

Abstract
summary
Background
Aurora kinase overexpression or amplifications are associated with high proliferation, poor prognosis, and therapeutic resistance in human tumors. AMG 900 is an investigational, oral, selective pan-Aurora kinase inhibitor.
Methods
This first-in-human trial included dose-escalation and dose-expansion phases (ClinicalTrials.gov: NCT00858377). Dose escalation evaluated the safety, tolerability, and pharmacokinetics of AMG 900 in advanced solid tumors and determined the maximum tolerated dose (MTD) with/without granulocyte colony-stimulating factor (G-CSF) prophylaxis. Dose expansion evaluated clinical activity in three tumor types: taxane- and platinum-resistant ovarian cancer, taxane-resistant triple-negative breast cancer (TNBC), and castration-resistant and taxane- or cisplatin/etoposide-resistant prostate cancer (CRPC). AMG 900 was administered 4 days on/10 days off at 1–50 mg/day during escalation and at the MTD with G-CSF during expansion.
Results
AMG 900 showed rapid absorption with fast clearance, supporting once-daily dosing. The MTD was 25 mg/day, increasing to 40 mg/day with G-CSF. Grade ≥ 3 treatment-related adverse events included neutropenia (37%), anemia (23%), leukopenia (14%), and thrombocytopenia (12%). During dose expansion, 3/29 (10.3%, 95% CI: 2.0%–28.0%) evaluable patients with ovarian cancer experienced partial response by central imaging per RECIST 1.1; median duration of response was 24.1 weeks (95% CI: 16.1–34.1). Seven patients (24.1%, 95% CI: 10.3%–43.5%) experienced partial response per Gynecologic Cancer InterGroup criteria; 5/9 patients positive for p53 expression responded to treatment. No objective responses were observed in patients with TNBC or CRPC per RECIST 1.1.
Conclusions
AMG 900 40 mg/day with G-CSF had manageable toxicity and demonstrated single-agent activity in patients with heavily pretreated, chemotherapy-resistant ovarian cancer.
Keywords: AMG 900, Antimitotic, Aurora kinase, pan-Aurora kinase inhibitor
Introduction
Aurora kinases are a family of serine/threonine protein kinases critical to cell division [1, 2]. Aurora kinases A and B are amplified and/or overexpressed in many malignancies, including ovarian, breast, and prostate cancers, and are associated with elevated tumor proliferation, poor prognosis, and resistance to treatment [3–5]. Inhibition of Aurora kinases is an active area of investigation for the treatment of various cancers [6–8].
Aurora kinases A and B have unique roles in the cell; although inhibitors of both proteins induce apoptosis, they do so through distinct mechanisms. Aurora A is important for centrosome maturation, spindle assembly, and meiotic maturation and metaphase I spindle orientation [1, 9]. Inhibition of Aurora A leads to transient spindle checkpoint-dependent mitotic arrest, followed by p53- and p21-dependent apoptosis [10]. Aurora B is part of the chromosomal passenger complex that is necessary for chromosome condensation and chromosome orientation on the mitotic spindle, as well as the final stages of cytokinesis [1, 9]. Inhibition of Aurora B disrupts the mitotic spindle checkpoint, preventing cytokinesis and leading to polyploidy and endo-reduplication followed by apoptotic cell death [10, 11].
Aurora kinase inhibitors have the potential to be efficacious in tumors that are resistant to agents that target tubulin (eg, taxanes, epothilones, vinca alkaloids) while avoiding the peripheral neuropathy that comes from disrupting tubulin dynamics in nondividing neural cells [7, 12]. Inhibition of both Aurora kinases A and B may have certain advantages, most notably reduced possibility of resistance as a result of hitting multiple antitumor targets, but because of their antimitotic mechanism of action, Aurora kinase inhibitors have been associated with hematologic toxicities such as neutropenia and febrile neutropenia [7, 13, 14] and have a narrow therapeutic window. Finding a dose and schedule that successfully inhibit tumor growth without excessive toxicity has therefore been challenging and has limited the successful development of this drug class [15].
AMG 900 is an investigational, orally administered, selective, small-molecule pan-Aurora kinase inhibitor that causes premature mitotic exit without cell division [16], leading to increased cell ploidy and tumor cell death through apoptosis or senescence [12]. In preclinical studies, AMG 900 inhibited the proliferation of cell lines from a variety of tumor types including several that were resistant to paclitaxel [16]. Similarly, in mouse models, AMG 900 inhibited growth of xenografts of diverse tumor types that were resistant to either docetaxel or paclitaxel [16]. In this first-in-human clinical study, we assessed the safety, tolerability, pharmacokinetics, and clinical activity of AMG 900 in patients with advanced solid tumors.
Methods
Study design
This was a phase 1, first-in-human, open-label study conducted in two parts. Part 1 was a sequential dose-escalation phase, separated into accelerated (one patient/cohort) and nonaccelerated (three to six patients/cohort) parts. Part 2 was a dose-expansion phase (Supplementary Fig. 1). The study was conducted at nine centers in Australia and the United States in accordance with International Conference on Harmonisation Good Clinical Practice regulations/guidelines. An independent ethics committee/institutional review board at each study center approved the protocol. All patients provided written informed consent. The primary objectives were to evaluate the safety, tolerability, and pharmacokinetics of AMG 900 in patients with advanced solid tumors; to determine the maximum tolerated dose (MTD; part 1 only); and to evaluate the clinical activity in patients with breast, ovarian, and prostate cancers (part 2 only). The secondary objectives included evaluation of change in tumor volume and tumor response. Exploratory objectives included biomarker analysis of archived tumor samples.
Study endpoints
The primary endpoints of this study were safety, including incidence of adverse events (AEs), dose-limiting toxicities (DLTs), and clinically significant changes in vital signs, weight, electrocardiograms (ECGs), and clinical laboratory tests; pharmacokinetics; and response rate in patients with taxane- and platinum-resistant ovarian cancer, taxane-resistant triple-negative breast cancer (TNBC), or castration-resistant and taxane- or cisplatin/etoposide-resistant prostate cancer (CRPC) by Response Evaluation Criteria In Solid Tumors (RECIST) and by Gynecologic Cancer InterGroup (GCIG) CA 125 criteria for patients with ovarian cancer (part 2 only). Secondary endpoints included change in tumor volume from baseline per volumetric computed tomography (CT) or magnetic resonance imaging (MRI), tumor response per RECIST as well as per GCIG CA 125 criteria in patients with ovarian tumors. Key exploratory endpoints included tumor biomarkers: TP53 mutation and p53 expression; levels of biomarkers Aurora A, Aurora B, p53, and p21, and Ki-67 (marker of cell proliferation) by immunohistochemistry (IHC); and Aurora A (AURKA) amplification by fluorescence in situ hybridization (FISH).
Patients
Patients were ≥ 18 years old with advanced solid tumors refractory to standard treatment, for which no standard therapy was available, or for which the patient refused standard therapy. Patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status ≤2; life expectancy >3 months; measurable disease by RECIST; and adequate hematologic, renal, and hepatic functions. In the dose-expansion phase, patients with three tumor types were enrolled: (1) platinum-resistant and taxane-resistant epithelial ovarian cancer (measureable per RECIST or GCIG criteria if nonmeasurable per RECIST); (2) taxane-resistant TNBC with ≥1 previous therapy for metastatic disease (measureable per RECIST); and (3) castration-resistant and taxane- or cisplatin/ etoposide-resistant stage 4 prostate cancer (measureable per RECIST) with high-grade neuroendocrine or anaplastic clinical or pathological features. These tumor types were selected based on high unmet need after chemotherapy failure, association of AURKA amplification or Aurora kinase overexpression with advanced clinical stage and poor prognosis, and evidence of clinical activity with Aurora kinase inhibitors in preclinical and clinical models [3–5, 7, 17–19]. Patients were excluded if they had active parenchymal brain metastases, prior bone marrow transplant, history or presence of hematologic malignancies, history of bleeding diathesis, or active peptic ulcer disease. The complete inclusion and exclusion criteria are provided in the Supplementary Methods.
Study procedures
The dose escalation tested dose levels from 1 to 50 mg/day on a schedule of 4 days on, 10 days off in an accelerated titration design; a cycle was defined as 14 days [20] (Supplementary Fig. 1). Dose escalation decisions were made based on the incidence of DLTs or grade 2 toxicities by the safety and data monitoring committee per protocol-specified guidelines. The safety and data monitoring committee included the medical monitor, global safety officer or designee, clinical study manager, biostatistician, study center investigators, and other functional area representatives. Initially, dose escalation involved 100% escalations with one patient per cohort until occurrence of a DLT or a grade 2 toxicity (hematologic or nonhematologic) in the first two treatment cycles (days 1–28) deemed at least possibly related to AMG 900. After a DLT or grade 2 toxicity was observed, cohorts were expanded and dose escalation continued using a standard 3 + 3 design (≤50% dose escalation steps after the first grade 2 toxicity or≤25% steps after a DLT). In the dose-escalation phase, patients received AMG 900 at 1, 2, 4, 8, 16, 24, 30, and (after de-escalation) 25 mg without G-CSF, as well as 30, 35, 40, and 50 mg with G-CSF support.
DLTs were evaluated in all patients who received ≥1 dose of AMG 900. A DLT was defined as a hematologic toxicity (febrile neutropenia, neutropenic infection, grade 4 neutropenia or grade ≥ 3 thrombocytopenia for >7 days, or grade 4 thrombocytopenia) or nonhematologic toxicity (grade ≥ 3 nausea, vomiting, or diarrhea despite treatment, grade 3 fatigue for >7 days or grade 4 fatigue, or any other grade ≥ 3 AE except alopecia) attributable to AMG 900 in the first two treatment cycles (days 1–28). A full definition of DLTs is provided in the Supplementary Methods.
Escalation continued until at least two DLTs were observed among the three to six patients in a cohort. This dose was defined as the lowest nontolerated dose (LNTD); the MTD was one dose level below the LNTD. If neutropenia or a neutropenia-related toxicity was dose limiting at the LNTD, a new cohort of three patients was enrolled at that level with prophylactic G-CSF support. Dose escalation continued until an MTD with prophylactic G-CSF was determined.
In the dose expansion, patients received AMG 900 at the MTD determined in part 1. Each of the expansion cohorts was to include approximately 14 patients. Patients continued AMG 900 until medication intolerance, disease progression, or withdrawal of consent.
AMG 900 was self-administered orally once daily in the morning for the first 4 days of every 14-day cycle (4 days on, 10 days off). Patients were required to have an empty stomach at the time of dosing and for 1 h postdose (no food or liquids except water 2 h predose). When applicable, prophylactic G-CSF (300 or 480 μg; dosage based on weight) was given subcutaneously once daily starting at least 24 h after the last dose of AMG 900 per local prescribing guidelines.
Assessments
Safety
The first patient of cohort 1 self-administered AMG 900 in the morning and was monitored in the clinic for at least 2 h for any signs of AEs. Subsequent patients self-administered AMG 900 at home and were seen in the clinic weekly for the first 9 weeks and every 2 weeks thereafter. Investigators or subinvestigators performed physical examinations, assessed vital signs, performed 12-lead electrocardiogram, reviewed concomitant medications, and assessed AEs at every visit. The Medical Dictionary for Regulatory Activities version 17.1 was used to code all AEs to a system organ class and a preferred term. The severity of each AE was graded using Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 [21].
Blood chemistry and coagulation (including blood urea nitrogen, creatinine, total bilirubin, alkaline phosphatase, aspartate aminotransferase, and alanine aminotransferase; and prothrombin time and partial thromboplastin time), urinalysis, and hematology (including white blood cells, hemoglobin, platelets, and neutrophils) were monitored closely throughout the study. More detail on laboratory tests is provided in the Supplementary Methods.
Pharmacokinetics
Plasma samples for AMG 900 pharmacokinetics were obtained predose and 1, 2, 3, 5, and 8 h after doses 1 and 2 during cycle 1; predose on day 2 of cycle 1; predose, 1 h, and 8 days after dose 1 of cycle 2; predose and 1 h after dose 1 of cycles 3 and 5; 1 h after dose 1 of cycle 4; and at the end-of-study visit. AMG 900 concentrations were determined in plasma using a validated liquid chromatography-tandem mass spectrometry assay run on an API 4000™ system (SCIEX, Framingham, MA, USA). Pharmacokinetic parameters, including maximum observed concentration (Cmax), time to Cmax (tmax), area under the plasma concentration-time curve (AUC), half-life (t1/2), and apparent clearance (CL/F) were estimated using noncompartmental analysis of individual AMG 900 plasma concentrations using Phoenix WinNonlin v.6.3 (Certara, Princeton, NJ).
Radiographic assessment and biomarkers
To assess tumor response/progression and change in tumor volume, CT or MRI scans were performed at screening, on day 28 (±3 days), at 8 weeks (±7 days), every 8 weeks (±7 days) for 6 months, and every 12 weeks thereafter. Patients who discontinued for reasons other than disease progression had an end-of-study scan. In patients with ovarian cancer, CA 125 levels were determined every 4 weeks. In patients with CRPC, prostate-specific antigen levels were determined every 4 weeks and bone scans were also performed to evaluate disease progression by appearance of new bone lesions. Scans were performed at screening, on day 28 (±3 days), at 8 weeks (±7 days), every 8 weeks (±7 days) for 6 months, and every 12 weeks (±7 days) thereafter. All scans were used to assess tumor response both in central review by RECIST 1.1 and local (investigator) review by RECIST 1.0; GCIG criteria were also used to assess ovarian tumors [22, 23].
Biomarkers were evaluated in archived tumor tissue samples (formalin-fixed, paraffin-embedded tumor samples) collected at the time of primary cancer diagnosis. Exons 2 to 11 of TP53 were analyzed for mutations by 454 sequencing at Transgenomic, Inc. (Omaha, NE). Aurora A, Aurora B, p53, p21, and Ki-67 protein expression were analyzed by IHC at HistoGeneX (Antwerp, Belgium). Amplifications of the AURKA gene were evaluated by FISH analysis (AURKA [20q13/20q11]) at Mosaic Laboratories, LLC (Lake Forest, CA).
Statistical analyses
A formal statistical hypothesis was not tested in this study. Sample size was determined empirically and was consistent with other initial human studies of this type. Descriptive statistics were calculated for select demographic, safety, pharmacokinetic, pharmacodynamic, imaging, biopsy, and biomarker parameters. Descriptive statistics included means, medians, standard deviations (SDs), and ranges for continuous variables, and frequency counts and percentages for categorical variables. Safety was assessed in all patients who received ≥1 dose of AMG 900. Tumor response was assessed in all patients with measurable disease per RECIST criteria who received ≥1 dose of AMG 900 and had reported baseline and postdose assessments of tumor burden. Response rate and median duration of response per RECIST were estimated for each tumor type along with 95% confidence intervals (CIs); Kaplan-Meier estimates with 80% CIs were provided for median progression-free survival (PFS).
Results
Patients and disposition
Between August 10, 2009 (first patient enrolled) and December 31, 2014 (data analysis cutoff), a total of 105 patients were enrolled, and all enrolled patients received ≥1 dose of AMG 900. The dose-escalation phase included 50 patients across 12 AMG 900 dose cohorts: 1 mg (n =1), 2 mg (n =1), 4 mg (n = 1), and 8 mg (n = 1) during the accelerated phase; and 16 mg (n = 3), 24 mg (n = 6), 30 mg (n = 6), and after deescalation, 25 mg (n = 7) in the nonaccelerated phase. Patients received the following doses with G-CSF support during the nonaccelerated phase: 30 mg (n = 6), 35 mg (n = 3), 40 mg (n = 11), and 50 mg (n = 4). The dose-expansion phase was conducted at 40 mg + G-CSF and included 55 patients across three cohorts: ovarian cancer (n = 29), TNBC (n = 14), and CRPC (n = 12). The ovarian cancer cohort was expanded from the planned 14 patients to 29 patients based on emerging efficacy data. Patient disposition is listed in Supplementary Table 1.
Overall, 72 patients (69%) were women, and the mean (SD) age was 59(12) years. The most common primary tumor types were epithelial ovarian (n = 31; 30%), breast (n = 20; 19%), prostate (n = 12; 11%), and colon (n = 9; 9%). Most patients (97%) had an ECOG performance status of 0 or 1, and patients were heavily pretreated with a median (range) of 4.0 (0–18) prior lines of chemotherapy. Baseline demographics and disease characteristics in the dose escalation, the dose expansion, and overall are listed in Table 1.
Table 1.
Baseline demographics and disease characteristics
| Dose escalation | Dose expansion | ||||
|---|---|---|---|---|---|
| Advanced solid tumorsa (n= 50) |
Ovarian cancer (n = 29) |
TNBC (n = 14) |
CRPC (n = 12) |
All (N = 105) |
|
| Sex, n (%) | |||||
| Male | 21 (42.0) | 0 | 0 | 12 (100) | 33 (31.4) |
| Female | 29 (58.0) | 29 (100) | 14 (100) | 0 | 72 (68.6) |
| Age, mean (SD), years | 57 (12) | 61(11) | 55 (11) | 68 (8) | 59 (12) |
| BMI, mean (SD), kg/m2 | 27(7) | 27 (6) | 27 (5) | 28 (3) | 27 (6) |
| Race/ethnicity, n (%) | |||||
| White | 35 (70.0) | 23 (79.3) | 12 (85.7) | 11 (91.7) | 81 (77.1) |
| Black | 2 (4.0) | 1 (3.4) | 2 (14.3) | 0 | 5 (4.8) |
| Hispanic | 6 (12.0) | 0 | 0 | 1 (8.3) | 7 (6.7) |
| Asian | 4 (8.0) | 3 (10.3) | 0 | 0 | 7 (6.7) |
| Otherb | 3 (6.0) | 2 (6.9) | 0 | 0 | 5 (4.8) |
| ECOG PS, n (%) | |||||
| 0 | 32 (64.0) | 16(55.2) | 6 (42.9) | 2 (16.7) | 56 (53.3) |
| 1 | 17 (34.0) | 13 (44.8) | 7 (50.0) | 9 (75.0) | 46 (43.8) |
| 2 | 1 (2.0) | 0 | 1 (7.1) | 1 (8.3) | 3 (2.9) |
| Number of prior lines of chemotherapy, median (range) | 3.5 (0–13) | 3.0 (1–18) | 5.5 (2–12) | 3.5 (1–9) | 4.0 (0–18) |
| Prior radiotherapy, n (%) | 24 (48.0) | 6 (20.7) | 10(71.4) | 7 (58.3) | 47 (44.8) |
CRPC castration-resistant prostate cancer, ECOGPS Eastern Cooperative Oncology Group performance status, TNBC triple-negative breast cancer
Tumor types included ovarian (n = 4), breast (n = 6), prostate (n = 2), colon (n = 9), rectal (n = 2), and small-cell lung cancers (n = 3), and other (n = 24)
“Other” includes two American Indian or Alaska native, one Afghan, one Sri Lankan, and one Middle Eastern
Across all cohorts, patients received a median (range) of 16.0(1–399) doses of AMG 900. The median (range) cumulative dose of AMG 900 was 480 (25–9775) mg. The median (range) of the average dose delivered across all cohorts for dose escalation and expansion was 35.6 (1–50) mg.
Safety and tolerability
Dose-escalation phase: DLTs and MTD
Overall, nine DLTs were reported in eight patients. There were no DLTs in the first four dose cohorts (1, 2, 4, and 8 mg). At 16 mg AMG 900, one patient experienced non-dose-limiting neutropenia, which led to initiation of the nonaccelerated portion of the dose-escalation phase. At 24 mg, one of six patients experienced a DLT of grade 4 neutropenia lasting >7 days. At 30 mg, two of six patients experienced DLTs of grade 4 thrombocytopenia lasting >7 days and grade 4 neutropenia lasting >7 days, respectively. At 25 mg, one of seven patients experienced two DLTs (grade 3 increases in alanine aminotransferase and aspartate aminotransferase). Consequently, 25 mg AMG 900 was determined to be the MTD without G-CSF.
Because neutropenia was dose limiting, dose escalation continued with G-CSF support, beginning with 30 mg AMG 900 + G-CSF. At this dose, one of six patients experienced a DLT of grade 4 thrombocytopenia. At 40 mg + G-CSF, one of 11 patients experienced a DLT of grade 4 febrile neutropenia. At 50 mg + G-CSF, two of four patients experienced DLTs of grade 4 thrombocytopenia lasting 2 days and grade 3 fatigue lasting >7 days, respectively. These results established the MTD with G-CSF and the recommended phase 2 dose as 40 mg AMG 900 + G-CSF.
Adverse events
Across the dose escalation and the dose expansion, treatment-related grade ≥ 3 AEs occurred in 61 patients (58%); the most common (>10% overall) were neutropenia (42%; n = 44), anemia (23%; n = 24), leukopenia (14%; n = 15), and thrombocytopenia (12%; n = 13; Table 2). Treatment-related grade≥4 AEs occurred in 31 patients (30%); the most common were neutropenia (29%; n = 30) and leukopenia (11%; n = 12). Diarrhea was the only nonhematologic grade ≥ 4 AE (1%; n = 1). Five patients (5%) had fatal AEs: cardiac arrest (n = 2; both patients TNBC cohort), tachycardia (n = 1; TNBC cohort), atelectasis (n =1; 50 mg + G-CSF cohort), and pulmonary embolism (n = 1; 25 mg cohort); none of the fatal AEs were considered treatment related as judged by the investigator.
Table 2.
Treatment-emergent AEs by grade and grade ≥ 3 treatment-related AEs by preferred term occurring in >1 patient overall
| Patients, n (%) | Part 1 | Part 2 | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose escalation | Dose expansion | ||||||||||||
| 1, 2, 4, 8 mg | 16 mg | 24 mg | 25 mg | 30 mg | 30 mg + G-CSF | 35 mg + G-CSF | 40 mg + G-CSF | 50 mg + G-CSF | OC | TNBC | CRPC | ||
| (n = 4) | (n = 3) | (n = 6) | (n = 7) | (n = 6) | (n = 6) | (n = 3) | (n = 11) | (n = 4) | (n = 29) | (n = 14) | (n = 12) | (N = 105) | |
| All treatment-emergent | 4 (100) | 3 (100) | 6 (100) | 7 (100) | 6 (100) | 6 (100) | 3 (100) | 11 (100) | 4 (100) | 29 (100) | 14 (100) | 12 (100) | 105 (100) |
| AEs | |||||||||||||
| Serious AEs | 0 | 0 | 1 (17) | 1 (14) | 3 (50) | 3 (50) | 1 (33) | 3 (27) | 2 (50) | 14 (48) | 7 (50) | 4 (33) | 39 (37) |
| AEs leading to discontinuation of AMG 900 | 0 | 0 | 1 (17) | 2 (29) | 4 (67) | 1 (17) | 0 | 2 (18) | 2 (50) | 5 (17) | 5 (36) | 3 (25) | 25 (24) |
| Grade ≥ 3 AEs | 1 (25) | 1 (33) | 5 (83) | 3 (43) | 6 (100) | 5 (83) | 2 (67) | 8 (73) | 3 (75) | 25 (86) | 11 (79) | 9 (75) | 79 (75) |
| Grade ≥ 4 AEs | 0 | 1 (33) | 3 (50) | 2 (29) | 6 (100) | 2 (33) | 1 (33) | 4 (36) | 2 (50) | 12 (41) | 5 (36) | 2 (17) | 40 (38) |
| Fatal AEs | 0 | 0 | 0 | 1 (14) | 0 | 0 | 0 | 0 | 1 (25) | 0 | 3 (21) | 0 | 5 (5) |
| All treatment-related AEs | 3 (75) | 3 (100) | 5 (83) | 5 (71) | 6 (100) | 6 (100) | 3 (100) | 10 (91) | 4 (100) | 28 (97) | 13 (93) | 12 (100) | 98 (93) |
| Grade ≥ 3 treatment-related AEs | 1 (25) | 1 (33) | 5 (83) | 2 (29) | 4 (67) | 3 (50) | 2 (67) | 6 (55) | 3 (75) | 22 (76) | 6 (43) | 6 (50) | 61 (58) |
| Neutropenia | 0 | 1 (33) | 4 (67) | 1 (14) | 4 (67) | 3 (50) | 1 (33) | 5 (46) | 1 (25) | 17 (59) | 4 (29) | 3 (25) | 44 (42) |
| Anemia | 1 (25) | 1 (33) | 0 | 2 (29) | 0 | 1 (17) | 0 | 0 | 1 (25) | 12 (41) | 4 (29) | 2 (17) | 24 (23) |
| Leukopenia | 0 | 1 (33) | 2 (33) | 1 (14) | 3 (50) | 2 (33) | 1 (33) | 1 (9) | 1 (25) | 3 (10) | 0 | 0 | 15 (14) |
| Thrombocytopenia | 0 | 0 | 0 | 0 | 1 (17) | 2 (33) | 0 | 1 (9) | 1 (25) | 6 (21) | 1 (7) | 1 (8) | 13 (12) |
| Febrile neutropenia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (18) | 1 (25) | 6 (21) | 1 (7) | 0 | 10 (10) |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (50) | 1 (3) | 0 | 2 (17) | 5 (5) |
| Diarrhea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (18) | 0 | 1 (3) | 1 (7) | 0 | 4 (4) |
| Hemoglobin decreased | 0 | 0 | 0 | 1 (14) | 0 | 0 | 0 | 0 | 0 | 2 (7) | 0 | 1 (8) | 4 (4) |
| Lymphopenia | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 1 (9) | 0 | 1 (3) | 0 | 1 (8) | 4 (4) |
| Nausea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 2 (17) | 3 (3) |
| White blood cell count decreased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (7) | 0 | 0 | 2 (2) |
| Vomiting | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 1 (3) | 0 | 0 | 2 (2) |
AE adverse event, CRPC castration-resistant prostate cancer, G-CSF granulocyte colony-stimulating factor, OC ovarian cancer, TNBC triple-negative breast cancer
At the time of the primary analysis (data cutoff December 31, 2014), 96 patients (91%) had discontinued AMG 900; the two most common reasons for discontinuation of AMG 900 were disease progression (n = 62; 59%) and AEs (n = 25; 24%). In 13 patients (12%), the AEs that led to discontinuation of AMG 900 were considered treatment related. These included thrombocytopenia (4 events); neutropenia (3 events); febrile neutropenia (3 events); fatigue (2 events); elevated liver enzymes (2 events); and anemia, neutropenic colitis, decreased hemoglobin, and nausea (1 event each). Overall, 51 patients (49%) had dose reductions or interruptions; these were due to AEs in 47 patients (45%). Nine patients (9%) were still receiving AMG 900 as of the primary analysis data cutoff date.
Pharmacokinetics
Overall, mean exposure (AUC24h) for AMG 900 following oral administration increased as the dose increased from 1 to 50 mg. The coefficient of variation (CV) values for AUC24h ranged from 14.5 to 60.4% for both cycle 1 day 1 and cycle 1 day 4. Median tmax was between 1 and 3 h postdose across the dose ranges of 2 to 30 mg AMG 900 monotherapy and 30 to 50 mg AMG 900 with G-CSF. Minimal accumulation (<2-fold based on AUC8h) was observed from cycle 1 day 1 to cycle 1 day 4 for all dose cohorts. The average Cmax at cycle 1 day 4 from the dose-expansion cohorts ranged from 906 to 1670 ng/mL, about 3- to 6-fold above the 50% inhibitory concentration for the in vivo inhibition of p-histone H3 (273 ng/mL). The CL/F after cycle 1 day 1 ranged from 1790 to 4330 mL/h for all dose cohorts and showed variability, with CV values ranging from 43.1 to 88.0%. The mean t1/2 after cycle 1 day 1 was approximately 6 to 9 h across all dose groups. Among patients who received 40 mg once daily in the dose expansion, exposure varied among cohorts, with a higher mean AUC24h observed in women in the ovarian cancer cohort (12,900 h-ng/mL) compared with TNBC (10,600 h·ng/mL) or CRPC (7700 h·ng/mL).
Efficacy
Eighty-three of the 105 patients (79%) were included in the assessment of change in tumor volume. The tumor volume results by CT or MRI assessment were generally consistent with the RECIST results. The maximum percentage reduction of 50.3% from baseline in the mean tumor volume was observed for two patients with ovarian cancer.
Although evaluation of clinical activity was not a primary objective of the dose-escalation phase, 42 of the 50 enrolled patients in the escalation phase were evaluable for tumor response. In the dose-escalation phase, one patient with ovarian cancer who received 30 mg AMG 900 had a partial response per RECIST 1.1 and GCIG criteria (central read). One additional patient with clear-cell endometrial cancer who received 30 mg + G-CSF had a partial response per RECIST 1.0 (local read) that was not confirmed per RECIST 1.1 (central read). Twenty-nine patients (58%) had stable disease (defined as best response of stable disease per RECIST version 1.1 at regularly scheduled scans); 12 patients (24%) had progressive disease. One patient with medullary thyroid cancer who had disease progression before enrollment received 24 mg AMG 900 and had stable disease for >3.5 years. Another patient with a germline retinoblastoma mutation and metastatic leiomyosarcoma received 40 mg AMG 900 and had stable disease for >3 years. Best overall response in the dose-escalation cohorts is presented in Table 3.
Table 3.
Best overall response in the dose-escalation and dose-expansion cohorts
| Dose-escalation cohorts | |||||||||
| Central Reada | |||||||||
| Tumor Response, | 1, 2, 4, 8 mg | 16 mg | 24 mg | 25 mg | 30 mg | 30 mg + G-CSF | 35 mg + G-CSF | 40 mg + G-CSF | 50 mg + G-CSF |
| Patients (%) | (n = 4) | (n = 3) | (n = 6) | (n = 7) | (n = 6) | (n = 6) | (n = 3) | (n = 11) | (n = 4) |
| Complete response | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Partial response | 0 | 0 | 0 | 0 | 1 (17)b | 0 | 0 | 0 | 0 |
| Stable disease | 2 (50) | 2 (67) | 5 (83) | 4 (57) | 3 (50) | 3 (50) | 1 (33) | 7 (64) | 2 (50) |
| Progressive disease | 2 (50) | 0 | 1 (17) | 1 (14) | 1 (17) | 3 (50) | 2 (67) | 2 (18) | 0 |
| Unable to evaluate | 0 | 1 (33) | 0 | 2 (29) | 1 (17) | 0 | 0 | 2 (18) | 2 (50) |
| Dose-expansion cohorts | |||||||||
| Central Reada | Local Readc | GCIG Criteriad | |||||||
| Tumor Response, | OC | TNBC | CRPC | OC | TNBC | CRPC | OC | ||
| Patients (%) | (n = 29) | (n = 14) | (n = 12) | (n = 29) | (n = 14) | (n = 12) | (n = 29) | ||
| Complete response | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Partial response | 3 (10) | 0 | 0 | 3 (10) | 1 (7) | 0 | 7 (24) | ||
| Stable disease | 21 (72) | 7 (50) | 9 (75) | 20 (69) | 5 (36) | 8 (67) | 16 (55) | ||
| Progressive disease | 3 (10) | 4 (29) | 1 (8) | 4 (14) | 5 (36) | 3 (25) | 3 (10) | ||
| Not evaluablee | 2 (7) | 3 (21) | 2 (17) | 2 (7) | 3 (21) | 1 (8) | 3 (10) | ||
| Progression-free survival, median (80% CI), weeks | 31.7 (23.7–47.6) | 7.6 (4.1–23.6) | 32.4 (15.7–39.6) | NA | NA | NA | NA | ||
CRPC castration-resistant prostate cancer, GCIG Gynecologic Cancer InterGroup, G-CSF granulocyte colony-stimulating factor, NA not available, OC ovarian cancer, RECIST response evaluation criteria in solid tumors, TNBC triple-negative breast cancer
Per RECIST 1.1
This patient also had a response by GCIG CA 125 criteria
Per RECIST 1.0
Per GCIG criteria: RECIST 1.1 and CA 125 [22]
Postbaseline scan not readable
In the dose-expansion phase, 27 of29 patients with ovarian cancer were evaluable for tumor response per RECIST 1.1. Three patients (10.3%) had a partial response and 21 (72.4%) had stable disease as their best response; 3 (10.3%) had progressive disease. The objective response rate (ORR; 95% CI) per RECIST 1.1 was 10.3% (2.0%–28.0%). The duration of partial response by RECIST 1.1 (and by GCIG criteria) in the three patients was 34.1 (65.6), 24.1 (74.9), and 16.1 (16.1) weeks. Best overall tumor responses by tumor type in the dose-expansion cohorts are summarized in Table 3. Maximum change in the sum of longest diameters in patients with ovarian cancer is presented in Fig. 1a.
Fig. 1.
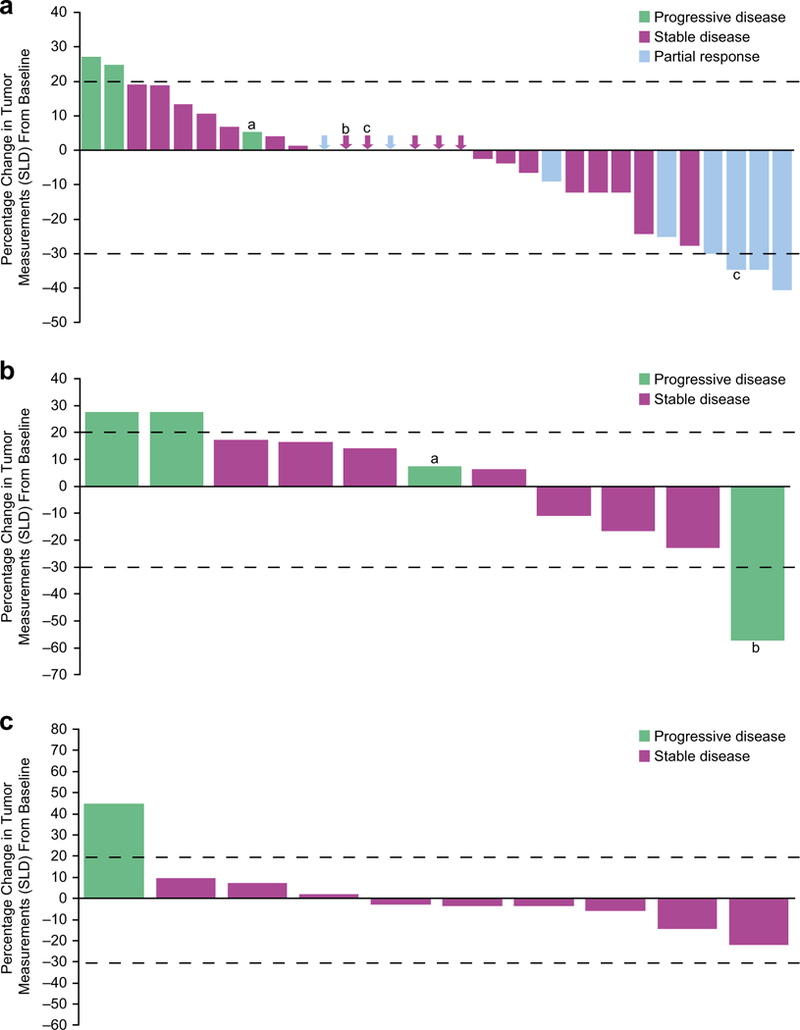
Maximum percentage tumor reduction and response (by RECIST 1.1) by individual patient for (a) all patients with ovarian tumors from the dose-escalation and -expansion phases. Central read data are available for 31 of 33 patients; plot includes four patients with ovarian cancer from the dose-escalation phase; aProgressive disease by new lesion; bStable disease by RECIST 1.1 (CA 125 was not available); cPatient still on treatment at data cutoff. Arrows represent the best response for patients whose tumor change was not visible because of being near 0. b Patients with triple-negative breast cancer in the dose-expansion phase. Central read data are available for 11 of 14 patients; aProgressive disease by new lesion; bProgressive disease of nontarget lesion. c Patients with castration-resistant prostate cancer in the dose-expansion phase. Central read data are available for 10 of 12 patients. Data cutoff: December 31, 2014. SLD = sum of longest diameters
Twenty-six of 29 patients with ovarian cancer were evaluable by GCIG criteria (RECIST 1.1 and CA 125). Seven (24.1%) had partial responses, 16 (55.2%) had stable disease, and 3 (10.3%) had progressive disease. The ORR (95% CI) was 24.1% (10.3%–43.5%). Of the seven patients with responses per CA 125, one had a response and normalization of CA 125 levels and six had responses without normalization of CA 125 levels.
Among the patients with ovarian cancer in the dose expansion, the Kaplan-Meier estimate for median (80% CI) PFS per RECIST 1.1 (central read) was 31.7 (23.7–47.6) weeks. Combining the ovarian cancer patients from both the dose-escalation (n = 4) and dose-expansion (n = 29) phases, the Kaplan-Meier estimate for median (80% CI) PFS was 34.1 (31.1–not estimable) weeks (Fig. 2a). Using GCIG criteria (RECIST 1.1 and CA 125), the median PFS was not reached; the lower limit of the 80% CI was 31.1 weeks (Fig. 2b).
Fig. 2.
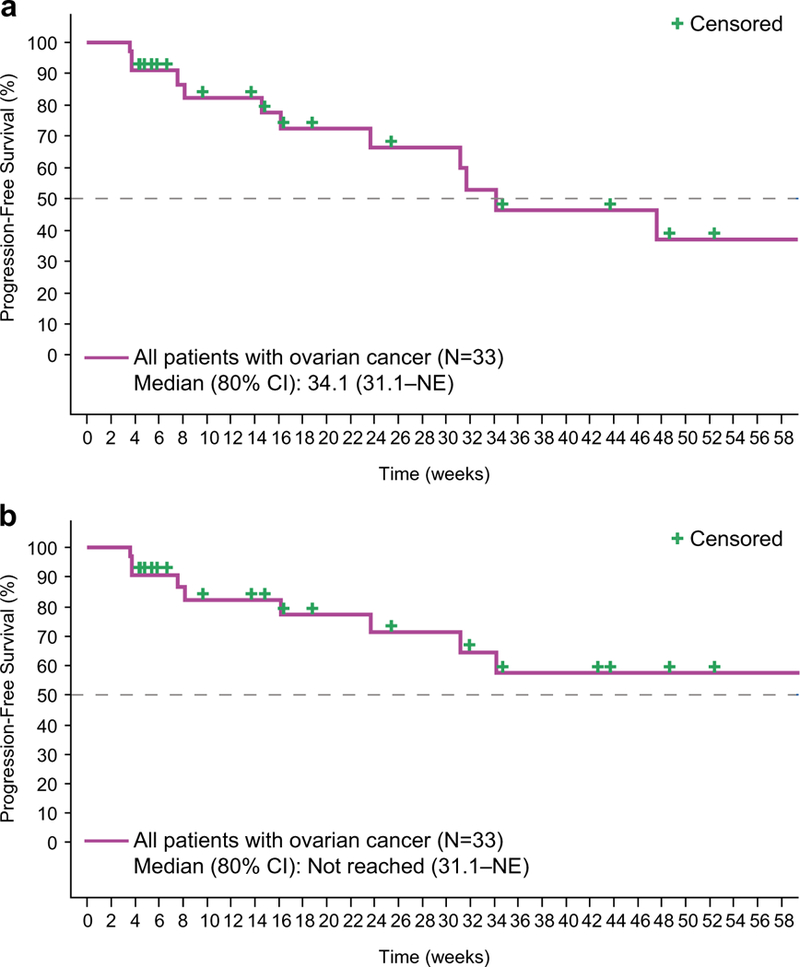
Progression-free survival in patients with ovarian cancer as determined by (a) RECIST 1.1 or by (b) Gynecologic Cancer InterGroup criteria (RECIST 1.1 and CA 125). NE = not estimable
Eleven of the 14 patients with TNBC and 10 of the 12 patients with CRPC were evaluable for tumor response. In patients with TNBC, 7 (50.0%) had stable disease as the best overall tumor response per RECIST 1.1 and 4 (28.6%) had progressive disease. One patient with TNBC who received 40 mg + G-CSF had a partial response per RECIST 1.0 (local read) that was not confirmed per RECIST 1.1 (central read). In patients with CRPC, 9 (75.0%) had stable disease and 1 (8.3%) had progressive disease. Maximum changes in the sum of longest diameters in patients with TNBC and CRPC are presented in Fig. 1b and c, respectively.
A total of 7 of 14 patients (50.0%) with TNBC and 5 of 12 patients (41.7%) with CRPC died or had progressive disease per RECIST 1.1 (central read). For patients with TNBC, median (80% CI) PFS was 7.6 (4.1–23.6) weeks. For patients with CRPC, median (80% CI) PFS was 32.4 (15.7–39.6) weeks.
Biomarkers
Sequencing of exons 2 to 11 of TP53 in the 67 patients available for analysis found ≥1 nucleotide aberration in each patient (100%); 53 patients (79%) had ≥1 nonsilent exon mutation. Preliminary data showed that in nine patients with ovarian cancer, five responders per GCIC criteria had positive p53 staining (>75% of cell nuclei) in tumor samples by IHC, whereas the four nonresponders showed negative p53 staining (<25% positive staining in cell nuclei). Of the 36 samples evaluable for AURKA amplification by FISH, one sample from a patient with ovarian cancer who had stable disease based on central and local read data had amplification, and one sample from a patient with ovarian cancer who had partial response based on central read and stable disease based on local read had borderline amplification. No correlations between protein expression and response were observed for the other markers evaluated (Aurora A, Aurora B, p21, or Ki-67).
Discussion
In this phase 1 study, AMG 900 had a manageable toxicity profile with G-CSF support and showed single-agent activity in heavily pretreated patients with taxane-resistant or -refractory tumors. One patient was still receiving AMG 900 as of September 26, 2017. The MTD of AMG 900 without G-CSF was established at 25 mg daily, for the schedule of 4 days on, 10 days off. DLTs were primarily neutropenia and thrombocytopenia; because neutropenia was dose limiting, dose escalation continued with G-CSF support. The MTD of AMG 900 with G-CSF support was 40 mg daily. Less than one-third of patients had treatment-related grade ≥ 4 AEs, and of the five deaths, none were considered treatment related. Although three of the deaths were cardiac related, two of these patients had previously received Adriamycin (doxorubicin hydrochloride), which may have increased their risk for cardiac-related death. Twelve percent of patients discontinued AMG 900 due to hematologic AEs. The most common treatment-related grade ≥ 3 AEs were neutropenia, anemia, leukopenia, and thrombocytopenia, which is consistent with the drug’s antimitotic mechanism of action. Clinical responses were observed in four patients with taxane- and platinum-resistant ovarian cancer. Together, these results indicate that pan-Aurora kinase inhibition with AMG 900 with G-CSF support could be delivered to patients at the dose and on the schedule used in this study. The use of AMG 900 in combination with G-CSF likely helped overcome the toxicities seen with prior Aurora kinase inhibitors and allowed higher doses to be explored [12].
The pharmacokinetics of AMG 900 was characterized by increased exposure with increasing dose and minimal accumulation after 4 days of dosing. Absorption was rapid with fast elimination that supported a daily dosing regimen. Exposure varied among patients in the expansion cohorts who received 40 mg once daily; higher AUC was observed in the ovarian cancer cohort than in the TNBC or CRPC cohorts, possibly as a result of lower body weight and body surface area. Patients in the ovarian cancer cohort had a higher incidence of grade ≥ 4 neutropenia than patients in the other cohorts, but they also had a higher rate of objective responses per RECIST 1.1.
Clinical responses to AMG 900 were observed in this group of heavily pretreated patients. Antitumor activity was particularly notable in the ovarian cancer cohort. Among the 29 patients with ovarian cancer in the dose-expansion phase, the ORR was 10.3% per RECIST 1.1 (central read) and 24.1% per GCIG criteria (central read); nearly three-quarters of patients had stable disease per RECIST 1.1. Importantly, disease control with AMG 900 in these patients was prolonged: the median PFS of 31.7 weeks with AMG 900 is considerably longer than the median PFS of approximately 14 weeks in patients with platinum-resistant recurrent ovarian cancer treated with chemotherapy alone in the AURELIA trial [24].
Limited antitumor activity was observed in patients with TNBC in the dose-expansion phase. No patients with TNBC had an objective response per RECIST 1.1 (central read). One patient had an objective response per RECIST 1.0 (local read). Half of the patients with TNBC had stable disease per RECIST 1.1 (central read) and median PFS was 7.6 weeks. Stable disease was the best response in patients with prostate cancer, indicating limited antitumor activity, but stable disease was in many cases quite prolonged; median PFS was 32.4 weeks.
In the exploratory biomarker analysis, all evaluable samples had TP53 mutations, consistent with the selection for this study of tumor types that typically show a high frequency of TP53 mutation. Presence of exon mutations (“loss of function” mutations) coincided with p53 expression by IHC, consistent with previous studies indicating an association between TP53 mutation and positive p53 IHC staining in ovarian tumors [25]; however, this biomarker was only assessed in a small group of patients. Nevertheless, these results support earlier data that showed that breast cancer cell lines with TP53 loss of function mutations had a greater sensitivity to AMG 900 compared with cell lines without such mutations [26]. No correlation was seen between AURKA FISH amplification and IHC for Aurora A and B.
Phase 1 and 2 trials of other Aurora kinase inhibitors have demonstrated efficacy in a variety of cancers, including solid tumors (eg, non-small-cell lung cancer, small-cell lung cancer, colorectal cancer, ovarian cancer, glioblastoma multiforme, neuroblastoma, prostate cancer, breast cancer) and hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphocytic leukemia, chronic myelogenous leukemia, myelofibrosis, multiple myeloma, B-cell lymphoma) [7–9, 27–31]. AEs associated with these inhibitors were broadly similar to those seen with AMG 900 [8, 9, 27, 29, 30]; however, there were a few differences. Some frequently occurring AEs or DLTs seen in several of these trials, such as mucositis and somnolence, were not prevalent in this study [7, 27, 29]. Most other trials did not assess Aurora kinase inhibitors in combination with G-CSF [7, 8, 27, 28, 30, 31].
As an adjunct to this study, we determined AURKA amplification levels by analyzing cell-free DNA in plasma collected from patients with recurrent ovarian cancer. We then compared those AURKA amplification levels to levels determined in the same patients at the time of initial diagnosis. We found that AURKA amplification in circulating tumor DNA from liquid biopsies was more frequent following multiple lines of therapy and therefore seems to be a late event in ovarian cancer progression (manuscript in preparation). Biomarker analyses in this study were conducted in archival tissue collected at the time of initial diagnosis rather than in fresh biopsies. The frequency of late events in tumor progression may therefore have been underestimated, and correlations between biomarker status and response may have been missed.
AMG 900 has a mechanism of action distinct from that of other antimitotics and has been shown to have antitumor activity in cell lines, mouse xenograft models [16, 32], and now in patients with taxane- and/or platinum-resistant or -refractory tumors. AMG 900 inhibited growth of TNBC cell lines and xenografts in combination with ixabepilone [32]. Aurora kinase inhibitors have demonstrated synergistic antitumor activity against multiple ovarian cancer cell lines when combined with CHEK1 inhibitors [33].
The tumor types evaluated in the dose-expansion phase were selected based on high unmet need after chemotherapy failure, association of AURKA amplification or Aurora kinase overexpression with advanced clinical stage and poor prognosis, and evidence of clinical activity with Aurora kinase inhibitors [3–5, 7, 17–19]. The combination of AMG 900 with G-CSF allowed the use of higher doses of AMG 900, potentially improving the therapeutic window relative to AMG 900 alone. Because this study was limited to patients with advanced solid tumors who could not be treated with standard therapy, investigators focused enrollment on patients with progressing disease. However, the rate of tumor progression was not measured in this study, limiting full assessment of antitumor activity. Furthermore, patients with TNBC were heavily pretreated. These patients typically progress rapidly, which limited our ability to dose the patients adequately to assess the activity of AMG 900 in that disease setting. Indeed, patients with TNBC had short median PFS.
The results of this study provide supportive evidence for the safety and tolerability of AMG 900 in heavily pretreated patients with chemotherapy-resistant ovarian, prostate, and breast tumors and efficacy in patients with taxane- and platinum-resistant ovarian tumors. AEs were manageable and single-agent activity was observed in refractory ovarian cancer. The clinical activity observed in ovarian cancer, together with previously presented preclinical data, suggests that AMG 900 at appropriate, tolerable doses in combination with other agents (eg, taxanes, doxorubicin, carboplatin, ixabepilone) may be a potential strategy for treating patients with ovarian cancer [32, 34].
Supplementary Material
Acknowledgements
This study was sponsored by Amgen Inc. Funding for writing of this manuscript was provided by Amgen Inc. Micah Robinson, PhD (Amgen Inc.), as well as Miranda Tradewell, PhD, James Balwit, MS, CMPP, and Rick Davis, MS (Complete Healthcare Communications, LLC; on behalf of Amgen Inc.) assisted with writing the manuscript.
Michael Carducci reports research funding from Amgen to his institution during the conduct of this study. Sara Hurvitz reports a grant from OBI Pharma during the conduct of this study, and grants/research support from Amgen Inc., Bayer, BI Pharma, Genentech, GSK, Lilly, Novartis, Pfizer, Roche, PUMA, Merrimack, Medivation, Dignitana, OBI Pharma, Biomarin, Cascadian, and Seattle Genetics outside the submitted work. Erik Rasmussen, Gloria Juan, Vincent Chow, and Gregory Friberg are employees of Amgen Inc. and hold Amgen stock. Florian Vogl was an employee of Amgen Inc. at the time this work was conducted and holds stock in Amgen Inc. Erick Gamelin was an employee of Amgen Inc. at the time this work was conducted. Jayesh Desai has received research grants from Novartis, Bionomics, and Roche/Genentech and has received fees for consulting from Amgen, Novartis, Bionomics, Lilly, and Eisai.
Footnotes
Compliance with ethical standards
Disclosure of potential conflicts of interest Montaser Shaheen, Ben Markman, Daruka Mahadevan, Dusan Kotasek, and Oscar Goodman declare they have no conflicts of interest.
Ethical approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent Informed consent was obtained from all individual participants included in the study.
Results of this study were previously presented in part at the American Society of Clinical Oncology Annual Meeting 2012, the 26th EORTC-NCI-AACR Symposium 2014, and the American Association for Cancer Research Annual Meeting 2016.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10637-018-0625-6) contains supplementary material, which is available to authorized users.
References
- 1.Carmena M, Earnshaw WC (2003) The cellular geography of Aurora kinases. Nat Rev Mol Cell Biol 4:842–854 [DOI] [PubMed] [Google Scholar]
- 2.Afonso O, Matos I, Pereira AJ, Aguiar P, Lampson MA, Maiato H (2014) Feedback control of chromosome separation by a midzone Aurora B gradient. Science 345:332–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, Borg A, Isola JJ (2000) Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 6:1833–1839 [PubMed] [Google Scholar]
- 4.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM (2005) Integrative genomic andproteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8:393–406 [DOI] [PubMed] [Google Scholar]
- 5.Ginestier C, Cervera N, Finetti P, Esteyries S, Esterni B, Adelaide J, Xerri L, Viens P, Jacquemier J, Charafe-Jauffret E, Chaffanet M, Birnbaum D, Bertucci F (2006) Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res 12: 4533–4544 [DOI] [PubMed] [Google Scholar]
- 6.D’Assoro AB, Haddad T, Galanis E (2015) Aurora-A kinase as a promising therapeutic target in cancer. Front Oncol 5:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falchook GS, Bastida CC, Kurzrock R (2015) Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin Oncol 42:832–848 [DOI] [PubMed] [Google Scholar]
- 8.Niu H, Manfredi M, Ecsedy JA (2015) Scientific rationale supporting the clinical development strategy for the investigational Aurora A kinase inhibitor alisertib in cancer. Front Oncol 5:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bavetsias V, Linardopoulos S (2015) Aurora kinase inhibitors: current status and outlook. Front Oncol 5:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaestner P, Stolz A, Bastians H (2009) Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol Cancer Ther 8:2046–2056 [DOI] [PubMed] [Google Scholar]
- 11.Girdler F, Gascoigne KE, Eyers PA, Hartmuth S, Crafter C, Foote KM, Keen NJ, Taylor SS (2006) Validating Aurora B as an anti-cancer drug target. J Cell Sci 119:3664–3675 [DOI] [PubMed] [Google Scholar]
- 12.Jackson JR, Patrick DR, Dar MM, Huang PS (2007) Targeted antimitotic therapies: can we improve on tubulin agents? Nat Rev Cancer 7:107–117 [DOI] [PubMed] [Google Scholar]
- 13.Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, Dogliotti L, Berardi R, Cappuzzo F, Tagawa ST, Sternberg CN, Jannuzzo MG, Mariani M, Petroccione A, de Wit R (2013) Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int 111:44–52 [DOI] [PubMed] [Google Scholar]
- 14.Seymour JF, Kim DW, Rubin E, Haregewoin A, Clark J, Watson P, Hughes T, Dufva I, Jimenez JL, Mahon FX, Rousselot P, Cortes J, Martinelli G, Papayannidis C, Nagler A, Giles FJ (2014) A phase 2 study of MK-0457 in patients with BCR-ABLT315I mutant chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Cancer J 4:e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asteriti IA, Di Cesare E, De Mattia F, Hilsenstein V, Neumann B, Cundari E, Lavia P, Guarguaglini G (2014) The Aurora-A inhibitor MLN8237 affects multiple mitotic processes and induces dose-dependent mitotic abnormalities and aneuploidy. Oncotarget 5: 6229–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P, Ross S, Cee VJ, Deak HL, Hodous BL, Nguyen HN, Olivieri PR, Romero K, Schenkel LB, Bak A, Stanton M, Dussault I, Patel VF, Geuns-Meyer S, Radinsky R, Kendall RL (2010) Preclinical evaluation of AMG 900, a novel potent and highly selective panaurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res 70:9846–9854 [DOI] [PubMed] [Google Scholar]
- 17.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, Dhir R, Nelson JB, de la Taille A, Allory Y, Gerstein MB, Perner S, Pienta KJ, Chinnaiyan AM, Wang Y, Collins CC, Gleave ME, Demichelis F, Nanus DM, Rubin MA (2011) Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 1:487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finetti P, Cervera N, Charafe-Jauffret E, Chabannon C, Charpin C, Chaffanet M, Jacquemier J, Viens P, Birnbaum D, Bertucci F (2008) Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res 68:767–776 [DOI] [PubMed] [Google Scholar]
- 19.Lee EC, Frolov A, Li R, Ayala G, Greenberg NM (2006) Targeting Aurora kinases for the treatment of prostate cancer. Cancer Res 66: 4996–5002 [DOI] [PubMed] [Google Scholar]
- 20.Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC (1997) Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst 89:1138–1147 [DOI] [PubMed] [Google Scholar]
- 21.US Department of Health and Human Services (2010) Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. National Institutes of Health; Available at: http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed 28 Feb 2017 [Google Scholar]
- 22.Rustin GJ, Vergüte I, Eisenhauer E, Pujade-Lauraine E, Quinn M, Thigpen T, du Bois A, Kristensen G, Jakobsen A, Sagae S, Greven K, Parmar M, Friedlander M, Cervantes A, Vermorken J, Gynecological Cancer InterGroup (2011) Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer 21:419–423 [DOI] [PubMed] [Google Scholar]
- 23.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247 [DOI] [PubMed] [Google Scholar]
- 24.Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, Sorio R, Vergote I, Witteveen P, Bamias A, Pereira D, Wimberger P, Oaknin A, Mirza MR, Follana P, Bollag D, Ray-Coquard I (2014) Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol 32:1302–1308 [DOI] [PubMed] [Google Scholar]
- 25.Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih IM, Kurman RJ (2011) Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol 24:1248–1253 [DOI] [PubMed] [Google Scholar]
- 26.Kalous O, Conklin D, Desai AJ, Dering J, Goldstein J, Ginther C, Anderson L, Lu M, Kolarova T, Eckardt MA, Langerod A, Borresen-Dale AL, Slamon DJ, Finn RS (2013) AMG 900, pan-Aurora kinase inhibitor, preferentially inhibits the proliferation of breast cancer cell lines with dysfunctional p53. Breast Cancer Res Treat 141:397–408 [DOI] [PubMed] [Google Scholar]
- 27.Cicenas J (2016) The Aurora kinase inhibitors in cancer research and therapy. J Cancer Res Clin Oncol 142:1995–2012 [DOI] [PubMed] [Google Scholar]
- 28.Schoffski P, Besse B, Gauler T, de Jonge MJ, Scambia G, Santoro A, Davite C, Jannuzzo MG, Petroccione A, Delord JP (2015) Efficacy and safety of biweekly i.v. administrations of the Aurora kinase inhibitor danusertib hydrochloride in independent cohorts of patients with advanced or metastatic breast, ovarian, colorectal, pancreatic, small-cell and non-small-cell lung cancer: a multi-tumour, multi-institutional phase II study. Ann Oncol 26:598–607 [DOI] [PubMed] [Google Scholar]
- 29.Cicenas J, Cicenas E (2016) Multi-kinase inhibitors, AURKs and cancer. Med Oncol 33:43. [DOI] [PubMed] [Google Scholar]
- 30.Borisa AC, Bhatt HG (2017) A comprehensive review on Aurora kinase: small molecule inhibitors and clinical trial studies. Eur J MedChem 140:1–19 [DOI] [PubMed] [Google Scholar]
- 31.Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J (2017) Aurora kinases: novel therapy targets in cancers. Oncotarget 8:23937–23954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bush TL, Payton M, Heller S, Chung G, Hanestad K, Rottman JB, Loberg R, Friberg G, Kendall RL, Saffran D, Radinsky R (2013) AMG 900, a small-molecule inhibitor of aurora kinases, potentiates the activity of microtubule-targeting agents in human metastatic breast cancer models. Mol Cancer Ther 12:2356–2366 [DOI] [PubMed] [Google Scholar]
- 33.Alcaraz-Sanabria A, Nieto-Jimenez C, Corrales-Sanchez V, Serrano-Oviedo L, Andres-Pretel F, Montero JC, Burgos M, Llopis J, Galan-Moya EM, Pandiella A, Ocana A (2017) Synthetic lethality interaction between Aurora kinases and CHEK1 inhibitors in ovarian cancer. Mol Cancer Ther 16:2552–2562 [DOI] [PubMed] [Google Scholar]
- 34.Kalous O, Conklin D, Manivong K, Wayne W, Hanestad K, Canon J, Loberg R, Friberg G, Gamelin E, Vogl FD, Juan G, Coxon A, Slamon D, Finn R, Payton M (2016) Preclinical characterization of AMG 900, a pan-aurora kinase inhibitor, alone and in combination with taxanes in ovarian cancer [abstract]. Cancer Res 76:3008 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.