mutations (Salk et al., 2018). Of the mutations that make up the 80% sensitivity achieved in our previous study, conventional NGS missed 25% (Maritschnegg et al., 2015). Currently, the most accurate NGS method is Duplex Sequencing (DS) (Salk et al., 2018), which uses double-stranded molecular barcodes for error correction and decreases the error rate of sequencing from 10−3 to <10−7 (Kennedy et al., 2014; Schmitt et al., 2012). We previously demonstrated that DS can detect ovarian cancer-derived mutations in DNA extracted from peritoneal fluid at frequencies as low as 1 tumor mutation per 25,000 normal genomes (Krimmel et al., 2016). This extreme sensitivity for mutation detection also led to the discovery of prevalent yet very-low-frequency (<0.01%) TP53 mutations in both the peritoneal fluid and peripheral blood from healthy women. These “biological background” mutations resembled TP53 mutations found in cancers, but appeared to result from the normal aging process. This observation was among the first of an emerging body of literature that has identified age-related, cancer-associated mutations within non-cancerous tissue (Risques and Kennedy, 2018).
In the present study, we combine the most sensitive reported sampling technique for ovarian cancer detection, uterine lavage (UL), with the highest-accuracy sequencing technology available, DS. High-grade serous ovarian carcinoma (HGSOC) is both the most common and the most deadly histological type, accounting for 70%–80% of ovarian cancer deaths (Bowtell et al., 2015). More than 98% of HGSOCs carry mutations in TP53, which makes this gene an ideal, cost-effective target for sequencing (Ahmed et al., 2010; The Cancer Genome Atlas Research Network, 2011; Vang et al., 2016). In addition, TP53 mutations are one of the earliest genetic events in HGSOC formation, as demonstrated by their presence in early serous epithelial proliferations found in the fallopian tubes (often termed p53 signatures), as well as serous tubal intraepithelial carcinomas (Chien et al., 2015; Kuhn et al., 2012; Kurman and Shih, 2010; Soong et al., 2019).
The primary goal of this study is to demonstrate the technical feasibility of using DS to deeply sequence TP53 from UL as a potential test for ovarian cancer detection. We capitalize on the accuracy of DS to identify true-positive cancer-derived mutations and to uniquely detect low-frequency, age-related mutations that may affect diagnostic performance. To better understand the extent and nature of these biological background mutations, we perform a detailed characterization of somatic TP53 mutations in multiple gynecologic tissues from women without ovarian cancer of ages spanning a century of human lifetime.
RESULTS
DS Detects Ovarian Cancer Mutations in ULs with High Sensitivity
We used DS to examine the coding region of TP53 in DNA extracted from the lavage cell pellet (Figures 1A and 1B) of 10 women with ovarian cancer and 11 controls under blinded conditions. Most of the lavages from women with ovarian cancer were included in the original study that reported 80% sensitivity for ovarian cancer detection with prior knowledge of tumor mutation (Maritschnegg et al., 2015; Table S1). We sought to determine whether similarly high sensitivity was possible without prior knowledge of the tumor mutation by using DS. DS uses special adapters with double-stranded molecular barcodes, which allow the identification of sequencing reads that are derived from both strands of each starting DNA molecule. Mutations are only scored if they are present in the majority of reads from both DNA strands, effectively eliminating sequencing and PCR artifacts (Figure 1B). The estimated error rate of DS is <1 in 10 million (Schmitt et al., 2012), which allows for extreme sensitivity and specificity of mutation detection when carrying out high-depth sequencing. To illustrate the superior accuracy of DS compared with standard NGS, an example of a UL sample (case 6) processed by both methods is shown in Figures 1C and 1D. Standard NGS entails alignment and variant calling from Illumina sequencing reads. Whereas every nucleotide position in the gene artifactually appears mutated in 0.1%–0.5% of molecules with standard sequencing, DS eliminates these tens of thousands of erroneous mutations to reveal the known tumor mutation at a mutant allele frequency (MAF) of 0.15%, a value that is very close to the frequency previously determined by ddPCR (0.12%; case 6 in Table S2; Maritschnegg et al., 2015).
Among the 10 lavages from women with ovarian cancer, we identified the expected tumor mutation (fuchsia bars, Figure 1E) in 8, matching the post hoc 80% sensitivity of the previous study. In the subset of these lavages that had been analyzed by conventional NGS and/or ddPCR, we confirmed tumor mutations at similar allele frequencies in most cases (Table S2). In addition to the tumor mutations, in nearly all of the lavages from women with and without ovarian cancer, we identified very-low-frequency TP53 mutations (blue bars, Figures 1E and 1F). To confirm that these mutations were not due to technical errors, two of the mutations identified in controls (lavages con2 and con7) were assessed by ddPCR (Table S2). This orthogonal assay demonstrated that these mutations, present at a comparable frequency of <0.1% by both assays, were authentic. As a further demonstration of the high sensitivity, accuracy, and precision of DS, we carried out a mixing experiment whereby cell lines bearing point mutations in TP53 and two other genes were spiked into a normal DNA sample at ratios from 1/100 to 1/100,000. Three replicates of this mixture were prepared and sequenced on different days at DS depths of up to 400,000x. All of the variants were identified in each replicate at expected frequencies (R2 of 0.95-0.98), demonstrating excellent reproducibility and accuracy (Figure S1).
Although TP53 background mutations were common, their MAF was always <1% (Figures 1E and 1F), which could be used as a threshold to optimally identify patients with ovarian cancers from patients who are cancer-free. In this, albeit small, pilot study, a 1% threshold yielded a sensitivity of 70% and a specificity of 100%. Furthermore, the maximum mutation frequency was significantly higher in cases than controls (p = 0.0005 for test of no difference in log maximum frequency between groups and no change after adjustment for age), and in the lavages in which the tumor mutation was identified, its frequency was at least 10-fold above the highest background mutation in that individual.
TP53 Mutations in UL Increase with Age
To better understand the basis of TP53 background mutations, we examined the association of their abundance with age. When patients were ordered by ascending age (Figures 1E and 1F), it appeared that older patients carried more mutations. However, the number of mutations found depends on the total number of nucleotides sequenced (Figure S2), which was variable across samples and tended to be higher in controls due to increased sequencing depths (Table S1). To compensate for this variation, for each sample we calculated the total TP53 mutation frequency by dividing the number of mutations identified in UL (including exons and flanking intronic sequences) by the total number of DS bases sequenced. For patients with cancer, we excluded the tumor mutation from this calculation to fairly reflect only TP53 background mutations. For patients with ovarian cancer, as well as cancer-free control patients, the TP53 mutation frequency significantly increased with age (Figure 2; p = 0.0006 for ovarian cancer, p = 0.001 for controls, Spearman’s correlation test). This trend was identical to prior observations of TP53 background mutation frequency in peritoneal fluid and peripheral blood (Krimmel et al., 2016).
Figure 2. The Frequency of TP53 Mutations in UL Increases with Age.

Frequency is calculated as the total number of unique TP53 mutations identified in each sample (including exons and flanking intronic regions) divided by the total number of DS nucleotides sequenced.
(A) UL samples from patients with ovarian cancer; n = 10, r = 0.89, p = 0.0006 by Spearman’s correlation test.
(B) UL samples from control patients without cancer; n = 11, r = 0.83, p = 0.001 by Spearman’s correlation test.
TP53 Mutations in UL Are Not Random, but Rather Are Positively Selected
The TP53 gene is a tumor suppressor, the genetic disruption of which facilitates cell proliferation in tumors, even when only one allele is mutated (Leroy et al., 2014). An age-associated increase in ultra-low-frequency TP53 background mutations could result from random, age-related mutagenic processes or, alternatively, from mutagenesis coupled with clonal selection of pathogenic variants. To distinguish between these possibilities, we performed a detailed analysis of traits of selection among the 112 age-associated TP53 background mutations collectively identified among all 21 patients (tumor mutations and intronic and UTR mutations excluded) (Figure 3; Table S3).
Figure 3. Evidence of Positive Selection in TP53 Background Mutations from ULs.

(A–D) Percentage of non-synonymous TP53 mutations (A), and percentage of TP53 mutations localized in CpG dinucleotides (B), in exons 5–8 (C), and in hotspot codons (D). For (A–D), TP53 mutations identified in UL from controls and cancer significantly exceed expected values without selection. *p < 0.05, **p < 0.0001 by binomial exact test, n = 79 for UL controls, and n = 33 for UL cancer. (E and F) Protein activity (E) and predicted pathogenicity (F) color-coded as five groups from Seshat data. Patients are sorted by ascending age. For each patient, TP53 mutation frequency is calculated as the number of mutations in the coding region divided by the total number of DS nucleotides sequenced in that region. Two cancer patients with unusually low sequencing depth and no identified TP53 mutations are not shown. Nearly all cases and controls carry mutations that have an impact on protein activity and predicted pathogenicity.
First, we calculated the proportion of non-synonymous TP53 mutations in ULs from cases and controls and compared it with the expected proportion when considering all of the possible mutations in the TP53 gene (n = 3,546). The percentage of non-synonymous mutations in both UL controls (90.5%) and cases (93.5%) was significantly higher than expected under no selection (76.6%) with p = 0.0035 and p = 0.031, respectively (Figure 3A, exact binomial test). The excess of non-synonymous mutations was not driven by a subset of outlier samples, but rather was uniformly observed across nearly all of the lavage samples (Figure S3A).
Second, we examined the metrics of selection related to the genic location of mutations. Background TP53 mutations were not randomly distributed along the gene but clustered in certain regions of biological significance. Nearly 25% of TP53 lavage background mutations occurred in the context of CpG dinucleotides, which is remarkable given the fact that these dinucleotides make up <5% of the coding region of TP53 (Figure 3B, p = 2.2 × 10−10 for controls and p = 1.4 × 10−5 for ovarian cancer mutations, by the exact binomial test). Mutations were also enriched in exons 5–8, which encode the DNA-binding domain of the protein (Figure 3C, p = 2.4 × 10−6 for controls and p = 4.5 × 10−4 for ovarian cancer mutations, by exact binomial test). The most significant enrichment, however, was observed in TP53 cancer-associated hotspot codons, which are the codons most recurrently mutated in cancer. We considered the nine most abundantly mutated codons in the Universal Mutation Database (UMD; April 2017 version) (Leroy et al., 2014). These codons encompass only 2.3% of the coding region of TP53, yet >25% of lavage background mutations clustered within these 27 bp (Figure 3D, p = 5.1 × 10−17 for controls and p = 2.4 × 10−9 for ovarian cancer mutations, by exact binomial test), and among these, all were non-synonymous. The biases for each characteristic were not driven by outliers but were distributed evenly across samples in both groups (Figure S3B-S3D). Cases and controls were not significantly different for any of these traits (using Fisher’s exact test or using generalized estimating equations [GEEs]) to account for the correlation between patients with and without adjustment for sequencing depth).
To assess the impact of these mutations on TP53 protein function, we took advantage of Seshat, a recently developed online tool for TP53 analysis that provides comprehensive mutational information, including prediction of impact on protein activity and pathogenicity (Tikkanen et al., 2018). We queried all background TP53 mutations identified in the 21 lavages and color-coded them according to 5 binned categories of protein activity and predicted pathogenicity. Nearly all of the samples carried at least one TP53 mutation that inactivated the protein totally or partially (Figure 3E) and/or was predicted to be pathogenic (Figure 3F). Cases and controls were not significantly different when comparing the proportion of mutations that inactivated protein activity (categories of “inactive,” “splice/truncated,” and “partially inactive” in Figure 3E) or the proportion of mutations with predicted pathogenicity (categories of “pathogenic,” “likely pathogenic,” and “possibly pathogenic” in Figure 3F) (Fisher’s exact test p values are 0.49 and 0.99, respectively). The unambiguous signature across six distinct metrics of positive selection (Figures 3A-3F) within the ultra-low-frequency TP53 mutations observed in all lavages, regardless of cancer status, indicate that these mutations expanded under strong selective pressure and are not the result of technical errors.
TP53 Mutations in UL Resemble Mutations in Cancer
We next compared the features of the selection of TP53 mutations identified in lavages to TP53 mutations from cancers. For this analysis, we used all of the cancer mutations present in the UMD cancer database (April 2017, n = 71,051). We deter-mined the percentage of these mutations that reside at CpG sites, cancer hotspots, and exons 5–8, as well as the percentage of mutations that affect protein activity (first 3 categories in Figure 3E) or are predicted to be pathogenic (first 3 categories in Figure 3F). For each trait, we compared the distribution of mutations in the theoretical absence of selection, in the 21 ULs, and in the cancer database (Figure 4A). Remarkably, for all of the traits, TP53 background mutations from ULs far more strongly resembled TP53 mutations in the cancer database than the pattern expected by random chance. This was true for TP53 mutations found in lavages from cases and controls (Figure S4), as expected due to the fact that mutations in both groups were not statistically different for these traits (Figure 3). We also used a feature of Seshat that categorizes TP53 mutations according to their frequency in the UMD database. Nearly all of the UL samples harbored TP53 mutations listed as “frequent” or “very frequent” in the database (Figure 4B). Again, cases and controls did not significantly differ in the proportion of mutations that were common in the cancer database (neither when comparing “very frequent” and “frequent” versus the rest nor when comparing all groups separately; Fisher’s exact test, p = 0.84 and p = 0.27, respectively).
Figure 4. TP53 Mutations in UL Are Very Similar to TP53 Mutations Found in Human Cancers.

(A) Traits of positive selection are compared between all of the possible mutations in the TP53 coding region (no selection; n = 3,546), TP53 background mutations found in UL (n = 112), and TP53 mutations reported in the UMD cancer database (n = 71,051).
(B) UL mutations in cases and controls color-coded by their abundance in the UMD TP53 database. For each sample, TP53 mutation frequency is calculated as the number of mutations in the coding region divided by the total number of DS nucleotides sequenced in that region. Most samples harbor TP53 mutations that are common in the database.
(C and D) TP53 mutation type (C) and TP53 mutation spectrum (D) are compared between mutations identified in UL from controls (n = 79), UL from cases (n = 33), and the UMD database (n = 71,051).
(E) TP53 mutation distribution map for UL (top) and the UMD cancer database (bottom). Bars quantify the frequency of mutations at each codon. Colored background reflects a 20-bp sliding window average of mutation density around each position. Gene exons and protein domains are indicated in the center section.
To further characterize background TP53 mutations in UL in comparison to those in cancers, we compared mutation type, spectrum, and gene location. Non-cancer-derived mutations in ULs from women with and without cancer were predominantly missense, similar to mutations in the database (Figure 4C), and displayed a mutational spectrum enriched in G > A and C > T transitions, comparable to cancer mutations (Figure 4D). The distribution of low-frequency TP53 background mutations from only 21 women along the length of the gene is a mirror image of the distribution of TP53 mutations from >71,000 different tumors included in the database (Figure 4E). Thus, the somatic TP53 mutations recovered from cells sloughed into the uterine cavity from normal healthy women are not random, but appear to emerge from an evolutionary process of mutation, selection, and clonal expansion akin to what takes place in tumors, but within normal tissue.
TP53 Mutations Are Common in Healthy Tissues from Middle-Aged Women
These striking results prompted us to consider what the tissue origin of the mutation-bearing cells in the ULs may be. To address this question, we sequenced TP53 from DNA obtained from pre-operative UL and peripheral blood, as well as multiple gynecological tissues collected postoperatively following total hysterectomy and bilateral salpingo-oophorectomy for symptomatic fibroids (benign leiomyomas) from two middle-aged women (Figure 5A; Table S4). DNA was extracted and processed for DS as before, except that samples were sequenced to a higher average depth (Table S4). We identified TP53 mutations in all of the samples from a 56-year-old woman and in all but 2 samples from a 46-year-old woman (Figure 5B). When we compared the mutation frequency across all of the samples, several interesting observations emerged. First, the UL from the 56-year-old had a mutation frequency that appeared disproportionally high, both when compared to that of most other tissues and when compared with the lavage of the 46-year-old. However, when compared to the mean values of ULs from control women of similar ages (50- to 56- and 40- to 46-year-olds) from the first part of the study, the frequencies by age were quite similar (Figure 5B).
Figure 5. TP53 Mutations in Normal Tissues and UL from Two Middle-Aged Women.

(A) Normal tissues collected included leukocytes, peritoneum, cervix, endometrium, myometrium, fallopian tube, ovary, and UL.
(B) TP53 mutation frequency for each sample calculated as the number of TP53 mutations in the coding region divided by the total number of DS nucleotides sequenced. (a) and (b) indicate two spatially separated samples from the same tissue. The blue bars correspond to the samples in this study; the orange bars correspond to the mean values for ULs from control women in the first study.
Moreover, the distribution of mutations according to each trait of positive selection (type, frequency in the cancer database, predicted activity and pathogenicity, exon clustering, CpG clustering, and enrichment for cancer hotspots) was comparable to the lavages previously analyzed (Figure S5). Both lines of reasoning support the conclusion that the elevated frequency of mutations in the UL of the 56-year-old woman is not artifactual and confirm the previously observed age effect.
For other tissues, however, we did not observe an obvious increase in TP53 mutation frequency between 46 and 56 years of age. There was substantial variability in the mutation content of different tissues and of different biopsies of the same tissue, which reflects either a stochastic effect or the imprecision of macrodissection for obtaining exactly comparable tissue samples (e.g., the depth of endometrium harvested, how distal the tubal fimbriae were cut). No single tissue stood out as obviously more mutation prone than another, nor could any tissue be identified as a dominant source of the mutations found in lavages. When we checked for mutations shared between tissues, we did not find any common mutation between UL and any of the rest of the tissues analyzed from the 49-year-old woman and only 1 mutation in the 56-year-old woman that was shared between UL and leukocytes (Figure S6).
TP53 Mutations Increase in Number and Cancer-like Features during Normal Human Aging
With the hope of observing a stronger aging mutational signal, we looked to tissue samples from greater extremes of age. While the procurement of such material was challenging, we obtained several gynecologic tissues at autopsy from a neonate who died from a congenital vascular malformation and from a 101-year-old female who died of natural causes (Table S4). Together with the middle-aged samples, this unique specimen collection represents the full breadth of a century of human lifespan.
Although the tissue types available were not fully identical across all four subjects, the pattern of TP53 mutations, nevertheless, yielded unique insights. To help more intuitively visualize this multiparametric data, in Figure 6 we annotated all of the mutations found among the different tissues of the four subjects as color-coded boxes for each feature of selection: red for “cancer-like,” blue for non-cancer-like. The number of columns of colored boxes per sample reflects the total number of mutations identified. When viewed in this format, it is apparent that TP53 mutations are not only more abundant with age but are also more “cancer-like.” Mutations found in older tissues are disproportionately observed in cancers and are predicted to inactivate the protein or be otherwise pathogenic. In contrast, mutations found in the newborn are rarely found in cancer, tend to preserve the protein activity, and are not predicted to be pathogenic.
Figure 6. Characterization of TP53 Mutations in Normal Tissues over a Century of Human Lifespan.

TP53 mutations identified by DS in leukocytes and gynecological tissues are indicated as columns within each tissue. Each mutation is characterized by four parameters: type (synonymous, non-synonymous, and hotspots), frequency in cancer, protein activity, and predicted pathogenicity. The last three parameters are color-coded, with red indicating “cancer-like” mutations and blue indicating benign mutations. Vertical black lines separate different biopsies from the same tissue. Within each biopsy, mutations are ordered left to right by decreasing cancer frequency. The biopsies that were sequenced but no mutations were identified are shown in gray. Note that the female newborn in the study had no blood collected. The leukocytes included here correspond to a similarly aged newborn male.
Different tissues and different biopsies within the same tissue showed substantial variability in both the number of mutations and their cancer-like features. In aggregate, fallopian tube epithelium appeared to be a “hot” tissue, with a high number of mutations and a high percentage of cancer-like mutations. However, in the 56-year-old, one fallopian tube sample harbored only a single synonymous mutation, which is consistent with the notion of “hot” and “cold” zones within a tissue. This was similarly seen in the two distinct endometrial biopsies of the centenarian.
In addition to a larger number of clones, with aging we would predict an increase in the size of clones, as would be reflected by a higher MAF of each variant found. However, in this study, some samples were sequenced at a lower depth, which may lead to outlier (low event count) biases in the calculation of MAFs, thus precluding a fair comparison between samples (Figure S7). Despite this caveat, 2 large clones were clearly seen within the peripheral blood leukocytes of the 101-year-old (Figure S7; Table S5; c.659A > G MAF: 1.2 × 10−2, and c.455C > T MAF: 4.5 × 10−3). The exact TP53 mutation that defined each of these clones was also detected at lower frequencies in peritoneal and endometrial samples from the same subject, revealing an apparent contribution of leukocyte DNAto those tissue samples (Figure S6).
In fact, this cross-tissue mutation sharing was common in the 101-year-old woman, suggesting that aged leukocytes may indeed harbor relatively large clones that recurrently contribute to the mutations found when sequencing other biopsies. Mutation sharing was less prevalent in middle-aged women. While very-low-frequency mutations are often hard to replicate due to the low precision of the measurement resulting purely from sampling statistics (not technical accuracy), it is important to keep in mind that certain mutations may also be recurrently identified simply because they are hotspots, and thus a common origin cannot automatically be assumed. For example, the hotspot mutation c.659A > G was identified in the large blood clone of the 101-year-old woman and in a myometrium sample and a fallopian tube biopsy of the 46-year-old woman (Figure S6). The processing of these particular samples was done on different days, making a cross-contamination explanation improbable.
As already considered, an important limitation of this study was the different depth of sequencing achieved across samples, due to the inherent variability in library preparations and differences in DNA availability. Because numerically more mutations will be identified in samples with more sequencing (Figure S8A), it is essential to compare samples based on their mutation frequency, which is a sequencing-normalized value calculated as the number of mutations divided by the number of total DS error-corrected nucleotides sequenced (Figure S8B). TP53 mutation frequency tended to be higher at older ages in the three tissue types shared by the neonate and the centenarian (leukocytes, peritoneum, and endometrium), although there was substantial variability across samples.
TP53 Mutations in Newborn Tissue Are Random, yet Become Positively Selected over a Lifetime
As further illustration of the increase of cancer-like mutations with aging, we divided all TP53 mutations into two binary categories: “common in cancer” and “not common in cancer,” with the former being defined by those classified as “frequent” or “very frequent” in the UMD cancer database. When plotted by age, the progressive enrichment for cancer-like mutations was easily apparent, especially in certain tissues such as fallopian tubes and leukocytes (Figure 7A).
Figure 7. Cancer-Associated TP53 Mutations Are Positively Selected during Normal Aging.

(A) Across a variety of human tissues, TP53 mutations accumulate with age and are progressively enriched for mutations commonly found in cancers. Tissues are color-coded. (a) and (b) indicate two biopsies from the same tissue.
(B) Traits of positive selection are compared between all of the possible mutations in the TP53 coding region (n = 3,546); TP53 mutations found in newborns (n = 19), middle-aged individuals (n = 85), and centenarian individuals (n = 38); and TP53 mutations reported in the UMD cancer database (n = 71,051).
(C and D) Distribution of TP53 mutation type (C) and mutation spectrum (D) for newborn, middle-aged, and centenarian mutations compared to the UMD cancer database.
We then examined the five traits of selection previously calculated for the UL study, but for TP53 mutations found in the newborn, middle-aged, and centenarian tissues (Figures 7B and S9). For mutations found in newborn tissue, all five traits yielded values consistent with random processes (e.g., absence of selection), yet in middle-aged and even more so in centenarian tissue, values reflected selection to an extent that neared that seen in mutations from tumors in the UMD database. Analysis of mutation type (Figure 7C) revealed a decrease in synonymous mutations with age (in fact, no synonymous mutations were identified in centenarian tissue; Table S5).
Lastly, regarding the mutation spectrum, we noted an interesting preponderance of C and G mutations in newborn tissue, which progressively shifted toward an increased representation of A and T mutations in centenarian tissue, which is more similar to the pattern in cancers. The significance of this shift is unknown as it could represent both biases in the nucleotide composition of the gene at selectable hotspots and differential age-associated mutagenic processes (Alexandrov et al., 2015), which disproportionately contribute to the clonal mutation burden of tumors because tumors mostly arise in the elderly.
TP53 Mutations in cfDNA and Peritoneal Fluid Follow the Same Patterns as Solid Tissue
To explore the abundance of TP53 mutations in liquid biopsies of clinical interest, we analyzed plasma-derived cell-free DNA (cfDNA) and peritoneal fluid from the 46-year-old woman. TP53 mutations were identified in both, with cancer-like features similar to what was observed for solid tissue biopsies, UL, and leukocytes (Figure S10). None of the mutations identified in cfDNA or peritoneal fluid overlapped with mutations detected in other samples from the same woman (Figure S6). Peritoneal fluid is routinely collected for disease staging during gynecologic surgery, and we previously demonstrated that it carries TP53 background mutations with cancer-like features (Krimmel et al., 2016), in agreement with our findings here. cfDNA had not been analyzed previously by DS. The fact that we identified one pathogenic mutation commonly found in cancers within cfDNA from a healthy woman (Figure S10; Table S5) raises important concerns over specificity in cancer-screening studies based on mutation detection in plasma.
DISCUSSION
We have demonstrated that DS can detect TP53 ovarian cancer mutations in UL, providing proof-of-principle for an innovative approach with the potential for ovarian cancer detection. Our combined approach of UL plus DS improves upon past mutation-based screening efforts through the use of a collection method that recovers cancer cells very close to the anatomical site of the tumor and an ultra-accurate DNA sequencing technology that can resolve exceptionally low-frequency mutations. We were able to achieve remarkable sensitivity and specificity without prior knowledge of the tumor mutation and using a MAF threshold for differentiating cancers from controls. The study, however, was small and patients had advanced ovarian cancer, important limitations that will need to be addressed with much larger prospective trials. These trials will be facilitated by the fact that UL is minimally invasive and can be practically integrated into routine gynecologic primary care (Maritschnegg et al., 2018).
However, the most profound finding of this work is not the biomarker performance itself, but the incidentally found mutational patterns that reflect a somatic evolutionary process that appears operative throughout much of human life in normal tissues. Specifically, we identified widespread low-frequency TP53 mutations that were heavily enriched for pathogenic variants. This enrichment reflects a process of natural selection that favors the survival and proliferation of cells with mutations that are identical to those observed in cancer, but as part of routine aging. The unambiguous selection signature is supported by multiple orthogonal metrics and cannot be explained by technical errors; both the biological and diagnostic implications are substantial.
One of the main reasons that cancer biomarkers fail to reach the clinic is their inability to achieve the extremely high specificity that is required for screening (Diamandis, 2012). This is critical for cancers with low incidence and that require an invasive procedure to follow up positive screening tests, such as the case with ovarian cancer (Drescher and Anderson, 2018). Harms due to false-positives and a lack of proven reduction in mortality are the reasons for the recent recommendation against the use of CA-125 and transvaginal sonography for screening asymptomatic women (Grossman et al., 2018). In recent years, mutation-based cancer screening from plasma or other body fluids has emerged as a promising method to detect cancer based on the supposition that cancer-associated mutations found in liquid biopsies are a specific indication of cancer somewhere in the body (Aravanis et al., 2017). Here, we demonstrate that cancer-associated mutations can be found in most normal tissues and, therefore, they are not cancer specific.
The detection of cancer-associated mutations in normal tissues is not entirely new (Risques and Kennedy, 2018). In 2014, 3 groups reported acute myeloid leukemia mutations found as minority subclones in the blood of ~10% of healthy elderly individuals—a phenomena dubbed clonal hematopoiesis of indeterminate potential (CHIP) (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). One year later, Martincorena et al. (2015) observed hundreds of tiny clones carrying cancer-associated driver mutations on sun-exposed eyelids, a finding recently replicated in normal aged esophagus (Martincorena et al., 2018). Cancer-associated mutations have been similarly reported in abnormal but non-cancerous tissues, including endometriosis (Anglesio et al., 2017; Suda et al., 2018) and benign dermal nevi (Shain et al., 2015). The use of laser capture microdissection in recent studies has revealed that as many as 1% of normal colorectal crypts of middle-aged individuals (Lee-Six et al., 2018) and >50% of normal endometrial glands of middle-aged women (Suda et al., 2018) carry mutations in cancer driver genes. A broader body of work related to quantifying the accumulation of neutral mutations in aging normal tissues (Abyzov et al., 2017; Welch et al., 2012; Yadav et al., 2016) has afforded important insights into the mechanisms of age-associated mutagenic processes.
The relation between mutations and cancer has been known for decades, yet the delay in appreciating their presence outside cancer can be largely attributed to technical limitations. The advent of NGS enabled surveying wide swaths of the genome and detection of mutations present clonally or as modest-size subclones. In the above studies, standard whole exome or multi-gene NGS was able to identify driver mutations because of unique scenarios in which clones were either relatively large (CHIP in a subset of very elderly individuals) (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014) or spatially coherent and comprising a sizeable percentage of cells when very small biopsies were taken (Lee-Six et al., 2018; Martincorena et al., 2015, 2018; Suda et al., 2018). With higher-accuracy NGS techniques able to resolve lower-frequency subclones, later studies have found that CHIP mutations are abundant in middle-aged adults (Acuna-Hidalgo et al., 2017; Young et al., 2016). Using ultra-accurate DS, we found extremely low-frequency cancer-associated TP53 mutations in both the blood and peritoneal fluid of women without cancer, and, in both sample types, the abundance of mutations increased with age (Krimmel et al., 2016). A subsequent study that used UL for endometrial cancer detection found pathogenic mutations in cancer driver genes in lavages of cases as well as controls (Nair et al., 2016), in agreement with the data reported here.
In addition to UL, we examined 10 different tissue or sample types from a unique cohort of individuals spanning more than a century of human lifespan and assessed the pattern of mutations found using multiple different metrics of selection. A significant finding was that not only do TP53 mutations increase in abundance with age but also the relative representation of random mutations versus cancer-associated mutations transitioned from almost entirely the former to almost entirely the latter from birth to the end of life. Moreover, the extent of mutation frequency varied considerably by sample type and tissue. Our research complements recent efforts to characterize the accumulation of random, unselected somatic mutations with aging (Blokzijl et al., 2016; Hoang et al., 2016) and suggests that these findings are likely only the tip of the iceberg. Further work is important to expand beyond the main limitations of this study, which include the analysis of mostly gynecological tissues from only four individuals at a coarse spacing along the aging continuum; imperfect matching of sample types for each subject due to the inherent challenges of tissue acquisition at the extremes of age; and focus on a single gene, albeit the one most commonly mutated in cancer.
The implications of our findings are important as a cautionary message for mutation-based cancer biomarkers. At the same time as we have shown that highly sensitive NGS methods are essential for maximal mutation detection, we have also illustrated a substantial specificity challenge related to biology, not technology, the extent of which has been under-appreciated. This is not limited to one or a few tissues; rather, it seems to be ubiquitous among the epithelial, mesenchymal, and hematopoietic cell lineages that we investigated. Moreover, cancer-associated mutations were found in liquid biopsies, including UL and cfDNA. This suggests that ongoing large-scale efforts to develop universal liquid biopsy cancer screening tests via deep sequencing of cfDNA need to be approached with great caution (Alix-Panabières and Pantel, 2016; Aravanis et al., 2017).
Despite biological background mutations and with the caveat of this being a quite small study from a biomarker perspective, our approach worked remarkably well as a minimally invasive cancer test. We identified 80% of tumor mutations, and 70% of those were above the 1% MAF threshold we used to distinguish cases from controls. The only tumor mutation missed below this threshold corresponded to a 42-year-old woman, one of the youngest in the study. Younger women tended to have fewer background mutations and mutations with lower MAFs, which suggest that, moving forward, sensitivity could probably be increased by using age-adjusted thresholds. In addition, specificity could likely be improved by uniform lavage collection at the luteal phase in premenopausal women, sequencing of peripheral blood to identify and exclude CHIP clones that may be present in lavage, and longitudinal assessment of mutations to identify MAF increases over time. Of note, we have demonstrated detection of intermediate- and late-stage cancers, but the most critical targets for screening are early-stage cancers because they are the most curable. In that regard, monitoring of high-risk populations, such as BRCA1 and BRCA2 carriers, may be the highest-impact near-term clinical implementation.
The sensitivity improvements lent by new sequencing technologies are forcing a far more nuanced genetic definition of what distinguishes a cancer cell from simply an old cell. Our results show that CHIP clones are merely one relatively easy to detect manifestation of a far broader phenomena that appears to extend to most, if not all, tissues in the body. From a biomarker perspective, the fact that those who are at greatest risk of cancer and for whom cancer screening holds the most benefit (older adults) are also the population with the most cancer-like age-associated background mutations is particularly inconvenient. Ongoing improvements will be needed to find ways to maximize specificity through careful MAF threshold calibration and combination with orthogonal biomarkers. Further investigation into the significance of biological background mutations from the perspective of human aging and biology is similarly warranted. While the notion that our somatic genomes are steadily evolving toward neoplasia with each passing decade may viewed as disheartening, an alternative perspective is that despite this, most people do not develop overt cancer in their lifetime. This serves as a reminder of just how much remains unknown about the body’s many complex mechanisms of tumor suppression, a toolkit that we can perhaps augment with future technologies for cancer prevention.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rosana Risques (rrisques@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAIL
We performed two complementary studies. The objective of the uterine lavage study was to determine the ability of DS to detect ovarian cancer through deep sequencing of TP53 mutations in uterine lavage. This study included 10 patients with high grade serous ovarian cancer (cases) and 11 with benign gynecological masses (controls). The objective of the normal tissue study was to characterize somatic TP53 mutations that accumulate during aging. This study included tissue from two newborn subjects (one newborn male only provided blood), two middle age women (ages 46 and 56 year-old) and one centenarian woman (101 year-old). Clinico-pathological information for all subjects is listed in Table S1.
Uterine Lavage
In the first study, we analyzed uterine lavages collected by a trans-cervical catheter from women undergoing procedures for suspected gynecological malignancies (Table S1). None of the women had endometriosis. Lavages were collected immediately pre-operatively as previously described (Maritschnegg et al., 2015). Lavage samples were centrifuged at 300x g for 10 minutes at room temperature and DNA was isolated from the cell pellet (QIAamp MinElute Kit, QIAGEN, Hilden, Germany). Patients were recruited in three institutions: Medical University of Vienna (Austria), Charles University Pilsen (Czech Republic) and University Hospitals Leuven (Belgium). Sample procurement was performed in accordance with the institutional review boards of the Medical University of Vienna (EK#1148/2011 and EK#1766/2013), the Catholic University Leuven (B322201214864/S54406) and the Medical Faculty Hospital Pilsen (No 502/2013).
Normal Tissue
In the second study, multiple gynecological tissues were collected per Table S4. Not all sample types were available for all subjects. Newborn and centenarian tissue was collected at autopsy, while tissue from middle age women was collected following hysterectomy. Peripheral blood was unavailable from the female newborn, so we sequenced peripheral blood from a similar aged neonatal male. No other tissue was collected from the newborn male other than blood. The two newborn autopsies were performed at Seattle Childrens’ Hospital and tissues were collected under research IRB #52304. The diagnoses were vein of Galen malformation (newborn female) and bronchopulmonary dysplasia (newborn male). The two middle age women had uterine leiomyoma and were operated on at the Medical University of Vienna. Samples were collected with informed consent and according to approved IRB EK# 1152/2014. Uterine lavage was collected with the same procedure as in the first study. For the 46 year-old woman, cfDNA was collected preoperatively and peritoneal lavage collected intraoperatively. For the centenarian woman, tissue was obtained via rapid autopsy from Tissue for Research Inc. and processed at the University of Washington under IRB waiver 2016-52304. All samples were collected using sterile new instruments between biopsies and frozen over liquid nitrogen immediately after collection and stored at −80°C until DNA extraction. To confirm normal histology, tissue biopsies immediately adjacent to the biopsy used for DNA extraction were embedded in OCT, sectioned, stained with H&E and reviewed by a pathologist (M.T.). In all samples, morphologic examination revealed only normal tissue, without inflammation, necrosis, hyperplasia or neoplasia.
METHOD DETAILS
Digital Droplet Polymerase Chain Reaction
Lavage DNA from 5 ovarian cancer cases and 2 controls was analyzed by ddPCR (Table S2). In ovarian cancer lavage, ddPCR amplified the tumor mutation whereas in benign lavage, the assay targeted two mutations previously identified by DS at frequencies below 0.1%. ddPCR was performed with the QX100 Droplet Digital PCR system (Bio-Rad Laboratories, Hercules, CA) using custom TaqMan SNP Genotyping Assays (Life Technologies, Carlsbard, CA) designed using Primer Express 3.0 software (ThermoFisher). 10-20ng of DNA were used in each reaction and samples were analyzed at least in duplicates. A positive control and a wild-type control were included in every run.
Duplex Sequencing
Duplex Sequencing was performed as previously described with minor modifications (Kennedy et al., 2014). Briefly for most samples, DNA was sonicated, end-repaired, A-tailed, and ligated with DS adapters using the KAPA HyperPrep library kit (Roche Sequencing, Pleasanton, CA). DNA from the two sets of normal tissues obtained from the middle-aged hysterectomy specimens were prepared with a prototype Duplex Sequencing kit (TwinStrand Biosciences, Seattle, WA). After initial amplification, 120 bp biotinylated oligonucleotide probes (Integrated DNA Technologies, Coralville, Iowa) were used to capture the coding region of TP53. Two successive rounds of captures were performed to ensure sufficient target enrichment, as previously described (Schmitt et al., 2015). Indexed libraries were pooled and sequenced on an Illumina HiSeq2500 or NextSeq500. Sequencing reads were aligned to hg19 then reads sharing a common molecular tag in both distinct strand orientations were grouped and assembled into an error-corrected Duplex Consensus Sequence as previously described (Kennedy et al., 2014).
The total number of Duplex nucleotides sequenced for each uterine lavage and tissue sample is listed in Tables S1 and S4. In aggregate, we sequenced 587,169,708 unique nucleotides, 319,576,913 of which corresponded to coding nucleotides. We targeted a median DS depth of ~1000x. Three tissue biopsies were excluded because of insufficient depth. Because Duplex reads correspond to original DNA molecules, DS depth reflects the total number of haploid genomes sequenced. For each sample, TP53 mutation frequency was calculated as the number of identified mutations divided by the total number of Duplex nucleotides sequenced. For each individual mutation, mutant allele frequency (MAF) was calculated as number of mutated Duplex bases divided by the total DS depth at a given nucleotide position. Mutations identified as SNPs in the 1000 genome database were excluded from mutation analysis. All mutations were manually reviewed with the Integrative Genome Viewer (IGV).
Characterization of TP53 mutations using Seshat and the UMD TP53 database
The final list of mutations from all samples in the study (uterine lavage study n = 166, normal tissue study n = 264) was converted into a Variant Call Format (VCF) file and submitted to Seshat (https://p53.fr/TP53-database/seshat), a web service that performs TP53 mutation annotation using data derived from the UMD TP53 database (Tikkanen et al., 2018). This database is the most updated and comprehensive repository of TP53 variants. From the Seshat output, the following variables were extracted: cDNA variant, HG19 Variant, Variant Classification, Frequency, Activity, Pathogenicity, Exon, Codon, CpG, Mutational Event and Variant Comment. These variables were used to annotate the DS pipeline-generated mutational calls in Table S3 (uterine lavage) and Table S5 (normal tissue). The human genomic reference hg19 (GRCH37) was used for data reporting. Mutations occurring in the coding region and adjacent splice sites were selected for mutational analysis (uterine lavage n = 112, normal tissue n = 180).
Mutations were annotated based on type (missense, nonsense, splice, indel, synonymous), mutation spectrum (each of the 12 possible nucleotide substitutions), localization to CpG dinucleotides, localization in exons 5 to 8 (encoding the protein’s DNA binding domain), localization to mutational hotspot (9 most common mutated codons in the UMD TP53 database: 175, 179, 213, 220, 245, 248, 249, 273, 282), frequency of the mutation in the cancer database, functional activity, and predicted pathogenicity. Functional activity was assessed by a transcriptional activity chart assay for 3,000 variants performed by Kato et al. (2003). Pathogenicity was based on multiple predictive algorithms included from dbNSFP (Liu et al., 2016) as well as functional activity (Tikkanen et al., 2018). For the last 3 variables (frequency in cancer database, activity, and pathogenicity), mutations were aggregated into 5 categories and 2 categories. Categories of frequency in cancer database included very frequent, frequent, not frequent, rare/unique, and never identified in human cancer. The first 2 categories were considered “common in cancer” and the last 3 categories “not common in cancer.” Categories of activity included inactive, splice/truncated, partially active, active, and synonymous/unknown. The first 3 categories were considered “impaired activity” and the last 2 categories “active/likely active.” Categories of pathogenicity included pathogenic, likely pathogenic, possibly pathogenic, benign, and variants of uncertain significance. The first 3 categories were considered “expected pathogenic” and the last 2 categories “unlikely pathogenic.”
TP53 cancer database mutational analysis
From the UMD TP53 database (April 2017 version), we selected the set of 71,051 mutations reported within human tumors of all types (mutations from cell lines, normal, and premalignant tissue were excluded). Then we determined the distribution of mutations in the following categories: CpG, hotspots, exons 5-8, activity, pathogenicity, mutation type, and mutation spectrum. These values were used as a comparator for TP53 mutations identified in uterine lavage and normal tissues.
TP53 mutations without selection
To assess the distribution of TP53 mutations in the absence of selection, we generated a list of all possible mutations in the gene coding region (n = 3,546) in silico. Then we submitted this list to Seshat to determine the distribution of mutations in the same categories as above. The values obtained represent the distribution of all possible TP53 mutations in the absence of selection and were used as a comparator for TP53 mutations identified in uterine lavage and normal tissue.
QUANTIFICATION AND STATISTICAL ANALYSIS
Correlations were tested with Spearman’s rank test due to high variability in the outcomes (non-normality). Comparisons by groups were performed with Fisher’s exact tests (for comparison between case and control groups) or with the exact binomial test for comparison of one group with a hypothesized null probability of a success (mutation). Generalized estimating equations (GEE) also was investigated to take into account possible correlation within patients (lack of independence among observations/mutations with a patient) for these comparisons, but the results from the exact tests that assume independence were more conservative and thus, are the ones reported. Further, taking into account the sequencing depth either as an adjustment in binomial models or an offset in Poisson count models did not affect the significance of results. Sensitivity and specificity were calculated as simple proportions. Adjustment for age was performed by fitting log (maximum frequency) in a linear model. All tests were two-sided at an alpha level (type 1 error rate) of 0.05. Statistical analyses were performed with SPSS and R.
DATA AND CODE AVAILABILITY
Code availability
Software for DS data analysis is available at https://github.com/loeblab/Duplex-Sequencing
Data availability
The accession number for the sequencing data reported in this paper is Sequence Read Archive BioProject ID: PRJNA503496.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Uterine lavage | Medical University of Vienna (Austria), Charles University Pilsen (Czech Republic) and University Hospitals Leuven (Belgium). | N/A |
| Newborn tissue | Autopsies from Seattle Childrens’ Hospital | N/A |
| Middle age tissue | Hysterectomies from Medical University of Vienna (Austria) | N/A |
| Centenarian tissue | Autopsy from Tissue for Research Inc. | N/A |
| Critical Commercial Assays | ||
| Custom TaqMan SNP Genotyping Assays for ddPCR |
Life Technologies | No.4331349 |
| Droplet Digital PCR system | BioRad | QX200 |
| Massively parallel DNA sequencer | Illumina | HiSeq 2500 |
| Massively parallel DNA sequencer | Illumina | NextSeq 550 |
| KAPA HyperPrep library Kit | Roche Sequencing | KK8505 |
| Deposited Data | ||
| TP53 Duplex Sequencing data | This article | PRJNA503496 |
| Oligonucleotides | ||
| Primers for ddPCR | Maritschnegg et al., 2015 | N/A |
| Duplex Sequencing primers and adapters | Kennedy et al., 2014 | N/A |
| Software and Algorithms | ||
| Duplex Sequencing Analysis | Kennedy et al., 2014 | https://github.com/loeblab/Duplex-Sequencing |
| Seshat | Tikkanen et al. 2018 | https://p53.fr/tp53-database/seshat |
| IGV | Broad Institute | https://software.broadinstitute.org/software/igv/ |
| Other | ||
| UMD TP53 database | TP53 website | https://p53.fr/tp53-database |
Highlights.
Ovarian cancer can be detected by ultra-accurate sequencing of uterine lavage DNA
However, low-frequency TP53 mutations also exist in normal tissue of healthy women
TP53 mutations are increasingly selected for with age, revealing somatic evolution
Age-associated, cancer-like mutations challenge specificity for cancer detection
ACKNOWLEDGMENTS
This work was supported in part by NIH grant R01CA181308 to R.A.R.; Mary Kay Foundation grant 045-15 to R.A.R.; Rivkin Center for Ovarian Cancer grant 567612 to R.A.R.; T32CA009515 to J.J.S.; R44CA221426 to J.J.S. and L.N.W.; CA077852 and CA193649 to L.A.L.; and Radiumhemmets Forskningsfonder 174261 to T.S. We thank members of the Loeb, Risques, Kennedy, and Swisher labs at the University of Washington for helpful discussions, as well as participating members of the LUSTIC and LUDOC clinical trials. Most important, we deeply thank the patients and families who volunteered to provide clinical samples, without which this research would not have been possible.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.109.
DECLARATION OF INTERESTS
J.J.S. and L.A.L. are founders and equity holders at TwinStrand Biosciences. J.J.S., C.C.V., L.N.W., and J.E.H. are employees and equity holders at TwinStrand Biosciences. P.S. is a founder and equity holder in Ovartec. R.Z. is a founder and equity holder in Oncolab GmbH. R.A.R. shares equity in NanoString Technologies and is the principal investigator in an NIH Small Business Innovation Research (SBIR) subcontract research agreement with TwinStrand Biosciences. Commercial uses of Duplex Sequencing are protected by multiple patents held or licensed by the University of Washington and TwinStrand Biosciences. Commercial uses of uterine lavage for cancer screening and diagnosis are protected by multiple patents licensed or held by the Medical University of Vienna and Ovartec.
REFERENCES
- Abyzov A, Tomasini L, Zhou B, Vasmatzis N, Coppola G, Amenduni M, Pattni R, Wilson M, Gerstein M, Weissman S, et al. (2017). One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin. Genome Res. 27, 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney LALM, Veltman JA, Gilissen C, and Hoischen A (2017). Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet 101, 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, Defazio A, et al. (2010). Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol 221, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, and Stratton MR (2015). Clock-like mutational processes in human somatic cells. Nat. Genet 47, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix-Panabières C, and Pantel K (2016). Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 6, 479–491. [DOI] [PubMed] [Google Scholar]
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T, et al. (2017). Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med 376, 1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravanis AM, Lee M, and Klausner RD (2017). Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell 168, 571–574. [DOI] [PubMed] [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. (2016). Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowtell DD, Bühm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, et al. (2015). Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 15, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Chien J, Sicotte H, Fan JB, Humphray S, Cunningham JM, Kalli KR, Oberg AL, Hart SN, Li Y, Davila JI, et al. (2015). TP53 mutations, tetraploidy and homologous recombination repair defects in early stage high-grade serous ovarian cancer. Nucleic Acids Res. 43, 6945–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamandis EP (2012). The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 10, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA Jr., and Bardelli A (2014). Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol 32, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drescher CW, and Anderson GL (2018). The Yet Unrealized Promise of Ovarian Cancer Screening. JAMA Oncol 4, 456–457. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman DC, Curry SJ, Owens DK, Barry MJ, Davidson KW, Doubeni CA, Epling JW Jr., Kemper AR, Krist AH, Kurth AE, et al. ; US Preventive Services Task Force (2018). Screening for Ovarian Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 319, 588–594. [DOI] [PubMed] [Google Scholar]
- Henderson JT, Webber EM, and Sawaya GF (2018). Screening for Ovarian Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 319, 595–606. [DOI] [PubMed] [Google Scholar]
- Hoang ML, Kinde I, Tomasetti C, McMahon KW, Rosenquist TA, Grollman AP, Kinzler KW, Vogelstein B, and Papadopoulos N (2016). Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 113, 9846–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, and Ishioka C (2003). Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 100, 8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SR, Schmitt MW, Fox EJ, Kohrn BF, Salk JJ, Ahn EH, Prindle MJ, Kuong KJ, Shen JC, Risques RA, and Loeb LA (2014). Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc 9, 2586–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde I, Bettegowda C, Wang Y, Wu J, Agrawal N, Shih IeM., Kurman R, Dao F, Levine DA, Giuntoli R, et al. (2013). Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci. Transl. Med 5, 167ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimmel JD, Schmitt MW, Harrell MI, Agnew KJ, Kennedy SR, Emond MJ, Loeb LA, Swisher EM, and Risques RA (2016). Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc. Natl. Acad. Sci. USA 113, 6005–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E, Kurman RJ, Vang R, Sehdev AS, Han G, Soslow R, Wang TL, and Shih IeM. (2012). TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinomax–evidence supporting the clonal relationship of the two lesions. J. Pathol 226, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurman RJ, and Shih IeM. (2010). The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am. J. Surg. Pathol 34, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, et al. (2017). High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun 8, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H, Ellis P, Osborne RJ, Sanders MA, Moore L, Georgakopoulos N, Torrente F, Noorani A, Goddard M, Robinson P, et al. (2018). The landscape of somatic mutation in normal colorectal epithelial cells. bioRxiv 10.1101/416800. [DOI] [PubMed] [Google Scholar]
- Leroy B, Anderson M, and Soussi T (2014). TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum. Mutat 35, 672–688. [DOI] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, and Boerwinkle E (2016). dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritschnegg E, Wang Y, Pecha N, Horvat R, Van Nieuwenhuysen E, Vergote I, Heitz F, Sehouli J, Kinde I, Diaz LA Jr., et al. (2015). Lavage of the Uterine Cavity for Molecular Detection of Müllerian Duct Carcinomas: A Proof-of-Concept Study. J. Clin. Oncol 33, 4293–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritschnegg E, Heitz F, Pecha N, Bouda J, Trillsch F, Grimm C, Vanderstichele A, Agreiter C, Harter P, Obermayr E, et al. (2018). Uterine and Tubal Lavage for Earlier Cancer Detection Using an Innovative Catheter: A Feasibility and Safety Study. Int. J. Gynecol. Cancer 28, 1692–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. (2018). Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair N, Camacho-Vanegas O, Rykunov D, Dashkoff M, Camacho SC, Schumacher CA, Irish JC, Harkins TT, Freeman E, Garcia I, et al. (2016). Genomic Analysis of Uterine Lavage Fluid Detects Early Endometrial Cancers and Reveals a Prevalent Landscape of Driver Mutations in Women without Histopathologic Evidence of Cancer: A Prospective Cross-Sectional Study. PLoS Med. 13, e1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risques RA, and Kennedy SR (2018). Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet 14, e1007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salk JJ, Schmitt MW, and Loeb LA (2018). Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet 19, 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, and Loeb LA (2012). Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 109, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, Fox EJ, Prindle MJ, Reid-Bayliss KS, True LD, Radich JP, and Loeb LA (2015). Sequencing small genomic targets with high efficiency and extreme accuracy. Nat. Methods 12, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al. (2015). The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med 373, 1926–1936. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2017). Cancer Statistics, 2017. CA Cancer J. Clin 67, 7–30. [DOI] [PubMed] [Google Scholar]
- Soong TR, Howitt BE, Horowitz N, Nucci MR, and Crum CP (2019). The fallopian tube, “precursor escape” and narrowing the knowledge gap to the origins of high-grade serous carcinoma. Gynecol. Oncol 152, 426–433. [DOI] [PubMed] [Google Scholar]
- Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, Yamawaki K, Adachi S, Takahashi T, Kase H, et al. (2018). Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 24, 1777–1789. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikkanen T, Leroy B, Fournier JL, Risques RA, Malcikova J, and Soussi T (2018). Seshat: A Web service for accurate annotation, validation, and analysis of TP53 variants generated by conventional and next-generation sequencing. Hum. Mutat 39, 925–933. [DOI] [PubMed] [Google Scholar]
- Vang R, Levine DA, Soslow RA, Zaloudek C, Shih IeM., and Kurman RJ (2016). Molecular Alterations of TP53 are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol 35,48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li L, Douville C, Cohen JD, Yen TT, Kinde I, Sundfelt K, Kjær SK, Hruban RH, Shih IM, et al. (2018). Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Sci. Transl. Med 10, eaap8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. (2012). The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, DeGregori J, and De S (2016). The landscape of somatic mutations in protein coding genes in apparently benign human tissues carries signatures of relaxed purifying selection. Nucleic Acids Res. 44, 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AL, Challen GA, Birmann BM, and Druley TE (2016). Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun 7, 12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code availability
Software for DS data analysis is available at https://github.com/loeblab/Duplex-Sequencing
Data availability
The accession number for the sequencing data reported in this paper is Sequence Read Archive BioProject ID: PRJNA503496.
The accession number for the sequencing data reported in this paper is Sequence Read Archive BioProject ID: PRJNA503496.