

Abstract
Triterpenoids are distributed widely in higher plants and are of interest because of their structural diversity and broad range of bioactivities. In particular, there is a very large literature on the propensity of a variety of triterpenoids to act as potential anticancer agents. In the present review, the anticancer potential is summarized for naturally occurring triterpenoids and their semi-synthetic derivatives, including examples of lupane-, oleanane-, ursane-, and cucurbitane-type pentacyclic triterpenoids, along with dammarane-type tetracyclic triterpenes including ginsenosides and their sapogenins and dichapetalins, which have been characterized as antitumor leads from higher plants. Preliminary structure-activity relationships and reported mechanisms of the antineoplastic-related activity are included. Prior studies for triterpenoids of plant origin are supportive of additional work being conducted on the more detailed biological and mechanistic evaluation for the progression of this type of natural products as possible cancer chemotherapeutic agents.
Keywords: natural products, triterpenoids, antitumor activity, synthetic modification, structure-activity relationships, mechanisms of anticancer activity
Graphical Abstract
Plant-derived triterpenoids may mediate their potent antitumor activity through inhibition of NF-κB and STAT3 or induction of autophagy, indicating a promise for the development of new anticancer drugs from these compounds.
Introduction
Higher plants have afforded four major well-established classes of approved cancer chemotherapeutic drugs, inclusive of compounds based on bisindole and camptothecin alkaloids, taxane diterpenoids, and podophyllotoxin lignans, with the cephalotaxine alkaloid, omacetaxine mepesuccinate, having been introduced clinically for this purpose more recently [1, 2]. Natural products from all classes of terrestrial and marine micro- and macro-organisms continue to play a vital role in drug discovery, inclusive of the search of new oncological agents. Indeed, of a total of 175 small molecules approved as anticancer drugs in western medicine from 1940–2014, approximately 50% were either directly obtained from micro- and macro-organisms or derived synthetically from naturally occurring lead molecules [2, 3].
The triterpenoids are a large group of over 20,000 natural products derived from C30 precursors, with over 100 different carbon skeletons, which are typically 6–6–6–5 tetracyclic and 6–6–6–6–5– or 6–6–6–6–6 pentacyclic substances [4, 5]. The occurrence of triterpenoids in different types of organisms has been subjected to frequent review, with several hundred new compounds of this type described by investigators all over the world each year [6]. While neither naturally occurring nor semi-synthetic triterpenoids have current use as anticancer agents in western medicine, bardoxolone methyl, a synthetic oleanane-type triterpenoid, has reached clinical trial recently for the potential treatment of cancer [7].
Several investigators have reviewed specifically various classes of naturally occurring triterpenoids as potential anticancer agents [e.g., 5, 8–16]. The interest of the present authors on this same issue has been stimulated as a result of 2 long-standing collaborative multidisciplinary projects funded sequentially by the U.S. National Cancer Institute, in which numerous triterpenoids isolated from tropical plants collected in several continents were evaluated biologically [17, 18]. A significant initial observation made in this work was the specific inhibitory activity in vitro of the pentacyclic triterpenoid, (+)-betulinic acid (1) (Fig. 1), against 3 different cancer patient-derived melanoma cell lines [19]. As part of the same study, follow-up in vivo work showed significant inhibition of tumor growth when four-week old athymic nude mice inoculated with MEL-1 human melanoma cells were treated with compound 1 (5 mg/kg, once four days, i.p.) for 6 weeks, and neither tumor metastases nor structural abnormalities were observed in mice [19].
Fig. 1.
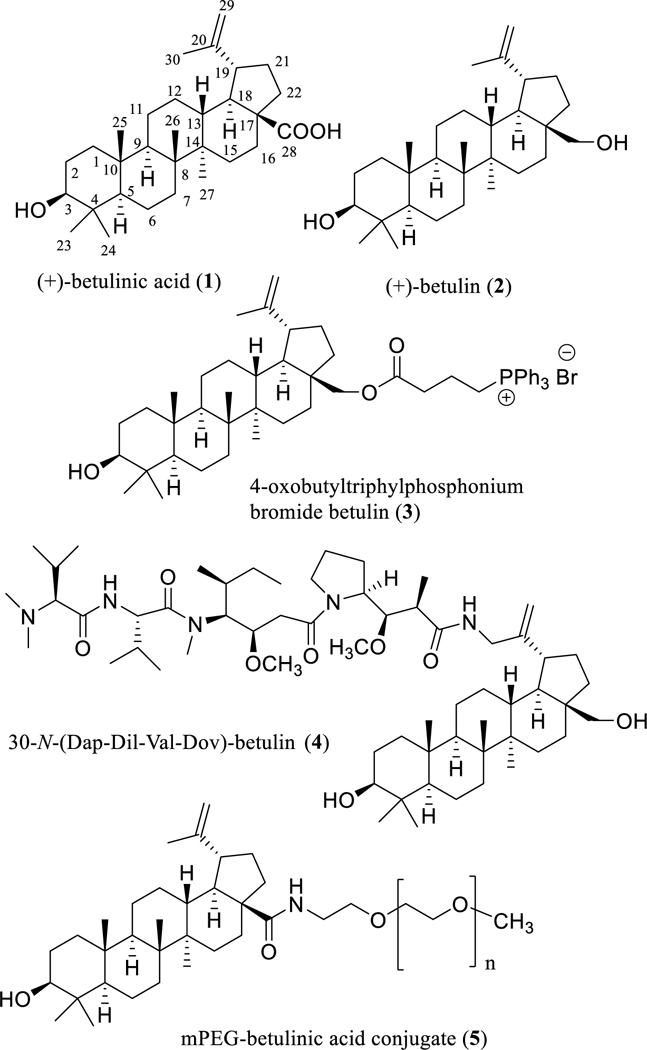
Structures of (+)-betulinic acid (1) and its natural and semi-synthetic derivatives (2–5) with potential anticancer activity (Dap: Dolaproine; Dil: Dolaisoleuine; Val: L-Valine; Dov: N,N-dimethyl-L-valine).
In the following portions of this review, our own recent experimental observations on biologically active plant-derived triterpenoids will be combined with descriptions of relevant work by other groups on triterpenoids having potential antineoplastic activity.
Lupane-type Triterpenoids
As part of a recent summary account, further preclinical studies were described in a book chapter that led eventually to the evaluation of (+)-betulinic acid (1) in phase I/II clinical trials to treat dysplastic melanocytic nevi. These clinical trials were conducted at the College of Medicine, University of Illinois at Chicago, Chicago, IL, USA, with the drug administered topically as a 20% ointment [20].
Following this development, there is presently quite a large contemporary body of literature on efforts to generate additional anticancer agents based on (+)-betulinic acid (1) and its close analogue, (+)-betulin (2) (Fig. 1), aimed at improving particularly both the antitumor efficacy and water solubility [e.g., 21–24]. It was realized in early work that the cytotoxic potency of (+)-betulinic acid (1) toward MEL-2 melanoma and KB human oral epidermoid carcinoma cells could be enhanced by altering the substituents at positions C-3, C-20, and C-28 [25]. As one synthetic approach, the inhibitory effects of (+)-betulin (2) were greatly enhanced on the proliferation of vincristine-resistant MCF-7 human breast cancer cells by introduction of a triphenylphosphonium cation conjugate at the C-28 position to produce 4-oxobutyltriphylphosphonium bromide betulin (3) (Fig. 1), which showed an IC50 value of 45 nM that was much lower than the positive controls, doxorubicin (IC50 0.3 μM) and vinblastine (IC50 9.2 μM) [26]. Another potent cytotoxic agent produced is the chimera derivative, betulastatin 3 [30-N-(Dap-Dil-Val-Dov)-betulin] (4, Fig. 1), which was prepared by bonding (+)-betulin (2, GI50 >10 μg/mL) to the Dov-Val-Dil-Dap unit of the anticancer compound, dolastatin 10, and showed a GI50 value of 0.04 μg/mL against DU-145 human prostate cancer cells [27].
Recently, an important approach that has been undertaken is production of the mPEG-betulinic acid conjugate, 5 (Fig. 1), a more water-soluble derivative of compound 1 that also augments the cytotoxic potency of the parent compound, (+)-betulinic acid (1) [28].
Oleanane-type Triterpenoids
(+)-Oleanolic acid (6) (Fig. 2) is a pentacyclic triterpenoid that exhibits promising antitumor efficacy and has been found to interact with multiple molecular targets [29]. It showed cytotoxic and anti-metastatic activities against A549 and H460 human non-small cell lung cancer cells by decreasing the expression of the angiogenic vascular endothelial growth factor (VEGF) and the development of melanoma-induced lung metastasis [30]. Also, it inhibited the proliferation of GBC-SD and NOZ human gallbladder cancer cells through the mitochondrial apoptosis pathway [31]. Tumor growth was suppressed significantly when 4 to 6-week-old athymic male nude mice inoculated with NOZ cells were treated with (+)-oleanolic acid (i.p., 75 or 150 mg/kg, once every two days) for 15 days, and no side effects were observed in the test mice used [31].
Fig. 2.
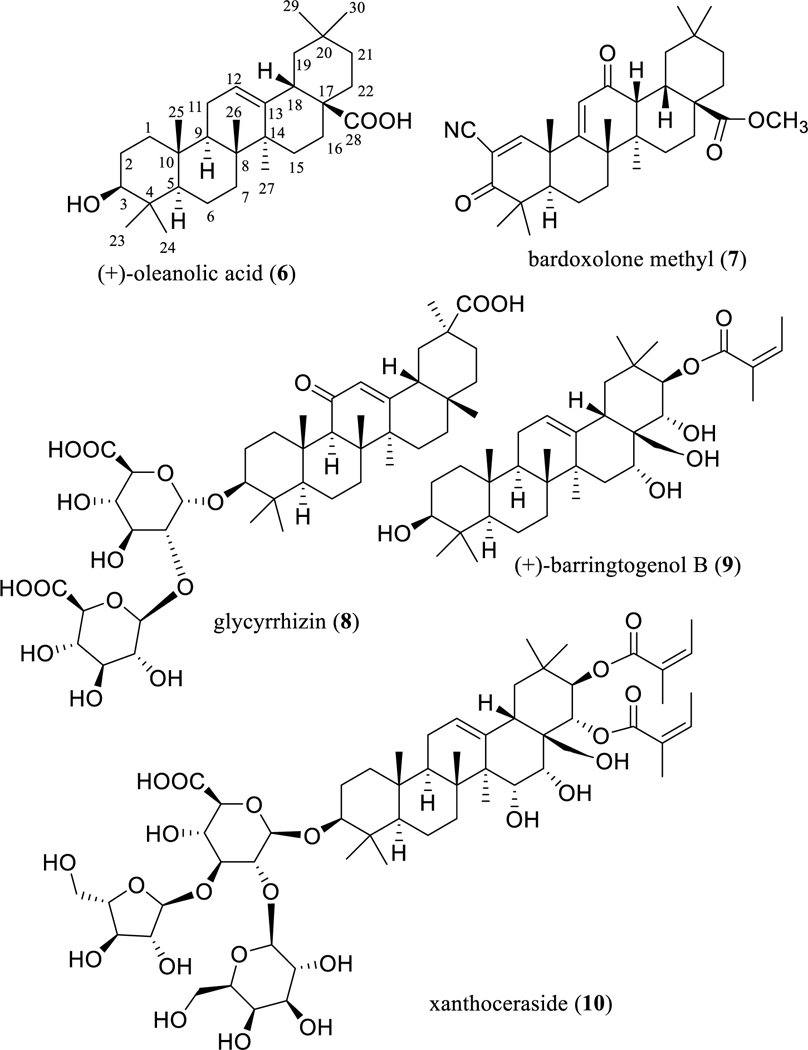
Structures of oleanane-type triterpenoids (6 and 8–10) and a semi-synthetic derivative (7) with potential anticancer activity.
In addition, (+)-oleanolic acid (6) was found to inhibit mitomycin C-resistant HT-29 human colon cancer cell growth, which was proposed to be mediated through selective inhibition of human aldo-keto reductase [32]. It also inhibited HepG2 and SMC7721 hepatoma cell growth by suppression of the PI3K/Aκt1/mTOR signaling pathway and as a result of triggered reactive oxygen species (ROS)-dependent autophagy [33]. Interestingly, (+)-oleanolic acid (6) has been found to increase MDA-MB-231 human breast cancer cell migration and hence show promising properties for wound healing, an effect that was mediated by mitogen-activated protein kinases [34].
In a very recent study, (+)-oleanolic acid (6) was found to induce MG63 and Saos-2 human osteosarcoma cell apoptosis by targeting mitochondria in a Notch signaling-dependent manner [35]. By acting via a diverse range of cellular mechanisms, this endorses the potential of (+)-oleanolic acid for further development as an antitumor agent.
To improve on the solubility and therapeutic efficacy of (+)-oleanolic acid (6), a novel dosage form has been developed and evaluated, namely, (+)-oleanolic acid-encapsulated multivesicular liposomes (OA-MVLs). In a side-by-side comparison, this liposomal form of (+)-oleanolic acid showed an enhanced tumor growth inhibitory effect with respect to compound 6, when male Kunming mice inoculated with murine hepatoma H22 cells were treated (i.p.) with (+)-oleanolic acid (6) or OA-MVLs (25, 50, or 100 mg/kg for each compound, once two days) for two weeks. The survival time of the tumor-bearing mice was increased by both treatments, and no body weight loss was observed in the OA-MVL treatment protocol, even at a high dose (100 mg/kg) [36].
As a result of structural modification at the C-3 and C-28 positions of (+)-oleanolic acid (6), the novel analogueCDDO-Me or RTA 402 (7) (Fig. 2) was synthesized [37]. This derivative inhibited potently production of nitric oxide induced by interferon-γ (IFN-γ) in mouse macrophages [37]. It also showed cytotoxicity against HepG2 human hepatoma and B16 2F10 mouse melanoma cells [38]. In addition, CDDO-Me (7) was found to redirect the activation profile of tumor-associated macrophages (TAMs), enhancers of tumor growth, development, and metastasis, from tumor-promoting to tumor-inhibiting, indicating a possible immunotherapeutic role in the treatment of cancer [39].
Increasingly, a number of investigations on CDDO-Me (7) have shown that this agent exhibited potent cytotoxicity toward various tumor cells to exert promising in vivo antitumor efficacy [40]. For example, it showed strong antiproliferative and proapoptotic activity against the MiaPaCa-2 and BxPC-3 human pancreatic ductal adenocarcinoma cell lines. Tumor growth was inhibited significantly when seven-week old Scid-Ncr female mice inoculated with MiaPaCa-2 cells were treated with CDDO-Me (7) (gavage, 7.5 mg/kg, daily, five days per week) for five weeks [41].
The antitumor efficacy of CDDO-Me (7) was found to be mediated by improving the immune response in cancer. Tumor growth was found to be inhibited significantly by CDDO-Me (7) when six- to eight-week old C57BL/6 female mice that were inoculated with EL-4 mouse thymoma cells and then vaccinated with dendritic cells transduced with full-length survivin were treated with CDDO-Me (7) (oral chow, 150 mg/kg, daily, four days per week) for three weeks [42].
In a phase I clinical trial study, patients with locally advanced or metastatic pancreatic adenocarcinoma were treated with gemcitabine (i.v., weekly) and CDDO-Me (7, orally, daily), with immunology evaluated before and after the two-week treatment. Neither toxicity attributed to compound 7 nor significant change of the proportion of myeloid-derived suppressor cells (MDSC) mainly responsible for immune suppression in cancer were observed in the patients. Combined CDDO-Me and gemcitabine treatment resulted in a significant increase in the T-cell responses of the patients to tetanus toxoid and phytohemagglutinin, indicating the therapeutic promise of this triterpenoid derivative in enhancing cancer immunotherapy [42].
A further phase I study on CDDO-Me in patients with advanced solid tumors or lymphomas has been reported, with its MTD, DLTs, and appropriate dose for phase II studies established [7, 43]. However, this clinical trial on CDDO-Me was suspended due to some serious adverse events evident, indicating that further structural modification is required to decrease the side effects observed for this triterpenoid derivative (7), for future development of an effective anticancer agent [44].
The sweet-tasting oleanane-type triterpene glycoside glycyrrhizin (8) (Fig. 2) is used as an ingredient in the food and beverage industry and is the major active constituent of Glycyrrhiza glabra L. (licorice) (Fabaceae) [45]. This and two other Glycyrrhiza species are official sources of licorice in the Chinese Pharmacopoeia, with G. glabra demonstrated as having anti-inflammatory, antioxidant, antiviral, hepatotoxic, and neuroprotective propensities [46].
Among recent reports on the antitumor potential of glycyrrhizin (8), its cytotoxicity toward HepG2 and MHCC97-H hepatocellular carcinoma cells was found to be mediated by autophagy induced from the inhibition of Aκt/mTOR signaling [47]. It is worth mentioning that Glycyrrhiza uralensis Fisch. containing glycyrrhizin (8) is used as a component of PHY906, which is a long-established four-herb formula used in Chinese traditional medicine to treat a number of gastrointestinal conditions and to improve the therapeutic indices of a number of standard anticancer agents [48].
In a murine colon xenograft model, PHY906 enhanced significantly the antitumor activity and reduced the toxicity of irinotecan (CPT-11), when tumor-bearing mice were treated by CPT-11 (i.p., 360 mg/kg, daily) with (500 mg/kg, orally, twice a day) or without PHY906 for four consecutive days [49].
In a phase II study conducted in the United States using PHY906 with capecitabine as second-line therapy in patients with advanced pancreatic cancer, it was concluded that this combination may provide a safe and feasible salvage therapy for such patients after failure using gemcitabine [50].
As an additional example of a group of oleanane-type triterpenoids, compounds related to barrigenol represent a small group of olean-12-enes hydroxylated at the C-3, C-15, and/or C-16, C-22 and/or C-21, and C-28 positions, which occur mainly in the plant families Apocynaceae, Lecythidaceae, Pittosporaceae, Sapindaceae, and Theaceae. In our continuing search for anticancer agents from tropical plants, a barrigenol-like triterpenoid, (+)-barringtogenol B (9) (Fig. 2), was isolated as a major cytotoxic component from an extract of the bark of Cyrilla racemiflora L. (Cyrillaceae), collected in Dominica [51]. This compound exhibited an IC50 value of 1.7 μM for HT-29 human colon cancer cells (the positive control, paclitaxel, showed an IC50 value of 0.8 nM) [51]. A preliminary structure-activity relationship (SAR) study showed that an angeloyl group attached at either C-21 or C-22 is necessary for barrigenol-like triterpenoids to mediate their cytotoxicity. The presence of a hydroxy group at the C-24 position enhances the cytotoxicity of some barrigenol-like triterpenoids, but introducing a hydroxy group at the C-15 position results in the potency being decreased [51].
Based on these SAR conclusions, xanthoceraside (10) (Fig. 2), a barrigenol-like triterpenoid isolated from the husks of Xanthoceras sorbifolia Bunge, with an angeloyl group substituted at C-21 and C-22, a hydroxy group connected at C-15, C-16, and C-28, and a tri-glycoside unit linked at the C-3 position, was found to be cytotoxic against A375.S2 human melanoma cells (IC50 5.71 μM) but non-cytotoxic against normal peripheral blood mononuclear cells (PBMC), indicating a selective cytotoxicity for 10. Mechanistic studies showed that xanthoceraside (10) induced A375.S2 cell apoptosis by activating caspase-3 and caspase-9 through the mitochondrial pathway that was induced by the downregulation of the IGF-1R/Raf/MEK/ERK cascade in A375.S2 cells [52]. These results indicate that the barrigenol-like triterpenoids may be worthy of further investigation for development as new anticancer agents.
Ursane-type Triterpenoids
There is an increasing interest in the potential antineoplastic propensities of the ursane-type triterpenoid, (+)-ursolic acid (11, Fig. 3) [e.g. 8, 10, 12, 15, 53], and a number of pertinent studies published in the last decade seem worthy of mention. (+)-Ursolic acid (11) suppressed the proliferation of androgen-independent DU145 and androgen-dependent LNCaP human prostate cancer cells through inhibition of NF-κB and signal transducer and activator of transcription-3 (STAT3) activation. Tumor growth was suppressed significantly when four-week old athymic BALB/c male nude mice inoculated with DU145 cells were treated with (+)-ursolic acid (i.p., 200 mg/kg, twice a week) for six weeks. No significant effects on body weight were observed in mice [53].
Fig. 3.
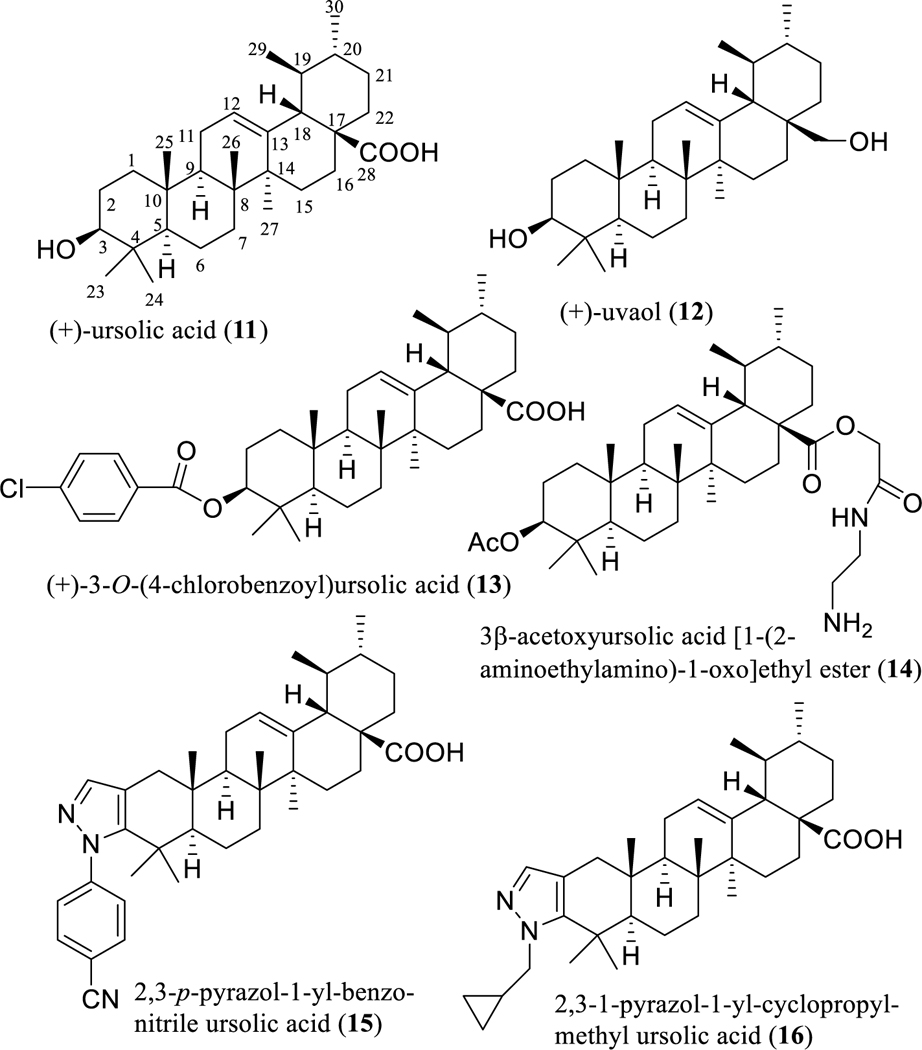
Structures of (+)-ursolic acid (11) and its semi-synthetic analogues (12–16) with potential anticancer activity.
In both in vitro and in vivo studies, (+)-ursolic acid (11) inhibited the proliferation of R-HepG2 doxorubicin-resistant human hepatoma cells by inducing apoptosis through the caspase-independent apoptosis-inducing factor (AIF) signaling pathway [54]. Tumor growth was suppressed significantly, when four- to six-week old female nude mice inoculated with R-HepG2 cells were treated with (+)-ursolic acid (orally, 50 or 75 mg/kg, daily) for two weeks. No significant effects on the body weight, liver, heart, and spleen of mice were observed [54].
Interestingly, (+)-ursolic acid (11) has been found to inhibit the growth of SKOV3 sphere human ovarian cancer stem-like cells (CSCs). In an in vivo study, tumor growth was inhibited significantly when five- to six-week old athymic BALB/c-nu female nude mice inoculated with SKOV3 sphere CSCs were treated with (+)-ursolic acid alone (i.p., 60 mg/kg, daily) or with (+)-ursolic acid (i.p., 60 mg/kg, daily) plus cisplatin (i.p., 2.5 mg/kg, daily) for two weeks [55].
In our collaborative work on tropical plants, Syzygium corticosum (Lour.) Merr. & L.M. Perry (Myrtaceae), collected in Vietnam, showed some promise in preliminary biological screening, and was thus subjected to activity-guided fractionation. Separation of the active extract from this species yielded a large amount of (+)-ursolic acid (11, with a yield of 0.2% w/w). This compound displayed a more potent NF-κB inhibitory activity (IC50 31 nM) than the positive control rocaglamide (IC50 70 nM). It also exhibited activity against MDA-MB-231 breast cancer cells (IC50 value of 5.9 μM) (the positive control, paclitaxel, showed an IC50 value of 17 nM) and was identified as the major cytotoxic principle of S. corticosum [56].
To characterize the functional groups necessary for mediation of its biological activity, two derivatives, (+)-uvaol (12) and (+)-3-O-(4-chlorobenzoyl)ursolic acid (13) (Fig, 3) were synthesized from (+)-ursolic acid (11). However, both semi-synthetic products (12 and 13) were found to be inactive, indicating the importance of the presence of both the C-3 hydroxy and C-28 carboxylic acid groups [56]. Following this observation, a preliminary structure-activity relationship (SAR) study using some semi-synthetic analogues indicated that the C-3 hydroxy and C-28 carboxylic acid groups and 19,20-dimethyl substitution are all essential for (+)-ursolic acid to mediate its cytotoxicity toward MDA-MB-231 breast cancer cells and NF-κB inhibitory activity [56].
Consistently, an earlier SAR study showed that the presence of a C-28 keto carbonyl group is important for pentacyclic triterpenes to inhibit mouse melanoma B16 2F2 cell growth by induction of cell differentiation [57]. Thus, the antiproliferative activity of (+)-ursolic acid (11) against all of A549 non-small cell lung, HeLa cervical, and MCF-7 breast human cancer cells was improved somewhat by modification at the C-28 position via esterification followed by amidation with amines. One of these semi-synthetic products, 3β-acetoxyursolic acid [1-(2-aminoethylamino)-1-oxo]ethyl ester (14) (Fig. 3), was found to exhibit discernible cytotoxicity, showing an IC50 value in the range 8–10 μM, while, the IC50 values of (+)-ursolic acid (11) were greater than 100 μM for all these three cancer cell lines used [58].
In addition, a series of novel pyrazole-fused (+)-ursolic acid derivatives was synthesized in a further structural modification of (+)-ursolic acid (11), by introduction of a pyrazole moiety at its C-2 and C-3 positions. Among these semi-synthetic derivatives, both a 2,3-p-pyrazol-1-yl-benzonitrile ursolic acid (15) (Fig. 3) and a 2,3–1-pyrazol-1-yl-cyclopropylmethyl ursolic acid (16) (Fig. 3) showed more potent cytotoxicity than either the other pyrazole-fused derivatives produced or the parent compound (11), toward a small panel of human cancer cell lines [59]. Surprisingly, compound 15 was found to induce HeLa human cervical cancer cell death through hyperstimulation of micropinocytosis, which can induce methuosis, a non-apoptotic type of cell death. This may provide a useful probe for the development of new cancer therapeutic agents to combat the multidrug resistance problem [59].
To improve on its water solubility for potential clinical applications, a carrier-free nanodrug by self-assembly of (+)-ursolic acid (11) has been developed, as a result of which the inhibitory effects on proliferation of A549 human lung cancer cells were found to be enhanced. Also, tumor growth was inhibited significantly, when six- to eight-week old BALB/c-nu female nude mice inoculated with A549 cells were treated with (+)-ursolic acid (11) or with (+)-ursolic acid-nanoparticles (i.p., 8 mg/kg, daily) for three weeks, and, in addition, the population of CD4+ T-cells in the mice was increased. These results indicate that this (+)-ursolic acid-nanoparticle carrier-free nanodrug has the potential for use in cancer immunotherapy [60].
Mechanistically, (+)-ursolic acid (11) mediates its antitumor potential through inhibition of NF-κB activation induced by carcinogenic agents with targets at cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D11 [61]. It also inhibits tumor growth through other promising mechanisms involving angiogenesis and metastasis [53]. In a phase I study to assess the multiple-dose tolerability, efficacy, and pharmacokinetics of a liposomal form of (+)-ursolic acid, patients with confirmed advanced solid tumors were administered with this drug candidate intravenously for 14 consecutive days of a 21-day treatment cycle. The (+)-ursolic acid liposome used was found to be safe and well-tolerated and showed improved potential for improving patient remission rates. Accordingly, a phase II study was recommended for this (+)-ursolic acid liposome drug candidate [62].
Cucurbitane-type Triterpenoids
Cucurbitacins are highly oxygenated cucurbitane-type tetracyclic triterpenoids that were identified initially from the plant family Cucurbitaceae and have been divided into twelve major structural categories [63]. Members of this group of natural products have been reported for their promising anticancer and anti-inflammatory activities [64], of which (+)-cucurbitacins B (17) and D (18) and (–)-cucurbitacins E (19) and I (20) have been investigated extensively for their cytotoxicity toward several human cancer cell lines (Fig. 4) [64, 65]. For example, (+)-cucurbitacin D (18) has been found to show potent cytotoxicity against a variety of human cancer cell lines, including human breast, central nervous system, colon, lung, oral epidermoid, and prostate cancer cells, with such activity mediated by induction of cell cycle arrest, mostly in the G2/M phase, and acting by modulating the JAK-STAT, Aκt-PKB, and MAPK pathways [63–65].
Fig. 4.
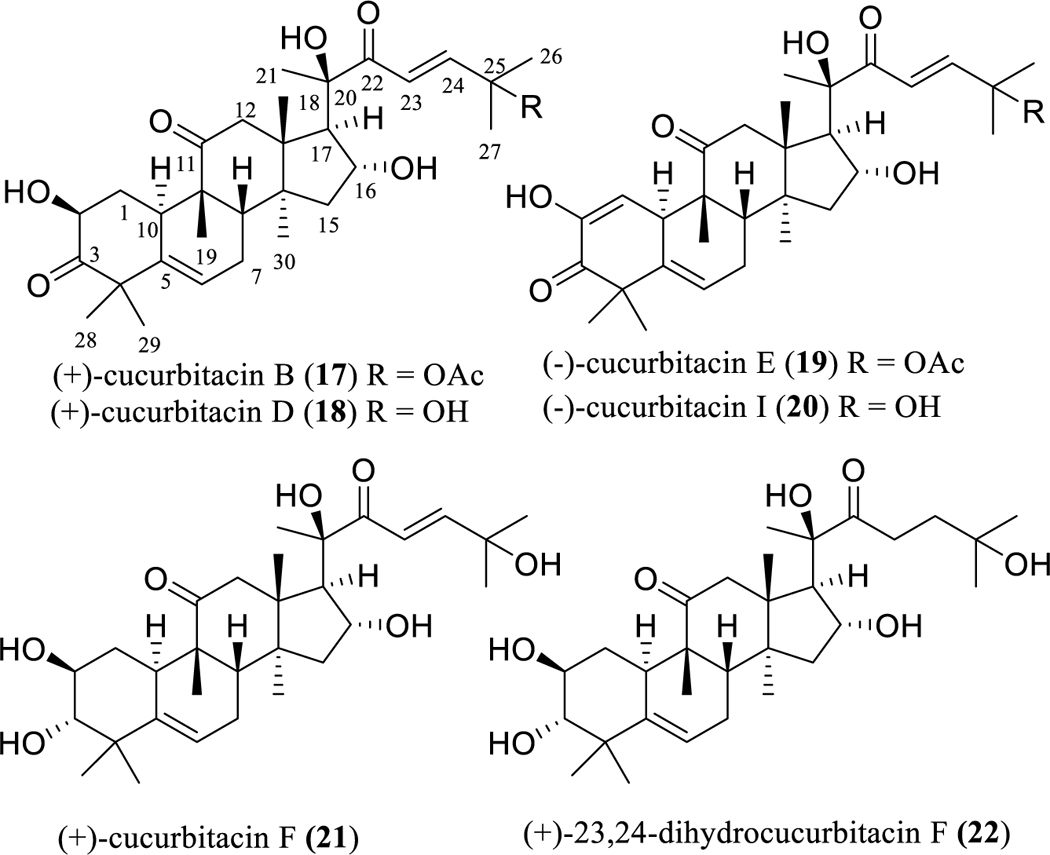
Structures of cucurbitacins (17–21) with potential anticancer activity and an inactive analogue (22).
In a study conducted in our own laboratory, (+)-cucurbitacin D (18) was characterized as a major cytotoxic component against HT-29 human colon cancer cells from both the fruits and stem bark of Elaeocarpus chinensis (Gardner & Champ.) Hook.f. ex Benth. (syn.: Friesia chinensis Gardner & Champ.) (Elaeocarpaceae), collected in Vietnam, and showed an IC50 value of 0.12 μM (the positive control, paclitaxel, showed an IC50 value of 6 nM) [66].
Analysis of the structures of these potently cytotoxic triterpenoids, including (+)-cucurbitacins B (17) and D (18) and (–)-cucurbitacins E (19) and I (20), shows that they all contain a C-2 hydroxy group, a ketocarbonyl group at the C-3 and C-11 positions, a double bond at the C-1 and/or C-5 positions, 16α,20β-dihydroxy groups, a C-22 enone group, and a C-25 hydroxy or acetoxy group, indicating that these structural moieties could be important for a given cucurbitacin to mediate cancer cell-line cytotoxicity. This has been supported by a study examining the chemical structures of 24 cucurbitacins and determining their resultant cytotoxicity against KB human oral epidermoid carcinoma cells, which showed that the presence of an α,β-unsaturated ketone and a C-25 acetoxy group, along with a free 16α-hydroxy group, are the most relevant structural features required for such activity [65].
An additional biological investigation showed that all of (+)-cucurbitacin D (18) and (–)-cucurbitacins E (19) and I (20) exhibited potent cytotoxicity against SW 1353 human chondrosarcoma cells, with the activity decreasing in the sequence 19, 20, and 18 [67]. This indicates that introduction of a C-1 enone group or replacement of C-25 hydroxy group with an acetoxy group could increase the cytotoxicity of (+)-cucurbitacin D (18) toward SW 1353 cells.
Consistent with these conclusions, (+)-cucurbitacin D (18) was found to exhibit around ten-fold more cytotoxic potency than (+)-cucurbitacin F (21) (Fig. 4) against all of the A549 non-small cell lung cancer, HCT-15 colon cancer, SK-MEL-2 melanoma, SKOV3 ovarian cancer, and XF 498 human central nervous system (CNS) human cancer cell lines. Furthermore, saturation of C-23 double bond in (+)-23,24-dihydrocucurbitacin F (22) (Fig. 4) resulted in loss of activity (IC50 >50 μg/mL) against both KB human solid tumor and P-388 murine leukemia cells, when compared with (+)-cucurbitacin F (21), which showed activity toward KB and P-388 cells, with IC50 values of 0.074 and 0.04 μg/mL, respectively [68, 69].
The antineoplastic potential of all (+)-cucurbitacins B (17) and D (18) and (–)-cucurbitacins E (19) and I (20) have been reviewed previously [64, 65]. Briefly discussed below are an update of more recent studies on the anticancer potentials of the representative compound, (+)-cucurbitacin D (18).
(+)-Cucurbitacin D (18) has been found to suppress the proliferation of Hep3B human hepatoma cells [70], as well neurofibromatosis type 2 (Nf2)-deficient mouse Sch10545 schwannoma and telomerase-immortalized benign Ben-Men-1 human meningioma cells [71]. Also, this agent induced apoptosis in the doxorubicin-resistant human MCF7/ADR breast cancer cells through inhibiting STAT3 and NF-κB signaling [72], and its inhibitory effects toward human T cell leukemia cells were found to be associated with autophagy [73].
Interestingly, (+)-cucurbitacin D (18) was found to mediate cytotoxicity toward MCF-7 human breast cancer cells through disrupting interactions between heat shock protein 90 (Hsp90) and two co-chaperones, Cdc37 and p23 [74]. Tumor growth was inhibited significantly when six-week old female athymic nude mice inoculated with CaSki human cervical cancer cells were treated with (+)-cucurbitacin D (18) (injected intratumorally, 1 mg/kg, three times a week) for four weeks [75].
All of (+)-cucurbitacins B (17) and D (18) and (–)-cucurbitacins E (19) and I (20) are potently cytotoxic, and, in general, these triterpenoids are strong STAT3 inhibitors and thus show selective inhibitory activity toward the JAK/STAT pathway [64, 65]. STAT3 is a promising target for the discovery of new anticancer drugs, and thus cucurbitacins 17–20 may accordingly prove useful in the treatment of human cancer targeting STAT3.
Dammarane-type Triterpenoids I: Ginsenoside Sapogenins
In Chinese traditional medicine, the roots of Panax ginseng C.A. Meyer (ginseng) (Araliaceae), known locally as “renshen”, are used as a complementary and alternative herbal supplement to support cancer chemotherapy [76]. The ginsenosides, containing mainly dammarane-type tetracyclic triterpene saponins, have been characterized as the major components of ginseng, and have attracted a great deal of attention owing to their promising bioactivities, including potential antitumor efficacy [77, 78]. As one of the major sapogenins of these ginsenosides, 20(S)-protopanaxadiol [20(S)-PPD, 23] (Fig. 5) has been well documented for its potent antitumor activity, and is known mechanistically to induce tumor cell apoptosis and to suppress the NF-κB, JNK, and MAPK/ERK signaling pathways. It also exhibits antimetastasis and antiangiogenesis activities, as well as synergistic effects with existing anticancer drugs [78].
Fig. 5.
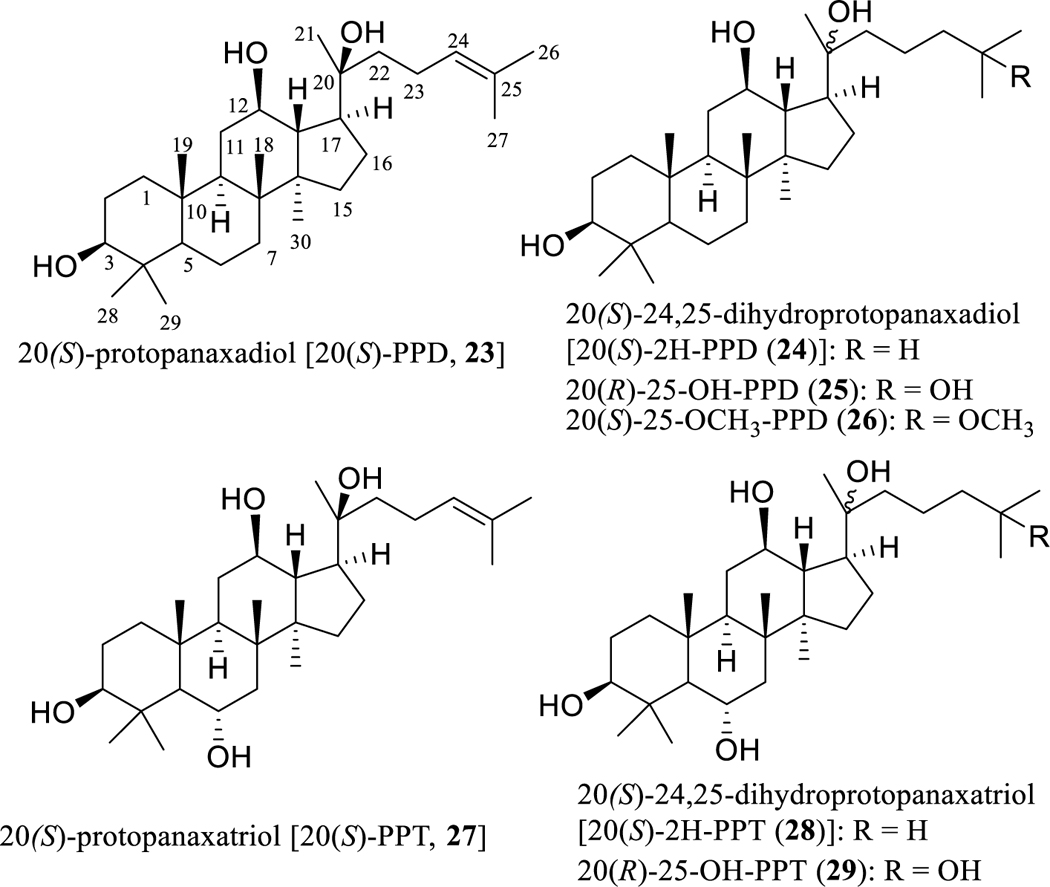
Structures of dammarane-type triterpenoids (23–29) with potential anticancer activity.
An in vitro study showed that 20(S)-PPD (23) exhibited only marginal activity toward a small panel of human cancer cell lines, with the IC50 values being in the range 20–80 μM [79]. In contrast, its growth inhibitory effect against MDA-MB-231 triple-negative human breast cancer cells was more highly evident (IC50 5.9 μM), which was comparable to the potency determined for paclitaxel (IC50 6.2 μM) [80]. This sapogenin (23) induced SF188 and U87MG human glioma cell apoptosis and autophagy through both caspase-dependent and -independent pathways [81], and it also inhibited Hep-2 human laryngeal carcinoma cell proliferation through apoptosis induction caused by down-regulation of the expression of the mTOR signaling pathway [82].
In an in vivo antitumor investigation, the sensitivity of radiotherapy to laryngeal carcinoma was found to be increased by 20(S)-PPD (23). When BALB/c female nude mice (18–22 g) bearing Hep-2 laryngeal tumors were treated with 23 (i.p., 20 mg/kg, once every two days), ionizing radiation (IR) (5 Gy), or 23 (i.p., 20 mg/kg, once every two days) plus IR (5 Gy) for two weeks, tumor growth was inhibited significantly in both the individual and combined treatments, and the combination (23 plus IR) treatment caused more substantial decreases of tumor volume and weight [83].
In addition, an anti-colon tumor efficacy mediated through the TRAIL pathway was observed for 20(S)-PPD (23), when four-week old athymic female nude mice inoculated with HCT-116-luc human colon cancer cells were treated with 23 (i.p., 25 or 50 mg/kg, once two every days) for four weeks [84].
It is worthy of note that 20(S)-PPD (23) significantly enhanced the antitumor action of 5-flurouracil (5-FU), when four- to six-week old BALB/c female nude mice inoculated with HCT-116-luc human colon cancer cells were treated (i.p.) with 5-FU (30 mg/kg) or compound 23 (15 or 30 mg/kg) plus 5-FU (30 mg/kg) once a week for six weeks [85].
Also, 20(S)-PPD (23) delayed the castration-resistant regrowth of LNCaP prostate tumors after androgen-deprivation therapy and inhibited castration-resistant 22Rv1 prostate tumor growth with endogenous expression of AR-FL and AR-Vs, when male nude mice inoculated with LNCaP or 22Rv1 were treated with 20(S)-PPD (23) (gavage, 40 mg/kg, daily, six days per week) for four weeks [86]. A very recent investigation on the role of 20(S)-PPD (23) in endometriosis (EMS) showed its anti-EMS activity, which was mediated possibly by control of estrogen-mediated autophagy regulation and improved natural killer (NK) cell cytotoxicity [87].
To increase its drug-like properties, nanosuspensions of 20(S)-PPD (23) have been prepared, and its oral bioavailability was found to be improved, when compared with the unmodified form of this compound [88]. An in vivo study showed that tumor growth was inhibited, when ICR mice inoculated with H22 murine sarcoma cells were treated with nanosuspensions of compound 23 (injected via the lateral tail vein, 20, 50, or 100 mg/kg, daily) for nine days [88].
To identify the structural requirements for the activity, a hydrogenated derivative, 20(S)-24,25-dihydroprotopanaxadiol [20(S)-2H-PPD (24), Fig. 5], was synthesized from 20(S)-PPD (23), and this synthetic analogue showed less potent cytotoxicity toward MDA-MB-231 human breast cancer cells than the parent compound, 23 [80]. However, an analogous compound, 20(R)-25-OH-PPD (25) (Fig. 5), isolated from the fruits of Panax ginseng, was found to show more potent cytotoxicity than 20(S)-PPD (23) toward a panel of human cancer cell lines. This cytotoxic sapogenin (25) was absorbed and distributed rapidly in the plasma and in the kidney, liver, spleen, and tumor tissues, after nude male mice bearing xenografts of human pancreatic tumors were treated (i.v. and oral) with this compound at doses of 10 and 20 mg/kg, respectively, indicating its relatively favorable pharmacokinetic properties [79, 89].
Interestingly, a methylated sapogenin, 20(S)-25-OCH3-PPD (26) (Fig. 5), isolated from the leaves of Panax notoginseng (Birkill) F.H. Chen ex C.H. Chow showed cytotoxicity toward SW620 human colon cancer cells, with the IC50 value being less than 5 μM. This sapogenin (26) induced LS174 and SW480 colon and A549 lung human cancer cell apoptosis by suppression of Wnt/β-catenin signaling [90], and it also inhibited t-HSC/Cl-6 murine hepatic stellate cell activation by inducing apoptosis and elevating the level of cellular glutathione, indicating that it exerts an antifibrosis effect on activated t-HSC/Cl-6 cells [91].
Both 20(R)-25-OH-PPD (25) and its close analogue, 20(R)-25-OCH3-PPD were found to inhibit significantly the growth of human BGC-823, SGC-7901, and MKN-28 gastric cancer cells [92]. These two compounds also inhibited HPAC and PANC-1 human pancreatic cancer cell growth [93]. Pancreatic tumor growth was inhibited, when four- to six-week old male athymic nude nu/nu mice inoculated by Panc-1 cells were treated (i.p.) with both compounds separately (1, 5, or 10 mg/kg, daily, five days/week) for six weeks [93].
Mechanistically, 20(R)-25-OH-PPD (25) and 20(R)-25-OCH3-PPD exhibited antipancreatic tumor efficacy partially through inhibition of the MDM2 oncogene and related pathways [93]. Mouse double minute 2 homolog (MDM2) protein is an important negative regulator of the p53 tumor suppressor and is regarded as a target for cancer chemotherapy. Thus, these two sapogenins show promise in terms of their potential anticancer activity targeting MDM2.
Importantly, 20(S)-25-OCH3-PPD (26) was found to be tolerated at doses up to 600 mg/kg, and no mortality and treatment-related toxicity were observed in Sprague-Dawley rats, when the rats were treated orally with this compound (daily, 150, 300 and 600 mg/kg) for 92 consecutive days [94]. Also, the water solubility of 20(S)-25-OCH3-PPD (26) was improved by synthetic derivatization reactions [95]. These support strongly the further development of 20(S)-25-OCH3-PPD (26) as a new anticancer agent.
Both 20(S)-PPD (23) and 20(S)-protopanaxatriol [20(S)-PPT, 27] (Fig. 5) are representative sapogenins of the ginsenosides, and these compounds were found to exhibit moderate cytotoxicity toward MDA-MB-231 human breast cancer cells. However, their hydrogenated derivatives, 20(S)-24,25-dihydroprotopanaxadiol [20(S)-2H-PPD (24)] and 20(S)-24,25-dihydroprotopanaxatriol [20(S)-2H-PPT (28)], were less potent than the parent compounds, 23 and 27, respectively [80], indicating the functional importance of a C-24/25 double bond in these compounds. In addition, 20(S)-PPD (23) was more active than 20(S)-PPT (27) against MDA-MB-231 cells [80], showing that introducing an α-hydroxy group at the C-6 position results in the cytotoxic potency of 20(S)-PPD (23) being decreased. This was supported by the cytotoxicity observed for 20(R)-25-OH-PPD (25) and 20(R)-25-OH-PPT (29) toward a panel of human cancer cell lines, including breast, glioma, lung, pancreatic, and prostate cancer cells, for which 25 showed weak activity, with IC50 values in the range 10–70 μM, but 29 did not (IC50>100 μM) [79].
Dammarane-type Triterpenoids II: Ginsenosides
In analogous work, the antitumor potential has been investigated extensively for the PPD-type ginseng saponins, including the 20(S)-ginsenosides Rh2 (G-Rh2, 30) and Rg3 (G-Rg3, 31), and ginsenosides Rh3 (G-Rh3, 32), and Rk2 (G-Rk2, 33) (Fig. 6) [76, 78, 79, 96]. Of these, G-Rh2 (30) was up to ten-fold more active than G-Rg3 (31) toward a panel of human cancer cells [79], indicating that introducing an additional sugar unit in the C-3 saccharide moiety decreases the cytotoxic potency of G-Rh2 (30).
Fig. 6.
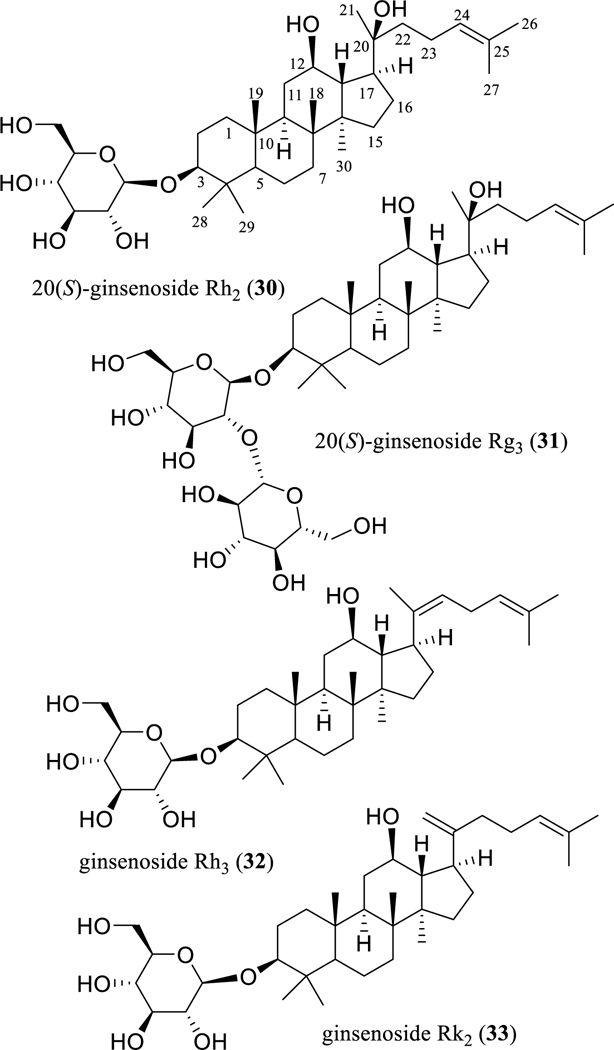
Structures of dammarane-type triterpenoid ginsenosides (30–33) with potential anticancer activity.
When 14 ginsenosides isolated from the steamed (heat-processed) leaves of Panax ginseng were evaluated for their cytotoxicity toward HL-60 human leukemia cells, G-Rh2 (30), G-Rh3 (32), and G-Rk2 (33) were found to be active, while 32 and 33 were the most potently active, showing IC50 values of 0.8 and 0.9 μM, respectively [97], indicating that a double bond at the C-20(21) or C-20(22) position enhances the cytotoxicity of G-Rh2 (30).
The antineoplastic potential of various ginsenosides has been reviewed previously [78], and thus presented immediately below is an update of this topic, with G-Rh2 (30) being used as a representative example.
In recent studies, G-Rh2 (30) was found to significantly suppress cell proliferation, invasion, and migration in HEC1A human endometrial cancer cells [98], and it also decreased the viability of U87MG and A172 human glioma cells [99]. It induced ROS (reactive oxygen species)-mediated ER stress-dependent apoptosis in H1299 human lung cancer cells [100] and induced apoptosis in KG-1a human leukemia cells [101].
In an in vivo study, hepatoma growth was inhibited significantly, when 18–22 g female mice inoculated with H22 murine hepatoma cells were gavaged with G-Rh2 (30) (3 or 4 mg/kg, daily) for ten days [102]. A further study showed that hepatoma growth was inhibited significantly, and the serum IL-2 levels, TNF-α production, T lymphocytes, CD4+/CD8+ ratio, and NK cell levels of mice were increased, when seven- to eight-week old male Kunming mice inoculated with H22 cells were gavaged with G-Rh2 (30) (5 or 10 mg/kg, daily) for 15 days. This indicates that G-Rh2 mediates its antitumor activity partially through modulation of the immune system [103]. No obvious toxicity was observed in the mice used in these in vivo studies [102, 103].
Mechanistically, G-Rh2 (30) exhibits its potential antitumor activity through modulating the Aκt and Wnt/β-catenin signaling pathways [99, 101]. It targets EZH2 (the polycomb repressive complex 2 component enhancer of zeste homologue 2), a potent histone methyltransferase that catalyzes the trimethylation of histone 3 and lysine 27 [104], down-regulates the expression of the inhibitors of apoptosis (IAP), and synergizes with Annexin A2 inactivation to promote apoptosis [105]. Both EZH2 and Annexin A2 are over-expressed in liver cancer, and thus G-Rh2 (30) has been regarded as a promising candidate agent for targeted liver cancer therapy [105].
Interestingly, G-Rh2 (30) was found to mediate its antitumor efficacy by modulating the immune response [103, 106]. G-Rh2 (30) triggered CD4+ and CD8a+ T-lymphocyte infiltration in tumor tissues and increased T-lymphocyte cytotoxicity. An enhanced antitumor immunological response contributes to preventing the recurrence of cancer, and, thus, G-Rh2 (30) could serve as an adjuvant for use with existing cancer chemotherapeutic agents [106].
Dichapetalin-type Triterpenoids
A final group of triterpenoids selected for inclusion in this review are compounds of the dichapetalin-type. These compounds constitute a small group of 13,30-cyclodammarano[4,3]pyran derivatives, with the first member, (+)-dichapetalin A (34) (Fig. 7), being reported from Dichapetalum madagascariense Poir. (Dichapetalaceae) in 1995. In this reference, the authors reported a greater susceptibility of (+)-dichapetalin A (34) against the L1210 murine leukemia cell line (EC90 <0.0001 μg/ml) than against the KB human oral epidermoid carcinoma cell line [107]. This trend was noted also by Tuchinda et al., for their isolate (+)-acutissmatriterpene E (35) (Fig. 7) from Phyllanthus acutissima Miq. (Phyllanthaceae), which was more potently cytotoxic for the P-388 murine lymphocytic leukemia cell line (IC50 0.005 μg/ml) than for the other human cancer cell lines evaluated, using ellipticine (IC50 0.2 μg/mL toward P-388 cells) as the positive control [108].
Fig. 7.
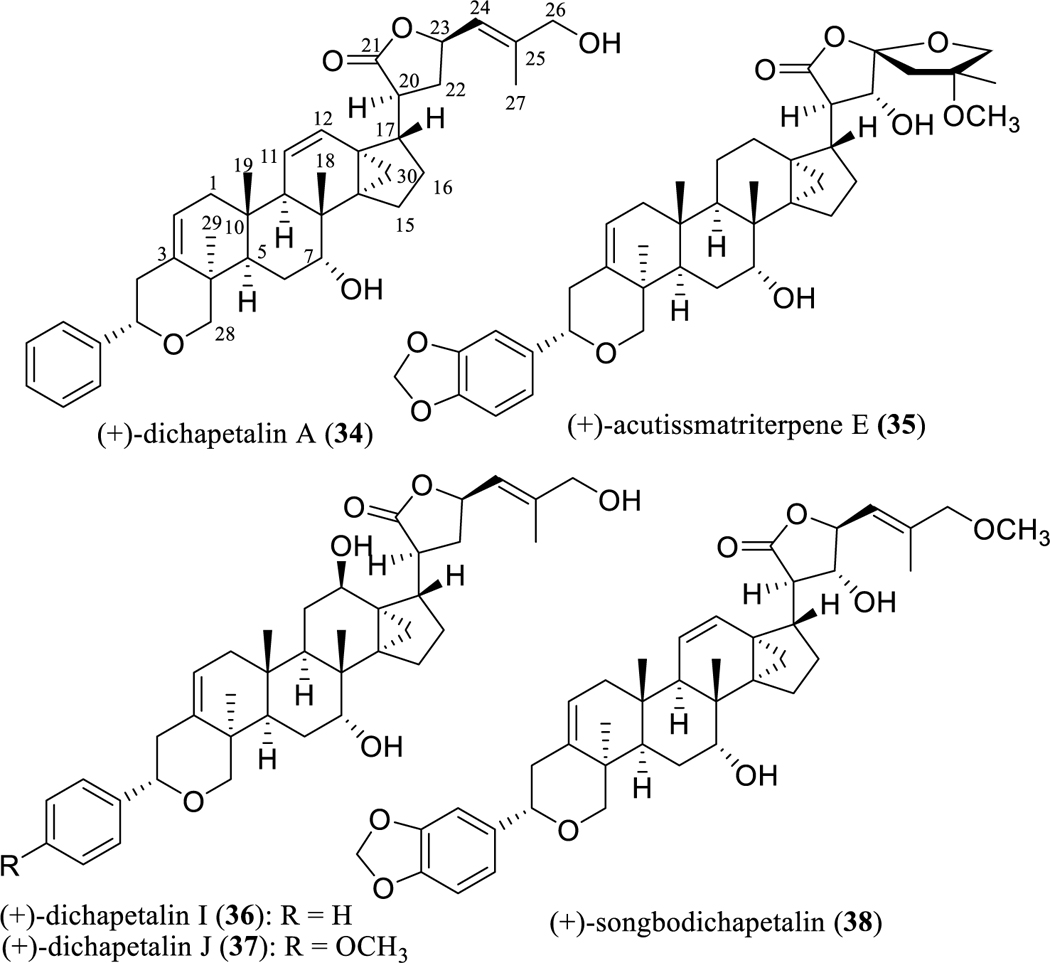
Structures of dichapetalin-type triterpenoids (34–37) with potential anticancer activity and their inactive analogue (38).
The interest of our group in this class of compounds was stimulated by the isolation of several cytotoxic dichapetalins from the stem bark of Dichapetalum gelonioides (Roxb.) Engl. (Dichapetalaceae) collected in the Philippines, among which (+)-dichapetalin A (34) exhibited its most potent cytotoxic effects toward the SW626 human ovarian cancer cell line (IC50 0.2 μg/ml). Also obtained in this study were the new (+)-dichapetalins I (36) and J (37) (Fig. 7), which again demonstrated their most potent cytotoxic activity for SW626 cells (IC50 0.5 and 0.4 μg/ml, respectively) [109].
However, when tested in a follow-up in vivo hollow fiber assay, (+)-dichapetalin A (34) was inactive when tested in mice at doses of 1, 2, 4, and 6 mg/kg (i.p. administration) for four different types of human cancer cell lines, including SW626 cells [109]. In a more recent investigation by our group, a non-cytotoxic dichapetalin A analogue, (+)-songbodichapetalin (38) (Fig. 7), was isolated from the aerial parts of Phyllanthus songboiensis N. N. Thin (Phyllanthaceae) collected in Vietnam [110].
Analysis of the dichapetalin derivatives and their cytotoxic activity against cancer cell lines indicates that a methylenedioxy group connected at the C-3′ and C-4′ position is not required for this type of activity, but the presence of a hydroxy group at the C-22 position can enhance cytotoxicity. In addition, a primary C-26 hydroxy group seems to play a key role in determining the cytotoxic potency of the dichapetalin-type triterpenes [108, 110]. These preliminary SAR conclusions indicate that the antitumor potential of the dichapetalins could be improved by synthetic modification. Owing to their inherently potent cytotoxic potency for certain cancer cell lines, this group of cyclodammarane-type triterpene lactones seems worthy of additional in vivo antineoplastic testing and mechanism-of-action studies.
Conclusions
Plant-derived triterpenoids were frequently encountered along with more promising compounds in early anticancer screening campaigns. However, this type of natural products were deemed to be of insufficient promise for further development or more in-depth biological investigation, because of their general lack of potency in inhibiting the growth of the panels of murine and human cancer cell lines. Fortunately, over the last nearly 20 years, there has been an undeniable uptake of interest in the antitumor activities of triterpenoids, which have been supported by promising effects observed in vivo and by supportive mechanism-of-action investigations. For example, (+)-ursolic acid (11), a pentacyclic triterpenoid distributing widely in plants, modulates several important inflammation associated signaling pathways, including NF-κB, STAT3, and TRAIL signalings, and has reached cancer clinical trials [111]. Several triterpenoids decrease the expression of specificity protein (Sp) transcription factors in cancer cells. They also induce ROS, an important proteasome-independent pathway for downregulation of Sp transcription factors, and activate or deactivate nuclear receptors and G-protein coupled receptors, which contribute to their antitumorigenic activity [112]. In this regard, the recent progress made on plant triterpenoids as potential cancer chemotherapeutic agents is analogous to that made on sesquiterpene lactones of plant origin [113].
Isolation chemistry work on a given cytotoxic plant lead tends to afford a suite of structurally related active compounds and permits an initial evaluation of structure-cytotoxicity relationships, which in turn may guide the subsequent synthesis of more potent analogues of a bioactive compound. New techniques of formulation, such as the production of liposomes and nanoparticules of triterpenoids, have enabled both the resultant water solubility and bioavailability of the leading compounds to be enhanced. A range of mechanistic effects on cancer cells have been shown for the triterpenoids, including inhibition of NF-κB and the induction of autophagy and/or the modulation of the human immune system. These mechanisms may contribute to limiting multidrug resistance, and thus, it may be confidently predicted that naturally occurring triterpenoids and their semi-synthetic derivatives will remain as promising leads for the development of new anticancer drugs in future years.
Acknowledgements
The experimental studies by our group mentioned in this article were supported by grants U19 CA52956 and P01 CA125066 funded by the National Cancer Institute, NIH, Bethesda, MD, USA. We are very grateful to many faculty colleagues, research staffs, postdoctoral fellows, and graduate students who have contributed to this work.
Abbreviations
- AIF
apoptosis-inducing factor ()
- CDDO-Me
methyl 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate or bardoxolone methyl
- Dap
dolaproine
- Dil
dolaisoleuine
- DLT
dose-limiting toxicities
- Dov
N,N-dimethyl-L-valine
- EMS
endometriosis
- ER
endoplasmic reticulum
- EZH2
the polycomb repressive complex 2 component enhancer of zeste homologue 2
- G-Rh2
20(S)-ginsenoside Rh2
- G-Rg3
20(S)-ginsenoside Rg3
- G-Rh3
ginsenoside Rh3
- G-Rk2
ginsenoside Rk2
- GI50
concentration that inhibits 50% cell growth
- Hsp90
heat shock protein 90
- IAP
inhibitors of apoptosis
- ICR
Institute of Cancer Research
- IFN-γ
interferon-γ
- i.p.
intraperitoneal
- i.v.
intravenous
- JNK
c-Jun NH2-terminal kinase
- MAPK/ERK
mitogen-activated protein kinase/extracellular signal-regulated kinase
- MDM2
mouse double minute 2 homolog
- MDSC
myeloid-derived suppressor cells
- MTD
maximum tolerated dose
- NF-κB
nuclear factor kappa B
- NK
natural killer
- NOZ
NOZ human gallbladder cancer cell line
- OA-MVL
oleanolic acid-encapsulated multivesicular liposome
- PPD
protopanaxadiol
- PPT
protopanaxatriol
- ROS
reactive oxygen species
- SAR
structure-activity relationship
- STAT3
signal transducer and activator of transcription-3
- TAM
tumor-associated macrophage
- TNF-α
tumor necrosis factor-α
- TRAIL
TNF-related apoptosis-inducing ligand
- Val
L-valine
- VEGF
vascular endothelial growth factor
Footnotes
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
* Dedicated to Professor Dr. Cosimo Pizza in recognition of his important contributions to natural product research on the occasion of his 70th birthday in 2019.
References
- [1].Cragg GM, Kingston DGI, Newman DJ. (Eds.). Anticancer Agents from Natural Products, 2nd Edition; CRC Press/Taylor & Francis; Boca Raton, FL, USA, 2012. [Google Scholar]
- [2].Butler MS, Robertson AAB, Cooper MA. Natural product and natural product derived drugs in clinical trials. Nat Prod Res 2014; 31: 1612–1661. [DOI] [PubMed] [Google Scholar]
- [3].Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 2016; 79: 629–661. [DOI] [PubMed] [Google Scholar]
- [4].Xu R, Fazio GC, Matsuda SPT. On the origins of triterpenoid skeletal diversity. Phytochemistry 2004; 65: 261–291. [DOI] [PubMed] [Google Scholar]
- [5].Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anti-Cancer Drugs 2009; 20: 880–892. [DOI] [PubMed] [Google Scholar]
- [6].Hill RA, Connolly JD. Triterpenoids. Nat Prod Rep 2017; 34: 90–122. [DOI] [PubMed] [Google Scholar]
- [7].Hong DS, Kurzrock R, Supko JG, He X, Naing A, Wheler J, Lawrence D, Eder JP, Meyer CJ, Ferguson DA, Mier J, Konopleva M, Konoplev S, Andreeff M, Kufe D, Lazarus H, Shapiro GI, Dezube BJ. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res 2012; 18: 3396–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baglin I, Mittaine-Offer AC, Nour M, Tan K, Cavé C, Lacaille-Dubois MA. A review of natural and modified betulinic, ursolic, and echinocystic acid derivatives as potential antitumor and anti-HIV agents. Mini Rev Med Chem 2003; 3: 525–539. [DOI] [PubMed] [Google Scholar]
- [9].Setzer WN, Setzer MC. Plant-derived triterpenoids as potential antineoplastic agents. Mini Rev Med Chem 2003; 3: 540–546. [DOI] [PubMed] [Google Scholar]
- [10].Laszczyk MN. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med 2009; 75: 1549–1560. [DOI] [PubMed] [Google Scholar]
- [11].Kuo RY, Qian K, Morris-Natschke SL, Lee KH. Plant-derived triterpenoids and analogues as antitumor and anti-HIV agents. Nat Prod Rep 2009; 26: 1321–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Salvador JAR, Moreira VM, Gonçlaves BMF, Leal AS, Jing Y. Ursane-type pentacyclic triterpenoids as useful platforms to discover anticancer drugs. Nat Prod Rep 2012; 29: 1463–1479. [DOI] [PubMed] [Google Scholar]
- [13].Shanmugam MK, Nguyen AH, Kumar AP, Tan BKH, Sethi G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett 2012; 320: 158–170. [DOI] [PubMed] [Google Scholar]
- [14].Kamble SM, Goyal SN, Patil CR. Multifunctional pentacyclic triterpenoids as adjuvants in cancer chemotherapy: A review. RSC Adv 2014; 4: 33370–33382. [Google Scholar]
- [15].Salvador JAR, Leal AS, Valdeira AS, Gonçlaves BMF, Alho DPS, Figueiredo SAC, Silvestre SM, Mendes VIS. Oleanane-, ursane-, and quinone methide freidelane-type triterpenoid derivatives: Recent advances in cancer treatment. Eur J Med Chem 2017; 142: 95–130. [DOI] [PubMed] [Google Scholar]
- [16].Peron G, Marzaro G, Dall’Acqua S. Known triterpenes and their derivatives as scaffolds for the development of new therapeutic agents for cancer. Curr Med Chem 2018; 25: 1259–1269. [DOI] [PubMed] [Google Scholar]
- [17].Kinghorn AD, Farnsworth NR, Soejarto DD, Cordell GA, Pezzuto JM, Udeani GO, Wani MC, Wall ME, Navarro HA, Kramer RA, Menendez AT, Fairchild CR, Lane KE, Forenza S, Vyas DM, Lam KS, Shu YZ. Novel strategies for the discovery of plant-derived anticancer agents. Pure Appl Chem 1999; 71: 1611–1618. [Google Scholar]
- [18].Kinghorn AD, Carcache de Blanco EJ, Lucas DM, Rakotondraibe HL, Orjala J, Soejarto DD, Oberlies NH, Pearce CJ, Wani MC, Stockwell BR, Burdette JE, Swanson SM, Fuchs JR, Phelps MA, Xu L, Zhang X, Shen YY. Discovery of anticancer agents of diverse natural origin. Anticancer Res 2016; 36: 5623–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pisha E, Chai H, Lee IS, Chagwedera TE, Farnsworth NR, Cordell GA, Beecher CWW, Fong HHS, Kinghorn AD, Brown DM, Wani MC, Wall ME, Hieken TJ, Das Gupta TK, Pezzuto JM. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat Med 1995; 1: 1046–1051. [DOI] [PubMed] [Google Scholar]
- [20].Henkin JM, Ren Y, Soejarto DD, Kinghorn AD. The Search for Anticancer Agents from Tropical Plants In: Progress in the Chemistry of Organic Natural Products, Vol. 107; Kinghorn AD, Falk H, Gibbons S, Kobayashi J, Asakawa Y, Liu JK, Eds.; Springer International, Cham, Switzerland, 2018; pp. 1–94. [DOI] [PubMed] [Google Scholar]
- [21].Zhang DM, Xu HG, Wang L, Li YJ, Sun PH, Wu XM, Wang GJ, Chen WM, Ye WC. Betulinic acid and its derivatives as potential antitumor agents. Med Res Rev 2015; 35: 1127–1155. [DOI] [PubMed] [Google Scholar]
- [22].Ali-Seyed M, Jantan I, Vijayaraghavan K, Bukhari SNA. Betulinic acid: Recent advances in chemical modifications, effective delivery, and molecular mechanisms of a promising anticancer therapy. Chem Biol Drug Des 2016; 87: 517–536. [DOI] [PubMed] [Google Scholar]
- [23].Pettit GR, Melody N, Hempelstall F, Chapuis JC, Groy TL, Williams L. Antineoplastic agents. 595. Structural modifications of betulin and the X-ray crystal structure of an unusual betulin amine dimer. J Nat Prod 2014; 77: 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ye Y, Zhang T, Yuan H, Li D, Lou H, Fan, P. Mitochondria-targeted lupane triterpenoid derivatives and their selective apoptosis-inducing anticancer mechanisms. J Med Chem 2017; 60: 6353–6363. [DOI] [PubMed] [Google Scholar]
- [25].Kim DSHL Pezzuto JM, Pisha E. Synthesis of betulinic acid derivatives with activity against human melanoma. Bioorg Med Chem Lett 1998; 8: 1707–1712. [DOI] [PubMed] [Google Scholar]
- [26].Tsepaeva OV, Nemtarev AV, Abdullin TI, Grigor’eva LR, Kuznetsova EV, Akhmadishina RA, Ziganshina LE, Cong HH, Mironov VF. Design, synthesis, and cancer cell growth inhibitory activity of triphenylphosphonium derivatives of the triterpenoid betulin. J Nat Prod 2017; 80: 2232–2239. [DOI] [PubMed] [Google Scholar]
- [27].Pettit GR, Melody N, Chapuis JC. Antineoplastic agents. 606. The betulastatins. J Nat Prod 2018; 81: 458–464. [DOI] [PubMed] [Google Scholar]
- [28].Seneja A, Sharma L, Dubey RD, Mintoo MJ, Singh A, Kumar A, Sangwan PL, Tasaduq SA, Singh G, Mondhe DM, Gupta PN. Synthesis, characterization and augmented anticancer potential of PEG-betulinic acid conjugate. Mat Sci Eng C 2017; 73: 616–626. [DOI] [PubMed] [Google Scholar]
- [29].Shanmugam MK, Dai X, Kumar AP, Tan BKH, Sethi G, Bishayee A. Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: Preclinical and clinical evidence. Cancer Lett 2014; 346: 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lúcio KA, Rocha GG, Moncão-Ribeiro LC, Fernandes J, Takiya CM, Gattass CR. Oleanolic acid initiates apoptosis in non-small cell lung cancer cell lines and reduces metastatis of a B16F10 melanoma model in vivo. PLoS One 2011; 6: e28596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li HF, Wang XA, Xiang SS, Hu YP, Jiang L, Shu YJ, Li ML, Wu XS, Zhang F, Ye YY, Weng H, Bao RF, Cao Y, Lu W, Dong Q, Liu YB. Oleanolic acid induces mitochondrial-dependent apoptosis and G0/G1 phase arrest in gallbladder cancer cells. Drug Des Dev Ther 2015; 9: 3017–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Takemura M, Endo S, Matsunaga T, Soda M, Zhao HT, El-Kabbani O, Tajima K, Iinuma M, Hara A. Selective inhibition of the tumor marker aldo-keto reductase family member 1B10 by oleanolic acid. J Nat Prod 2011; 74: 1201–1206. [DOI] [PubMed] [Google Scholar]
- [33].Shi Y, Song Q, Hu D, Zhuang X, Yu S, Teng D. Oleanolic acid induced autophagic cell death in hepatocellular carcinoma cells via PI3K/Aκt/mTOR and ROS-dependent pathway. Korean J Physiol Pharmacol 2016; 20: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bernabé-García Á, Armero-Barranco D, Liarte S, Ruzafa-Martínez M, Ramos-Morcillo AJ, Nicolás FJ. Oleanolic acid induces migration in Mv1Lu and MDA-MB-231 epithelial cells involving EGF receptor and MAP kinases activation. PLoS One 2017; 12: e0172574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu Y, Shu B, Tian Y, Wang G, Wang Y, Wang J, Dong Y. Oleanolic acid induces osteosarcoma cell apoptosis by inhibition of Notch signaling. Mol Carcinog 2018; 57: 896–902. [DOI] [PubMed] [Google Scholar]
- [36].Luo Y, Liu Z, Zhang X, Huang J, Yu X, Li J, Xiong D, Sun X, Zhong Z. Effect of a controlled-release drug delivery system made of oleanolic acid formulated into multivesicular liposomes on hepatocellular carcinoma in vitro and in vivo. Int J Nanomed 2016; 11: 3111–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro FG, Suh N, Wang Y, Sporn MB, Gribble GW. Synthetic oleanane and ursane triterpenoids with modified rings A and C: A series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem 2000; 43: 4233–4246. [DOI] [PubMed] [Google Scholar]
- [38].Wang TT, Liu Y, Chen L. Synthesis and cytotoxic activity of nitric oxide-releasing isosteviol derivatives. Bioorg Med Chem Lett 2014; 24: 2202–2205. [DOI] [PubMed] [Google Scholar]
- [39].Ball MS, Shipman EP, Kim H, Liby KT, Pioli PA. CDDO-Me redirects activation of breast tumor associated macrophages. PLoS One 2016; 11: e0149600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang YY, Zhe H, Zhao R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol Cancer 2014; 13: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gao X, Deeb D, Liu Y, Liu P, Zhang Y, Shaw J, Gautam SC. CDDO-Me inhibits tumor growth and prevents recurrence of pancreatic ductal adenocarcinoma. Int J Oncol 2015; 47: 2100–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, Sporn MB, Liby KT, Rawal B, Lee JH, Gabrilovich DI. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res 2010; 16: 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang YY, Yang YX, Zhe H, He ZX, Zhou SF. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des Dev Ther 2014; 8: 2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cragg GM, Grothaus PG, Newman DJ. New horizons for old drugs and drug leads. J Nat Prod 2014; 77: 703–723. [DOI] [PubMed] [Google Scholar]
- [45].Schmid C, Dawid C, Peters V, Hofmann T. Saponins from European licorice roots (Glycyrrhiza glabra). J Nat Prod 2018; 81: 1734–1744. [DOI] [PubMed] [Google Scholar]
- [46].Li K, Ji S, Song W, Kuang Y, Lin Y, Tang S, Cui Z, Qiao X, Yu S, Ye M. Glycybridins A–K, bioactive phenolic compounds from Glycyrrhiza glabra. J Nat Prod 2017; 80: 334–346. [DOI] [PubMed] [Google Scholar]
- [47].Zhang X, Yang H, Yue S, He G, Qu S, Zhang Z, Ma B, Ding R, Peng W, Zhang H, Yang Z, Dou K, Tao K, Li X. The mTOR inhibition in concurrence with ERK1/2 activation is involved in excessive autophagy induced by glycyrrhizin in hepatocellular carcinoma. Cancer Med 2017; 6: 1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu SH, Cheng YC. Old formula, new Rx: The journey of PHY906 as cancer adjuvant therapy. J Ethnopharmacol 2012; 140: 614–623. [DOI] [PubMed] [Google Scholar]
- [49].Kummar S, Copur MS, Rose M, Wadler S, Stephenson J, O’Rourke M, Brenckman W, Tilton R, Liu SH, Jiang Z, Su T, Cheng YC, Chu E. A phase I study of the Chinese herbal medicine PHY906 as a modulator of irinotecan-based chemotherapy in patients with advanced colorectal cancer. Clin Colorect Cancer 2011; 10: 85–96. [DOI] [PubMed] [Google Scholar]
- [50].Saif MW, Li J, Lamb L, Kaley K, Elligers K, Jiang Z, Bussom S, Liu SH, Cheng YC. First-in-human Phase II trial of the botanical formulation PHY906 with capecitabine as second-line therapy in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol 2014; 73: 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ren Y, VanSchoiack A, Chai HB, Goetz M, Kinghorn AD. Cytotoxic barrigenol-like triterpenoids from an extract of Cyrilla racemiflora housed in a repository. J Nat Prod 2015; 78: 2440–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jiao Q, Zou L, Liu P, Xu Q, Zhang Y, Yu Y, Zou L, Chi T, Ji X. Xanthoceraside induces apoptosis in melanoma cells through the activation of caspases and the suppression of the IGF-1R/Raf/MEK/ERK signaling pathway. J Med Food 2014; 17: 1070–1078. [DOI] [PubMed] [Google Scholar]
- [53].Shanmugam MK, Rajendran P, Li F, Nema T, Vali S, Abbasi T, Kapoor S, Sharma A, Kumar AP, Ho PC, Hui KM, Sethi G. Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice. J Mol Med 2011; 89: 713–727. [DOI] [PubMed] [Google Scholar]
- [54].Yang L, Liu X, Lu Z, Chan JYW, Zhou L, Fung KP, Wu P, Wu S. Ursolic acid induces doxorubicin-resistant HepG2 cell death via the release of apoptosis-inducing factor. Cancer Lett 2010; 298: 128–138. [DOI] [PubMed] [Google Scholar]
- [55].Zhang J, Wang W, Qian L, Zhang Q, Lai D, Qi C. Ursolic acid inhibits the proliferation of human ovarian cancer stem-like cells through epithelial-mesenchymal transition. Oncology Rep 2015; 34: 2375–2384. [DOI] [PubMed] [Google Scholar]
- [56].Ren Y, Anaya-Eugenio GD, Czarnecki AA, Ninh TN, Yuan C, Chai HB, Soejarto DD, Burdette JE, Carcache de Blanco EJ, Kinghorn AD. Cytotoxic and NF-κB and mitochondrial transmembrane potential inhibitory pentacyclic triterpenoids from Syzygium corticosum and their semi-synthetic derivatives. Bioorg Med Chem 2018; 26: 4452–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hata K, Hori K, Takahashi S. Differentiation- and apoptosis-inducing activities by pentacyclic triterpenes on a mouse melanoma cell line. J Nat Prod 2002; 65: 645–648. [DOI] [PubMed] [Google Scholar]
- [58].Tian T, Liu X, Lee ES, Sun J, Feng Z, Zhao L, Zhao C. Synthesis of novel oleanolic acid and ursolic acid in C-28 position derivatives as potential anticancer agents. Arch Pharm Res 2017; 40: 458–468. [DOI] [PubMed] [Google Scholar]
- [59].Sun L, Li B, Su X, Chen G, Li Y, Yu L, Li L, Wei W. An ursolic acid derived small molecule triggers cancer cell death through hyperstimulation of macropinocytosis. J Med Chem 2017; 60: 6638–6648. [DOI] [PubMed] [Google Scholar]
- [60].Fan L, Zhang B, Xu A, Shen Z, Guo Y, Zhao R, Yao H, Shao JW. Carrier-free, pure nanodrug formed by the self-assembly of an anticancer drug for cancer immune therapy. Mol Pharm 2018; 15: 2466–2478. [DOI] [PubMed] [Google Scholar]
- [61].Shishodia S, Majumdar S, Banerjee S, Aggarwal BB. Ursolic acid inhibits nuclear factor-κB activation induced by carcinogenic agents through suppression of IκBα kinase and p65 phosphorylation: Correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res 2003; 63: 4375–4383. [PubMed] [Google Scholar]
- [62].Qian Z, Wang X, Song Z, Zhang H, Zhou S, Zhao J, Wang H. A phase I trial to evaluate the multiple-dose safety and antitumor activity of ursolic acid liposomes in subjects with advanced solid tumors. BioMed Res Int 2015; 809714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chen JC, Chiu MH, Nie RL, Cordell GA, Qiu SX. Cucurbitacins and cucurbitane glycosides: Structures and biological activities. Nat Prod Rep 2005; 22: 386–399. [DOI] [PubMed] [Google Scholar]
- [64].Lee DH, Iwanski GB, Thoennissen NH. Cucurbitacin: Ancient compound shedding new light on cancer treatment. ScientificWorldJournal 2010; 10: 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ríos JL, Andúlar I, Escandell JM, Giner RM, Recio MC. Cucurbitacins as inducers of cell death and a rich source of potential anticancer compounds. Curr Pharm Des 2012; 18: 1663–1676. [DOI] [PubMed] [Google Scholar]
- [66].Pan L, Yong Y, Deng Y, Lantvit DD, Ninh TN, Chai H, Carcache de Blanco EJ, Soejarto DD, Swanson SM, Kinghorn AD. Isolation, structure elucidation, and biological evaluation of 16,23-epoxycucurbitacin constituents from Elaeocarpus chinensis. J Nat Prod 2012; 75: 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Abbas S, Vincourt JB, Habib L, Netter P, Greige-Gerges H, Magdalou J. The cucurbitacins E, D and I: Investigation of their cytotoxicity toward human chondrosarcoma SW 1353 cell line and their biotransformation in human liver. Toxicol Lett 2013; 216: 189–199. [DOI] [PubMed] [Google Scholar]
- [68].Fang X, Phoebe CH, Pezzuto JM, Fong HHS, Farnsworth NR, Yellin B, Hecht SM. Plant anticancer agents. XXXIV. Cucurbitacins from Elaeocarpus dolichostylus. J Nat Prod 1984; 47: 988–993. [DOI] [PubMed] [Google Scholar]
- [69].Kim DK, Choi SH, Lee JO, Ryu SY, Park DK, Shin DH, Jung JH, Pyo SK, Lee KR, Zee OP. Cytotoxic constituents of Sorbaria sorbifolia var. stellipila. Arch Pharm Res 1997; 20: 85–87. [DOI] [PubMed] [Google Scholar]
- [70].Takahashi N, Yoshida Y, Sugiura T, Matsuno K, Fujino A, Yamashita U. Cucurbitacin D isolated from Trichosanthes kirilowii induces apoptosis in human hepatocellular carcinoma cells in vitro. Int Immunopharmacol 2009; 9: 508–513. [DOI] [PubMed] [Google Scholar]
- [71].Spear SA, Burns SS, Oblinger JL, Ren Y, Pan L, Kinghorn AD, Welling DB, Chang LS. Natural compounds as potential treatments of NF2-deficient schwannoma and meningioma: Cucurbitacin D and goyazensolide. Otol Neurotol 2013; 34: 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ku JM, Kim SR, Hong SH, Choi HS, Seo HS, Shin YC, Ko SG. Cucurbitacin D induces cell cycle arrest and apoptosis by inhibiting STAT3 and NF-κB signaling in doxorubicin-resistant human breast carcinoma (MCF7/ADR) cells. Mol. Cell Biochem 2015; 409: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nakanishi T, Song Y, He C, Wang D, Morita K, Tsukada J, Kanazawa T, Yoshida Y. Autophagy is associated with cucurbitacin D-induced apoptosis in human T cell leukemia cells. Med Oncol 2016; 33: 30. [DOI] [PubMed] [Google Scholar]
- [74].Hall JA, Seedarala S, Rice N, Kopel L, Halaweish F, Blagg BSJ. Cucurbitacin D is a disruptor of the HSP90 chaperone machinery. J Nat Prod 2015; 78: 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sikander M, Hafeez BB, Malik S, Alsayari A, Halaweish FT, Yallapu MM, Chauhan SC, Jaggi M. Cucurbitacin D exhibits potent anti-cancer activity in cervical cancer. Sci Rep 2016; 6: 36594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ruan J, Zheng C, Qu L, Liu Y, Han L, Yu H, Zhang Y, Wang T. Plant resources, 13C-NMR spectral characteristic and pharmacological activities of dammarane-type triterpenoids. Molecules 2016; 21: 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Patel S, Rauf A. Adaptogenic herb ginseng (Panax) as medical food: Status quo and future prospects. Biomed Pharmacother 2017; 85: 120–127. [DOI] [PubMed] [Google Scholar]
- [78].Chen XJ, Zhang XJ, Shui YM, Wan JB, Gao JL. Anticancer activities of protopanaxadiol-and protopanaxatriol-type ginsenosides and their metabolites. Evid Based Compl Altern Med 2016; 2016: 5738694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang W, Zhao Y, Rayburn ER, Hill DL, Wang H, Zhang R. In vitro anticancer activity and structure-activity relationships of natural products isolated from fruits of Panax ginseng. Cancer Chemother Pharmacol 2007; 59: 589–601. [DOI] [PubMed] [Google Scholar]
- [80].Kwak JH, Park JY, Lee D, Kwak JY, Park EH, Kim KH, Park HJ, Kim HY, Jang HJ, Ham J, Hwang GS, Yamabe N, Kang KS. Inhibitory effects of ginseng sapogenins on the proliferation of triple negative breast cancer MDA-MB-231 cells. Bioorg Med Chem Lett 2014; 24: 5409–5412. [DOI] [PubMed] [Google Scholar]
- [81].Liu GY, Bu X, Yan H, Jia WWG. 20S-Protopanaxadiol-induced programmed cell death in glioma cells through caspase-dependent and -independent pathways. J Nat Prod 2007; 70: 259–264. [DOI] [PubMed] [Google Scholar]
- [82].Teng B, Jiang J, Zhao L, Gao J, Chen J, Liu Z, Wang H, Lu B. Ginsenoside PPD’s antitumor effect via down-regulation of mTOR revealed by super-resolution imaging. Molecules 2017; 22: 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Teng B, Zhao L, Gao J, He P, Li H, Chen J, Feng Q, Yi C. 20(S)-Protopanaxadiol (PPD) increases the radiotherapy sensitivity of laryngeal carcinoma. Food Funct 2017; 8: 4469–4477. [DOI] [PubMed] [Google Scholar]
- [84].Zhang Z, Li Z, Wu X, Zhang CF, Calway T, He TC, Du W, Chen J, Wang CZ, Yuan CS. TRAIL pathway is associated with inhibition of colon cancer by protopanaxadiol. J Pharmacol Sci 2015; 127: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang CZ, Zhang Z, Wan JY, Zhang CF, Anderson S, He X, Yu C, He TC, Qi LW, Yuan CS. Protopanaxadiol, an active ginseng metabolite, significantly enhances the effects of fluorouracil on colon cancer. Nutrients 2015; 7: 799–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Cao B, Qi Y, Yang Y, Liu X, Xu D, Guo W, Zhan Y, Xiong Z, Zhang A, Wang AR, Fu X, Zhang H, Zhao L, Gu J, Dong Y. 20(S)-Protopanaxadiol inhibition of progression and growth of castration-resistant prostate cancer. PLoS One 2014; 9: e111201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhang B, Zhou WJ, Gu CJ, Wu K, Yang HL, Mei J, Yu JJ, Hou XF, Sun JS, Xu FY, Li DJ, Jin LP, Li MQ. The ginsenoside PPD exerts anti-endometriosis effects by suppressing estrogen receptor-mediated inhibition of endometrial stromal cell autophagy and NK cell cytotoxicity. Cell Death Dis 2018; 9: 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Han M, Ma L, Yu X, Li Z, Guo Y, Wang X. A nanoparticulate drug-delivery system for 20(S)-protopanaxadiol: Formulation, characterization, increased oral bioavailability and anti-tumor efficacy. Drug Deliv 2016; 23: 2410–2418. [DOI] [PubMed] [Google Scholar]
- [89].Hao M, Wang W, Zhao Y, Zhang R, Wang H. Pharmacokinetics and tissue distribution of 25-hydroxyprotopanaxadiol, an anti-cancer compound isolated from Panax ginseng, in athymic mice bearing xenografts of human pancreatic tumors. Eur J Drug Metab Pharmacokinet 2011; 35: 109–113. [DOI] [PubMed] [Google Scholar]
- [90].Bi X, Zhao Y, Fang W, Yang W. Anticancer activity of Panax notoginseng extract 20(S)-25-OCH3-PPD: Targeting β-catenin signaling. Clin Exp Pharmacol Physiol 2009; 36: 1074–1078. [DOI] [PubMed] [Google Scholar]
- [91].Wu YL, Wan Y, Jin XJ, OuYang BQ, Bai T, Zhao YQ, Nan JX. 25-OCH3-PPD induces the apoptosis of activated t-HSC/Cl-6 cells via c-FLIP-mediated NF-κB activation. Chem Biol Interact 2011; 194: 106–112. [DOI] [PubMed] [Google Scholar]
- [92].Zhao C, Su G, Wang X, Zhang X, Guo S, Zhao Y. Antitumor activity of ginseng sapogenins, 25-OH-PPD and 25-OCH3-PPD, on gastric cancer cells. Biotechnol Lett 2016; 38: 43–50. [DOI] [PubMed] [Google Scholar]
- [93].Wang W, Rayburn ER, Zhao Y, Wang H, Zhang R. Novel ginsenosides 25-OH-PPD and 25-OCH3-PPD as experimental therapy for pancreatic cancer: Anticancer activity and mechanisms of action. Cancer Lett 2009; 278: 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Li W, Zhang X, Xin Y, Xuan Y, Liu J, Li P, Zhao Y. Oral subchronic toxicity evaluation of a novel antitumor agent 25-methoxydammarane-3,12,20-triol from Panax notoginseng in Sprague-Dawley rats. Regul Toxicol Pharmacol 2016; 77: 240–251. [DOI] [PubMed] [Google Scholar]
- [95].Zhou WX, Sun YY, Yuan WH, Zhao YQ. Water-soluble derivatives of 25-OCH3-PPD and their anti-proliferative activities. Steroids 2017; 121: 32–39. [DOI] [PubMed] [Google Scholar]
- [96].Loizzo MR, Menichini F, Tundis R. Recent Insights into the Emerging Role of Triterpenoids in Cancer Therapy: Part I Studies in Natural Products Chemistry, Vol. 40, Atta-ur-Rahman, Ed., Elsevier Amsterdam, Netherlands, 2013, p. 1–31. [Google Scholar]
- [97].Tung NH, Song GY, Van Minh C, Van Kiem P, Jin LG, Boo HJ, Kang HK, Kim YH. Steamed ginseng-leaf components enhance cytotoxic effects on human leukemia HL-60 cells. Chem Pharm Bull 2010; 58: 1111–1115. [DOI] [PubMed] [Google Scholar]
- [98].Kim JH, Kim M, Yun SM, Lee S, No JH, Suh DH, Kim K, Kim YB. Ginsenoside Rh2 induces apoptosis and inhibits epithelial-mesenchymal transition in HEC1A and Ishikawa endometrial cancer cells. Biomed Pharmacother 2017; 96: 871–876. [DOI] [PubMed] [Google Scholar]
- [99].Li KF, Kang CM, Yin XF, Li HX, Chen ZY, Li Y, Zhang Q, Qiu YR. Ginsenoside Rh2 inhibits human A172 glioma cell proliferation and induces cell cycle arrest status via modulating Aκt signaling pathway. Mol Med Rep 2018; 17: 3062–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ge G, Yan Y, Cai H. Ginsenoside Rh2 inhibited proliferation by inducing ROS mediated ER stress dependent apoptosis in lung cancer cells. Biol Pharm Bull 2017; 40: 2117–2124. [DOI] [PubMed] [Google Scholar]
- [101].Chen Y, Liu ZH, Xia J, Li XP, Li KQ, Xiong W, Li J, Chen DL. 20(S)-ginsenoside Rh2 inhibits the proliferation and induces the apoptosis of KG-1a cells through the Wnt/β-catenin signaling pathway. Oncol Rep 2016; 36: 137–146. [DOI] [PubMed] [Google Scholar]
- [102].Lv Q, Rong N, Liu LJ, Xu XL, Liu JT, Jin FX, Wang CM. Antitumoral activity of (20R)- and (20S)-ginsenoside Rh2 on transplanted hepatocellular carcinoma in mice. Planta Med 2016; 82: 705–711. [DOI] [PubMed] [Google Scholar]
- [103].Chen F, Sun Y, Zheng SL, Qin Y, McClements DJ, Hu JN, Deng ZY. Antitumor and immunomodulatory effects of ginsenoside Rh2 and its octyl ester derivative in H22 tumor-bearing mice. J Funct Foods 2017; 32: 382–390. [Google Scholar]
- [104].Li Q, Li B, Dong C, Wang Y, Li Q. 20(S)-Ginsenoside Rh2 suppresses proliferation and migration of hepatocellular carcinoma cells by targeting EZH2 to regulate CDKN2A-2B gene cluster transcription. Eur J Pharmacol 2017; 815: 173–180. [DOI] [PubMed] [Google Scholar]
- [105].Wang YS, Lin Y, Li H, Li Y, Song Z, Jin YH. The identification of molecular target of (20S)-ginsenoside Rh2 for its anti-cancer activity. Sci Rep 2017; 7: 12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wang M, Yan SJ, Zhang HT, Li N, Liu T, Zhang YL, Li XX, Ma Q, Qiu XC, Fan QY, Ma BA. Ginsenoside Rh2 enhances the antitumor immunological response of a melanoma mice model. Oncol Lett 2017; 13: 681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Achenbach H, Asunka SA, Weibel R, Addae-Mensah I, Oppong IV. Dichapetalin A, a novel plant constituent from Dichapetalum madagascariense with potential antineoplastic activity. Nat Prod Lett 1995; 7: 93–100. [Google Scholar]
- [108].Tuchinda P, Kornsakulkarn J, Pohmakotr M, Kongsaeree P, Prabpai S, Yoosook C, Kasisit J, Napaswad C, Sophasan S, Reutrakul V. Dichapetalin-type triterpenoids and lignans from the aerial parts of Phyllanthus acutissima. J Nat Prod 2008; 71: 655–663. [DOI] [PubMed] [Google Scholar]
- [109].Fang L, Ito A, Chai HB, Mi Q, Jones WP, Madulid DR, Oliveros MB, Gao Q, Orjala J, Farnsworth NR, Soejarto DD, Cordell GA, Swanson SM, Pezzuto JM, Kinghorn AD. Cytotoxic constituents from the stem bark of Dichapetalum gelonioides collected in the Philippines. J Nat Prod 2006; 69: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Ren Y, Yuan C, Deng Y, Kanagasabai R, Ninh TN, Tu VT, Chai HB, Soejarto DD, Fuchs JR, Yalowich JC, Yu J, Kinghorn AD. Cytotoxic and natural killer cell stimulatory constituents of Phyllanthus songboiensis. Phytochemistry 2015; 111: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Shanmugam MK, Dai X, Kumar AP, Tan BKH, Sethi G, Bishayee A. Ursolic acid in cancer prevention and treatment: Molecular targets, pharmacokinetics and clinical studies. Biochem Pharmacol 2013; 85: 1579–1587. [DOI] [PubMed] [Google Scholar]
- [112].Safe SH, Prather PL, Brents LK, Chadalapaka G, Jutooru I. Unifying mechanisms of action of the anticancer activities of triterpenoids and synthetic analogs. Anticancer Agents Med Chem 2012; 12: 1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ren Y, Yu J, Kinghorn AD. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr Med Chem 2016; 23: 2397–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]