

Abstract
Many of the genes disrupted in autism are identified as histone-modifying enzymes and chromatin remodelers, most prominently those that mediate histone methylation/demethylation. However, the role of histone methylation enzymes in the pathophysiology and treatment of autism remains unknown. To address this, we used mouse models of haploinsufficiency of the Shank3 gene (a highly penetrant monogenic autism risk factor), which exhibits prominent autism-like social deficits. We found that histone methyltransferases EHMT1 and EHMT2, as well as histone lysine 9 dimethylation (specifically catalyzed by EHMT1/2), were selectively increased in the prefrontal cortex (PFC) of Shank3-deficient mice and autistic human postmortem brains. Treatment with the EHMT1/2 inhibitor UNC0642 or knockdown of EHMT1/2 in PFC induced a robust rescue of autism-like social deficits in Shank3-deficient mice, and restored NMDAR-mediated synaptic function. Activity-regulated cytoskeleton-associated protein (Arc) was identified as one of the causal factors underlying the rescuing effects of UNC0642 on NMDAR function and social behaviors in Shank3-deficient mice. UNC0642 treatment also restored a large set of genes involved in neural signaling in PFC of Shank3-deficient mice. These results suggest that targeting histone methylation enzymes to adjust gene expression and ameliorate synaptic defects could be a potential therapeutic strategy for autism.
INTRODUCTION
Autism is a devastating neurodevelopmental disorder characterized by impaired social interaction and repetitive behaviors. Complicated synaptic, transcriptional, and epigenetic mechanisms have been implicated in the disease1–4. Interestingly, many prominent autism risk factors are identified as histone-modifying enzymes that mediate histone methylation/demethylation5.
Lysine (K) residues on histone proteins can be mono-, di- or tri-methylated by specific histone methyltransferases (HMTs), which are associated with gene activation or repression6, 7. H3K4 methyltransferases KMT2C (MLL3), ASH1L and SETD5, H3K4 demethylase KDM5B, H3K9 demethylases KDM3A and KDM4B, H3K27 demethylases KDM6A and KDM6B are all found to be disrupted in autism5. Analyses of postmortem frontal cortex from autism spectrum disorder (ASD) patients have found the altered trimethylated H3K4 landscape8. H3K9 methyltransferase EHMT1 is causally linked to intellectual disability in humans9, 10. Reduced exploration, increased anxiety and altered social behavior are found in heterozygous EHMT1 knockout mice11. However, the potential of targeting histone methylation enzymes to treat autism remains largely unknown.
In this study, we sought to explore the therapeutic potential of targeting histone methylation for autism by using mouse models that exhibit the prominent “core symptoms” of autism12. Haploinsufficiency of the Shank3 gene that encodes a synaptic scaffolding protein at glutamatergic synapses13 contributes to ~1% of all ASD cases14–17. Thus, the Shank3-deficient mice that recapitulate pronounced autism-like social preference deficits18–20 were selected in this study for the discovery of potential targets for autism treatment.
We focused on prefrontal cortex (PFC), a neural circuitry that plays an essential role in mediating social cognition21. It has been found that glutamatergic neurons in PFC are severely impaired in autistic children22 and Shank3-deficient non-human primate23. Here we sought to identify the HMTs that are altered in the PFC of Shank3-deficient mice, and reveal the rescuing effects of pharmacological agents targeting these HMTs on autistic behavioral and synaptic deficits in this autism model.
MATERIALS AND METHODS
Animals, Human Postmortem Tissues, and Reagents
All animal studies were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the State University of New York at Buffalo. Shank3+/ΔC mice expressing C-terminal (exon 21) deleted Shank3 (Jackson Labs, Bar Harbor, ME) were generated and maintained as previously described18, 24. Because only heterozygous deletion or loss-of-function mutation in the Shank3 gene has been linked to human autism and intellectual disability15, 16, and female Shank3+/ΔC mice lack autism-like social deficits, only male heterozygous Shank3+/ΔC mice (5–8 weeks old), and age- and sex-matched wild-type littermates, were used in this study. Homozygous male and female Shank3e4–9 mice with the loss of major isoforms of the Shank3 gene resulting from the deletion of N-terminal exons 4–9 (kindly provided by Dr. Yong-Hui Jiang at Duke University), were also used. Frozen human postmortem tissues (Brodmann’s Area 9) were provided by NIH NeuroBioBank. See Supplementary Materials and Methods for details.
Quantitative Real-time RT-PCR and Western Blotting
All biochemical analysis were performed as previously described25, 26. See Supplementary Materials and Methods for details.
Behavioral Testing
Behavioral studies, including social preference test, social approach test, Rota-rod test, self-grooming test, locomotion test and elevated plus maze test, were preformed to examine the social behavior, motor coordination, repetitive behavior, locomotor activity and anxiety-like behavior18, 19. Detailed information is included in Supplementary Methods.
Viral Gene Transfer
The shRNA oligonucleotide targeting mouse Ehmt1 (CGCTATGATGATGATGAATAA), Ehmt2 (CCGAGAGAGTTCATAGCTCTT), or Arc (GAGGAGGAGATCATTCAGTAT) sequence was inserted to the lentiviral vector pLKO.3G (Addgene), which contains an eGFP reporter. The virus production was performed as previously described18, 26. Arc CRISPR activation lentiviral particle (Santa Cruz Biotech., sc-419184-LAC) was delivered to PFC for Arc overexpression. See Supplementary Materials and Methods for details.
Immunohistochemistry
Immunostaining was carried out using the standard free-floating methods. Details on staining, image acquisition and quantification are included in Supplementary Methods.
Electrophysiological Recordings in slices.
Whole-cell voltage-clamp recording technique was used to measure synaptic currents in layer V pyramidal neurons of prefrontal cortical slices, as previously described 19, 26–28. See Supplementary Materials and Methods for details.
RNA Sequencing and analysis
Detailed information on data acquisition and analysis is included in Supplementary Materials and Methods.
Chromatin Immunoprecipitation (ChIP)
ChIP samples were prepared and ChIP assay was performed as previously described19, 26, 29. Details are included in Supplementary Materials and Methods.
Primary Neuronal Culture
Rat PFC cultures were prepared as described previously27. See Supplementary Materials and Methods for details.
Statistics
All data were expressed as the mean ± sem. No sample was excluded from the analysis. The sample size was based on power analyses and was similar to those reported in previous works18, 19, 26, 28. Each set of the experiments was replicated at least 3 times. There is no variance difference between the groups that are being statistically compared. Experiments with two groups were analyzed statistically using unpaired Student’s t-tests. Experiments with more than two groups were subjected to one-way ANOVA, two-way ANOVA, two-way repeated measure ANOVA (rmANOVA), or three-way ANOVA, followed by post hoc Bonferroni’s multiple comparisons tests with correction.
RESULTS
Histone K9 dimethylation is specifically altered in the PFC of Shank3-deficient mice and autistic human postmortem brains.
To reveal the role of histone methylation in autism pathophysiology, we first examined lysine methyltransferases (KMTs) that control H3K9, H3K4 or H3K27 methylation. Heterozygous Shank3+/ΔC mice (male, 5–6 weeks old), that have lost full-length Shank3 expression and synaptic localization of Shank3 due to the deletion of C-terminal (exon 21) Shank318 were used as a model of Shank3 haploinsufficency. Among the 10 KMTs tested, the transcription of euchromatic histone methyltransferases Ehmt1 and Ehmt2 was selectively unregulated (~40.7% and ~23.3% increase, respectively; P = 0.013 and P = 0.0058, t-test) in the PFC of Shank3+/ΔC mice, compared to WT mice (Fig. 1a). No changes were found on the mRNA level of the chromatin remodeler Chd8 (an autism risk factor5) (Fig. 1a). Along with the significantly increased EHMT1 and EHMT2 protein expression in the PFC of Shank3+/ΔC mice (Fig. 1b, ~23.7% and ~28.3% increase, respectively; P = 0.019 and P = 0.0097, t-test), the level of H3K9 dimethylation (H3K9me2), which is specifically catalyzed by EHMT1/2 and is associated with gene silencing30, 31, was also significantly elevated (Fig. 1c, ~38.1% increase; P = 0.028, t-test), while the level of H3K4me3 and H3K27me3 was largely unchanged (Fig. 1c). Immunostaining of H3K9me2 and NeuN (a neuronal marker) showed that most PFC neurons exhibited the significantly elevated H3K9me2 signaling (Fig. 1d and 1e, ~129.2% increase, P < 0.0001, t-test). In the dorsal striatum or ventral tegmental area (VTA) of Shank3+/ΔC mice, Ehmt1 and Ehmt2 mRNA levels were not altered (Fig. S1a and 1b). Consistently, immunohistochemical studies found that H3K9me2 level in striatum (Fig. S1c), dentate gyrus (Fig. S1d) and CA1 (Fig. S1e) was largely unchanged in Shank3+/ΔC mice.
Figure 1. EHMT1/2 and H3K9me2 levels are specifically elevated in the PFC of Shank3+/ΔC mice and autistic human patients.

(a) Quantitative real-time RT-PCR data on the mRNA level of 10 histone methyltransferases catalyzing H3K9, H3K27 or H3K4 methylation, and Chd8 in the frontal cortical tissue from WT and Shank3+/ΔC mice (Ehmt1/2: n = 13–16/group; others: n = 6–8/group, * P < 0.05, t-test). (b, c) Quantitation and representative immunoblots of EHMT1 and EHMT2 protein levels (b) and H3K9me2, H3K4me3 and H3K27me3 levels (c) in the nuclear fraction of prefrontal cortical tissue from WT and Shank3+/ΔC mice (n = 9–10/group, * P < 0.05, ** P < 0.01, t-test). (d) Representative confocal images of immunohistochemical staining of H3K9me2 and NeuN in PFC of WT and Shank3+/ΔC mice. (e) Plots showing the level of H3K9me2 in PFC of WT and Shank3+/ΔC mice. (n = 15–30 slices/5 animals; *** P < 0.001, t-test). (f) ChIP assay data showing β-catenin binding at Ehmt1 and Ehmt2 promoter regions in PFC lysates from WT vs. Shank3+/ΔC mice (n = 7–8/group, * P < 0.05, t-test). Top: schematic graph showing the location of primer covering the TCF-LEF binding motif (labeled with vertical gray lines). (g) qPCR data on the mRNA level of EHMT1, EHMT2 and several other histone methyltransferases catalyzing H3K9, H3K27 or H3K4 methylation in Brodmann’s Area 9 (BA9) of postmortem tissue from autistic human patients and healthy controls (n = 7–10/group, * P < 0.05, t-test). (h) Quantitation and representative immunoblots of EHMT1, EHMT2, H3K9me2 and β-catenin levels in the nuclear fraction of BA9 from autistic human patients and healthy controls (n = 6–9/group, * P < 0.05, ** P < 0.01, t-test). Data are expressed as mean ± sem.
We next investigated the potential molecular mechanism underlying the elevation of nuclear proteins EHMT1/2 by the loss of synaptic protein Shank3. Our previous studies have found that Shank3 interacts with β-catenin19, a dual function protein regulating both cell-cell adhesion and gene transcription. Loss of Shank3 results in the translocation of β-catenin from synapses into the nucleus19, where β-catenin could form complexes with transcription factor TCF/LEF to activate target genes32. Multiple TCF/LEF binding sites were identified at the promoter region of Ehmt1 and Ehmt2 (Fig. 1f). Using chromatin immunoprecipitation (ChIP) assays with primers against TCF/LEF binding sites on Ehmt1 and Ehmt2, we found that β-catenin occupancy at Ehmt1 and Ehmt2 promoters was significantly increased in PFC of Shank3+/ΔC mice (Fig. 1f, ~48.5% and ~31.6% increase, respectively; P = 0.031 and P = 0.033, t-test), suggesting that Shank3 deficiency results in Ehmt1/2 upregulation via β-catenin.
To find out the validity and translational value of our findings in the Shank3-deficient mouse model of autism, we compared EHMT1/2 mRNA levels in the postmortem PFC tissues (Brodmann’s area 9) from autism patients vs. age and sex-matched control subjects. Autism patients had a significantly higher level of EHMT2 mRNA (Fig. 1g, ~89.6% increase, P = 0.018, t-test), but not other tested histone methyltransferases. Elevated levels of EHMT2 protein (~97.9% increase, P = 0.038, t-test) and euchromatic H3K9me2 (~54.8% increase, P = 0.027, t-test) were also observed in BA9 region of ASD patients (Fig. 1h). Consistently, EHMT2 was found to be significantly elevated in the peripheral blood cells of Japanese autism subjects33. In addition, we observed an increased β-catenin protein level in the nuclear fraction of postmortem PFC tissues from autistic patients (Fig. 1h, ~85.1% increase, P = 0.009, t-test). The accumulated β-catenin in the nucleus could form complexes with transcription factor TCF/LEF to activate the expression of genes32, including EHMT2. Taken together, these data suggest that altered histone methylation in PFC, particularly abnormally high level of the repressive H3K9me2, occurs in autism.
EHMT1/2 inhibition or knockdown in PFC rescues autism-like social deficits and restores NMDAR function in Shank3-deficient mice.
Given the elevated EHMT1/2 expression in Shank3+/ΔC mice, we next examined the therapeutic potential of EHMT1/2 inhibitors. Systemic administration of UNC0642 (1 mg/kg, i.p., once daily for 3 days), a highly potent, selective and brain-permeable inhibitor of EHMT1 and EHMT234, significantly reduced the elevated level of H3K9me2 in the PFC of Shank3+/ΔC mice (Fig. 2a, ~41.9% decrease, F1,17 (treatment) = 13.38, P = 0.0019, two-way ANOVA). In the 3-chamber social preference assay, a brief UNC0642 treatment (1 mg/kg, i.p., 3x) of Shank3+/ΔC mice significantly increased the investigation time on the social stimulus (Fig. 2b and 2c, ~49.2% increase, F1,94 (interaction) = 10.83, P = 0.001, three-way ANOVA), as well as the preference for the social stimulus over the non-social object (Fig. 2d, ~117.0% increase, F1,47 (interaction) = 26.2, P < 0.0001, two-way ANOVA). In the social approach assay, UNC0642 treatment also restored the interaction time with the social stimulus in Shank3+/ΔC mice (Fig. 2e and 2f, ~57.6% increase, F1,37 (interaction) = 15.5, P < 0.001, two-way ANOVA). Importantly, the therapeutic effect of a single round UNC0642 treatment on social deficits in Shank3+/ΔC mice lasted for ~15 days post-injection, and repeated administration of UNC0642 still significantly improved social preference (Fig. 2g), as well as the social interaction time (Fig. S2a and S2b). Consistent with this time frame, the reducing effect of UNC0642 treatment (i.p., 3x) on global H3K9me2 level in Shank3+/ΔC mice was present at 8–10 days, but not 20 days, post-treatment (Fig. S2c and S2d). A lower dose of UNC0642 treatment (0.25 mg/kg, i.p., 3x) of Shank3+/ΔC mice failed to restore the social preference deficits (Fig. S2e), in agreement with its lack of effect on H3K9me2 (Fig. S2f), thus UNC0642 (1 mg/kg) was used in following studies. Systemic administration of BIX01294 (1.0 mg/kg, s.c., 3x), another specific EHMT1/2 inhibitor35, only induced a transient improvement of social preference in Shank3+/ΔC mice (Fig. S2g), which is probably because of the low potency of this compound36.
Figure 2. EHMT1/2 inhibition or knockdown induces robust and prolonged rescue of autism-like social deficits in Shank3-deficient mice.

(a) Immunoblots and quantitation of the level of H3K9me2 in the nuclear fraction of frontal cortical tissue from WT or Shank3+/ΔC mice injected (i.p.) with the EHMT1/2 inhibitor UNC0642 (UNC, 1 mg/kg, 3x) or saline. (n = 5–6/group, * P < 0.05, ** P < 0.01, two-way ANOVA). (b) Representative heat maps illustrating the time spent in different locations of the 3 chambers in the social preference tests of WT or Shank3+/ΔC mice treated with UNC0642 (1 mg/kg, i.p., 3x) or saline. Locations of social (SOC) and non-social (NS) stimuli are labeled with the circles. (c, d) Plots showing the time spent investigating either the SOC or NS stimulus (c) or the preference index (d) from 3-chamber sociability testing of WT or Shank3+/ΔC mice treated with UNC0642 or saline (n = 11–14/group, c: * P < 0.05, *** P < 0.001, saline vs. UNC; +++ P < 0.001, SOC vs. NS, three-way ANOVA; d: *** P < 0.001, two-way ANOVA). (e) Representative heat maps illustrating the time spent in different locations of the apparatus from the social approach tests of WT or Shank3+/ΔC mice treated with UNC0642 or saline. Locations of SOC stimuli are labeled with the circles. (f) Scatter plots showing the social interaction time in social approach tests of all groups (n = 9–11/group, * P < 0.05, ** P < 0.01, two-way ANOVA). (g) Plots of social preference index from the 3-chamber sociability testing of Shank3+/ΔC mice treated with 2 rounds of UNC0642 (1 mg/kg, i.p., 3x) or saline at different time points (n = 8–11/group, * P < 0.05, *** P < 0.001, pre- vs. post-injection, two-way rmANOVA). (h) Immunoblots and quantitation of EHMT1, EHMT2 and H3K9me2 in WT mice with the stereotaxic injection of shRNA lentiviruses against EHMT1 and EHMT2 or a scrambled shRNA lentivirus to the medial PFC (n = 3–4/group, * P < 0.05, ** P < 0.01, t-test). Inset: Image showing the lentiviral expression in PFC. (i, j) Plots of the time spent investigating either the SOC or NS stimulus (i) and the social preference index (j) from 3-chamber sociability testing of WT or Shank3+/ΔC mice with the prior injection of EHMT1/2 shRNA or a scrambled shRNA lentivirus into PFC (n = 8–10/group, i: ** P < 0.01, scrambled vs. EHMT1/2 shRNA; +++ P < 0.001, SOC vs. NS, three-way ANOVA; j: ** P < 0.01, two-way ANOVA). Data are presented as mean ± sem.
To find out whether the therapeutic effects of UNC0642 are mediated by EHMT1/2 inhibition in the PFC, we generated shRNA lentiviruses against EHMT1 and EHMT2, and stereotaxically injected them into medial PFC of Shank3+/ΔC mice. In vivo knockdown of EHMT1 and EHMT2 expression, as well as a significant reduction of H3K9me2, was confirmed in viral-infected PFC tissue from WT or Shank3+/ΔC mice (Fig. 2h, 26.9%−37.0% decrease; Fig. S3). In 3-chamber social preference assay, Shank3+/ΔC mice with PFC injection of EHMT1/2 shRNA exhibited the significantly increased social interaction time (Fig. 2i, ~67.2% increase, F1,72 (interaction) = 6.0, P = 0.017, three-way ANOVA) and social preference (Fig. 2j, ~188.0% increase, F1,32 (interaction) = 7.0, P = 0.013, two-way ANOVA), compared to those injected with a scrambled shRNA. In WT mice, EHMT1/2 knockdown did not change the social interaction time (Fig. 2i) or social preference (Fig. 2j). Taken together, these results indicated that EHMT1/2 inhibition or knockdown could lead to a robust rescue of autism-like social deficits in Shank3-deficient mice.
Next, we assessed the safety and potential side effects associated with UNC0642 treatment (1 mg/kg, i.p., 3x) by performing a general health screening and other behavioral assays. In blood chemistry and hematological analyses of UNC0642-treated animals, all the indicators for liver and kidney functions, as well as lipid and protein metabolism, were within normal ranges (Suppl. Table 1). UNC0642 treatment did not have an anxiogenic effect as measured by elevated plus maze tests (Fig. S4a). Repetitive grooming in Shank3+/ΔC mice was largely unaffected by UNC0642 treatment (Fig. S4b). Motor coordination as measured by rotarod tests and general movement as measured by locomotion tests were not affected by UNC0642 treatment (Fig. S4c and S4d). Loss of body weight, a sign of general health deficiency, was not observed in UNC0642-treated animals (Fig. S4e). All these data suggest the general safety of the UNC0642 regimen used in our studies, consistent with a lack of toxicity of UNC0642 treatment at a higher dose and longer duration (5 mg/kg, i.p. 5 days37).
To determine whether the therapeutic effect of UNC0642 is generally applicable in Shank3-associated autism conditions, we examined another Shank3-deficient mouse model, Shank3e4−9 mice, which have lost the major Shank3 isoforms because of the deletion of N-terminal (exon 4–9) Shank320. Levels of EHMT1 and EHMT2 proteins were significantly increased in the PFC of homozygous Shank3e4−9 mice, compared to WT mice (Fig. 3a, ~42.6% and ~23.8% increase, respectively, P = 0.046, P = 0.031, t-test). A significant increase of H3K9me2 was also detected in the PFC of Shank3e4−9 mice (Fig. 3b, ~49.3% increase), which was normalized by the systemic administration of UNC0642 (1 mg/kg, i.p., 3x, Fig. 3b, ~34.7% decrease, F2,15 = 6.60, P = 0.009, one-way ANOVA). In the 3-chamber social preference assay, UNC0642 treatment of Shank3e4−9 mice significantly increased the investigation time on the social stimulus (Fig. 3c and 3d, 42.3%−61.0% increase, F3,72 (interaction) = 9.48, P < 0.0001, two-way ANOVA), as well as the preference for the social stimulus over the non-social object (Fig. 3e, 152.6%−184.2% increase, F3,36 = 14.0, P < 0.0001, one-way ANOVA). The rescuing effect of UNC0642 on social deficits in Shank3e4−9 mice was still present at day 6 post-treatment (Fig. 3d and 3e), reminiscent of what was found in Shank3+/ΔC mice.
Figure 3. Shank3e4−9 mice exhibit H3K9me2 elevation and autism-like social deficits, which are rescued by EHMT1/2 inhibition.

(a) Quantitation and representative immunoblots of EHMT1 and EHMT2 protein levels in the nuclear fraction of prefrontal cortical tissue from WT and Shank3e4−9 mice (n = 6/group, * P < 0.05, t-test). (b) Immunoblots and quantitation of the level of H3K9me2 in the nuclear fraction of frontal cortical tissue from WT or Shank3e4−9 mice injected (i.p.) with UNC0642 (UNC, 1 mg/kg, 3x) or saline (n = 6/group, * P < 0.05, one-way ANOVA). (c) Representative heat maps illustrating the time spent in different locations of 3-chamber social preference tests of Shank3e4−9 mice treated with UNC0642 or saline. (d, e) Plots showing the time spent investigating either the SOC or NS stimulus (d) or the preference index (e) from 3-chamber sociability testing of Shank3e4−9 mice treated with UNC0642 or saline (n = 9–11/group, d: * P < 0.05, *** P < 0.001, saline vs. UNC; +++ P < 0.001, SOC vs. NS, two-way ANOVA; e: *** P < 0.001, two-way ANOVA). Data are presented as mean ± sem.
To determine the synaptic basis for the amelioration of autism-like behaviors by EHMT1/2 inhibition, we focused on the glutamatergic system, which is strongly linked to the pathophysiology of autism associated with Shank3 deficiency18, 20, 24, 38. NMDAR-mediated excitatory postsynaptic currents (EPSC) were measured in deep layer PFC pyramidal neurons, which were mostly affected in autistic children22, to examine the impact of UNC0642 treatment on NMDAR function. As shown in Fig. 4a, the input/output curves of NMDAR-EPSC induced by a series of stimuli of different intensities were significantly diminished in saline-injected Shank3+/ΔC mice (~28.0%−30.2% decrease), and UNC0642 treatment (1 mg/kg, i.p., 3x) restored NMDAR-EPSC to the control level (F1,219 (genotype x treatment) = 14.86, P < 0.0001, three-way ANOVA). In contrast, evoked AMPAR-EPSC was unaffected by Shank3 deficiency or UNC0642 treatment (Fig. 4b), and the amplitude and frequency of spontaneous AMPAR-EPSC showed no change across the genotypes and treatment groups (Fig. 4c and 4d). Consistent with the effect of UNC0642 on NMDAR function in PFC, knockdown of EHMT1/2 with the shRNA lentivirus in PFC of Shank3+/ΔC mice also led to the significant recovery of NMDAR-EPSC (Fig. 4e, F1,16 (interaction) = 11.49, P = 0.0037, two-way ANOVA). We further tested whether UNC0642 treatment could affect NMDAR synaptic function in other brain regions, such as striatum (STR). H3K9me2 protein level in STR was reduced by UNC0642 treatment (Fig. S5a), but NMDAR-EPSC was not affected by Shank3 deficiency or UNC0642 treatment (Fig. S5b).
Figure 4. EHMT1/2 inhibition or knockdown restores NMDAR function in PFC of Shank3-deficient mice.

(a, b) Input-output curves of NMDAR-EPSC (a) and AMPAR-EPSC (b) in PFC pyramidal neurons from WT or Shank3+/ΔC mice treated with UNC0642 (UNC, 1 mg/kg, 3x) or saline (NMDA: n = 9–12 cells/3–4 mice each group, * P < 0.05, ** P < 0.01; AMPA: n = 9–12 cells/3 mice each group, P > 0.05, three-way rmANOVA). Recordings were performed at 4–5 days post-injection. Inset: representative NMDAR-EPSC and AMPAR-EPSC traces. (c, d) Summary graph (c) and cumulative distribution plots (d) of spontaneous EPSC (sEPSC) amplitudes and frequencies in WT or Shank3+/ΔC mice treated with UNC0642 or saline. Inset (d): representative sEPSC traces. (e) Input-output curves of NMDAR-EPSC in PFC pyramidal neurons from Shank3+/ΔC mice with the prior injection of EHMT1/2 shRNA or a scrambled shRNA lentivirus into PFC (n = 8–10 cells/5 mice each group, * P < 0.05, *** P < 0.001, two-way rmANOVA). Inset: representative NMDAR-EPSC traces. Data are presented as mean ± sem.
The synaptic plasticity gene Arc is one of the key molecules mediating the rescuing effects of UNC0642 in Shank3-deficient mice.
Next, we sought to identify the molecular determinants mediating the rescuing effects of EHMT1/2 inhibition on autism-like synaptic and behavioral deficits in Shank3-deficient mice. We speculate that the transcription of certain genes important for neuronal signaling is downregulated in Shank3+/ΔC mice due to the abnormally high level of repressive H3K9me2, and UNC0642 treatment restores these genes expression by inhibiting H3K9me2 enrichment at their promoters. To test this, we focused on the risk factors for autism, schizophrenia or intellectual disability that are involved in regulating synaptic structure and function, including ankyrin 2 (Ank2), Cortactin-binding protein 2 (Cttnbp2), cortactin (Cttn) and activity-regulated cytoskeleton-associated protein (Arc)5, 39–42. As shown in Fig. 5a, the mRNA level of Arc was significantly lost in the PFC of Shank3+/ΔC mice, compared to WT mice (~52.2% decrease), which was markedly restored by UNC0642 treatment (~170.7% increase, F1,31 (interaction) = 9.07, P = 0.0051, two-way ANOVA). Meanwhile the mRNA levels of Ank2, Cttnbp2 and Cttn were not altered by Shank3 deficiency or UNC0642 treatment. Moreover, Arc protein expression was attenuated by Shank3 deficiency (Fig. 5b, ~58.7% decrease), and reversed by UNC0642 treatment (~125.7% increase, F1,24 (interaction) = 12.66, P = 0.016, two-way ANOVA). Additionally, UNC0642 treatment was unable to recover the mRNA level and protein level of Shank3 (Fig. 5a and 5b).
Figure 5. The synaptic plasticity gene Arc mediates the rescuing effects of UNC0642 in Shank3-deficient mice.
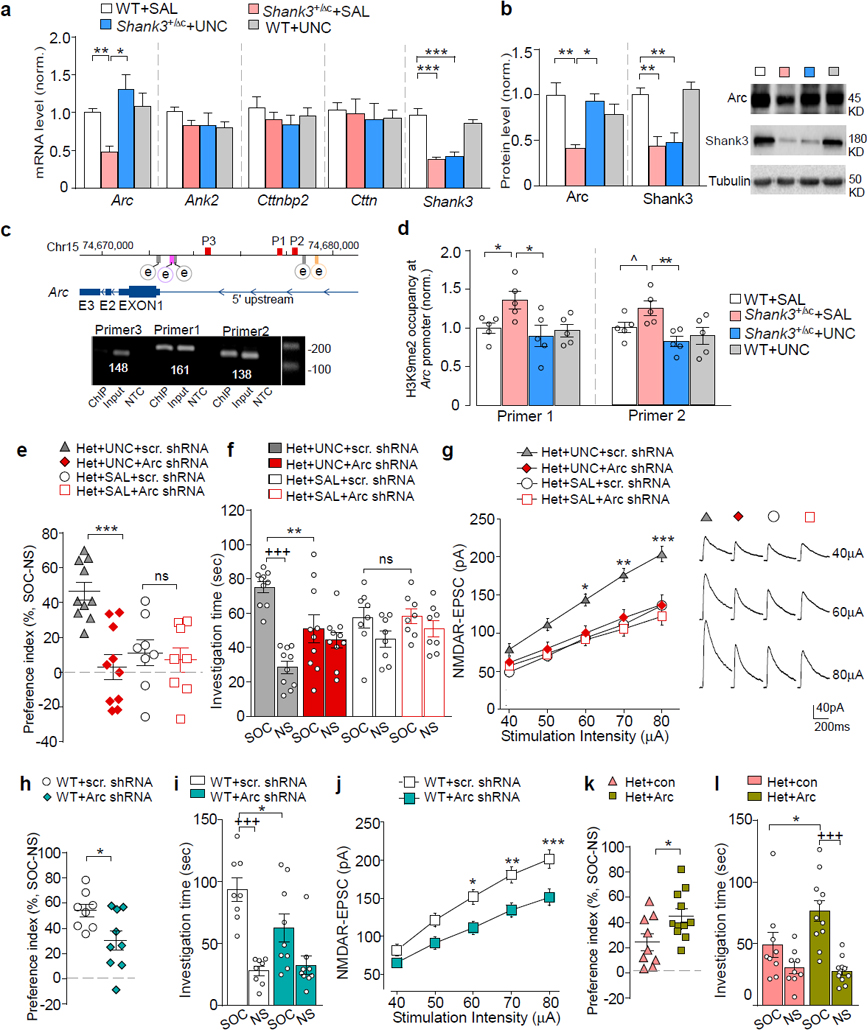
(a) qPCR data showing the mRNA level of Ank2, Cttnbp2, Cttn, and Arc in PFC from WT or Shank3+/ΔC mice treated with UNC0642 (1 mg/kg, i.p., 3x) or saline (n = 7–10/group, * P < 0.05, ** P < 0.01, two-way ANOVA). (b) Immunoblots and quantitation of Arc and Shank3 proteins in PFC from UNC0642- or saline-treated WT or Shank3+/ΔC mice (n = 6–8 /group, * P < 0.05, ** P < 0.01, two-way ANOVA). (c) Diagram showing the locations of Arc transcription enhancers (e), SRE (gray), Zeste-like elements (purple), SARE (orange), and primers (P1, P2, P3) used in ChIP assays (red). (d) ChIP assay data showing the enrichment of H3K9me2 at the Arc promoter region in PFC from WT or Shank3+/ΔC mice treated with UNC0642 or saline (n = 5 /group, ^ P < 0.1, * P < 0.05, two-way ANOVA). (e, f) Plots of social preference index (e) or the time spent investigating either the social (SOC) or non-social (NS) stimulus (f) from 3-chamber sociability testing of UNC0642- or saline-treated Shank3+/ΔC mice with the prior infection of Arc or a scrambled shRNA lentivirus (n = 8–10/group, e: *** P < 0.001, two-way ANOVA; f: +++ P < 0.001 SOC vs. NS, ** P < 0.01 scrambled vs. Arc shRNA, ns: not significant, three-way ANOVA). (g) Summarized input/output curves of NMDAR-EPSC in UNC0642- or saline-treated Shank3+/ΔC mice with the prior injection of Arc shRNA or a scrambled shRNA lentivirus into PFC (n = 8–9 cells/3 mice for each group, * P < 0.05, ** P < 0.01, *** P < 0.001, three-way rmANOVA). Right: Representative NMDAR-EPSC traces. (h, i) Plots of social preference index (h) or the time spent investigating either SOC or NS stimulus (i) from 3-chamber sociability testing of WT mice with the prior infection of Arc shRNA or a scrambled shRNA lentivirus (n = 8–9/group) (h: * P < 0.05, t-test; i: +++ P < 0.001, SOC vs. NS, * P < 0.05, scrambled vs. Arc shRNA, two-way ANOVA). (j) Summarized input/output curves of NMDAR-EPSC in WT mice with the prior injection of Arc shRNA or a scrambled shRNA lentivirus into PFC (n = 8–9 cells/3–5 mice for each group, * P < 0.05, ** P < 0.01, *** P < 0.001, two-way rmANOVA). (k, l) Plots of social preference index (k) or the time spent investigating either SOC or NS stimulus (l) from 3-chamber sociability testing of Shank3+/ΔC mice with the prior infection of Arc activation lentivirus or a control virus (n = 9–11/group) (k: * P < 0.05, t-test; l: +++ P < 0.001, SOC vs. NS, * P < 0.05, con vs. Arc, two-way ANOVA). Data are presented as mean ± sem.
To find out whether the alteration of Arc transcription by Shank3 deficiency or UNC0642 treatment is due to the change in repressive H3K9me2 at the Arc promoter, we performed ChIP assays using PFC tissue. Arc transcriptional regulation depends on several enhancer elements upstream of transcription start sites (TSS), including serum response elements (SRE) at 0.9 kb, 1.5 kb and 6.5 kb43, 44, Zeste-like elements at 1.4 kb43, and distal enhancer synaptic activity-responsive element (SARE) at 7 kb45. H3K9me2 occupancy around these active enhancer elements was identified (Fig. 5c). Compared with WT mice, H3K9me2 enrichment at two loci of the Arc promoter was significantly increased in Shank3+/ΔC mice (Fig. 5d), and UNC0642 treatment reversed it to the wild-type level. These results suggest that the transcriptional regulation of Arc is dependent on histone K9 dimethylation.
We further examined whether the restoration of Arc expression was necessary for the rescuing effects of UNC0642 in Shank3-deficient mice. An shRNA lentivirus was generated to knock down Arc expression. Significantly lower expression of Arc was observed in viral-infected cortical cultures and PFC tissues from viral-injected WT or Shank3+/ΔC mice (Fig. S6). In Shank3+/ΔC mice with the stereotaxic injection of Arc shRNA lentivirus into the PFC, UNC0642 treatment (1 mg/kg, i.p., 3x) failed to restore social preference (Fig. 5e) and social interaction time (Fig. 5f, F1,72 (treatment) = 0.72, P = 0.40, three-way ANOVA) in the 3-chamber sociability test. Arc knockdown and Shank3 deficiency did not have additive effects on social deficits (Fig. 5e and 5f). Arc knockdown in Shank3+/ΔC mice also prevented the UNC0642-induced recovery of NMDAR-EPSC in PFC pyramidal neurons (Fig. 5g, F1,141 (treatment x virus) = 6.02, P = 0.016, three-way ANOVA), and failed to induce additional reduction of NMDAR-EPSC (Fig. 5g). These data suggest the necessity of Arc in UNC0642-induced rescue of social deficits and NMDAR hypofunction in Shank3-deficient mice.
Next, we tested the impact of manipulating Arc expression on social behaviors and NMDAR function. WT mice injected with Arc shRNA to the PFC exhibited the significantly diminished social preference (Fig. 5h, ~43.8% decrease, P =0.026, t-test) and social interaction time (Fig. 5i, ~36.8% decrease, F1,30 (interaction) = 4.23, P = 0.049, two-way ANOVA). Moreover, NMDAR-EPSC in PFC pyramidal neurons was significantly smaller in WT mice injected with Arc shRNA lentivirus than the scrambled control virus (Fig. 5j, 24.8%−26.8% decrease, F1,15 (interaction) = 15.08, P = 0.015, two-way ANOVA). These data suggest that lowering the expression of Arc in wild-type mice is sufficient to induce autism-like social deficits and NMDAR hypofunction. Consistent with this, Arc knockout mice also exhibit disrupted social abilities46. On the other hand, Shank3+/ΔC mice with overexpression of Arc in the PFC (Fig. S6f), exhibited the significantly elevated social preference (Fig. 5k, ~103.8% increase, P = 0.010, t-test) and social interaction time (Fig. 5l, ~62.1% increase, F1,36 (interaction) = 4.5, P = 0.041, two-way ANOVA), suggesting that elevating the expression of Arc in Shank3-deficient mice is capable of rescuing autism-like social deficits.
Arc interacts with many synaptic proteins, including receptor channels and other synapse function genes (Fig. 6a), so we further examined whether Arc-interacting proteins might also contribute to the rescuing effects of UNC0642 in Shank3-deficient mice. Quantitative PCR and Western blots indicated that NMDAR subunits (GluN1, GluN2A, GluN2B) and AMPAR subunits (GluA1, GluA2) were unchanged in Shank3+/ΔC mice, but UNC0642 treatment elevated Grin1 and Grin2a mRNA (~94.9% and ~84.5% increase, respectively, Fig. 6b, Grin1: F1,36 (treatment) = 9.89, P = 0.0033; Grin2a: F1,35 (treatment) = 8.94, P = 0.0051; two-way ANOVA) and protein levels (~58.8% and ~46.1% increase, respectively, Fig. 6c, Grin: F1,36 (treatment) = 15.06, P = 0.0005; Grin2a: F1,28 (treatment) = 28.57, P < 0.0001; two-way ANOVA). ChIP assays identified H3K9me2 occupancy at Grin1 (~2500 bp from TSS), Grin2a (~6800bp from TSS) and Grin2b (~2400bp from TSS) promoters (Fig. 6d). UNC0642 treatment significantly decreased H3K9me2 occupancy at Grin1 and Grin2a promoters in Shank3+/ΔC mice (~36.4% and ~30.0% decrease, respectively; Grin1: F1,20 (treatment) = 7.35, P = 0.014; Grin2a: F1,16 (treatment) = 5.97, P = 0.027; two-way ANOVA) without a significant effect on the Grin2b promoter (Fig. 6e).
Figure 6. Arc-interacting proteins contribute to the rescuing effects of UNC0642 in Shank3-deficient mice.

(a) Arc-interacting protein networks derived from CYTOSCAPE. (b, c) qPCR and immunoblotting data showing the mRNA (b) or protein (c) level of Grin1 (GluN1), Grin2a (GluN2A), Gria2b (GluN2B), Gria1 (GluA1), Gria2 (GluA2), and Shank3 (Shank3) in WT or Shank3+/ΔC mice treated with UNC0642 or saline (n = 6–11/group, * P < 0.05, ** P < 0.01, *** P < 0.001, two-way ANOVA). (d) Diagram showing the location of primers used in ChIP assays (red). (e) ChIP assay data showing the enrichment of H3K9me2 at Grin1, Grin2a and Grin2b promoters in PFC from WT or Shank3+/ΔC mice treated with UNC0642 or saline (n = 5–6/group * P < 0.05, ** P < 0.01, two-way ANOVA). (f) qPCR data showing the mRNA level of Camk2b, Camk2a, Cyfip1, Syngap1 and Homer1 in WT or Shank3+/ΔC mice treated with UNC0642 or saline (n = 6–8/group, * P < 0.05, *** P < 0.001, two-way ANOVA). Data are presented as mean ± sem.
Other than NMDARs, we also examined synapse function genes in the Arc “interactome” that are associated with autism and intellectual disability, including Camk2a, Camk2b, Cytoplasmic FMR1-interacting protein 1 (Cyfip1), synaptic Ras GTPase-activating protein 1 (Syngap1) and Homer15, 47–49. As shown in Fig. 6f, most of the tested genes were not significantly changed in Shank3+/ΔC mice with or without UNC0642 treatment, while Homer1 was significantly reduced in Shank3+/ΔC mice (~35.7% decrease) and restored by UNC0642 treatment (~100.4% increase, F1,26 (treatment) = 18.17, P = 0.0002, two-way ANOVA).
UNC0642 treatment restores the expression of a large set of genes in the PFC of Shank3-deficient mice.
In order to determine whether UNC0642 treatment exerts genome-wide effects on Shank3+/ΔC mice, we performed RNA sequencing to analyze the gene expression profile in the PFC of WT and Shank3+/ΔC mice treated with saline (Het+SAL) or UNC0642 (Het+UNC) (Fig. 7a). Compared to WT mice, 434 genes showed a down-regulated change in expression (> 1.2 fold and p < 0.05, Suppl. Table 2) and 383 genes showed an up-regulated change in expression (> 1.2 fold and P < 0.05, Suppl. Table 3) in saline-treated Shank3+/ΔC mice. Among these altered genes, 58 overlapped with ASD susceptibility genes (Suppl. Table 4) including Homer1.
Figure 7. UNC0642 treatment induces genome-wide restoration of genes involved in neural signaling in PFC of Shank3-deificient mice.

(a) Volcano plot of differentially expressed genes between wild-type (WT), Shank3+/ΔC mice treated with saline (Het+SAL) or UNC0642 (1 mg/kg, i.p., 3x) (Het+UNC). Het+SAL vs WT: fold change > 1.2 and P < 0.05; Het+UNC vs Het+SAL: fold change > 1.1 and P < 0.05; Het+UNC vs WT: P < 0.05. (b) Venn diagram displaying the overlap between genes down-regulated in the PFC of Shank3+/ΔC mice (Het+SAL vs. WT) and up-regulated by UNC0642 treatment (Het+UNC vs. Het+SAL) (top), as well as genes up-regulated in SAL and down-regulated in UNC (bottom). (c, f) Heatmaps of the 107 genes down-regulated (c) and the 84 genes up-regulated (f) in saline-treated Shank3+/ΔC mice (SAL) and normalized in UNC0642-treated Shank3+/ΔC mice (UNC). (d, g) Functional protein classification analysis for the 107 down-regulated genes (d) and the 84 up-regulated genes (g). (e, h) Enrichment analysis for the 107 down-regulated genes (e) and the 84 up-regulated genes (h).
In UNC0642-treated Shank3+/ΔC mice, many of these down-regulated genes were elevated to near control levels, compared to saline-treated Shank3+/ΔC mice (n=107 genes, > 1.1 fold and P < 0.05, Fig. 7b, Suppl. Table 2), or WT mice (n=414 genes, −1.3 < fold change < 1.3 and P > 0.05, Suppl. Table 5). Moreover, many of these up-regulated genes were reduced to near control levels by UNC0642 treatment, compared to saline-treated Shank3+/ΔC mice (n=84 genes, > 1.1 fold and P < 0.05, Fig. 7b, Suppl. Table 3), or WT mice (n=350 genes, −1.3 < fold change < 1.3 and P > 0.05, Suppl. Table 6). We further selected several differentially expressed genes from the top list of RNAseq results, and confirmed with qPCR experiments that they were indeed down-regulated in Shank3+/ΔC mice and recovered by UNC treatment (Fig. S7c).
Heatmaps generated with the expression values for the 107 down-regulated genes (Fig. 7c) and the 84 up-regulated genes (Fig. 7f) showed that saline-treated Shank3+/ΔC samples clustered separately from WT samples, and UNC0642-treated Shank3+/ΔC samples were closer to those from WT samples than saline-treated Shank3+/ΔC samples. Functional protein classification analysis indicated that many of the 107 down-regulated genes (Fig. 7d) and the 84 up-regulated genes (Fig. 7g) fell into the categories including signaling molecules, nuclei acid binding proteins, and enzyme modulators.
Gene Ontology Biological Process analyses revealed that the 107 down-regulated genes are mostly involved in cytoskeleton organization, positive regulation of transcription, cell adhesion, phosphorylation regulation and developmental processes (Fig. 7e), while the 84 up-regulated genes are largely involved in regulation of cell morphogenesis, axon development, and protein kinase activity (Fig. 7h). Protein-protein interaction (PPI) network modeling indicated that 73 out of the 107 down-regulated genes and 49 out of the 84 up-regulated genes showed high PPI connectivity within and between several functional clusters including signaling molecules, receptors, transcription factors, proteases, and cell adhesion molecules (Fig. S7a and Fig. S7b). These results suggest that UNC0642 treatment can affect the expression of a large set of genes in Shank3-deficient mice, which may collectively contribute to the therapeutic effects of UNC0642 on social deficits.
DISCUSSION
Genetic screening has identified many synaptic, transcriptional and chromatin remodeling genes as autism risk factors5. However, their interconnections and functional roles in autism are largely unknown. Here we have discovered that aberrant histone methylation, particularly the abnormally high levels of EHMT1/2 and H3K9me2, occurs in the PFC of Shank3-deficient mice, resulting in the disrupted transcription of a number of genes, including synaptic plasticity gene Arc and its interacting partners. EHMT1/2 inhibition restores Arc expression, as well as many other genes involved in the regulation of synaptic function, transcription and development, leading to the recovery of NMDAR function in PFC pyramidal neurons and the prolonged rescue of autism-like social deficits (Fig. S8).
Histone methylation plays a key role in maintaining transcriptional homeostasis via the activation and repression of gene transcription6, 7. Disrupted function of EHMT1 or EHMT2 has been implicated in neurodevelopmental disorders9, 10, 37. De novo copy number variants (CNVs) were found to affect EHMT1 in schizophrenia patients, and EHMT1 has been implicated as a schizophrenia susceptibility gene40. Forebrain neuron-specific EHMT1 or EHMT2 knockout mice exhibit abnormal cognition and adaptive behaviors that resemble mental retardation syndrome50. Our results suggest that the elevation of EHMT1/2 and H3K9me2 in PFC may play a key role in transcriptional dysregulation in autism. Region-specific genetic and epigenetic changes have been found in autistic brains51, which may be due to the distinct transcriptional programs in different cell types52. Since deep cortical layer (layer V) pyramidal neurons show the most prominent deficits in autistic children22, we mainly focused on these neurons in electrophysiological recordings. Upper cortical layer 2/3 neurons have also been implicated in psychiatric disorders53, 54, and their functional changes associated with autistic phenotypes await to be further examined.
EHMT1/2-mediated H3K9me2 deposition is associated with the transcriptional regulation of genes involved in synaptic networks29, 55, 56. In Drosophila, EHMT1 directly regulates components in postsynaptic signaling complexes at the postsynaptic density57. One of the key molecular targets identified in this study is Arc, a prominent synaptic plasticity gene regulating cognitive processes such as learning and memory consolidation58–60. While Arc is involved in regulating AMPA receptor trafficking61–63, Arc overexpression in neuronal cultures also induces an increase of GluN1 puncta63, consistent with our finding that knockdown of Arc in WT mice induces NMDAR hypofunction. Interestingly, genetic studies have found enriched mutations among glutamatergic postsynaptic proteins comprising Arc and NMDAR complexes in schizophrenia39. Arc knockout mice exhibit a full spectrum of schizophrenia-like behaviors including impaired social ability46, suggesting that Arc is linked to the regulation of social behaviors. In this study, we have revealed that knockdown of Arc in Shank3-deficient mice prevents UNC0642 from restoring NMDAR function and social preference, implicating the key role of Arc in autism treatment by EHMT1/2 inhibition.
The transcriptional level of Arc gene is regulated by synaptic activity58. Direct suppression of neuronal activity by TTX significantly decreases Arc protein, while elevation of neuronal activity by bicuculline or BDNF enhances Arc expression62, 64, 65. Transcriptional regulation of the Arc gene is through several transcription enhancer elements located at proximal and distal promoter regions43–45. Our ChIP data have discovered an alteration of H3K9me2 occupancy around the distal enhancer element of the Arc gene by Shank3 deficiency or EHMT1/2 inhibition, which correlates well with the alteration of Arc gene expression.
Given the interaction of Arc with glutamate receptors and other synaptic proteins, restoration of Arc expression by UNC0642 treatment of Shank3-deficient mice may facilitate the recovery of the NMDAR-Arc complex at the synaptic membrane. An additional contributing factor to overcome NMDAR hypofunction is the removal of repressive H3K9me2 to elevate the transcription of NMDAR subunits by EHMT1/2 inhibition. Indeed, we have found a UNC0642-induced transcriptional increase of Grin1, a target of EHMT235, and Grin2a in Shank3-deficient mice. These data are consistent with our previous finding that NMDAR hypofunction in Shank3+/ΔC mice is not due to the loss of NMDAR expression, but due to the disrupted synaptic delivery of NMDARs resulting from aberrant actin regulators18. It is likely that UNC0642 rescues NMDAR function via multiple mechanisms. One is to facilitate NMDAR trafficking by rescuing actin regulators and synaptic scaffolding proteins, such as Arc and Homer1. The other is to boost the overall expression of NMDAR subunits to compensate for the diminished NMDAR trafficking.
The UNC0642-induced recovery of Homer1, a Shank3 binding partner and one of the autism risk genes47, suggests that metabotropic glutamate receptor signaling may also play a role in the behavioral rescue66. Furthermore, genome-wide studies have revealed the alteration of additional genes involved in a multitude of neuronal functions in Shank3+/ΔC mice. By targeting EHMT1/2-mediated H3K9me2, we are able to restore a number of genes with a broad range of biological functions, which may collectively contribute to the therapeutic effects of UNC0642 (Fig. S7). The down-regulated genes in Shank3+/ΔC mice revealed by RNAseq could be the direct effect of the increased H3K9me2 levels, while the up-regulated genes could be indirectly caused by the loss of silencers/repressors resulting from increased H3K9me2.
Taken together, these results have not only revealed a novel molecular mechanism underlying the pathophysiology of autism, but also suggested a potential intervention avenue for the treatment of this disease. While epigenetic drugs offer the advantage of having broad, multi-functional actions and being able to normalize a network of genes important for neuronal functions, it is important to be cautious about potential off-target side effects. Given the heterogeneity of autism, the general applicability of this treatment strategy also awaits to be further tested.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Xiaoqing Chen for excellent technical support. We acknowledge the support of University at Buffalo’s Genomics and Bioinformatics Core and the New York State Center of Excellence in Bioinformatics and Life Sciences. This work was supported by Nancy Lurie Marks Family Foundation and National Institutes of Health (MH112237, MH108842) to Z. Y.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY
Genomic data will be deposited in a public repository. The access number and the dataset will be available for access.
REFERENCES:
- 1.Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med 2013; 19(6): 685–694. [DOI] [PubMed] [Google Scholar]
- 2.Loke YJ, Hannan AJ, Craig JM. The Role of Epigenetic Change in Autism Spectrum Disorders. Front Neurol 2015; 6: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012; 338(6114): 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485(7397): 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515(7526): 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129(4): 823–837. [DOI] [PubMed] [Google Scholar]
- 7.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39(3): 311–318. [DOI] [PubMed] [Google Scholar]
- 8.Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry 2012; 69(3): 314–324. [DOI] [PubMed] [Google Scholar]
- 9.Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet 2006; 79(2): 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LE et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012; 91(1): 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balemans MC, Huibers MM, Eikelenboom NW, Kuipers AJ, van Summeren RC, Pijpers MM et al. Reduced exploration, increased anxiety, and altered social behavior: Autistic-like features of euchromatin histone methyltransferase 1 heterozygous knockout mice. Behav Brain Res 2010; 208(1): 47–55. [DOI] [PubMed] [Google Scholar]
- 12.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 2010; 11(7): 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 1999; 23(3): 569–582. [DOI] [PubMed] [Google Scholar]
- 14.Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism 2013; 4(1): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A et al. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet 2001; 69(2): 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007; 39(1): 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet 2014; 10(9): e1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep 2015; 11(9): 1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin L, Ma K, Wang ZJ, Hu Z, Matas E, Wei J et al. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nature neuroscience 2018; 21(4): 564–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet 2011; 20(15): 3093–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci 2006; 7(4): 268–277. [DOI] [PubMed] [Google Scholar]
- 22.Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med 2014; 370(13): 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Tu Z, Xu H, Yan S, Yan H, Zheng Y et al. Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res 2017; 27(10): 1293–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N et al. Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J Neurosci 2013; 33(47): 18448–18468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ZJ, Martin JA, Mueller LE, Caccamise A, Werner CT, Neve RL et al. BRG1 in the Nucleus Accumbens Regulates Cocaine-Seeking Behavior. Biol Psychiatry 2016; 80(9): 652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei J, Xiong Z, Lee JB, Cheng J, Duffney LJ, Matas E et al. Histone Modification of Nedd4 Ubiquitin Ligase Controls the Loss of AMPA Receptors and Cognitive Impairment Induced by Repeated Stress. J Neurosci 2016; 36(7): 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS et al. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry 2011; 16(2): 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 2012; 73(5): 962–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maze I, Covington HE 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 2010; 327(5962): 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293(5532): 1074–1080. [DOI] [PubMed] [Google Scholar]
- 31.Kouzarides T Chromatin modifications and their function. Cell 2007; 128(4): 693–705. [DOI] [PubMed] [Google Scholar]
- 32.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell 2009; 17(1): 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balan S, Iwayama Y, Maekawa M, Toyota T, Ohnishi T, Toyoshima M et al. Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHTM2 and WIZ, in Japanese autism subjects. Mol Autism 2014; 5(1): 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang XP et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem 2013; 56(21): 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 2007; 25(3): 473–481. [DOI] [PubMed] [Google Scholar]
- 36.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA et al. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem 2010; 53(15): 5844–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y, Lee HM, Xiong Y, Sciaky N, Hulbert SW, Cao X et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat Med 2017; 23(2): 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron 2013; 78(1): 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014; 506(7487): 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 2012; 17(2): 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014; 506(7487): 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall J, Trent S, Thomas KL, O’Donovan MC, Owen MJ. Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biol Psychiatry 2015; 77(1): 52–58. [DOI] [PubMed] [Google Scholar]
- 43.Pintchovski SA, Peebles CL, Kim HJ, Verdin E, Finkbeiner S. The serum response factor and a putative novel transcription factor regulate expression of the immediate-early gene Arc/Arg3.1 in neurons. J Neurosci 2009; 29(5): 1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waltereit R, Dammermann B, Wulff P, Scafidi J, Staubli U, Kauselmann G et al. Arg3.1/Arc mRNA induction by Ca2+ and cAMP requires protein kinase A and mitogen-activated protein kinase/extracellular regulated kinase activation. J Neurosci 2001; 21(15): 5484–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawashima T, Okuno H, Nonaka M, Adachi-Morishima A, Kyo N, Okamura M et al. Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proc Natl Acad Sci U S A 2009; 106(1): 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manago F, Mereu M, Mastwal S, Mastrogiacomo R, Scheggia D, Emanuele M et al. Genetic Disruption of Arc/Arg3.1 in Mice Causes Alterations in Dopamine and Neurobehavioral Phenotypes Related to Schizophrenia. Cell Rep 2016; 16(8): 2116–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelleher RJ 3rd, Geigenmuller U, Hovhannisyan H, Trautman E, Pinard R, Rathmell B et al. High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS One 2012; 7(4): e35003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kury S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am J Hum Genet 2017; 101(5): 768–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pathania M, Davenport EC, Muir J, Sheehan DF, Lopez-Domenech G, Kittler JT. The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl Psychiatry 2014; 4: e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 2009; 64(5): 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474(7351): 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S et al. Functional organization of the transcriptome in human brain. Nature neuroscience 2008; 11(11): 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Page SC, Hamersky GR, Gallo RA, Rannals MD, Calcaterra NE, Campbell MN et al. The schizophrenia- and autism-associated gene, transcription factor 4 regulates the columnar distribution of layer 2/3 prefrontal pyramidal neurons in an activity-dependent manner. Molecular psychiatry 2018; 23(2): 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwon HB, Kozorovitskiy Y, Oh WJ, Peixoto RT, Akhtar N, Saulnier JL et al. Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nature neuroscience 2012; 15(12): 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benevento M, Iacono G, Selten M, Ba W, Oudakker A, Frega M et al. Histone Methylation by the Kleefstra Syndrome Protein EHMT1 Mediates Homeostatic Synaptic Scaling. Neuron 2016; 91(2): 341–355. [DOI] [PubMed] [Google Scholar]
- 56.Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL et al. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci 2012; 32(16): 5440–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kramer JM, Kochinke K, Oortveld MA, Marks H, Kramer D, de Jong EK et al. Epigenetic regulation of learning and memory by Drosophila EHMT/G9a. PLoS Biol 2011; 9(1): e1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci 1999; 2(12): 1120–1124. [DOI] [PubMed] [Google Scholar]
- 59.Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG et al. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 1995; 14(2): 433–445. [DOI] [PubMed] [Google Scholar]
- 60.Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 2006; 52(3): 437–444. [DOI] [PubMed] [Google Scholar]
- 61.Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron 2006; 52(3): 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006; 52(3): 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 2006; 52(3): 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Korb E, Wilkinson CL, Delgado RN, Lovero KL, Finkbeiner S. Arc in the nucleus regulates PML-dependent GluA1 transcription and homeostatic plasticity. Nat Neurosci 2013; 16(7): 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saha RN, Wissink EM, Bailey ER, Zhao M, Fargo DC, Hwang JY et al. Rapid activity-induced transcription of Arc and other IEGs relies on poised RNA polymerase II. Nat Neurosci 2011; 14(7): 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vicidomini C, Ponzoni L, Lim D, Schmeisser MJ, Reim D, Morello N et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol Psychiatry 2017; 22(5): 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.