

Abstract
Mouse models of the human porphyrias have proven useful for investigations of disease pathogenesis and to facilitate the development of new therapeutic approaches. To date, mouse models have been generated for the major porphyrias, with the exception of X-linked protoporphyria (XLP) and the ultra rare 5-aminolevulinic acid dehydratase deficiency porphyria (ADP). Mouse models have been generated for all three autosomal dominant acute hepatic porphyrias, acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP). The AIP mice, in particular, provide a useful investigative model as they have been shown to have acute biochemical attacks when induced with the prototypic porphyrinogenic drug, phenobarbital. In addition to providing important insights into the disease pathogenesis of the neurological impairment in AIP, these mice have been valuable for preclinical evaluation of liver-targeted gene therapy and RNAi-mediated approaches. Mice with severe HMBS deficiency, which clinically and biochemically mimic the early-onset homozygous dominant AIP (HD-AIP) patients, have been generated and were used to elucidate the striking phenotypic differences between AIP and HD-AIP. Mice modeling the hepatocutaneous porphyria, porphyria cutanea tarda (PCT), made possible the identification of the iron-dependent inhibitory mechanism of uroporphyrinogen III decarboxylase (UROD) that leads to symptomatic PCT. Mouse models for the two autosomal recessive erythropoietic porphyrias, congenital erythropoietic porphyria (CEP) and erythropoeitic protoporphyria (EPP), recapitulate many of the clinical and biochemical features of the severe human diseases and have been particularly useful for evaluation of bone marrow transplantation and hematopoietic stem cell (HSC)-based gene therapy approaches. The EPP mice have also provided valuable insights into the underlying pathogenesis of EPP-induced liver damage and anemia.
Keywords: mouse models, porphyrias, acute hepatic pophyrias, erythropoietic porphyrias, inborn errors of heme biosynthesis
1. Introduction
The inherited porphyrias are metabolic disorders resulting from the defective activity of specific enzymes in the heme biosynthetic pathway [Fig. 1]. Of the eight major hereditary porphyrias, seven are autosomal dominant or recessive disorders due to loss-of-function (LOF) mutations that lead to enzyme deficiencies, while X-linked protoporphyria (XLP) is caused by gain-of-function (GOF) mutations in the erythroid-specific 5-aminolevulinic acid synthase 2 (ALAS2) gene [Table 1] [1, 2]. Thus far, a human disorder associated with deficiency of the housekeeping 5-aminolevulinc acid synthase (ALAS1) has not been reported.
Figure 1.
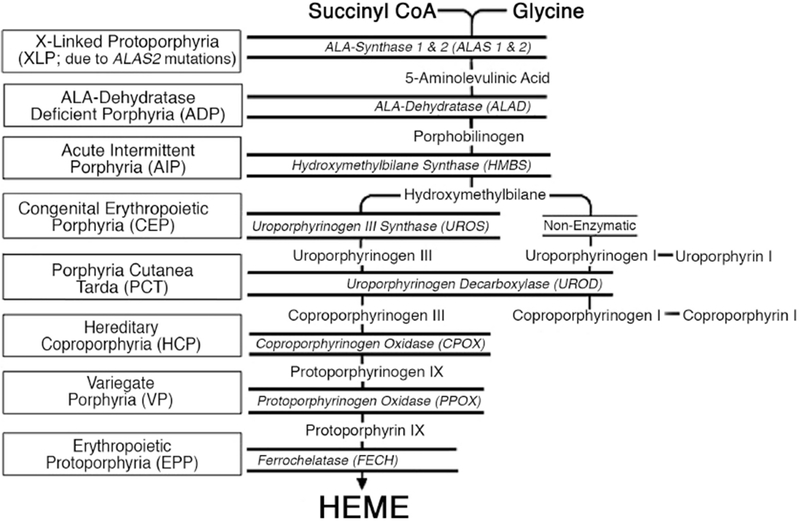
Schematic of the human heme biosynthetic pathway and the porphyrias resulting from the indicated enzymatic defect. Heme is synthesized from succinyl CoA and glycine through eight enzymatic steps. The heme biosynthetic enzymes are italicized, and the resulting porphyrias are shown in boxes. Note that there are two isozymes for the first enzyme, 5-aminolevulinic acid synthase (ALAS): a housekeeping enzyme, ALAS1, encoded by a gene that is under negative feedback regulation of the end product heme, and an erythroid-specific enzyme, ALAS2, that is regulated by iron response proteins and erythroid transcription factors.
Table 1.
Classification of the porphyrias and available porphyria mouse models
| Classification | Gene | LOF/ | Inheritance** | Mouse Model |
|---|---|---|---|---|
| Acute Hepatic Porphyrias (AHPs) | ||||
| Acute Intermittent Porphyria | Hydroxymethylbilane Synthase (HMBS) | LOF | AD | T1/T2 R167Q+/+*** |
| Hereditary Coproporphyria | Coproporphyrinogen Oxidase (CPOX) | LOF | AD | Cpox+/W373X |
| Variegate Porphyria | Protoporphyrinogen Oxidase (PPOX) | LOF | AD | PPOXR59W+/− |
| ALA-Dehydratase Deficiency Porphyria | ALA-Dehydratase (ALAD) | LOF | AR | N/A |
| Hepatocutaneous Porphyria | ||||
| Porphyria Cutanea Tarda Type 1 | - | - | Sporadic | Iron/ALA/PCB |
| Porphyria Cutanea Tarda Type 2 | Uroporphyrinogen Decarboxylase (UROD) | LOF | AD | Urod+/−/Hfe+/− |
| Erythropoietic Cutaneous Porphyrias | ||||
| Congenital Erythropoietic Porphyria | Uroporphyrinogen Synthase (UROS) | LOF | AR | UrosP248Q/P248Q UrosC73R/C73R |
| Erythropoietic Protoporphyria | Ferrochelatase (FECH) | LOF | AR | Fechm1Pas/m1Pas FechΔExon10 Emi/mPas1 |
| X-linked Protoporphyria | ALA-Synthase 2 (ALAS2) | GOF | X-L | N/A |
N/A, not available; ALA, aminolevulinic acid; PCB, polychlorinated biphenyls
LOF = Loss-of-Function; GOF = Gain-of-Function
AD = Autosomal Dominant; AR = Autosomal Recessive; X-L = X-Linked
R167Q+/+mice model homozygous dominant AIP
Porphyrias are classified as hepatic or erythropoietic, depending on whether the excess production of porphyin precursors and/or porphyrins occur primarily in the liver or in the erythron [1, 2]. The hepatic porphyrias include three autosomal dominant porphyrias, acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP), and the ultra rare autosomal recessive 5-aminolevulinic acid dehydratase deficient porphyria (ADP). These diseases are characterized by acute neurovisceral attacks that are precipitated by factors that induce heme biosynthesis, including cytochrome P450 (CYP450)-inducing drugs, hormonal changes, and fasting [2, 3]. Porphyria cutanea tarda (PCT) is considered to be a hepatocutaneous porphyria, as it presents with cutaneous lesions but the primary site of porphyrin accumulation is the liver. Erythropoietic porphyrias include congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and XLP, and present with cutaneous photosensitivity that leads to skin fragility and blisters or pain and swelling [1, 2].
To date, mouse models have been generated for all of the major hereditary porphyrias, with the exception of XLP and the ultra rare ADP [Table 1]. Most were generated via gene targeting, while a mouse model for EPP and HCP were identified in N-ethyl-N-nitrosourea (ENU) mutagenesis screens [4, 5]. As heme is essential to life, homozygous knockout mice for any of the heme biosynthetic genes are embryonic lethal [4, 6–8]. Spontaneous mouse models have not been identified for any of the porphyrias thus far. However, larger animals with various porphyrias have been identified, including cats with AIP and CEP [9, 10] and EPP in cows [11]. While some of the porphyrias can be chemically induced in mice [e.g. Griseofulvan induces pathological changes similar to those in EPP-associated liver damage [12, 13]], this review focuses on genetically modified mouse models.
Here, each of the porphyric mouse models are briefly described, highlighting the similarities in clinical and biochemical phenotypes with their respective human counterparts. The major contributions of these porphyric mouse models towards furthering our understanding of disease pathogenesis and for preclinical evaluation of novel therapeutic approaches are highlighted.
2. Methods
A pubmed search was performed using the following terms: acute intermittent porphyria mouse model, variegate porphyria mouse model, hereditary coproporphyria mouse model, aminolevulinic acid dehydratase deficiency porphyria mouse model, acute porphyria mouse model, hepatic porphyria mouse model, porphyria cutanea tarda mouse model, congenital erythropoeitic porphyria mouse model, erythropoeitic protoporphyria mouse model, and X-linked protoporphyria mouse model. Publications describing the generation and characterization of the mouse models, as well as those that showcase the usefulness of the porphyric mice for understanding disease pathogenesis and/or for preclinical evaluation of novel therapeutic approaches were selected, and the findings were summarized.
3. Acute Hepatic Porphyrias (AHPs)
The AHPs include three autosomal dominant porphyrias: AIP, VP, and HCP, which are due to half-normal activities of hydroxymethylbilane synthase (HMBS), protoporphyrinogen oxidase (PPOX), and coproporphyrinogen III oxidase (CPOX), respectively, and the ultra rare autosomal recessive ADP, caused by loss-of-function aminolevulinic acid dehydratase (ALAD) mutations (Table 1). All four AHPs present with acute neurovisceral attacks, which typically start with crippling abdominal pain and may progress to include tachycardia, hypertension, motor weakness, and seizures [1, 2]. The attacks are triggered by various porphyrinogenic factors, including fasting, CYP450-inducing drugs, and hormonal fluctuations, all of which induce the hepatic expression of ALAS1, the first and rate-limiting enzyme of the heme biosynthetic pathway [2, 3]. When hepatic ALAS1 is induced, the respective enzyme deficiency becomes rate-limiting, leading to insufficient heme production and depletion of the ‘free’ heme pool. This leads to derepression and further induction of hepatic ALAS1, resulting in the marked elevation of the putative neurotoxic porphyrin precursors, 5-aminolevulinic acid (ALA) and porphobilinogen (PBG) [1, 2].
To date, mouse models have been developed for the three autosomal dominant acute porphyrias, AIP, HCP, and VP. The AIP mouse model has been particularly useful for studies of disease pathogenesis and for evaluating novel therapeutic approaches.
3.1. ATP Mouse Model: T1/T2 mice
Homozygous HMBS knockout mice were embryonic lethal, while heterozygotes with half-normal activity were viable but did not accumulate urinary ALA when administered the prototypic porphyrinogenic drug, phenobarbital (PB) [7]. Therefore, compound heterozygous mice with ~30% of normal HMBS activity were generated and designated T1/T2 mice [7]. The T1 hypomorphic allele was generated by insertion of the Neomycin gene under the transcriptional control of the phosphoglycerate kinase promoter in antisense orientation in exon 1, leading to disruption of the house-keeping Hmbs isoform. The T2 allele had a competitive splice acceptor site in intron 1. Mice homozygous for the T1 allele were viable and had ~55% of normal HMBS activity. T2 homozygotes were embryonic lethal, while T2 heterozygotes had ~57% of normal HMBS activity [7].
The T1/T2 mice have baseline hepatic Alas1 mRNA expression and urinary and plasma ALA and PBG concentrations that are slightly increased (2 to 6-fold) compared to normal. When administered PB, hepatic Alas1 mRNA expression is markedly increased, leading to massive accumulation of plasma and urinary ALA and PBG, thereby biochemically mimicking a porphyric acute attack [7]. These mice do not develop overt neurologic manifestations of an acute attack. Rather, they develop chronic and progressive motor axonal degeneration, similar to changes seen in patients with severe attacks, by 6 months of age [7, 14]. This is accompanied by neuromotor impairment, including decreased rotarod and muscular performances. Of note, the axonal motor degeneration occurred regardless of whether the mice were induced with PB or not [14].
3.1.1. Investigations of Disease Pathogenesis in T1/T2 mice
The pathogenesis of the acute neurovisceral attacks has been long debated, particularly whether the accumulated porphyrin precursors, ALA and/or PBG, are neurotoxic, or if heme deficiency in nervous tissues leads to neurologic dysfunction [15]. In particular, ALA is thought to be neurotoxic, as it is elevated in all four acute hepatic porphyrias (whereas PBG is not elevated in ADP) and it is a structural analogue of the inhibitory neurotransmitter, γ-aminobutyric acid (GABA). ALA has also been shown to exhibit pro-oxidative properties in vitro [16–19]. The fact that liver transplantation completely stopped the recurrent acute attacks in patients with AIP clearly demonstrates that the liver is the main site of pathology [20]. Conversely, when livers of AIP patients with recurrent attacks were ‘domino’ transplanted into non-porphyric patients, these patients developed acute porphyric attacks [21]. While these studies and others [22–24] strongly support that ALA and/or PBG are neurotoxic and are the main mediators of the attacks, studies in the T1/T2 mice suggest that other mechanisms may also contribute.
As noted above, the T1/T2 mice develop axonal degeneration without exposure to porphyrinogenic factors, hence with only slight elevations of plasma and urinary ALA and PBG [14]. Thus, it has been proposed that neuronal heme deficiency may be involved in the development of the porphyric neuropathy [14]. Related to this, it was shown that the T1/T2 mice have lower heme saturation and enzymatic activity of Cyp1a1 in the brain following treatment with β-naphthoflavone, a Cyp1a1-inducer, compared to normal mice [25].
More recent studies have demonstrated mitochondrial energetic defects in the T1/T2 mice, suggesting that this, too, may play a role in disease pathogenesis. In the livers of PB-induced T1/T2 mice, the activities of the TCA cycle and downstream respiratory chain complexes were markedly decreased, presumably due to a large amount of succinyl-CoA being shuttled away from the TCA cycle to be used as substrate for the massively induced ALAS1 enzyme [26]. Enzymatic activities of the respiratory chain complexes were also decreased in skeletal muscles and brains of the PB-induced T1/T2 mice, leading to decreased ATP production in muscle [27]. Mitochondrial impairments in the brain and muscle were postulated to be due to the neurotoxic effects of ALA overproduced by the liver, although in the brain, the activity of heme-dependent respiratory enzymes were decreased, suggesting that heme deficiency may also contribute [27].
Additionally, these mice have been useful for the identification of potential biomarkers for AIP [28], for the assessment of porphyrinogenicity of certain drugs [29], and to investigate alterations in glucose metabolism induced by fasting in AIP livers [30].
3.1.2. Preclinical Studies in T1/T2 mice
The most effective standard treatment for the acute attacks is intravenous administration of hemin, which provides exogenous heme for the negative feedback inhibition of hepatic ALAS1 [31]. Although patients generally respond well, its effect is slow, and frequent administration is associated with side effects such as phlebitis and iron overload [32]. While liver transplantation is curative, it is a high-risk procedure associated with limited organ availability. Therefore, alternative therapeutic approaches for the AHPs that are more effective, faster-acting, and safe are desirable.
Due to the fact that the T1/T2 mice can be induced into a biochemical acute attack with PB, these mice have been very useful for preclinical evaluation of various therapeutic strategies, including hepatocyte transplantation. Yin et al. showed that engraftment of < 3% of normal hepatocytes into the T1/T2 livers reduced the PB-induced elevations of plasma ALA and PBG by ~50% [33]. These disproportional findings suggest that the porphyrin precursors readily cross cellular membranes and are efficiently metabolized by the transplanted cells.
The effectiveness of liver-targeted gene therapy strategies for AIP, including adeno-viral [34, 35] and adeno associated viral (AAV)-mediated approaches, has been evaluated in the T1/T2 mice. In particular, Yasuda and Unzu demonstrated that gene transfer of the humanHMBS cDNA via AAV2/8- and AAV2/5-vectors, respectively, provided complete and sustained protection against the induced biochemical acute attacks and improved neuromotor function in the T1/T2 mice [36, 37]. Notably, transduction of only 5-10% of hepatocytes was sufficient to completely prevent the PB-induced acute attacks [36]. AAV2/5-mediated gene therapy was further evaluated in Phase I Clinical Trials and proved to be safe and well-tolerated in human AIP patients, but no appreciable decreases in urinary ALA and PBG were achieved, presumably due to inadequate HMBS expression in the liver [38]. Thus, recent efforts have been directed towards optimizing the therapeutic vector to achieve high transgene expression, either by 1) incorporating human ALAS drug responsive enhancing sequences (ADRES) into the promoter or 2) modifying the HMBS cDNA to include the p.I291M and p.N340S variants, which synergistically increase kinetic stability of the HMBS enzyme [39, 40]. The ADRES, previously identified in the distal 5’-flanking region of the human ALAS1 gene, have been shown to activate transcriptional activity of ALAS1 in response to porphyrinogenic drugs [41]. Administration of an AAV2/8 vector harboring two tandem ADRES immediately upstream of the promoter (AAV2/8-ADRES-HMBS) led to markedly increased hepatic HMBS activities in the T1/T2 mice, particularly upon exposure to factors that precipitate attacks, including porphyrinogenic drugs, fasting, and estrogen [40]. Importantly, compared to a ‘control’ AAV vector lacking the ADRES elements (AAV2/8-HMBS), the vector dose of AAV2/8-ADRES-HMBS could be reduced by 10-fold to achieve the same therapeutic effect. Treatment with an AAV2/8 vector that included the p.I291M and p.N340S variants in its HMBS transgene (AAV-HMBS-I291M/N304S) also resulted in markedly elevated hepatic HMBS activities in the T1/T2 mice, representing a 3-fold decrease in elfective vector dose relative to the AAV2/8-HMBS ‘control’ vector [39].
Recently, the T1/T2 mice have been used for preclinical studies of a lipid nanoparticle (LNP)-encapsulated short interfering RNA (siRNA) specifically targeting hepatic Alas1 (Alas1-siRNA). Intravenous administration of the Alas1-siRNA effectively prevented and treated induced biochemical attacks in these mice and did not perturb the activities of representative hemoproteins [24]. Importantly, when administered during an induced attack, the Alas1-siRNA reduced the markedly elevated plasma ALA and PBG levels more rapidly and effectively than did a single intravenous hemin infusion. Subsequent efforts developed a N-acetylgalactosamine (GalNAc)-conjugated Alas1-siRNA (GalNAc-Alas1) that can be administered subcutaneously and demonstrated its efficacy in the T1/T2 mice [42]. Recent Phase I/II Clinical trials for a subcutaneously administered GalNAc-ALAS1 have indicated this approach to be safe and effective in human AIP patients (R.J.D., unpublished data).
3.2. VP Mouse Model: PPOXR59W+/− Mice
The prevalence of VP is highest in South Africa due to an ancestral founder mutation, c.175C>T (p.R59W), that was brought to the Cape by a Dutch couple in 1680. A mouse model of VP carrying the p.R59W mutation was generated via gene targeting by Medlock et al [43]. Heterozygous mice (R.59W+/−) had approximately half-normal PPOX activity and accumulated porphyrins in a pattern similar to human VP patients. However, administration of phenobarbital (PB) did not result in clinical manifestations of an acute attack or elevation of urinary ALA or PBG (M.Y., personal communication).
3.3. HCP Mouse Model: Cpox+/W373X Mice
Recently, a mouse line carrying the c.1118G>A (p.W373X) mutation in its Cpox gene was identified in a genome-wide ENU study aimed to identify novel regulators of erythropoiesis [4]. Heterozygous mice (Cpox+/W373X) had half-normal hepatic Cpox mRNA and presented with a mild microcytic anemia. Homozygous mice were not viable and died in uetro by embryonic day 9.5.
Female Cpox+/W373X mice had slightly elevated (3 to 4-fold) urinary and fecal porphyrins, with an increased coproporphyrin III:I ratio in faeces, and normal baseline urinary PBG. Male heterozygotes, on the contrary, had normal porphyrin profiles and were not further characterized. Porphyrinogenic stimuli, including fasting (15 hr/day, two consecutive days) and PB administration (75, 80, 85, and 90 mg/kg/day; 4 consecutive days), did not induce apparent acute neurovisceral symptoms or significantly increase urinary PBG in Cpox+/W373X females [4].
3.4. Homozygous Dominant AHPs
Biallelic loss-of-function mutations in HMBS, CPOX, and PPOX genes lead to rare conditions known as homozygous dominant (HD)-AIP, HD-HCP, and HD-VP, respectively [22, 44–51]. Unlike the autosomal dominant forms, these diseases present in infancy with severe and progressive neurological impairment and/or marked cutaneous manifestations with growth retardation and short stature. Of interest, with the exception of one HD-HCP patient [46], acute neurovisceral attacks have not been reported in patients with the HD-AHPs.
HD-AIP patients have less than 4% residual HMBS activity and constitutively elevated urinary ALA and PBG. In addition to gross psychomotor retardation, patients have early-onset ataxia, nystagmus, and dystonia [22]. Patients typically have short stature and die in childhood. Interestingly, of the five HD-AIP patients reported to date, four had c.500G>A (p.R167Q), c.499C>T (p.R167W), or c.518G>A (p.R173Q) mutations: one was homozygous for p.R167W [22], two siblings were heterozygous for p.R167Q and p.R167W [49], while one was heterozygous for p.R167W and p.R173Q [44]. In all cases, their parents were asymptomatic AIP heterozygotes.
A mouse model for HD-AIP has been generated and characterized in an effort to understand the striking differences in clinical presentation between human AIP and HD-AIP. Mouse models for HD-HCP and HD-VP have not been generated to date.
3.4.1. HD-AIP Mouse Model: R167Q+/+ Mice and Investigations of Disease Pathogenesis
Hmbs knock-in mice carrying the ‘common’ human HD-AIP mutations, p.R167Q and p.R173W, were generated by gene targeting. Mice homozygous for p.R173W were embryonic lethal, while p.R167Q homozygotes (R167Q+/+) survive to adulthood and have a normal lifespan [52]. R167Q+/+ mice are severely runted and display profound neurological abnormalities similar to the human HD-AIP patients, including early-onset ataxia, delayed neuromotor development, and impaired rotarod performance. These mice also recapitulate the biochemical features of human HD-AIP, having only ~5% of normal HMBS activity and constitutively elevated plasma and urinary ALA and PBG in the absence of porphyrinogenic factors [52]. Central nervous system (CNS) histology was grossly normal, but CNS myelination was delayed and overall myelin volume was decreased. When compared with the T1/T2 mice, a striking biochemical difference was that the R167Q+/+ mice had much higher elevations of ALA and PBG in their CNS tissues. The finding that the T1/T2 mice accumulated high levels of porphyrin precursors in their livers and plasma following PB induction, but not their CNS tissues, suggested that ALA and PBG do not readily cross the blood-brain-barrier [52]. Taken together, these studies suggest that the severe HD-AIP neurological phenotype results from decreased myelination and the accumulation of locally produced neurotoxic porphyrin precursors within the CNS. Importantly, these findings suggest that liver-directed therapies for heterozygous AIP, including hemin, RNAi-mediated silencing of hepatic ALAS1, and liver transplantation, are likely not effective for HD-AIP, as they do not address these CNS issues.
4. Porphyria Cutanea Tarda (PCT)
PCT, the most common human porphyria, is characterized by the development of blisters on sun-exposed skin. It is unique among the inherited porphyrias in that it occurs both in a Type 1 sporadic form, in the absence of an UROD mutation, and in a Type 2 familial form, in which an UROD mutation is inherited in an autosomal dominant pattern (Table 1). Clinically, the two forms are indistinguishable, although age of disease onset is significantly lower in Type 2 familial patients [53]. The disease becomes active when patients are exposed to predisposing factors that lead to hepatic iron overload, including hemochromatosis, excessive alcohol consumption, estrogens, and viral infections such as hepatitis C and HIV [1–3]. While the half-normal activity of UROD predisposes to PCT, additional susceptibility factors are required to activate disease in Type 2 familial patients, like the Type 1 sporadic patients. Patients with active disease display marked porphyrinuria, predominantly consisting of uroporphyrin and 7-carboxylate porphyrin. Mouse models for PCT have played a particularly important role in defining the underlying pathogenic mechanism of this hepatocutenous porphyria.
4.1. Mouse Models for Type 1 Sporadic and Type 2 Familial PCT and Investigations of Disease Pathogenesis
To model Type 2 autosomal dominant familial PCT, Urod knockout mice were generated by gene targeting. Much like human UROD heterozygotes, mice heterozygous for the Urod-null allele (Urod+/−) did not display porphyrinuria unless predisposing factors were present [54]. Administration of a single intraperitoneal dose of iron, combined with ALA to bypass the rate-limiting step of heme biosynthesis (i.e. ALAS1), led to marked porphyrinuria by 3 weeks. Subsequently, these mice were bred against mice null for the hemochromatosis (Hfe) allele to produce mice heterozygous for the Urod-null allele and homozygous null for the hemochromatosis (Hfe) allele (Urod+/−/Hfe−/−). Urod+/−/Hfe−/− mice accumulated high levels of hepatic iron and developed porphyuria at 14 weeks of age without ALA supplementation or other exogenous precipitating factors, establishing that iron alone is sufficient to cause the porphyric phenotype in mice [54]. Assessment of hepatic UROD enzymatic activity showed that, contrary to the Urod+/−/Hfe +/+ mice with intact Hfe which had about half-normal UROD activity, the Urod+/−/Hfe−/− mice had only ~14% of WT. Notably, the marked decrease in UROD activity occurred without changes to UROD protein levels, indicating that the catalytic activity of UROD was abolished. These findings confirmed previous reports that an iron-dependent inhibitor of UROD is formed in symptomatic Type 2 familial PCT [55–58].
In a follow up study, Phillips et al. demonstrated that a similar iron-dependent mechanism of hepatic UROD inhibition is observed in a mouse model for Type 1 sporadic PCT, generated by administrating iron, ALA, and polychlorinated biphenyls (PCB) to C57BL/6 wildtype mice [59]. Analyses of liver cytosolic extracts isolated from the sporadic PCT and familial PCT (Urod+/−/Hfe−/−) mice identified a competitive inhibitor of recombinant human UROD that proved to be uroporphomethane, an iron-oxidized product of uroporphyrinogen [59]. Thus, these studies established that Type 2 familial PCT and Type 1 sporadic PCT share a common pathogenic mechanism, namely, the iron-dependent formation of an UROD inhibitor in the liver that leads to transient decrease of hepatic UROD activity and disease activation.
5. Erythropoietic Porphyrias
The erythropoietic porphyrias include two autosomal recessive disorders, CEP and EPP, which are caused by loss-of-function mutations in the uroporphyrinogen III synthase (UROS) and ferrochelatase (FECH) genes, respectively. XLP is an X-linked disease caused by gain-of-function mutations in the ALAS2 gene (Table 1). These disorders are characterized by cutaneous photosensitivity that results from the massive accumulation of photoreactive porphyrins within the bone marrow and circulating erythrocytes, which eventually are deposited in the skin.
5.1. CEP
Major clinical features of human CEP include marked cutaneous photosensitivity with blistering, hemolytic anemia, and erythrodontia, which is reddish-brownish discoloration of teeth [1, 2]. Clinical severity can vary, from non-immune hydrops fetalis in utero to late-onset disease with only mild cutaneous photosensitivity in adulthood. Genotype/phenotype correlations predict disease severity, as the activity of the encoded UROS enzyme deficiency generally correlates with clinical severity. The most common presentation is severe, transfusion-dependent CEP due to homozygosity for the c.217T>C (p.C73R) mutation [60]. Biochemically, CEP patients have markedly elevated erythrocyte and urinary uroporphyrin I (Uro I), and coproporphyrin I (Copro I) isomers, which are phototoxic, non-physiologic substrates that are not utilized to synthesize heme.
5.1.1. CEP Mouse Models: UROSP248Q/P248Q and UROSC73R/C73R Mice
Previously, it was shown that Uros knockout mice were homozygous lethal [6]. Therefore, Ged et al. generated a homozygous knock-in mouse model carrying the human severe UROS mutation, c.743C>A (p.P248Q; designated UROSP248Q/P248Q) [61]. Bishop et al. generated mice with the most common and severe human genotype, p.C73R/p.C73R (UROSC73R/C73R), and mice with later-onset genotypes, p.V99L/p.V99L (UROSV99L/V99L) and p.C73R/p.V99L (UROSC73R/V99L) [62]. Much like their human counterparts, the UROSP248Q/P248Q and UROSC73R/C73R mice had severe disease, with ≤1% of normal UROS activity in their erythrocytes and urinary Uro I concentrations that were increased >990-fold compared to that in normal mice. Both mouse models developed a profound hemolytic anemia and had an abundance of fluorocytes in their bone marrow and peripheral blood [61, 62]. Splenomegaly and erythrodontia were also apparent. A moderate skin photosensitivity was documented in the UROSP248Q/P248Q mice [61].
In contrast, the UROSC73R/V99L and UROSV99L/V99L mice had ~11 and 19% of normal erythrocyte UROS activity, respectively [62]. The UROSC73R/V99L mice had a very mild anemia, while adult UROSV99L/V99L mice were hematologically normal. Importantly, these findings suggested that only ~10% of wildtype UROS activity may be necessary to correct the pathology in CEP patients.
5.1.2. Preclinical Studies in UROSP248Q/P248Q Mice
For non-transfusion CEP patients, efforts to keep the hematocrit above 35 with fresh erythrocyte transfusions has proven to be effective [63]. To date, the only curative treatment for transfusion-dependent CEP patients is bone marrow transplantation (BMT), which not only is a high-risk procedure, but also requires a suitable donor. Therefore, efforts have been directed to develop novel therapeutic approaches for this disease. Both the UROSP248Q/P248Q and UROSC73R/C73R are excellent models to evaluate new therapies for CEP, as they recapitulate most of the clinical and biochemical features of the severe human disease.
Robert-Richard et al. demonstrated that ex vivo lentiviral-mediated gene transfer of the human UROS cDNA into hematopoietic stem cells (HSC), followed by re-injection into the UROSP248Q/P248Q mice, achieved complete and long-term enzymatic, metabolic, and phenotypic correction of disease [64]. Of note, transduction of only ~40% of HSC was sufficient to achieve this effect, due to the survival advantage of the corrected cells over non-transduced cells. That the corrected cells have a survival advantage over non-transduced cells is consistent with the finding that UROS-deficient erythrocytes have a shorter half-life and increased osmotic fragility compared to UROS-sufficient erythrocytes [65].
Blouin et al. showed in the UROSP248Q/P248Q mice that administration of Bortezomib, a clinically approved proteasome inhibitor, resulted in decreased UroI levels in circulating erythrocytes and urine and reversion of skin photosensitivity, although it did not correct the anemia [66]. The biochemical and phenotypic correction was shown to occur through rescue of the P248 mutant protein from premature proteosomal degradation. These studies suggested the therapeutic potential of proteasome inhibitors and/or pharmacological chaperones to treat CEP patients with certain UROS mutations, such as p.P248Q and the common p.C73R, which retain intrinsic catalytic activity but are mis-folded and targeted to the proteasome degradation pathway. In fact, Urquiza et al. recently demonstrated that ~75% of UROS mutations reported to date lead to decreased stability of the enzyme and premature proteosomal degradation [67], and therefore, are candidates for such therapeutic approaches. As long-term treatment with proteasome inhibitors such as Bortezomib may be difficult, due to high rates of acquired drug resistance and cytotoxicity such as peripheral neuropathy [68, 69], a pharmacological chaperone with better safety profile would be highly advantageous.
To this end, Urquiza and colleagues screened a library of compounds and identified ciclopirox (CPX), a FDA-approved synthetic antimicrobial, as a pharmacological chaperone that stabilizes and restores the activity of the UROS enzyme, both in vitro and ex vivo [67]. Notably, they demonstrated that CPX binds UROS at an allosteric site that is distant from the active site, and therefore, does not interfere with the enzyme’s catalytic function. Daily oral administration of sub-toxic doses CPX to the UROSP248Q/P248Q mice via their diet resulted in significant reduction of erythrocyte, hepatic, and urinary Uro I and Copro I porphyrin concentrations, as well as decreased splenomegaly, suggesting improved hemolysis. Erythrocyte PPIX levels were increased, supporting restoration of heme biosynthesis in the treated mice [67]. The effectiveness and safety of this therapeutic approach in human CEP patients has yet to be determined.
5.2. EPP
EPP, the most common childhood porphyria, is primarily characterized by extremely painful photosensitivity that is accompanied by marked elevation of free protoporphyrin IX (PPIX) in plasma and erythrocytes [1, 2]. A mild microcytic anemia occurs in 20 to 60% of patients [70], but the etiology of the anemia is unclear. 10-20% of patients experience cholelithiasis, while ~5% of patients develop liver failure due to hepatobiliary involvement, requiring liver transplantation [71]. Since the accumulated PPIX released from erythroid cells is lipophilic, it accumulates in the liver and is secreted into the bile. The hepatic accumulation of PPIX leads to cholestasis and hepatobiliary fibrosis, but the precise underlying pathogenic mechanism leading to liver failure in EPP is not fully understood. Studies in an EPP mouse model have suggested possible mechanisms that lead to severe hepatobiliary disease.
5.2.1. EPP Mouse Model: Fechm1Pas Mice
The first genetically modified mouse model for EPP, described by Tutois et al., was identified in an ENU mutagenesis study and designated Fechm1Pas mice [5]. Boulechfar et al. later determined the molecular lesion to be a c.293T>A substitution (p.M98K) of the Fech gene [72]. Heterozygous mice were clinically and biochemically normal, while homozygous mice (Fechm1Pas/m1Pas) displayed biochemical and clinical features mimicking a severe form of human EPP. They had ~3 to 6% of FECH activity and ~25-fold increase in erythrocyte protoporphyrin IX (PPIX) levels compared to normal [5]. Clinical findings included mild to moderate photosensitivity under normal husbandry conditions and marked normocytic hemolytic anemia by 3 to 6 months of age.
These mice also displayed severe hepatobiliary involvement, with apparent jaundice and enlarged livers and spleens from the early days of life onwards [5]. PPIX was markedly accumulated in the liver, associated with elevated plasma transaminases and hyperlipidemia [73]. Histopathologic examination of the livers showed characteristic PPIX deposits predominantly in bile canaliculi, but also in hepatocytes, and lobular and portal macrophages, and in the lumen of small bile ducts, similar to findings in human EPP patients with severe hepatic disease. Bile duct proliferation and fibrosis with portoportal bridging, sclerosing cholangitis, and hepatolithiasis were also noted [74, 75].
5.2.2. Investigations of Disease Pathogenesis Using Fechm1Pas/m1Pas mice
Previously, it was suggested that EPP-induced liver injury results from the cytotoxic effects of PPIX and obstruction of bile canaliculi by PPIX [76, 77]. In support of this, studies in the Fechm1Pas/m1Pas mice showed that the extent of hepatocyte damage correlated with the amount of intralobular PPIX deposition and with the proportion of portal tracts containing small bile ducts with PPIX deposition [74]. However, more recent studies found that bile flow was increased in the Fechm1Pas/m1Pas mice compared to normal [78]. Additionally, bile composition was significantly altered in these mice, with marked increases in cytotoxic bile acids and decreased lipid content [75, 78]. As PPIX was shown to bind strongly to phospholipids (PL) in vitro, it was proposed that the large efflux of PPIX into the canaliculi leads to PL depletion and the formation of lipid-free cytotoxic bile, which then causes damage to bile duct epithelium, and, consequentially, biliary fibrosis and sclerosing cholangitis [75].
Whereas the originally described Fechm1Pas/m1Pas mice were on a BALB/cByJCrl genetic background, these mice have since been backcrossed onto C57BL/6JCrl and SJL/JOrlCrl backgrounds [79]. Compared to the BALB/cBYlCrl Fechm1Pas/m1Pas mice, the SJL/JorlCrl Fechm1Pas/m1Pas mice had mild anemia and hepatobiliary disease, while C57BL/6JCr1 Fechm1Pas/m1Pas mice had a moderate anemia and severe hepatocyte damage associated with markedly elevated PPIX levels in the cytosol of hepatocytes and Kupffer cells [79]. Notably, the C57BL/6J Fechm1Pas/m1Pas mice had little PPIX in the bile canaliculi and were protected from the severe bile duct epithelial cell damage, presumably due to reduced efflux of PPIX and reduced synthesis and export of cytotoxic bile acids [75]. The marked differences in the severity of anemia and hepatobiliary disease of the Fechm1Pas/m1Pas mice on these genetic backgrounds suggest that modifier genes are likely involved in the variable expression of anemia and liver disease in EPP.
Studies by Han et al. proposed that heme-regulated elF2α kinase (HRI) may be an important modifier gene that contributes to the clinical severity of EPP and other hematologic disorders [80]. HRI is a heme-regulated protein kinase that phosphorylates the α-subunit of the eukaryotic translational initiation factor 2 (eIF2α). Phosphorylation of eIF2α leads to global cessation of protein synthesis. In heme-deficient states, hepatic HRI expression is activated, thus promoting eIF2α phosphorylation and cessation of protein synthesis to protect against excess globin production. Compared to BALB/cBYlCrl Fechm1Pas/m1Pas mice with intact HRI (Fechm1Pas/m1Pas/HRI+/+). BALB/cBY1Cr1 Fechm1Pas/m1Pas mice homozygous null for HRI (Fechm1Pas/m1Pas/HRI−/−) had 30-fold higher PPIX, a more severe anemia, and markedly more severe hepatocellular toxicity and cutaneous photosensitivity [80].
To date, the pathogenesis of the microcytic hypochromic anemia in EPP is not fully understood. While EPP patients often have laboratory tests indicative of iron-deficiency (e.g. microcytic hypochromic anemia with low serum iron, decreased ferritin and transferrin saturation), there are conflicting reports on the therapeutic benefit of iron supplementation in these patients. Some studies report improvement of cutaneous photosensitivity and increased hemoglobin levels following iron supplementation, suggesting that the exogenous iron was efficiently incorporated into the excess PPIX to form heme, and consequentially, PPIX levels were decreased [81]. However, the majority of studies show that iron supplementation exacerbates photosensitivity and/or PPIX accumulation [82–84], consistent with the fact that iron-sufficient conditions induce ALAS2 translation via the iron response element (IRE) and promote erythroid heme biosynthesis, thereby resulting in increased PPIX production.
While initial description of the BALB/cByJCrl Fechm1Pas/m1Pas mice included a normocytic hemolytic anemia [5], more recent studies have demonstrated these mice to have a microcytic hypochromic anemia with absence of sideroblasts, little or no hemolysis, and no bone marrow or spleen erythroid hyperplasia [79, 85]. Lyoumi et al. demonstrated that the microcytic anemia in the BALB/cByJCrl Fechm1Pas/m1Pas mice is not associated with iron deficiency, as serum ferritin levels, hepcidin mRNA levels, and total body iron amounts were normal [85]. However, iron distribution was altered in the Fechm1Pas/m1Pas mice, with reduced iron amounts in liver, kidney, and heart, and increased splenic iron compared to normal. Of particular interest, serum transferrin levels were elevated by 2-fold in the Fechm1Pas/m1Pas mice, while serum iron levels were normal, resulting in low transferrin saturation levels that mimicked an iron-deficient state [85]. Taken together, these studies support that iron supplementation is not beneficial for the microcytic anemia associated with FECH deficiency.
5.2.3. Preclinical Studies in Fechm1Pas/m1Pas Mice
Recent clinical studies have demonstrated that an α-melanocyte stimulating hormone analogue effectively protects against the photosensitivity in EPP patients by darkening their skin [86]. However, the only curative therapy to date is bone marrow transplantation (BMT). The EPP mice have been used extensively for preclinical studies of BMT and HSC-mediated gene therapy approaches.
Transplantation of normal bone marrow cells into 3-4 week Fechm1Pas/m1Pas mice mice dramatically decreased erythrocyte PPIX accumulation but only partially corrected the liver disease [87]. Notably, BMT into neonates led to complete correction of erythrocyte PPIX levels and skin photosensitivity and almost entirely prevented liver damage [88].
When FECH-deficient HSCs were transduced with retroviral vectors expressing the human FECH enzyme and re-implanted into adult Fechm1Pas/m1Pas mice, complete metabolic correction and reversal of skin photosensitivity were achieved only if the transduced cells were pre-selected prior to implantation [89, 90]. Ex vivo gene transfer using lenti-viral vectors precluded the need for pre-selection, but high transduction rates (> 50%) associated with myloablative pre-conditioning were necessary, as there wasn’t a natural selective advantage of corrected cells over FECH-deficient cells [91]. Therefore, Richard et al. developed a bicistronic SIN-lentiviral vector co-expressing the human FECH cDNA and methylguanine DNA methyltransferase (MGMT) G156A mutant, which provides resistance against alkylating agents [temozolomide or bis-chloroethylnitrosourea (BCNU)] in transduced cells, thereby permitting drug-mediated in vivo selection of corrected cells [92]. Administration of this vector, followed by moderate doses of BCNU into adult Fechm1Pas/m1Pas mice led to progressive expansion of transduced cells (up to 95%) and the complete correction of PPIX levels and photosensitivity. Further, the investigators developed a bi-promoter lentiviral vector in which the expression of FECH is driven by the erythroid-specific chimeric HS40 enhancer/ankyrin promoter, while the expression of the MGMT G156A mutant is driven by the ubiquitous human PGK promoter. The erythroid-restricted expression of FECH was sufficient to achieve full correction of PPIX and photosensitivity [92]. Of note, HSC gene therapy in adult Fechm1Pas/m1Pas mice did not correct the liver disease, indicating that early intervention may be necessary to fully prevent this condition. Thus far, there have not been clinical trials for gene therapy approaches in human EPP patients.
5.2.3. EPP Mouse Model: Fech Exon 10 Deletion Mice
A mouse with deletion of Fech exon 10, a mutation identified in human EPP, was generated by Magness et al [8]. Homozygous mice were embryonic lethal, while heterozygous mice had ~37% of normal FECH activity in their livers, presumably due to a dominant-negative effect of the mutant allele on the wildtype allele. These mice were clinically mild compared to the Fechm1Pas/m1Pas mice, with erythrocyte PPIX levels only ~9-fold higher than normal controls and no liver disease. A mild photosensitivity, manifested as edema and necrosis on depilated skin, was observed only after 48 to 72 hours of light exposure.
5.2.4. EPP Mouse Model: Partially Humanized Emi/mPas1 Mice
Over 90% of patients with EPP have a pathogenic mutation on one FECH allele and the c.315-48C (IVS3-48T>C) ‘low expression’ allele on the other. This change leads to the increased use of an alternative 3’-splice site that is 63 nt upstream of the normal one, leading to an aberrant transcript with 63 additional nucleotides. Two in-frame stop codons in this additional sequence target this aberrant transcript for nonsense-mediated decay. As a result, the ‘low expression allele’ has −30% of normal FECH activity. Individuals heterozygous for a pathogenic FECH mutation without the ‘low expression’ allele are asymptomatic, and therefore, correction of the low expression allele alone is therapeutic. To facilitate the development of novel therapeutic approaches for EPP, Barman-Aksozen et al. generated mice that harbor the human FECH intron 3 with the IVS3-48T>C polymorphism (designated Emi mice) [93]. Homozygous Emi mice were embryonic lethal, therefore, Emi mice were bred against the Fechm1Pas/m1Pas mice to generate Emi/mPas1 mice. Emi/mPas1 mice were even more severely affected than the Fechm1Pas/m1Pas mice, with 6 to 8–folder higher erythrocyte PPIX levels, increased prenatal mortality, more pronounced hepatosplenomegaly, and overall higher bilirubin levels. Within minutes of exposure of shaved skin to 400-420 nm light (1.69-2.3 J/cm2), behavior indicative of pain was observed and erythema became evident by 2-3 hours post-exposure.
RT-PCR analyses revealed that the partially humanized Emi allele led to unexpected splicing events, with the majority of transcripts completely lacking exon 3. Only a small number of transcripts used the aberrant splice site that is 63 nucleotides upstream of the normal one, as described in humans carrying the ‘low expression’ allele [93]. Presumably, the exon 3-lacking transcripts were stable and resulted in non-functional protein, leading to lethality of homozygous Emi mice and a very severe EPP phenotype in the Emi/m1Pas mice.
6. Discussion
To date, mouse models have been generated for all eight major human porphyrias, with the exception of XLP and the ultra-rare ADP. These mouse models of the human porphyrias have clearly provided relevant and important information on the pathophysiology of their human counterparts as well as the opportunity to provide proof-of-concept and preclinical data for various therapeutic strategies. For the acute porphyrias, the ‘inducible’ T1/T2 AIP mice have been particularly useful for preclinical evaluation of liver-directed gene therapy and RNAi-mediated approaches that specifically knock down hepatic ALAS1 mRNA expression [24, 35–37, 89, 90]. A subcutaneously administered RNAi therapeutic is currently in Phase III Clinical Trials and is emerging as a safe and effective alternative treatment for human AHP patients. If important modifying genes that predispose or protect against the acute attacks are discovered, they could be evaluated in these mice. While human AIP, VP, and HCP patients have half-normal activities of their respective enzymes, HMBS, PPOX, and CPOX, it is notable that mice with 50% reduction of these enzymes do not display PB-induced acute biochemical attacks [4, 7, 43]. For AIP, decreasing the HMBS activity to ~30% of normal, as was the case in the T1/T2 mice, resulted in an ‘inducible’ phenotype [7, 14]. Therefore, it is likely that further reduction of CPOX and PPOX activities are necessary to produce HCP and VP mice, respectively, that display acute biochemical attacks. The recently generated HD-AIP mice, which capture many of the clinical and biochemical features of the human disease, have provided important insights as to why autosomal dominant AIP and HD-AIP have vastly different manifestations. To date, there is no effective treatment for HD-AIP. These mice facilitate studies aimed to develop new therapeutic approaches for HD-AIP.
Mouse models for PCT have played a particularly important role in delineating the pathogenic mechanism of this hepatocutaneous porphyria. In addition to facilitating the understanding of the role of iron overload in the etiology of PCT, they made possible the identification of the specific inhibitor of UROD that forms in the liver during active disease [54, 59]. In the future, these mice may serve in preclinical studies to evaluate newer approaches to efficiently treat PCT.
Mouse models for the erythropoietic porphyrias, particularly the UROSP248Q/P248Q mice for CEP and the Fechm1Pas/m1Pas for EPP, clinically and biochemically mimic the severe forms of their respective human diseases and have been valuable for preclinical evaluation of BMT and/or HSC gene therapy approaches. The Fechm1Pas/m1Pas mice have also provided important insights into the underlying pathogenic mechanisms of the liver damage and hematologic abnormalities in EPP. The fact that the Fechm1Pas/m1Pas mice have vastly different clinical severity depending on their genetic background provides a unique opportunity to identify potential modifying genes for EPP. Efforts to generate a partially humanized EPP mouse model carrying the common ‘low expression’ allele were hampered by unexpected missplicing events that led to skipping of exon 3 in the majority of Fech transcripts, rather than use of the −63 nt aberrant splice site that is normally used in human patients with this allele. While this limits the use of this mouse model for evaluating therapies aimed to correct the aberrant splicing caused by the ‘low expression’ allele, these mice may be useful for assessing the impact of various therapies on the marked cutaneous photosensitivity in EPP, given their pronounced photosensitive phenotype.
In summary, mouse models of the human porphyrias have contributed significantly to furthering our understanding of disease pathogenesis and have facilitated the development of new therapeutic approaches for these disorders. Recent advances in gene editing technology, in particular the clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 system, has made generation of mouse models much more rapid and affordable, while offering the capacity to alter multiple genes simultaneously. Furthermore, this system is applicable to various animal species, including non-human primates, which are not readily amenable to conventional genetic manipulation techniques. Thus, going forward, additional porphyric animal models in mice and other species, perhaps with more complex genetic modifications, are likely to be generated using these newer technologies.
Acknowledgements
This work was supported in part by the Department of Genetics and Genomic Sciences at the Icahn School of Medicine at Mount Sinai, and by the NIH-supported Porphyrias Consortium (U54 DK083909), which is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN) of the National Institutes of Health. RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through collaboration between NCATS and the NIDDK.
Conflicts of Interest
MY and RJD are past recipients of research grants from Alnylam Pharmaceuticals and Recordati Rare Diseases. They are co-inventors of a patent licensed to Alnylam Pharmaceuticals for RNAi therapy of the AHPs. RJD is a consultant for Alnylam Pharmaceuticals, Mitsubishi Tanabe Pharma Development America and Recordati Rare Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Makiko Yasuda, Email: makiko.yasuda@mssm.edu.
Robert J. Desnick, Email: Robert.desnick@mssm.edu.
References
- [1].Anderson KE, Sassa S, Bishop DF, Desnick RJ, Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias, in: Scriver C, Beaudet A, Sly W, Valle D (Eds.), The Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, New York, 2001, pp. 2961–3062. [Google Scholar]
- [2].Puy H, Gouya L, Deybach JC, Porphyrias, Lancet 375 (2010) 924–937. [DOI] [PubMed] [Google Scholar]
- [3].Bissell DM, Anderson KE, Bonkovsky HL, Porphyria, N. Engl. J. Med 377 (2017) 2101. [DOI] [PubMed] [Google Scholar]
- [4].Conway AJ, Brown FC, Fullinfaw RO, Kile BT, Jane SM, Curtis DJ, A mouse model of hereditary coproporphyria identified in an ENU mutagenesis screen, Dis. Model Mech 10 (2017) 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tutois S, Montagutelli X, Da Silva V, Jouault H, Rouyer-Fessard P, Leroy-Viard K, Guenet JL, Nordmann Y, Beuzard Y, Deybach JC, Erythropoietic protoporphyria in the house mouseA recessive inherited ferrochelatase deficiency with anemia, photosensitivity, and liver disease, J. Clin. Invest 88 (1991) 1730–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bensidhoum M, Larou M, Lemeur M, Dierich A, Costet P, Raymond S, Daniel JY, de Verneuil H, Ged C, The disruption of mouse uroporphyrinogen III synthase (uros) gene is fully lethal, Transgenics 2 (1998) 275–280. [Google Scholar]
- [7].Lindberg RL, Porcher C, Grandchamp B, Ledermann B, Burki K, Brandner S, Aguzzi A, Meyer UA, Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria, Nat. Genet 12 (1996) 195–199. [DOI] [PubMed] [Google Scholar]
- [8].Magness ST, Maeda N, Brenner DA, An exon 10 deletion in the mouse ferrochelatase gene has a dominant-negative effect and causes mild protoporphyria, Blood 100 (2002) 1470–1477. [DOI] [PubMed] [Google Scholar]
- [9].Clavero S, Bishop DF, Giger U, Haskins ME, Desnick RJ, Feline congenital erythropoietic porphyria: two homozygous UROS missense mutations cause the enzyme deficiency and porphyrin accumulation, Mol. Med 16 (2010) 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Clavero S, Bishop DF, Haskins ME, Giger U, Kauppinen R, Desnick RJ, Feline acute intermittent porphyria: a phenocopy masquerading as an erythropoietic porphyria due to dominant and recessive hydroxymethylbilane synthase mutations, Hum. Mol. Genet 19 (2010) 584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ruth GR, Schwartz S, Stephenson B, Bovine protoporphyria: the first nonhuman model of this hereditary photosensitizing disease, Science 198 (1977) 199–201. [DOI] [PubMed] [Google Scholar]
- [12].Gschnait F, Konrad K, Honigsmann H, Denk H, Wolff K, Mouse model for protoporphyria. I. The liver and hepatic protoporphyrin crystals, J. Invest. Dermatol 65 (1975) 290–299. [DOI] [PubMed] [Google Scholar]
- [13].Takeshita K, Takajo T, Hirata H, Ono M, Utsumi H, In vivo oxygen radical generation in the skin of the protoporphyria model mouse with visible light exposure: an L-band ESR study, J. Invest. Dermatol 122 (2004) 1463–1470. [DOI] [PubMed] [Google Scholar]
- [14].Lindberg RL, Martini R, Baumgartner M, Erne B, Borg J, Zielasek J, Ricker K, Steck A, Toyka KV, Meyer UA, Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria, J. Clin. Invest 103 (1999) 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Meyer UA, Schuurmans MM, Lindberg RL, Acute porphyrias: pathogenesis of neurological manifestations, Semin. Liver Dis 18 (1998) 43–52. [DOI] [PubMed] [Google Scholar]
- [16].Demasi M, Penatti CA, DeLucia R, Bechara EJ, The prooxidant effect of 5-aminolevulinic acid in the brain tissue of rats: implications in neuropsychiatric manifestations in porphyrias, Free Radic. Biol. Med 20 (1996) 291–299. [DOI] [PubMed] [Google Scholar]
- [17].Felitsyn N, McLeod C, Shroads AL, Stacpoole PW, Notterpek L, The heme precursor delta-aminolevulinate blocks peripheral myelin formation, J. Neurochem 106 (2008) 2068–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Neal R, Yang P, Fiechtl J, Yildiz D, Gurer H, Ercal N, Pro-oxidant effects of delta-aminolevulinic acid (delta-ALA) on Chinese hamster ovary (CHO) cells, Toxicol. Lett 91 (1997) 169–178. [DOI] [PubMed] [Google Scholar]
- [19].Onuki J, Chen Y, Teixeira PC, Schumacher RI, Medeiros MH, Van Houten B, Di Mascio P, Mitochondrial and nuclear DNA damage induced by 5-aminolevulinic acid, Arch. Biochem. Biophys 432 (2004) 178–187. [DOI] [PubMed] [Google Scholar]
- [20].Soonawalla ZF, Orug T, Badminton MN, Elder GH, Rhodes JM, Bramhall SR, Elias E, Liver transplantation as a cure for acute intermittent porphyria, Lancet 363 (2004) 705–706. [DOI] [PubMed] [Google Scholar]
- [21].Dowman JK, Gunson BK, Bramhall S, Badminton MN, Newsome PN, Liver transplantation from donors with acute intermittent porphyria, Ann. Intern. Med 154 (2011) 571–572. [DOI] [PubMed] [Google Scholar]
- [22].Solis C, Martinez-Bermejo A, Naidich TP, Kaufmann WE, Astrin KH, Bishop DF, Desnick RJ, Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias, Arch. Neurol 61 (2004) 1764–1770. [DOI] [PubMed] [Google Scholar]
- [23].Yasuda M, Erwin AL, Liu LU, Balwani M, Chen B, Kadirvel S, Gan L, Fiel MI, Gordon RE, Yu C, Clavero S, Arvelakis A, Naik H, Martin LD, Phillips JD, Anderson KE, Sadagoparamanujam VM, Florman SS, Desnick RJ, Liver Transplantation for Acute Intermittent Porphyria: Biochemical and Pathologic Studies of the Explanted Liver, Mol. Med 21 (2015) 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yasuda M, Gan L, Chen B, Kadirvel S, Yu C, Phillips JD, New MI, Liebow A, Fitzgerald K, Querbes W, Desnick RJ, RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice, Proc. Natl. Acad. Sci. U.S.A 111 (2014) 7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meyer RP, Lindberg RL, Hoffmann F, Meyer UA, Cytosolic persistence of mouse brain CYP1A1 in chronic heme deficiency, Biol. Chem 386 (2005) 1157–1164. [DOI] [PubMed] [Google Scholar]
- [26].Homedan C, Laafi J, Schmitt C, Gueguen N, Lefebvre T, Karim Z, Desquiret-Dumas V, Wetterwald C, Deybach JC, Gouya L, Puy H, Reynier P, Malthiery Y, Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model, Int. J. Biochem. Cell Biol 51 (2014) 93–101. [DOI] [PubMed] [Google Scholar]
- [27].Homedan C, Schmitt C, Laafi J, Gueguen N, Desquiret-Dumas V, Lenglet H, Karim Z, Gouya L, Deybach JC, Simard G, Puy H, Malthiery Y, Reynier P, Mitochondrial energetic defects in muscle and brain of a Hmbs−/− mouse model of acute intermittent porphyria, Hum. Mol. Genet 24 (2015) 5015–5023. [DOI] [PubMed] [Google Scholar]
- [28].Serrano-Mendioroz I, Sampedro A, Mora MI, Mauleon I, Segura V, Enriquez de Salamanca R, Harper P, Sardh E, Corrales FJ, Fontanellas A, Vitamin D-binding protein as a biomarker of active disease in acute intermittent porphyria, J. Proteomics 127 (2015) 377–385. [DOI] [PubMed] [Google Scholar]
- [29].Ruspini SF, Zuccoli JR, Lavandera JV, Martinez MDC, Oliveri LM, Gerez EN, Batlle A, Buzaleh AM, Effects of volatile anaesthetics on heme metabolism in a murine genetic model of Acute Intermittent Porphyria. A comparative study with other porphyrinogenic drugs, Biochim. Biophys. Acta 1862 (2018) 1296–1305. [DOI] [PubMed] [Google Scholar]
- [30].Collantes M, Serrano-Mendioroz I, Benito M, Molinet-Dronda F, Delgado M, Vinaixa M, Sampedro A, Enriquez de Salamanca R, Prieto E, Pozo MA, Penuelas I, Corrales FJ, Barajas M, Fontanellas A, Glucose metabolism during fasting is altered in experimental porphobilinogen deaminase deficiency, Hum. Mol. Genet 25 (2016) 1318–1327. [DOI] [PubMed] [Google Scholar]
- [31].Bonkovsky HL, Healey JF, Lourie AN, Gerron GG, Intravenous heme-albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools, Am. J. Gastroenterol 86 (1991) 1050–1056. [PubMed] [Google Scholar]
- [32].Goetsch CA, Bissell DM, Instability of hematin used in the treatment of acute hepatic porphyria, N. Engl. J. Med 315 (1986) 235–238. [DOI] [PubMed] [Google Scholar]
- [33].Yin Z, Wahlin S, Ellis EC, Harper P, Ericzon BG, Nowak G, Hepatocyte transplantation ameliorates the metabolic abnormality in a mouse model of acute intermittent porphyria, Cell Transplant. 23 (2014) 1153–1162. [DOI] [PubMed] [Google Scholar]
- [34].Johansson A, Nowak G, Moller C, Blomberg P, Harper P, Adenoviral-mediated expression of porphobilinogen deaminase in liver restores the metabolic defect in a mouse model of acute intermittent porphyria, Mol. Ther 10 (2004) 337–343. [DOI] [PubMed] [Google Scholar]
- [35].Unzu C, Sampedro A, Mauleon I, Gonzalez-Aparicio M, Enriquez de Salamanca R, Prieto J, Aragon T, Fontanellas A, Helper-dependent adenoviral liver gene therapy protects against induced attacks and corrects protein folding stress in acute intermittent porphyria mice, Hum. Mol. Genet 22 (2013) 2929–2940. [DOI] [PubMed] [Google Scholar]
- [36].Unzu C, Sampedro A, Mauleon I, Alegre M, Beattie SG, de Salamanca RE, Snapper J, Twisk J, Petry H, Gonzalez-Aseguinolaza G, Artieda J, Rodriguez-Pena MS, Prieto J, Fontanellas A, Sustained enzymatic correction by rAAV-mediated liver gene therapy protects against induced motor neuropathy in acute porphyria mice, Mol. Ther 19 (2011) 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yasuda M, Bishop DF, Fowkes M, Cheng SH, Gan L, De snick RJ, AAV8-mediated gene therapy prevents induced biochemical attacks of acute intermittent porphyria and improves neuromotor function, Mol. Ther 18 (2010) 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].D’Avola D, Lopez-Franco E, Sangro B, Paneda A, Grossios N, Gil-Farina I, Benito A, Twisk J, Paz M, Ruiz J, Schmidt M, Petry H, Harper P, de Salamanca RE, Fontanellas A, Prieto J, Gonzalez-Aseguinolaza G, Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria, J. Hepatol 65 (2016) 776–783. [DOI] [PubMed] [Google Scholar]
- [39].Serrano-Mendioroz I, Sampedro A, Serna N, Salamanca RE, Sanz-Parra A, Corrales F, Berraondo P, Millet O, Fontanellas A, Bioengineered PBGD variant improves the therapeutic index of gene therapy vectors for acute intermittent porphyria, Hum. Mol. Genet (2018). [DOI] [PubMed] [Google Scholar]
- [40].Serrano-Mendioroz I, Sampedro A, Alegre M, Enriquez de Salamanca R, Berraondo P, Fontanellas A, An Inducible Promoter Responsive to Different Porphyrinogenic Stimuli Improves Gene Therapy Vectors for Acute Intermittent Porphyria, Hum. Gene Ther 29 (2018) 480–491. [DOI] [PubMed] [Google Scholar]
- [41].Podvinec M, Handschin C, Looser R, Meyer UA, Identification of the xenosensors regulating human 5-aminolevulinate synthase, Proc. Natl. Acad. Sci. U.S.A 101 (2004) 9127–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chan A, Liebow A, Yasuda M, Gan L, Racie T, Maier M, Kuchimanchi S, Foster D, Milstein S, Charisse K, Sehgal A, Manoharan M, Meyers R, Fitzgerald K, Simon A, Desnick RJ, Querbes W, Preclinical Development of a Subcutaneous ALAS1 RNAi Therapeutic for Treatment of Hepatic Porphyrias Using Circulating RNA Quantification, Mol. Ther. Nucleic Acids 4 (2015) e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Medlock AE, Meissner PN, Davidson BP, Corrigall AV, Dailey HA. A mouse model for South African (R59W) variegate porphyria: construction and initial characterization, Cell Mol. Biol. (Noisy-le-grand) 48 (2002) 71–78. [PubMed] [Google Scholar]
- [44].Beukeveld GJ, Wolthers BG, Nordmann Y, Deybach JC, Grandchamp B, Wadman SK, A retrospective study of a patient with homozygous form of acute intermittent porphyria, J. Inherit. Metab. Dis 13 (1990) 673–683. [DOI] [PubMed] [Google Scholar]
- [45].Doss MO, Gross U, Lamoril J, Kranl C, Jacob K, Doss M, da Silva V, Freesemann AG, Deybach JC, Sepp N, Nordmann Y, Compound heterozygous hereditary coproporphyria with fluorescing teeth, Ann. Clin. Biochem 36 (1999) 680–682. [DOI] [PubMed] [Google Scholar]
- [46].Grandchamp B, Phung N, Nordmann Y, Homozygous case of hereditary coproporphyria, Lancet 2 (1977) 1348–1349. [DOI] [PubMed] [Google Scholar]
- [47].Hasanoglu A, Balwani M, Kasapkara CS, Ezgu FS, Okur I, Tumer L, Cakmak A, Nazarenko I, Yu C, Clavero S, Bishop DF, Desnick RJ, Harderoporphyria due to homozygosity for coproporphyrinogen oxidase missense mutation H327R, J. Inherit. Metab. Dis 34 (2011) 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hift RJ, Meissner PN, Todd G, Kirby P, Bilsland D, Collins P, Ferguson J, Moore MR, Homozygous variegate porphyria: an evolving clinical syndrome, Postgrad. Med. J 69 (1993) 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Llewellyn DH, Smyth SJ, Elder GH, Hutchesson AC, Rattenbury JM, Smith MF, Homozygous acute intermittent porphyria: compound heterozygosity for adjacent base transitions in the same codon of the porphobilinogen deaminase gene, Hum. Genet 89 (1992) 97–98. [DOI] [PubMed] [Google Scholar]
- [50].Picat C, Delfau MH, de Rooij FW, Beukeveld GJ, Wolthers BG, Wadman SK, Nordmann Y, Grandchamp B, Identification of the mutations in the parents of a patient with a putative compound heterozygosity for acute intermittent porphyria, J. Inherit. Metab. Dis 13 (1990) 684–686. [DOI] [PubMed] [Google Scholar]
- [51].Schmitt C, Gouya L, Malonova E, Lamoril J, Camadro JM, Flamme M, Rose C, Lyoumi S, Da Silva V, Boileau C, Grandchamp B, Beaumont C, Deybach JC, Puy H, Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic harderoporphyria, Hum. Mol. Genet 14 (2005) 3089–3098. [DOI] [PubMed] [Google Scholar]
- [52].Yasuda M, Gan L, Chen B, Yu C, Zhang J, Gama-Sosa MA, Pollak DD, Berger S, Phillips JD, Edelmann W, Desnick RJ, Homozygous hydroxymethylbilane synthase knockout mice provide pathogenic insights into the severe neurological impairments present in human homozygous dominant acute intermittent porphyria, Hum. Mol. Genet (2019) In press 10.1093/hmg/dd2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wickliffe JK, Abdel-Rahman SZ, Lee C, Kormos-Hallberg C, Sood G, Rondelli CM, Grady JJ, Desnick RJ, Anderson KE, CYP1A2*1F and GSTM1 alleles are associated with susceptibility to porphyria cutanea tarda, Mol. Med 17 (2011) 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Phillips JD, Jackson LK, Bunting M, Franklin MR, Thomas KR, Levy JE, Andrews NC, Kushner JP, A mouse model of familial porphyria cutanea tarda, Proc. Natl. Acad. Sci. U.S.A 98 (2001) 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cantoni L, Graziani A, Rizzardini M, Saletti MC, Porphyrinogenic effect of hexachlorobenzene and 2,3,7,8-tetrachlorodibenzo-para-dioxin: is an inhibitor involved in uroporphyrinogen decarboxylase inactivation? IARC Sci. Publ (1986) 449–456. [PubMed] [Google Scholar]
- [56].Elder GH, Urquhart AJ, De Salamanca RE, Munoz JJ, Bonkovsky HL, Immunoreactive uroporphyrinogen decarboxylase in the liver in porphyria cutanea tarda, Lancet 2 (1985) 229–233. [DOI] [PubMed] [Google Scholar]
- [57].Franklin MR, Phillips JD, Kushner JP, Uroporphyria in the uroporphyrinogen decarboxylase-deficient mouse: Interplay with siderosis and polychlorinated biphenyl exposure, Hepatology 36 (2002) 805–811. [DOI] [PubMed] [Google Scholar]
- [58].Smith AG, Francis JE, Chemically-induced formation of an inhibitor of hepatic uroporphyrinogen decarboxylase in inbred mice with iron overload, Biochem. J 246 (1987) 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP, A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda, Proc. Natl. Acad. Sci. U.S.A 104 (2007) 5079–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Frank J, Wang X, Lam HM, Aita VM, Jugert FK, Goerz G, Merk HF, Poh-Fitzpatrick MB, Christiano AM, C73R is a hotspot mutation in the uroporphyrinogen III synthase gene in congenital erythropoietic porphyria, Ann. Hum. Genet 62 (1998) 225–230. [DOI] [PubMed] [Google Scholar]
- [61].Ged C, Mendez M, Robert E, Lalanne M, Lamrissi-Garcia I, Costet P, Daniel JY, Dubus P, Mazurier F, Moreau-Gaudry F, de Verneuil H, A knock-in mouse model of congenital erythropoietic porphyria, Genomics 87 (2006) 84–92. [DOI] [PubMed] [Google Scholar]
- [62].Bishop DF, Clavero S, Mohandas N, Desnick RJ, Congenital erythropoietic porphyria: characterization of murine models of the severe common (C73R/C73R) and later-onset genotypes, Mol. Med 17 (2011) 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Piomelli S, Poh-Fitzpatrick MB, Seaman C, Skolnick LM, Berdon WE, Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment with high-level transfusions, N. Engl. J. Med 314 (1986) 1029–1031. [DOI] [PubMed] [Google Scholar]
- [64].Robert-Richard E, Moreau-Gaudry F, Lalanne M, Lamrissi-Garcia I, Cario-Andre M, Guyonnet-Duperat V, Taine L, Ged C, de Verneuil H, Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells, Am. J. Hum. Genet 82 (2008) 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Millot S, Delaby C, Moulouel B, Lefebvre T, Pilard N, Ducrot N, Ged C, Letteron P, de Franceschi L, Deybach JC, Beaumont C, Gouya L, De Verneuil H, Lyoumi S, Puy H, Karim Z, Hemolytic anemia repressed hepcidin level without hepatocyte iron overload: lesson from Gunther disease model, Haematologica 102 (2017) 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Blouin JM, Duchartre Y, Costet P, Lalanne M, Ged C, Lain A, Millet O, de Verneuil H, Richard E, Therapeutic potential of proteasome inhibitors in congenital erythropoietic porphyria, Proc. Natl. Acad. Sci. U.S.A 110 (2013) 18238–18243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Urquiza P, Lain A, Sanz-Parra A, Moreno J, Bernardo-Seisdedos G, Dubus P, Gonzalez E, Gutierrez-de-Juan V, Garcia S, Erana H, San Juan I, Macias I, Ben Bdira F, Pluta P, Ortega G, Oyarzabal J, Gonzalez-Muniz R, Rodriguez-Cuesta J, Anguita J, Diez E, Blouin JM, de Verneuil H, Mato JM, Richard E, Falcon-Perez JM, Castilla J, Millet O, Repurposing ciclopirox as a pharmacological chaperone in a model of congenital erythropoietic porphyria, Sci. Transl. Med 10 (2018). [DOI] [PubMed] [Google Scholar]
- [68].Mujtaba T, Dou QP, Advances in the understanding of mechanisms and therapeutic use of bortezomib, Discov. Med 12 (2011) 471–480. [PMC free article] [PubMed] [Google Scholar]
- [69].Roeten MSF, Cloos J, Jansen G, Positioning of proteasome inhibitors in therapy of solid malignancies, Cancer Chemother. Pharmacol 81 (2018) 227–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lecha M, Puy H, Deybach JC, Erythropoietic protoporphyria, Orphanet J. Rare Dis 4 (2009) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Casanova-Gonzalez MJ, Trapero-Marugan M, Jones EA, Moreno-Otero R, Liver disease and erythropoietic protoporphyria: a concise review, World J. Gastroenterol 16 (2010) 4526–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Boulechfar S, Lamoril J, Montagutelli X, Guenet JL, Deybach JC, Nordmann Y, Dailey H, Grandchamp B, de Verneuil H, Ferrochelatase structural mutant (Fechm1Pas) in the house mouse, Genomics 16 (1993) 645–648. [DOI] [PubMed] [Google Scholar]
- [73].Bloks VW, Plosch T, van Goor H, Roelofsen H, Baller J, Havinga R, Verkade HJ, van Tol A, Jansen PL, Kuipers F, Hyperlipidemia and atherosclerosis associated with liver disease in ferrochelatase-deficient mice, J. Lipid Res 42 (2001) 41–50. [PubMed] [Google Scholar]
- [74].Libbrecht L, Meerman L, Kuipers F, Roskams T, Desmet V, Jansen P, Liver pathology and hepatocarcinogenesis in a long-term mouse model of erythropoietic protoporphyria, J. Pathol 199 (2003) 191–200. [DOI] [PubMed] [Google Scholar]
- [75].Lyoumi S, Abitbol M, Rainteau D, Karim Z, Bernex F, Oustric V, Millot S, Letteron P, Heming N, Guillmot L, Montagutelli X, Berdeaux G, Gouya L, Poupon R, Deybach JC, Beaumont C, Puy H, Protoporphyrin retention in hepatocytes and Kupffer cells prevents sclerosing cholangitis in erythropoietic protoporphyria mouse model, Gastroenterology 141 (2011) 1509–1519. [DOI] [PubMed] [Google Scholar]
- [76].Cox TM, Erythropoietic protoporphyria, J. Inherit. Metab. Dis 20 (1997) 258–269. [DOI] [PubMed] [Google Scholar]
- [77].Meerman L, Erythropoietic protoporphyria. An overview with emphasis on the liver, Scand. J. Gastroenterol Suppl. (2000) 79–85. [PubMed] [Google Scholar]
- [78].Meerman L, Koopen NR, Bloks V, Van Goor H, Havinga R, Wolthers BG, Kramer W, Stengelin S, Muller M, Kuipers F, Jansen PL, Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria, Gastroenterology 117 (1999) 696–705. [DOI] [PubMed] [Google Scholar]
- [79].Abitbol M, Bernex F, Puy H, Jouault H, Deybach JC, Guenet JL, Montagutelli X, A mouse model provides evidence that genetic background modulates anemia and liver injury in erythropoietic protoporphyria, Am. J. Physiol. Gastrointest. Liver Physiol 288 (2005) G1208–1216. [DOI] [PubMed] [Google Scholar]
- [80].Han AP, Fleming MD, Chen JJ, Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia, J. Clin. Invest 115 (2005) 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gordeuk VR, Brittenham GM, Hawkins CW, Mukhtar H, Bickers DR, Iron therapy for hepatic dysfunction in erythropoietic protoporphyria, Ann. Intern. Med 105 (1986) 27–31. [DOI] [PubMed] [Google Scholar]
- [82].Baker H, Erythropoietic protoporphyria provoked by iron therapy, Proc. R. Soc. Med 64 (1971) 610–611. [PMC free article] [PubMed] [Google Scholar]
- [83].McClements BM, Bingham A, Callender ME, Trimble ER, Erythropoietic protoporphyria and iron therapy, Br. J. Dermatol 122 (1990) 423–424. [DOI] [PubMed] [Google Scholar]
- [84].Milligan A, Graham-Brown RA, Sarkany I, Baker H, Erythropoietic protoporphyria exacerbated by oral iron therapy, Br. J. Dermatol 119 (1988) 63–66. [DOI] [PubMed] [Google Scholar]
- [85].Lyoumi S, Abitbol M, Andrieu V, Henin D, Robert E, Schmitt C, Gouya L, de Verneuil H, Deybach JC, Montagutelli X, Beaumont C, Puy H, Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice, Blood 109 (2007) 811–818. [DOI] [PubMed] [Google Scholar]
- [86].Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ, Afamelanotide for Erythropoietic Protoporphyria, N. Engl. J. Med 373 (2015) 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Fontanellas A, Mazurier F, Landry M, Taine L, Morel C, Larou M, Daniel JY, Montagutelli X, de Salamanca RE, de Verneuil H, Reversion of hepatobiliary alterations By bone marrow transplantation in a murine model of erythropoietic protoporphyria, Hepatology 32 (2000) 73–81. [DOI] [PubMed] [Google Scholar]
- [88].Duchartre Y, Petit N, Moya C, Lalanne M, Dubus P, Verneuil H, Moreau-Gaudry F, Richard E, Neonatal bone marrow transplantation prevents liver disease in a murine model of erythropoietic protoporphyria, J. Hepatol 55 (2011) 162–170. [DOI] [PubMed] [Google Scholar]
- [89].Fontanellas A, Mazurier F, Belloc F, Taine L, Dumain P, Morel C, Ged C, de Verneuil H, Moreau-Gaudry F, Fluorescence-based selection of retrovirally transduced cells in congenital erythropoietic porphyria: direct selection based on the expression of the therapeutic gene, J. Gene Med 1 (1999) 322–330. [DOI] [PubMed] [Google Scholar]
- [90].Pawliuk R, Bachelot T, Wise RJ, Mathews-Roth MM, Leboulch P, Long-term cure of the photosensitivity of murine erythropoietic protoporphyria by preselective gene therapy, Nat. Med 5 (1999) 768–773. [DOI] [PubMed] [Google Scholar]
- [91].Richard E, Mendez M, Mazurier F, Morel C, Costet P, Xia P, Fontanellas A, Geronimi F, Cario-Andre M, Taine L, Ged C, Malik P, de Verneuil H, Moreau-Gaudry F, Gene therapy of a mouse model of protoporphyria with a self-inactivating erythroid-specific lentiviral vector without preselection, Mol. Ther 4 (2001) 331–338. [DOI] [PubMed] [Google Scholar]
- [92].Richard E, Robert E, Cario-Andre M, Ged C, Geronimi F, Gerson SL, de Verneuil H, Moreau-Gaudry F, Hematopoietic stem cell gene therapy of murine protoporphyria by methylguanine-DNA-methyltransferase-mediated in vivo drug selection, Gene Ther 11 (2004) 1638–1647. [DOI] [PubMed] [Google Scholar]
- [93].Barman-Aksozen J, P CW, Bansode VB, Koentgen F, Trub J, Pelczar P, Cinelli P, Schneider-Yin X, Schumperli D, Minder EI, Modeling the ferrochelatase c.315-48C modifier mutation for erythropoietic protoporphyria (EPP) in mice, Dis. Model Mech 10 (2017) 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]