

Abstract
Previous studies have shown that the cardiolipin (CL)-deficient yeast mutant, crd1Δ, has decreased levels of acetyl-CoA and decreased activities of the TCA cycle enzymes aconitase and succinate dehydrogenase. These biochemical phenotypes are expected to lead to defective TCA cycle function. In this study, we report that signaling and anaplerotic metabolic pathways that supplement defects in the TCA cycle are essential in crd1Δ mutant cells. The crd1Δ mutant is synthetically lethal with mutants in the TCA cycle, retrograde (RTG) pathway, glyoxylate cycle, and pyruvate carboxylase 1. Glutamate levels were decreased, and the mutant exhibited glutamate auxotrophy. Glyoxylate cycle genes were up-regulated, and the levels of glyoxylate metabolites succinate and citrate were increased in crd1Δ. Import of acetyl-CoA from the cytosol into mitochondria is essential in crd1Δ, as deletion of the carnitine-acetylcarnitine translocase led to lethality in the CL mutant. β-oxidation was functional in the mutant, and oleate supplementation rescued growth defects. These findings suggest that TCA cycle deficiency caused by the absence of CL necessitates activation of anaplerotic pathways to replenish acetyl-CoA and TCA cycle intermediates. Implications for Barth syndrome, a genetic disorder of CL metabolism, are discussed.
Keywords: β-oxidation, cardiolipin, carnitine shuttle, glyoxylate cycle, RTG pathway, TCA cycle
1. INTRODUCTION
Cardiolipin (CL) is synthesized in the mitochondrial inner membrane and interacts with many mitochondrial proteins and protein complexes [1, 2]. The yeast Saccharomyces cerevisiae has been a powerful tool for studying CL function. Yeast mutants lacking CL are viable, although they exhibit growth defects during stressful conditions, e.g. in response to elevated temperature [3, 4]. The CL synthase mutant, crd1Δ, which does not synthesize CL, exhibits numerous defects in mitochondrial function, including bioenergetics [5, 6], protein import, membrane potential homeostasis [7], and assembly of outer membrane complexes [8]. In addition to mitochondrial membrane and bioenergetic functions, recent reports indicate that CL plays a key role in iron homeostasis and energy metabolism. The crd1Δ mutant exhibits defective Fe-S biogenesis resulting in decreased activity of Fe-S enzymes [9]. These include two that function in the TCA cycle, aconitase and succinate dehydrogenase. Furthermore, acetyl-CoA levels are decreased in the crd1Δ mutant as a result of a defect in the pyruvate dehydrogenase (PDH) bypass pathway [10]. These studies predict that the TCA cycle is impaired in CL-deficient cells.
The TCA cycle is an amphibolic pathway that is critical for oxidation of acetyl-CoA and for the production of reducing equivalents used by oxidative phosphorylation complexes to generate ATP. In yeast cells actively growing on glucose, the TCA cycle is required for the generation of reducing equivalents as well as for the production of metabolites that support biosynthesis of amino acids, porphyrin, and pyrimidines, which are essential building blocks of macromolecules required for cellular growth [11, 12]. When TCA metabolites are limiting, signaling and metabolic anaplerotic pathways may be utilized to replenish them. One such pathway is the mitochondrial retrograde (RTG) pathway, a signal transduction mechanism whereby mitochondria communicate with the nucleus [13, 14]. The RTG pathway modulates the expression of nuclear genes required to replenish key TCA cycle metabolites, including α-ketoglutarate [15]. As α-ketoglutarate is a direct precursor for the synthesis of glutamate, the RTG pathway plays a key role in glutamate biosynthesis, and all three RTG mutants (rtg1Δ, rtg2Δ, and rtg3Δ) are glutamate auxotrophs [16–18]. Sufficient glutamate production is essential, as glutamate and its downstream metabolite, glutamine, provide all of the nitrogen used in biosynthetic reactions [19].
In addition to regulating gene expression through the RTG pathway, metabolic routes are utilized to replenish TCA cycle intermediates. These include the glyoxylate cycle, carboxylation of pyruvate to oxaloacetate (via pyruvate carboxylase), β-oxidation, and the carnitine shuttle (Fig. 1). In cells with dysfunctional mitochondria, there is a concomitant increase in peroxisome biogenesis and the expression of genes involved in these anaplerotic pathways, which can compensate for a defective TCA cycle by producing acetyl-CoA, succinate, and citrate [15].
FIGURE 1. Metabolic pathways for replenishing TCA cycle intermediates.
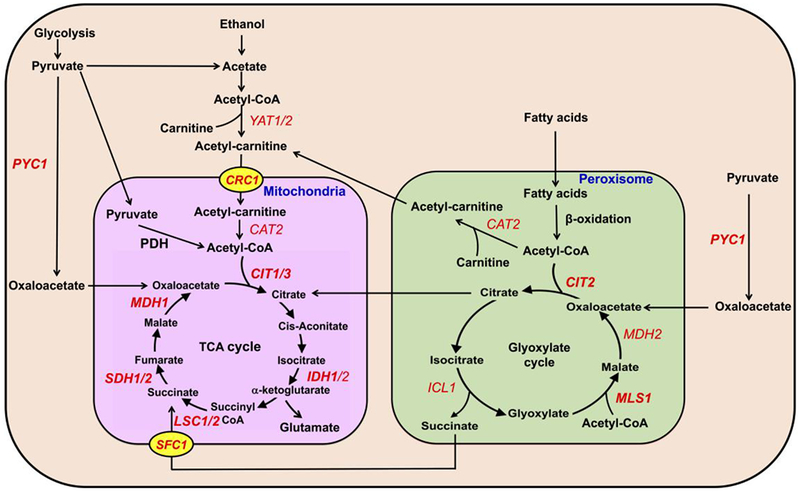
The TCA cycle carries out the oxidation of acetyl-CoA for the production of reducing equivalents. Additionally, TCA cycle intermediates are precursors for other biosynthetic pathways (not shown). Anaplerotic pathways such as pyruvate carboxylase (PYC1) and the glyoxylate cycle can generate oxaloacetate, succinate, and citrate, which can directly replenish TCA cycle intermediates diminished by utilization in biosynthetic pathways. Peroxisomal and cytosolic acetyl-CoA derived from β-oxidation and acetate may be imported into the mitochondria by the carnitine shuttle through the carnitine acetylcarnitine translocase (CRC1). Genes that are synthetically lethal or sick with crd1Δ are in bold. For simplicity, the glyoxylate cycle is depicted inside of the peroxisome, but several enzymes are cytosolic [21].
Acetyl-CoA in the peroxisome can enter the glyoxylate cycle, which has been described as a “modified TCA cycle” [20]. The glyoxylate cycle incorporates two units of acetyl-CoA into one unit of the four-carbon dicarboxylic acid, succinate (Fig. 1). This stepwise process is mediated by five enzymes, which are located both inside and outside of the peroxisome [21]. These include malate dehydrogenase (Mdh2), citrate synthase (Cit2), aconitase (Aco1), isocitrate lyase (Icl1), and malate synthase (Mls1) (Fig. 1). The reactions catalyzed by the first three enzymes listed above also occur in the TCA cycle, while the last two enzymatic reactions are unique to the glyoxylate cycle. At the start of the cycle, an acetyl-CoA unit is condensed with oxaloacetate (via Cit2) to form citrate, which undergoes isomerization (via Aco1) to isocitrate. Icl1 cleaves the six-carbon isocitrate to form succinate and glyoxylate. Succinate, the net product of the glyoxylate cycle, is transported into mitochondria by the succinate-fumarate transporter (Sfc1) [22], where it is used to replenish intermediates of the TCA cycle. Alternatively, glyoxylate is condensed with an acetyl-CoA unit by Mls1 to generate malate, which is oxidized by Mdh2 to form oxaloacetate that can be recycled for another round of the glyoxylate cycle [21]. Most of the yeast glyoxylate cycle mutants, including icl1Δ, mls1Δ, cit2Δ, aco1Δ, and mdh2Δ, are viable and do not exhibit growth defects when grown on glucose [23]. Oxaloacetate can also be synthesized from pyruvate by pyruvate carboxylase [24, 25]. It can then enter the mitochondrial matrix and be utilized in the TCA cycle, or it can be converted to citrate by the glyoxylate cycle and then exported to the mitochondria (Fig. 1).
Acetyl-CoA can also be produced from β-oxidation of fatty acids, which occurs only in peroxisomes in yeast [26]. Acetyl-CoA produced from β-oxidation can be utilized in the glyoxylate cycle to synthesize metabolites required for the TCA cycle, or it can be transported out of peroxisomes and into mitochondria through the carnitine shuttle [27–29]. In this pathway, acetyl-CoA is converted to acetylcarnitine by the carnitine acetyltransferase, Cat2 in peroxisomes, or Yat1/Yat2 in the cytoplasm. Acetylcarnitine is transported into mitochondria by the inner-membrane carnitine-acetylcarnitine translocase (Crc1) and converted back to carnitine and acetyl-CoA by mitochondrial Cat2 [29, 30]. Mitochondrial acetyl-CoA can then enter the TCA cycle (Fig. 1).
In wild-type yeast cells, most anaplerotic pathway mutations are not lethal. However, in the current study, we demonstrate for the first time that the mitochondrial retrograde pathway, glyoxylate cycle, and carnitine shuttle are essential for the viability of cells lacking CL, which are deficient in TCA cycle enzymes and acetyl-CoA synthesis.
2. MATERIAL AND METHODS
2.1. Yeast strains and growth media -
The S. cerevisiae strains used in this study are listed in Table 1. Single deletion mutants were obtained from the yeast knockout deletion collection (Invitrogen). Synthetic complete medium contained adenine (20.25 mg/liter), arginine (20 mg/liter), histidine (20 mg/liter), leucine (60 mg/liter), lysine (200 mg/liter), methionine (20 mg/liter), threonine (300 mg/liter), tryptophan (20 mg/liter), uracil (20 mg/liter), yeast nitrogen base without amino acids (Difco), and either glucose (2%) (YNBD), or galactose (2%) (YNBG) as a carbon source. Synthetic dropout medium contained all ingredients mentioned above but lacked the indicated amino acids. Sporulation medium contained potassium acetate (1%), glucose (0.05%), and the essential amino acids required for sporulation. Complex medium contained yeast extract (1%), peptone (2%), and glucose (2%) (YPD). Solid medium was prepared by adding 2% agar. Cells used for carnitine, β-oxidation, and enzyme assays were grown on minimal glucose medium (YNBD), inoculated at an A600 of 0.2 in YNB with 0.3% glucose medium, and grown for 8 hours. Finally, the strains were inoculated into rich oleate medium (YPO; 1% yeast extract, 2% peptone, 0.12% oleic acid, 0.2% Tween 80) at an A600 of 0.05 and grown for 16 hours.
TABLE 1.
Yeast strains used in this study.
| Strains | Genotype | Source or Ref. |
|---|---|---|
| FGY3 | MATa, ura3-52, lys2-801, ade2-101, trpl-Δ1, his3-Δ200, leu2- Δ1 | [4] |
| FGY2 | MATa, ura3-52, lys2-801, ade2-101, trpl-Δ1, his3-Δ200, leu2- Δ1, crd1Δ::URA3 | [4] |
| BY4741 | MATa, his3Δ1, leu2Δ0, ura3Δ0, met15Δ0 | Invitrogen |
| BY4742 | MATα, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0 | Invitrogen |
| CG922-a | MATa, lys2-801, ade2-101, trp1Δ1, his3Δ200, leu2Δ1, crd1Δ::URA3 | Invitrogen |
| CG923-α | MATα, lys2-801, ade2-101, trp1Δ1, his3Δ200, leu2Δ1, crd1Δ::URA3 | Invitrogen |
| crd1Δ | MATa, his3Δ1, len2Δ0, met15Δ0, ura3Δ0, crd1Δ::KanMX4 | Invitrogen |
| mls1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mls1Δ::KanMX4 | Invitrogen |
| cit2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cit21Δ::KanMX4 | Invitrogen |
| crc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, crc1Δ::KanMX4 | Invitrogen |
| sfc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sfc1Δ::KanMX4 | Invitrogen |
| cit1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cit1Δ::KanMX4 | Invitrogen |
| cit3Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cit3Δ::KanMX4 | Invitrogen |
| idh1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, idh1Δ::KanMX4 | Invitrogen |
| lsc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lsc1Δ::KanMX4 | Invitrogen |
| lsc2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lsc2Δ::KanMX4 | Invitrogen |
| sdh1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sdh1Δ::KanMX4 | Invitrogen |
| sdh2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sdh2Δ::KanMX4 | Invitrogen |
| mdh1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mdh1Δ::KanMX4 | Invitrogen |
| pyc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pyc1Δ::KanMX4 | Invitrogen |
| pyc2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pyc2Δ::KanMX4 | Invitrogen |
| rtg1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rtg1Δ::KanMX4 | Invitrogen |
| rtg2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rtg2Δ::KanMX4 | Invitrogen |
| rtg3Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rtg3Δ::KanMX4 | Invitrogen |
| mls1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mls1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| cit2Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cit21Δ::KanMX4, crd1Δ::KanMX4 | This study |
| crc1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, crc1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| sfc1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sfc1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| cit1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cit1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| cit3Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cit3Δ::KanMX4, crd1Δ::KanMX4 | This study |
| idh1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, idh1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| lsc1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lsc1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| lsc2Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lsc2Δ::KanMX4, crd1Δ::KanMX4 | This study |
| sdh1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sdh1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| sdh2Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sdh2Δ::KanMX4, crd1Δ::KanMX4 | This study |
| mdh1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mdh1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| pyc1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pyc1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| pyc2Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pyc2Δ::KanMX4, crd1Δ::KanMX4 | This study |
| rtg1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rtg1Δ::KanMX4, crdlΔ::KanMX4 | This study |
| rtg2Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rtg2Δ::KanMX4, crd1Δ::KanMX4 | This study |
| rtg3Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, rtg3Δ:KanMX4, crd1Δ::KanMX4 | This study |
2.2. Construction of double mutants and assessment of synthetic genetic interaction -
The crd1Δ::URA3 mutant was crossed with mutants of the opposite mating type obtained from the yeast deletion collection. The heterozygous diploids were selected on dropout medium lacking methionine and lysine, sporulated, and tetrads were dissected. Synthetic interaction between crd1Δ and deletion mutants was determined by examining the growth of the double mutant compared to isogenic parent strains and wild-type cells on YPD.
2.3. Sample preparation, metabolite extraction, NMR Spectroscopy analysis and quantification –
Cells were cultured in YNBG until stationary phase. Cells were collected, and metabolites were extracted by vortexing with 70% ethanol. After centrifugation, supernatants were filtered by Amicon® Ultra-0.5 mL centrifugal filters (Millipore) to remove large particles (more than 3 kDa). Samples were dried under vacuum (Savant DNA 120) and metabolites were analyzed by NMR spectroscopy as mentioned elsewhere [31]. Briefly, samples were prepared by adding D2O and reference buffer to the dried samples. The 1H NMR spectroscopy was conducted at 600 MHz in an Agilent spectrophotometer (Agilent Technologies, Santa Clara, CA) at 25°C, using the nuclear overhauser effect spectroscopy (NOESY) pulse sequence with presaturation. Metabolite identification and quantification were conducted using Chenomx NMR Suite 7.6 software (Chenomx Inc., Edmonton, Canada).
2.4. β-oxidation assay –
Cells grown on oleate as the sole carbon source were washed with water and resuspended in phosphate buffered saline (PBS) to an A600 of 2.5. Aliquots of 20 μl of cell suspension were used for fatty acid β-oxidation measurements in 200 μl of medium containing PBS plus 10 μM [1-14C]-palmitate or [l-14C]-octanoate. Reactions were allowed to proceed for 6 or 12 min at 30°C, followed by termination of reactions by adding 100 μl of 1.3 M perchloric acid. Radiolabeled CO2 was trapped overnight in 500 μl of 2 M NaOH. The 14C-labelled β-oxidation products were subsequently collected after extracting the acidified material with chloroform/methanol/heptane as described [32] and quantified in a liquid scintillation counter. The β-oxidation capacity of wild-type cells grown on oleate in each experiment was taken as a reference (100%).
2.5. Carnitine acetyltransferase (Cat2) assay -
Oleate-grown cells (20 ml) were washed twice and pelleted. The cell homogenate was prepared by disrupting the cell pellet in 500 μl of 0.9% NaCl with 200 μl of glass beads for 5 min by vortexing at 4°C. Cell debris was removed by centrifugation for 1 min at 13,000 rpm. For measurement of carnitine acetyltransferase, the assay mixture (final volume of 100 μl) was composed of 40 mM HEPES (2 M; pH 7.4), 0.1% triton-x-100, 1 mM carnitine, 60 μM acetyl-CoA, 200,000 dpm [1-C14]-acetyl-CoA, and 40 μl of homogenate. The reaction mixture was incubated for 10 min at 28°C. The reaction was stopped with 500 μl of ice-cold ethanol and acetylcarnitine was separated from acetyl-CoA with an AG11-X8 column. The column was washed twice with 500 μl ethanol and [1-C14]-acetylcarnitine was measured.
2.6. Carnitine-acetylcarnitine translocase (CACT) assay -
Carnitine acetylcarnitine translocase activity was measured in spheroplasts prepared from wild-type or mutant cells grown on oleate. Activity measurements were performed in a medium containing 1.2 M sorbitol, 50 mM KPi pH 7.5, 1 mM EDTA, 200,000 d.p.m. [1-14C] acetylcarnitine (5 μM) and digitonized (20 μg/ml) spheroplasts (100 μg protein). This digitonin concentration selectively permeabilizes the plasma membrane, as demonstrated by complete release of the cytosolic marker enzyme phosphoglucose isomerase (PGI), whereas intracellular membranes of mitochondria and peroxisomes remain intact [33]. Reactions were allowed to proceed for 10 min at 28°C, and subsequently stopped by the addition of 100 μl 1.3 M perchloric acid. Radiolabeled CO2 was trapped overnight in 500 μl of 2 M NaOH.
2.7. Carnitine measurement -
Oleate-grown cells (20 ml) were washed twice and disrupted by vigorously vortexing for 30 min at 4°C with ~200 μl of glass beads in an end volume of 400 μl. Cell debris (75 μl) was mixed and subsequently deproteinized with 500 μl acetonitrile and centrifuged for 15 min at 12,000 rpm. The resulting supernatant was dried under nitrogen at 45°C and subsequently derivatized in 100 μl butanol-HCl for 15 min at 60°C. Samples were dried under nitrogen at 45°C and re-dissolved in 140 μl acetonitrile. Free carnitine was measured as described by [34].
2.8. Quantitative PCR (qPCR) -
Cultures (10 ml) were grown to the stationary growth phase, cells were harvested, and total RNA was isolated using the RNeasy Plus Mini kit from Qiagen. Complementary DNA (cDNA) was synthesized using a Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science) according to the manufacturer’s recommendations. qPCRs were performed in a 25 μl volume using Brilliant III Ultra-Fast SYBR Green qPCR Master Mix (Agilent Technologies, Santa Clara, CA). The primers for qPCR are listed in Table 2. PCRs were initiated at 95°C for 10 min for denaturation followed by 40 cycles consisting of 30 s at 95°C and 60 s at 55°C. RNA levels were normalized to the ACT1 gene encoding actin. Relative values of mRNA transcripts are shown as fold change relative to indicated controls. Primer sets were validated according to the Methods and Applications Guide from Agilent Technologies. Optimal primer concentrations were determined, and primer specificity of a single product was monitored by a melt curve following the amplification reaction. All primers were validated by measurement of PCR efficiency and have calculated reaction efficiencies between 90 and 110%.
TABLE 2.
Primers used for qPCR analyses.
| Gene | Primer | Sequence |
|---|---|---|
| ACT1 | Forward | GATTCTGAGGTTGCTGCTTTG |
| Reverse | TTGACCCATACCGACCATGA | |
| TDH1 | Forward | AAGGGTACTGTTTCCCATGAC |
| Reverse | CAAGTTAGCTGGGTCTCTTTCT | |
| CIT2 | Forward | TCGTTATATGGCTCAGCGTAAG |
| Reverse | CCAGGTGCTACCTCGTATATTG | |
| ICL1 | Forward | GGTGGGACGCAATGTTCTAT |
| Reverse | CTGTTGGAAGTCTGGGTAGTTAG | |
| MLS1 | Forward | GGCCAACTTGCCCACTATTA |
| Reverse | CAAAGATGGAAGCGCTGATTG | |
| MDH2 | Forward | GTCCCAGTGATGGTTTCTAACA |
| Reverse | TGACACCCATGATCCTTCTTTC |
2.9. Acetyl-CoA determination -
Cells were grown to the logarithmic growth phase and a sample (3 ml) of the culture was centrifuged at 1700 g. Pellets were resuspended in 1 ml ddH2O and centrifuged for 1 min at 9300 g. The resulting cell pellet was subjected to cell lysis and the lysed samples were rapidly quenched with 130 μl of 45:45:10 acetonitrile / methanol / H2O + 0.1% glacial acetic acid and spiked with 10 μmol l−1 glutaryl-CoA as an internal standard. The resuspended extract was incubated on ice with intermittent vortexing for 15 min. An equal molar volume of ammonium hydroxide was added post incubation to neutralize the acetic acid, and each extract was centrifuged for 3 min at 15,700 g, transferred to a new 1.5 ml microcentrifuge tube and centrifuged for 5 min at 15,700 g. The clarified extract (10 μl) was injected for HPLC-MS/MS analysis.
3. RESULTS
3.1. crd1Δ cells exhibit TCA cycle defects –
Previous reports of decreased acetyl-CoA and defective activities of Fe-S-requiring TCA cycle enzymes in crd1Δ cells have established an intriguing link between CL and the TCA cycle, leading to the prediction that further disruption of the TCA cycle in crd1Δ would exacerbate the growth defects observed in these cells. To test this possibility, TCA cycle mutants were screened for synthetic lethality with crd1Δ. Consistent with the prediction, lsc1Δcrd1Δ, lsc2Δcrd1Δ, sdh1Δcrd1Δ, sdh2Δcrd1Δ, and mdh1Δcrd1Δ mutants showed synthetic lethality at 37°C (Fig. 2A). Double mutants of crd1Δ cells with mutations in CIT1, CIT3, and IDH1, which are redundant enzymes, exhibited a less severe (synthetically sick) phenotype at 37°C. Glutamate, which is synthesized from α-ketoglutarate, was significantly decreased in crd1Δ cells (Fig. 2B). Consistent with this, growth of crd1Δ at 39°C was restored by supplementation of glutamate (Fig. 2C). These phenotypes are consistent with a defective TCA cycle in the crd1Δ mutant.
FIGURE 2. Metabolic deficiencies in crd1Δ.
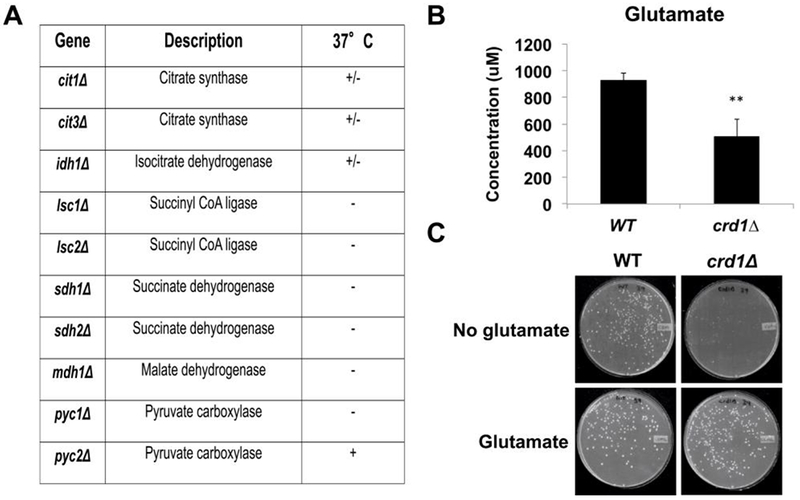
(A) Genetic interaction between crd1Δ and TCA cycle mutants. Cells were pre-cultured in YPD overnight, serially diluted, plated on YNBD, and incubated at 37°C for 4 days. (B) Glutamate levels were determined by NMR spectroscopy and quantified by CHENOMX NMR suite (n=3). (C) Glutamate supplementation rescues the temperature-sensitive phenotype of crd1Δ. Cells were pre-cultured overnight in YPD at 30°C, washed and plated onto synthetic medium with or without 0.05% glutamate and incubated at 39°C for 4 days. (**, p < 0.01)
3.2. The RTG pathway is essential for growth of crd1Δ cells –
Decreased activity of the TCA cycle in crd1Δ cells suggested that the RTG pathway may be required to ameliorate the glutamate deficiency. The requirement for the RTG pathway is demonstrated by synthetic lethality of crd1Δ cells with rtg1Δ, rtg2Δ, and rtg3Δ mutants at 37°C (Fig. 4A).
3.3. The glyoxylate cycle and pyruvate carboxylase 1 are essential in crd1Δ cells –
As discussed above, the glyoxylate cycle is an anaplerotic pathway that synthesizes TCA cycle intermediates independent of the TCA cycle enzymes. Interestingly, glyoxylate cycle genes were up-regulated in crd1Δ cells. MLS1, which encodes malate synthase, was significantly up-regulated about two-fold in the BY4741 genetic background. In the FGY3 genetic background [3], in which CL deficiency leads to a more severe phenotype than in BY4741, mRNA levels of CIT2, ICL1, in addition to MLS1, were elevated (Fig. 3). Consistent with increased expression of these enzymes, the glyoxylate cycle metabolites succinate and citrate were elevated as well (Fig. 5). Deletion of glyoxylate genes was found to be lethal in crd1Δ cells. The mls1Δ mutant was synthetically lethal with crd1Δ cells even at the optimal growth temperature of 30°C, as reflected by the inability to form colonies from single cells at this temperature (Fig. 4B). Other mutants of the glyoxylate cycle, including cit2Δ and sfc1Δ, were synthetically lethal with crd1Δ at 37°C (Fig. 4C). Pyruvate carboxylase, an anaplerotic enzyme that produces oxaloacetate in the cytoplasm, was also determined to be essential in crd1Δ cells, as pyclΔ was synthetically lethal with crd1Δ at 37°C, although crd1Δpyc2Δ was viable (Fig. 2A).
FIGURE 3. crd1Δ exhibits increased expression of glyoxylate cycle genes.
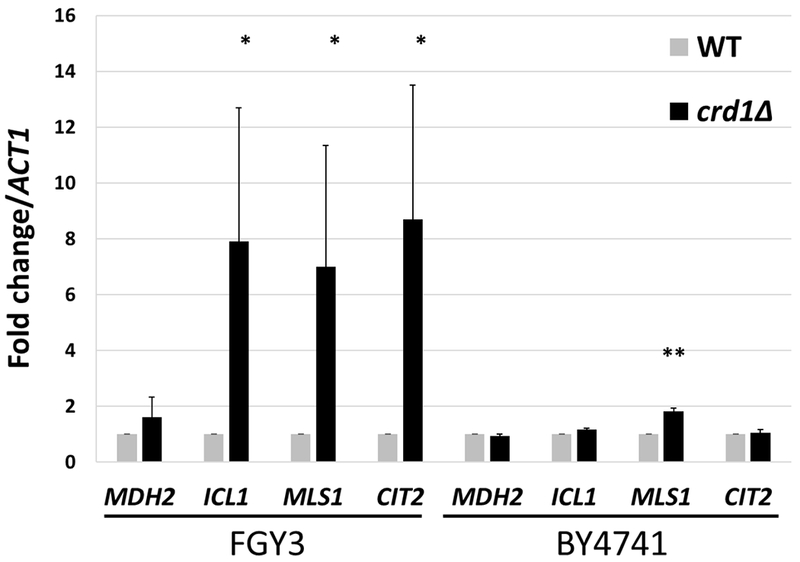
Total RNA was extracted from WT and crd1Δ cells, in FGY3 and BY4741 strain backgrounds, grown to stationary phase in YPD at 30°C. mRNA levels of glyoxylate cycle genes were determined by real-time PCR with MDH2 as a control. Expression was normalized to the mRNA levels of the internal control ACT1. Values are mean ± S.D. from three independent experiments with technical duplicates. (*, p < 0.05; **, p < 0.01).
FIGURE 5. Succinate and citrate levels are increased in crd1Δ.
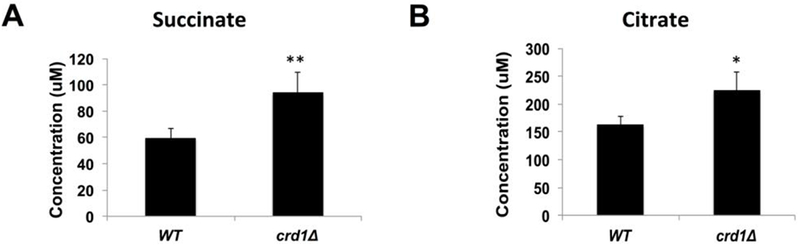
Succinate and citrate levels in WT and crd1Δ were analyzed by NMR spectroscopy and quantified by CHENOMX NMR suite (n=3). (*, p < 0.05; **, p < 0.01).
FIGURE 4. crd1Δ is synthetically lethal with mutants in the RTG pathway, glyoxylate cycle, and carnitine shuttle.
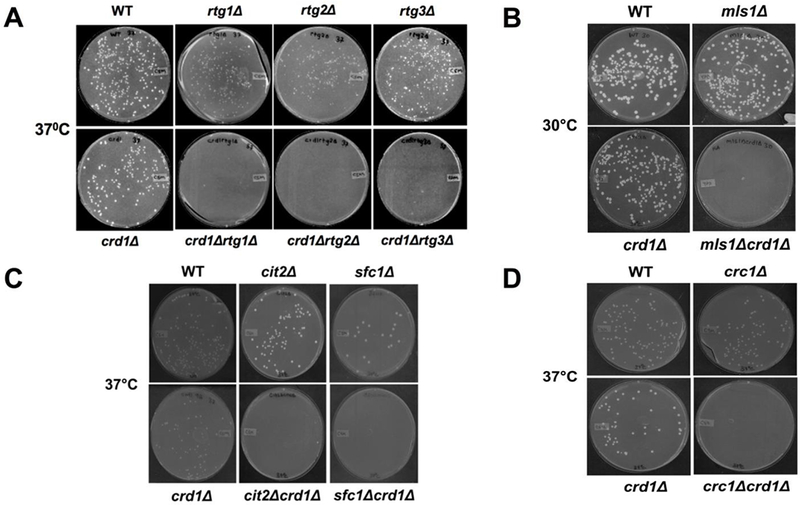
Genetic interaction between crd1Δ and rtg mutants (A), glyoxylate mutants, (B and C), and carnitine shuttle mutant crc1Δ (D) was determined. For experiments in A, C, and D, cells were pre-cultured in YPD overnight, serially diluted, plated on YNBD, and incubated at 37°C for 4 days. For the experiment in B, crd1Δmls1Δ cells recovered as tiny colonies from tetrad dissection were pre-cultured in YPD overnight, serially diluted, plated on YPD, and incubated at 30°C for 4 days.
3.4. Carnitine-acetylcarnitine translocase is essential in crd1Δ cells –
Acetyl-CoA that is produced from acetate in the cytosol and fatty acid oxidation in the peroxisome must be transported across the mitochondrial membrane by the carnitine shuttle in order to be available for mitochondrial metabolism. Genetic interaction studies indicated that the carnitine-acetylcarnitine translocase mutant crc1Δ exhibited synthetic lethality with crd1Δ at 37°C (Fig. 4D), indicating that CL-deficient cells require the carnitine shuttle to maintain mitochondrial acetyl-CoA levels. In agreement with this, growth of crd1Δ at elevated temperature was restored by supplementation of carnitine or acetylcarnitine (Fig. 6A). Unlike the glyoxylate cycle, which was up-regulated in crd1Δ cells, activities of carnitine acetyltransferase and carnitine-acetylcarnitine translocase in the mutant were similar to those of wild-type cells (Fig. 6B, C). Carnitine levels were marginally, but not significantly, reduced (~14%) in crd1Δ cells (Fig. 6D). These findings suggest that crd1Δ cells depend on carnitine-mediated acetyl-CoA transport from the cytoplasm to support mitochondrial metabolism.
FIGURE 6. Carnitine rescues growth of crd1Δ cells.
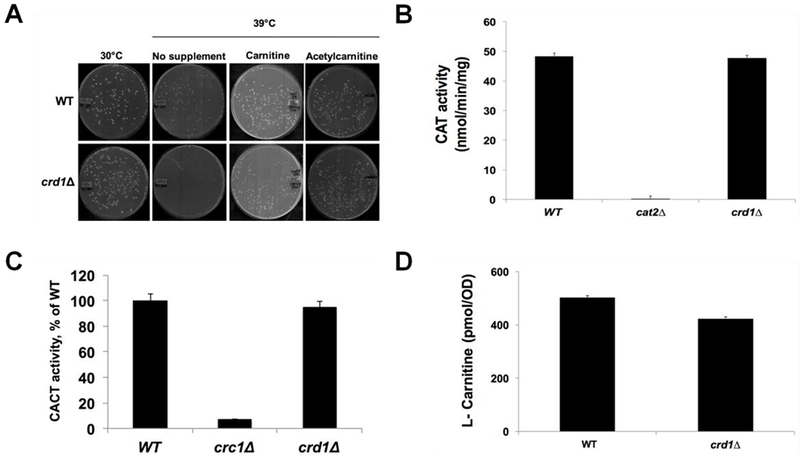
(A) Supplementation of carnitine and acetylcarnitine restored the growth of crd1Δ at elevated temperature. Cells were pre-cultured in YNBD overnight, serially diluted, and plated on YNBD with or without carnitine or acetylcarnitine (500 mg/1). Plates were incubated at 30°C or 39°C for 4 days. (B) Carnitine acetyltransferase activity and (C) carnitine-acetylcarnitine translocase activity are not altered in crd1Δ. Wild-type and crd1Δ cells were grown in YPO and enzymatic activity was assayed (n=3). (D) Carnitine levels are marginally decreased in crd1Δ. Wild-type and crd1Δ cells were grown in YPO. Total intracellular carnitine levels were determined by tandem mass spectrometry (n=3).
3.5. β-oxidation can restore growth of crd1Δ –
Acetyl-CoA can be generated through β-oxidation of fatty acids in addition to synthesis from pyruvate and acetate. β-oxidation activity in crd1Δ cells was similar to that of wild-type (Fig. 7A), and supplementation of oleate rescued crd1Δ temperature sensitivity (Fig. 7B). Our previous study indicated that acetyl-CoA levels in crd1Δ cells are decreased to about 11% of wild-type levels when grown on glucose at 39°C [10]. In contrast, levels of acetyl-CoA at 39°C in crd1Δ cells supplemented with oleate were about 74% of levels in wild-type cells (Fig. 7C). These findings suggest that energy deficits in crd1Δ cells can be rescued by β-oxidation.
FIGURE 7. β-oxidation can restore acetyl-CoA levels in crd1Δ cells.
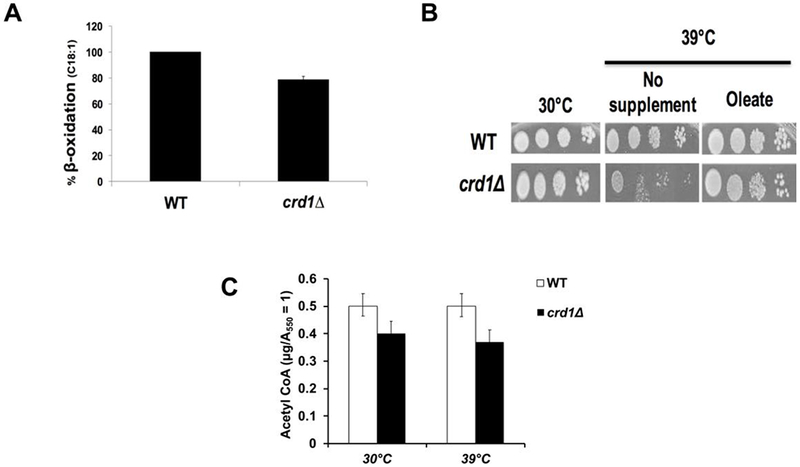
(A) β-oxidation activity of crd1Δ cells compared to WT (100%). Cells were grown on YPO and incubated with [1-14C]-fatty acids. 14CO2 was quantified to determine β-oxidation activity (n=3). (B) Supplementation of oleate restored the growth of crd1Δ at elevated temperature. Cells were pre-cultured in YPD overnight, serially diluted, and plated on YPD with oleate (0.1%) + tergitol (1%). Plates were incubated at 30°C or 39°C for 4 days. (C) Wild-type and crd1Δ cells were grown at the indicated temperatures in YPD with oleate (0.1%) + tergitol (1%) until cells reached an A550 of 1. Cells were pelleted, and acetyl-CoA was extracted. Data shown are mean ± S.D. (n = 6).
4. DISCUSSION
In this study, we report for the first time that yeast cells lacking the phospholipid CL exhibit TCA cycle-associated defects that necessitate utilization of the mitochondrial retrograde RTG signaling pathway as well as activities of metabolic anaplerotic pathways, including the glyoxylate cycle, pyruvate carboxylase, and the carnitine shuttle for viability.
In yeast, the two main routes to replenish intermediates of the TCA cycle are the glyoxylate cycle and oxaloacetate synthesis from pyruvate carboxylase [35]. The importance of the glyoxylate cycle and pyruvate carboxylase to CL-deficient cells is underscored by genetic interactions of crd1Δ with mutants in Mls1, Sfc1, Cit2, and Pyc1 (Figs. 2A, 4B, 4C), up-regulated expression of glyoxylate cycle genes (Fig. 3), and increased levels of citrate and succinate (Fig. 5). Several mechanisms may lead to increased citrate and succinate in crd1Δ cells. Up-regulation of the glyoxylate pathway, as shown in Fig. 3, is expected to increase levels of these intermediates. Decreased aconitase and succinate dehydrogenase activities, as previously reported in crd1Δ cells [9], could lead to increased citrate and succinate [36, 37]. Finally, activation of the RTG pathway, which is required for viability of crd1Δ cells (Fig. 4A), has been shown to be essential for accumulation of these metabolites [36].
The carnitine shuttle transports acetyl-CoA produced in the cytosol or peroxisomes into mitochondria. The finding that a mutant in the carnitine-acetylcarnitine translocase (Crc1) is synthetically lethal with crd1Δ (Fig. 4D) and that carnitine rescues the growth defect of the crd1Δ mutant (Fig. 6A) suggests that CL-deficient cells require cytosolic supplementation of acetyl-CoA for TCA cycle activity. However, the mitochondrial source of acetyl-CoA production, dehydrogenation of pyruvate through PDH, is unaffected in the crd1Δ mutant [10]. Therefore, the need for cytosolic acetyl-CoA supplementation suggests that crd1Δ cells require higher amounts of mitochondrial acetyl-CoA. The implication of this finding is that PDH activity cannot supply enough acetyl-CoA to support crd1Δ growth. This is consistent with a previous study indicating that addition of CL to mitochondria isolated from crd1Δ substantially increased PDH activity [10]. The finding that oleic acid supplementation rescued growth of crd1Δ cells (Fig. 7B) and increased acetyl-CoA levels in the mutant from 11% [10] to 74% of wild-type levels (Fig. 7C) suggests that β-oxidation produces sufficient acetyl-CoA to alleviate the deficiencies of the mutant when exogenous substrate is available.
The current study may have implications for the severe genetic disorder, Barth syndrome, which results from mutations in the CL remodeling enzyme, tafazzin [38, 39]. Loss of tafazzin results in decreased levels of CL as well as increased levels of monolysocardiolipin [40]. Notably, these biochemical phenotypes have been observed across the evolutionary spectrum in tafazzin-deficient cells, from yeast to humans. Analysis of the crd1Δ mutant contributes insight specifically into the biochemical phenotype of loss of CL, which occurs in tafazzin-deficient cells. Importantly, the NMR-metabolomics study that showed decreased glutamate in the crd1Δ mutant (Fig. 2B) also revealed a significant decrease in glutamate levels in the yeast tafazzin-deficient mutant, taz1Δ (data not shown). The clinical presentation of Barth syndrome includes cardiomyopathy and skeletal muscle weakness, exercise intolerance, as well as lactic acidosis and other metabolic abnormalities [41–43]. Unpublished findings also suggest that BTHS patients have low levels of specific amino acids [43]. Importantly, the disorder is characterized by wide disparities in clinical phenotypes, strongly suggesting that the outcome of CL deficiency is exacerbated by physiological modifiers [44]. The present study demonstrates that in yeast, CL deficiencies lead to abnormalities in the TCA cycle that require anaplerotic pathways to replenish key metabolites. This suggests the possibility that anaplerotic pathways may be physiological modifiers of Barth syndrome, and may also identify possible routes whereby metabolic deficiencies may be supplemented.
Highlights.
Yeast cardiolipin-deficient (crd1Δ) cells exhibit TCA cycle deficiencies.
Mitochondrial retrograde signaling is essential for growth of crd1Δ.
Anaplerotic metabolic pathways are required for crd1Δ viability.
Carnitine-acetylcarnitine translocase is essential for crd1Δ growth.
β-oxidation of supplemented fatty acids can rescue growth of crd1Δ cells.
ACKNOWLEDGMENTS
We thank Dr. Krishna Rao Maddipati for help with acetyl-CoA measurement and Dr. Satya Kiran Koya for help with qPCR statistical analysis. This work was supported by the National Institutes of Health grant HL117880 to M.L.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- [1].Houtkooper RH, Vaz FM, Cardiolipin, the heart of mitochondrial metabolism, Cell Mol Life Sci, 65 (2008) 2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H, Cardiolipin stabilizes respiratory chain supercomplexes, J Biol Chem, 278 (2003) 52873–52880. [DOI] [PubMed] [Google Scholar]
- [3].Jiang F, Gu Z, Granger JM, Greenberg ML, Cardiolipin synthase expression is essential for growth at elevated temperature and is regulated by factors affecting mitochondrial development, Mol Microbiol, 31 (1999) 373–379. [DOI] [PubMed] [Google Scholar]
- [4].Jiang F, Rizavi HS, Greenberg ML, Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources, Mol Microbiol, 26 (1997) 481–491. [DOI] [PubMed] [Google Scholar]
- [5].Koshkin V, Greenberg ML, Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria, Biochem J, 364 (2002) 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Raja V, Greenberg ML, The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype, Chem Phys Lipids, 179 (2014) 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, Pfanner N, Greenberg ML, Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function, J Biol Chem, 275 (2000) 22387–22394. [DOI] [PubMed] [Google Scholar]
- [8].Gebert N, Joshi AS, Kutik S, Becker T, McKenzie M, Guan XL, Mooga VP, Stroud DA, Kulkarni G, Wenk MR, Rehling P, Meisinger C, Ryan MT, Wiedemann N, Greenberg ML, Pfanner N, Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome, Curr Biol, 19 (2009) 2133–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Patil VA, Fox JL, Gohil VM, Winge DR, Greenberg ML, Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis, J Biol Chem, 288 (2013) 1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Raja V, Joshi AS, Li G, Maddipati KR, Greenberg ML, Loss of Cardiolipin Leads to Perturbation of Acetyl-CoA Synthesis, J Biol Chem, 292 (2017) 1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Engelking LR, Textbook of veterinary physiological chemistry, Edition Third. ed., Academic Press/Elsevier, Amsterdam ; Boston, 2015. [Google Scholar]
- [12].Owen OE, Kalhan SC, Hanson RW, The key role of anaplerosis and cataplerosis for citric acid cycle function, J Biol Chem, 277 (2002) 30409–30412. [DOI] [PubMed] [Google Scholar]
- [13].Gangloff SP, Marguet D, Lauquin GJ, Molecular cloning of the yeast mitochondrial aconitase gene (ACO1) and evidence of a synergistic regulation of expression by glucose plus glutamate, Mol Cell Biol, 10 (1990) 3551–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu Z, Butow RA, A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function, Mol Cell Biol, 19 (1999) 6720–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Epstein CB, Waddle JA, Hale W.t., Dave V, Thornton J, Macatee TL, Garner HR, Butow RA, Genome-wide responses to mitochondrial dysfunction, Mol Biol Cell, 12 (2001) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jia Y, Rothermel B, Thornton J, Butow RA, A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus, Mol Cell Biol, 17 (1997) 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liao X, Butow RA, RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus, Cell, 72 (1993) 61–71. [DOI] [PubMed] [Google Scholar]
- [18].Liu Z, Butow RA, Mitochondrial retrograde signaling, Annu Rev Genet, 40 (2006) 159–185. [DOI] [PubMed] [Google Scholar]
- [19].Magasanik B, Kaiser CA, Nitrogen regulation in Saccharomyces cerevisiae, Gene, 290 (2002) 1–18. [DOI] [PubMed] [Google Scholar]
- [20].Kornberg HL, Madsen NB, The metabolism of C2 compounds in microorganisms. 3. Synthesis of malate from acetate via the glyoxylate cycle, Biochem J, 68 (1958) 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kunze M, Pracharoenwattana I, Smith SM, Hartig A, A central role for the peroxisomal membrane in glyoxylate cycle function, Biochim Biophys Acta, 1763 (2006) 1441–1452. [DOI] [PubMed] [Google Scholar]
- [22].Palmieri L, Lasorsa FM, De Palma A, Palmieri F, Runswick MJ, Walker JE, Identification of the yeast ACR1 gene product as a succinate-fumarate transporter essential for growth on ethanol or acetate, FEBS Letters, 417 (1997) 114–118. [DOI] [PubMed] [Google Scholar]
- [23].Lee YJ, Jang JW, Kim KJ, Maeng PJ, TCA cycle-independent acetate metabolism via the glyoxylate cycle in Saccharomyces cerevisiae, Yeast, 28 (2011) 153–166. [DOI] [PubMed] [Google Scholar]
- [24].Flikweert MT, Van Der Zanden L, Janssen WM, Steensma HY, Van Dijken JP, Pronk JT, Pyruvate decarboxylase: an indispensable enzyme for growth of Saccharomyces cerevisiae on glucose, Yeast, 12 (1996) 247–257. [DOI] [PubMed] [Google Scholar]
- [25].Pronk JT, Yde Steensma H, Van Dijken JP, Pyruvate metabolism in Saccharomyces cerevisiae, Yeast, 12 (1996) 1607–1633. [DOI] [PubMed] [Google Scholar]
- [26].Kunau WH, Dommes V, Schulz H, beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress, Prog Lipid Res, 34 (1995) 267–342. [DOI] [PubMed] [Google Scholar]
- [27].Hettema EH, Tabak HF, Transport of fatty acids and metabolites across the peroxisomal membrane, Biochim Biophys Acta, 1486 (2000) 18–27. [DOI] [PubMed] [Google Scholar]
- [28].van der Klei IJ, Veenhuis M, Yeast peroxisomes: function and biogenesis of a versatile cell organelle, Trends Microbiol, 5 (1997) 502–509. [DOI] [PubMed] [Google Scholar]
- [29].van Roermund CW, Elgersma Y, Singh N, Wanders RJ, Tabak HF, The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions, EMBO J, 14 (1995) 3480–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van Roermund CW, Hettema EH, van den Berg M, Tabak HF, Wanders RJ, Molecular characterization of carnitine-dependent transport of acetyl-CoA from peroxisomes to mitochondria in Saccharomyces cerevisiae and identification of a plasma membrane carnitine transporter, Agp2p, EMBO J, 18 (1999) 5843–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Geamanu A, Gupta SV, Bauerfeld C, Samavati L, Metabolomics connects aberrant bioenergetic, transmethylation, and gut microbiota in sarcoidosis, Metabolomics, 12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heikoop JC, van Roermund CW, Just WW, Ofman R, Schutgens RB, Heymans HS, Wanders RJ, Tager JM, Rhizomelic chondrodysplasia punctata. Deficiency of 3-oxoacylcoenzyme A thiolase in peroxisomes and impaired processing of the enzyme, J Clin Invest, 86 (1990) 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Verleur N, Hettema EH, van Roermund CW, Tabak HF, Wanders RJ, Transport of activated fatty acids by the peroxisomal ATP-binding-cassette transporter Pxa2 in a semi-intact yeast cell system, Eur J Biochem, 249 (1997) 657–661. [DOI] [PubMed] [Google Scholar]
- [34].Vreken P, van Lint AE, Bootsma AH, Overmars H, Wanders RJ, van Gennip AH, Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects, J Inherit Metab Dis, 22 (1999) 302–306. [DOI] [PubMed] [Google Scholar]
- [35].Haarasilta S, Oura E, On the activity and regulation of anaplerotic and gluconeogenetic enzymes during the growth process of baker’s yeast. The biphasic growth, Eur J Biochem, 52 (1975) 1–7. [DOI] [PubMed] [Google Scholar]
- [36].Lin AP, Anderson SL, Minard KI, McAlister-Henn L, Effects of excess succinate and retrograde control of metabolite accumulation in yeast tricarboxylic cycle mutants, J Biol Chem, 286 (2011) 33737–33746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lin AP, Hakala KW, Weintraub ST, McAlister-Henn L, Suppression of metabolic defects of yeast isocitrate dehydrogenase and aconitase mutants by loss of citrate synthase, Arch Biochem Biophys, 474 (2008) 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D, A novel X-linked gene, G4.5. is responsible for Barth syndrome, Nat Genet, 12 (1996) 385–389. [DOI] [PubMed] [Google Scholar]
- [39].Xu Y, Kelley RI, Blanck TJ, Schlame M, Remodeling of cardiolipin by phospholipid transacylation, J Biol Chem, 278 (2003) 51380–51385. [DOI] [PubMed] [Google Scholar]
- [40].Hauff KD, Hatch GM, Cardiolipin metabolism and Barth Syndrome, Prog Lipid Res, 45 (2006) 91–101. [DOI] [PubMed] [Google Scholar]
- [41].Barth PG, Wanders RJ, Vreken P, Janssen EA, Lam J, Baas F, X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060), J Inherit Metab Dis, 22 (1999) 555–567. [DOI] [PubMed] [Google Scholar]
- [42].Christodoulou J, McInnes RR, Jay V, Wilson G, Becker LE, Lehotay DC, Platt BA, Bridge PJ, Robinson BH, Clarke JT, Barth syndrome: clinical observations and genetic linkage studies, Am J Med Genet, 50 (1994) 255–264. [DOI] [PubMed] [Google Scholar]
- [43].Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI, Steward CG, Barth syndrome, Orphanet J Rare Dis, 8 (2013) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Johnston J, Kelley RI, Feigenbaum A, Cox GF, Iyer GS, Funanage VL, Proujansky R, Mutation characterization and genotype-phenotype correlation in Barth syndrome, Am J Hum Genet, 61 (1997) 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]