

Abstract
Next‐generation sequencing (NGS) technology is currently used to establish mutational profiles in many heterogeneous diseases. The aim of this study was to evaluate the mutational spectrum in Taiwanese patients with colorectal cancer (CRC) to help clinicians identify the best treatment method. Whole‐exome sequencing was conducted in 32 surgical tumor tissues from patients with CRC. DNA libraries were generated using the Illumina TruSeq DNA Exome, and sequencing was performed on the Illumina NextSeq 500 system. Variants were annotated and compared to those obtained from publicly available databases. The analysis revealed frequent mutations in APC (59.38%), TP53 (50%), RAS (28.13%), FBXW7 (18.75%), RAF (9.38%), PIK3CA (9.38%), SMAD4 (9.38%), and SOX9 (9.38%). A mutation in TCF7L2 was also detected, but at lower frequencies. Two or more mutations were found in 22 (68.75%) samples.
The mutation rates for the WNT, P53, RTK‐RAS, TGF‐β, and PI3K pathways were 78.13%, 56.25%, 40.63%, 18.75%, and 15.63%, respectively. RTK‐RAS pathway mutations were correlated with tumor size (P = 0.028). We also discovered 23 novel mutations in NRAS, PIK3CA, SOX9, APC, SMAD4, MSH3, MSH4, PMS1 PMS2, AXIN2, ERBB2, PIK3R1, TGFBR2, and ATM that were not reported in the COSMIC, The Cancer Genome Atlas, and dbSNP databases. In summary, we report the mutational landscape of CRC in a Taiwanese population. NGS is a cost‐effective and time‐saving method, and we believe that NGS will help clinicians to treat CRC patients in the near future.
Keywords: colorectal cancer, gene mutation, next‐generation sequencing, pathway mutation
1. INTRODUCTION
Globally, colorectal cancer (CRC) is one of the most common human cancers and the fourth leading cause of cancer‐related death among males and females, with an estimated 1.4 million new cases and 694 000 deaths from the disease annually.1 In Taiwan, CRC ranked as the fourth leading cause of death, accounting for 14 965 cases diagnosed in 2012. CRC has increased significantly from 1990, with a growth rate of more than 2% per year worldwide. The likelihood of developing CRC is strongly correlated with old age, male gender, smoking, drinking alcohol, lack of exercise, being overweight, the consumption of red and/or processed meat, and a history of diabetes.2, 3
Epidermal growth factor receptor (EGFR) has been recognized as an effective anticancer target during the last few years. Monoclonal antibodies used to block EGFR in combination with chemotherapy or radiation have yielded improved outcomes in CRC patients with extended RAS wild‐type tumors. Mutations in the RAS and BRAF genes are harmful to anti‐EGFR therapy in metastatic CRC (mCRC).4 RAS and BRAF oncogene mutations are mutually exclusive and occur in 36.97% and 4.24% of CRC patients, respectively, as described in our previous work.5 Thus, identifying the unique genomic profiles and molecular phenotypes could help effectively establish the best treatment method in patients with anti‐EGFR therapy resistance.
CRC is one of the most interesting fields of next‐generation sequencing (NGS) application. The number of studies employing the NGS technique continues to increase. The Cancer Genome Atlas (TCGA) project studied more than 224 CRC cases and showed that 24 genes, including APC, TP53, SMAD4, PIK3CA, and KRAS, contained significant mutations. Three genes (ARID1A, SOX9, and FAM123B/WTX) were frequently mutated.6 Ashktorab et al analyzed 63 Iranian patients using targeted exome sequencing and found higher mutation rates of MSH3, MSH6, APC, and PIK3CA and hypothesized a larger role for these genes in CRC. They suggested the adoption of a specific informed genetic diagnostic protocol and tailored therapy in this population.7 Because patients with RAS wild‐type CRC can be non‐responders to EGFR‐targeted therapy, Geibler et al analyzed cell lines and tumor specimens to identify prediction markers by NGS, EGFR methylation and expression, and E‐cadherin expression. The authors revealed ATM mutations and low E‐cadherin expression as novel supportive predictive markers.8 Adua et al analyzed primary tumor and liver metastasis samples from 7 KRAS wild‐type patients and compared the genotypes of 22 genes associated with anti‐EGFR before and after chemotherapy. The results showed marked genotypic differences between pre‐ and post‐treatment samples, which were likely attributable to tumor cell clones selected by therapy.9 Gong et al analyzed 315 cancer‐related genes and introns of 28 frequently rearranged genes in 138 mCRC cases using FoundationOne. They identified a novel KRAS mutation (R68S) associated with an aggressive phenotype. The authors reported that ERBB2‐amplified tumors may benefit from anti‐HER2 therapy, and hypermutated tumors or tumors with high tumor mutational burden with MSI‐H or POLE mutation may benefit from anti‐PD‐1 therapy.10
This study examined genetic alterations in CRC in a Taiwanese population. We performed whole‐exome sequencing (WES) to detect the mutational status in all human protein‐coding genes using fresh frozen tissue from 32 Taiwanese patients with CRC.
2. MATERIALS AND METHODS
2.1. Study patients and tumor samples
This study was approved by the China Medical University Hospital Institutional Review Board. A summary of all patient characteristics is provided in Table 1. Patients ranged in age from 35 to 90 years, with a median age of 62 years. DNA was extracted using a QIAamp® DNA Micro Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer's instructions. Extracted DNA was immediately stored at −20°C until further processing. DNA concentration was measured by the Qubit dsDNA Assay Kit (Life Technologies, Carlsbad, CA, USA).
Table 1.
Clinical features of 32 colorectal cancer patients
| Characteristic | n (Frequency) |
|---|---|
| Age (years) | |
| Average: 60.47 | Range: 35‐90 |
| Sex | |
| Male | 20 |
| Female | 12 |
| Differentiation | |
| Low | 2 |
| Middle | 28 |
| Middle to Low | 2 |
| AJCC stage | |
| I | 4 |
| IIA | 15 |
| IIIB | 5 |
| IIIC | 2 |
| IVA | 1 |
| IVB | 4 |
| NA | 1 |
| Regional lymph node metastasis | |
| N0 | 19 |
| N1 | 4 |
| N2 | 7 |
| NA | 2 |
| Site | |
| Rectum | 8 |
| Colon | 24 |
2.2. WES and data analysis
DNA libraries were prepared using the Illumina TruSeq Exome Library Prep Kit and sequenced on the Illumina NextSeq 500 platform. Base calling and quality scoring were performed by an updated implementation of Real‐Time Analysis in NextSeq500. Bcl2fastq Conversion Software was used to demultiplex data and convert BCL files into FASTQ files. Sequenced reads were trimmed for low‐quality sequences and aligned to the human reference genome (hg 19) using Burrows‐Wheeler Alignment.11 Finally, single nucleotide polymorphisms and small insertion and deletion mutations were called in individual samples by the Genome Analysis Toolkit and VarScan using default settings.12, 13 We then performed ANNOVAR to functionally annotate genetic variants.14 The following criteria were used to select confident somatic single nucleotide variants: mutant allele frequency >5%, global minor allele frequency <1%, or NA (comparing the ExAC and 1000 Genome Databases data), eliminating known harmless variants present in ClinVar or the in‐house polymorphism database, and predicted to be pathogenic by all three software programs (SIFT, PolyPhen‐2, and CADD).
2.3. Statistical analysis
Comparisons between clinicopathological features and the status of critical pathway mutations in CRC were performed using Fisher's exact test. Two‐sided P‐values < 0.05 were considered statistically significant.
3. RESULTS
3.1. WES analysis and coverage
Using massive parallel sequencing on a NextSeq platform, we generated a mean of 157 M raw reads per sample, of which 141 M were aligned to the human reference genome (hg19; Table 2). The mean depth of the target regions for the 32 samples was 119× (range 34.79‐197.53×). The coverage of the target regions exceeded 97.97%. Figure 1 is an overview of our approach used to identifying variants.
Table 2.
Alignment and coverage statistics for 32 colorectal cancer patients
| Patient ID | Total raw reads | Total effective reads | Reads mapped to genome | Average sequencing depth on target | Coverage on target (%) |
|---|---|---|---|---|---|
| 16 | 82 864 708 | 66 661 376 | 66 657 979 | 47.14 | 98.44 |
| 25 | 69 110 948 | 56 550 852 | 56 544 621 | 40.16 | 98.00 |
| 36 | 68 965 280 | 56 546 730 | 56 539 081 | 38.51 | 98.10 |
| 50 | 356 294 022 | 326 553 966 | 326480947 | 188.98 | 99.01 |
| 56 | 75 141 654 | 60 803 428 | 60 794 779 | 43.41 | 97.97 |
| 62 | 71 243 396 | 58 278 776 | 58 270 698 | 40.07 | 98.12 |
| 71 | 70 086 092 | 57 388 574 | 57 381 580 | 41.24 | 98.05 |
| 89 | 63 437 554 | 50 916 024 | 50 913 067 | 37.87 | 98.35 |
| 93 | 59 001 856 | 47 743 596 | 47 736 346 | 34.79 | 98.03 |
| 98 | 269 310 102 | 24 7274 550 | 247 189 402 | 197.53 | 99.37 |
| 99 | 63 078 404 | 51 065 200 | 51 058 021 | 37.07 | 98.10 |
| 103 | 66 173 134 | 52 911 932 | 52 907 746 | 39.03 | 98.31 |
| CC01 | 202 308 880 | 182 302 644 | 182 249 487 | 148.28 | 98.93 |
| CC02 | 196 162 260 | 179 076 152 | 179036670 | 137.71 | 98.86 |
| CC03 | 149 966 094 | 138 301 996 | 138 263 040 | 124.07 | 99.16 |
| CC04 | 188 762 344 | 175 316 188 | 175 287 324 | 154.65 | 99.17 |
| CC05 | 174 170 480 | 161 466 102 | 161 439 317 | 143.79 | 98.92 |
| CC06 | 163 747 730 | 151 903 466 | 151 881 413 | 128.67 | 99.13 |
| CC07 | 180 821 438 | 167 186 452 | 167 155 256 | 133.86 | 99.00 |
| CC08 | 174 412 902 | 161 772 158 | 161 747 641 | 146.76 | 99.17 |
| CC10 | 178 559 504 | 160 173 326 | 160 136 434 | 148.31 | 99.15 |
| CC11 | 202 264 106 | 182 800 322 | 182 757 243 | 168.55 | 98.94 |
| CC12 | 203 133 950 | 183 665 658 | 183 629 660 | 164.16 | 98.92 |
| CC13 | 195 342 238 | 176 816 668 | 176 779 527 | 163.11 | 99.14 |
| CC14 | 215 392 940 | 192 504 740 | 192 468 467 | 176.64 | 98.96 |
| CC15 | 186 503 740 | 168 736 670 | 168 699 555 | 150.49 | 99.13 |
| CC16 | 188 775 628 | 173 447 948 | 173 418 659 | 160.24 | 99.20 |
| CC17 | 189 597 468 | 174 502 714 | 174 458 692 | 157.42 | 99.21 |
| CC18 | 179 218 892 | 164 454 320 | 164 426 639 | 153.55 | 98.96 |
| CC20 | 179 435 082 | 165 011 368 | 164 988 404 | 155.39 | 98.95 |
| CC21 | 195 883 102 | 179 886 726 | 179 858 958 | 168.34 | 99.21 |
| CC24 | 173 198 708 | 159 597 160 | 159 569 988 | 153.26 | 98.92 |
| Average | 157 261 395 | 141 613 056 | 141 585 208 | 119.47 | 98.78 |
Figure 1.
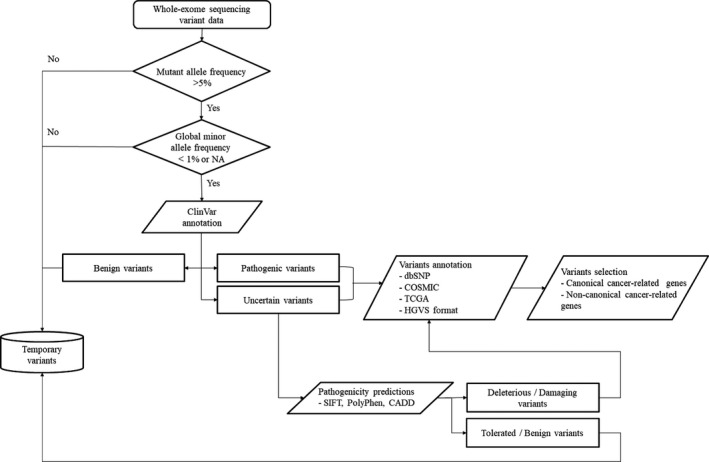
Overview of our approach used to identify variants
3.2. CRC‐associated oncogene variants
3.2.1. RAS mutations
Overall, RAS mutations were present in 28.13% of our CRC patients (Figure 2). The most common RAS mutations were KRAS mutations in exon 2 (codons 12 and 13), including G12V (44.44%), G12C (11.11%), and G13D (11.11%). Beyond the well‐established point mutations in codons 12 and 13 of exon 2 of KRAS, we identified mutations in codon 117 of exon 4 (K117N, 11.11%) and codon 146 of exon 4 (A146T, 11.11%). One mutation (11.11%) in codon 68 (exon 3) of NRAS was also detected; this was a novel alteration (R68I). The non‐synonymous variant at locus 115256508 had a C‐to‐A change mapped in the small GTP‐binding protein domain, with an allele fraction of 21.19% (total reads 118, variant count 25) (Figure S1A). Together, these non‐KRAS exon 2 mutations constituted 33.33% of all RAS mutations (Figure 3).
Figure 2.
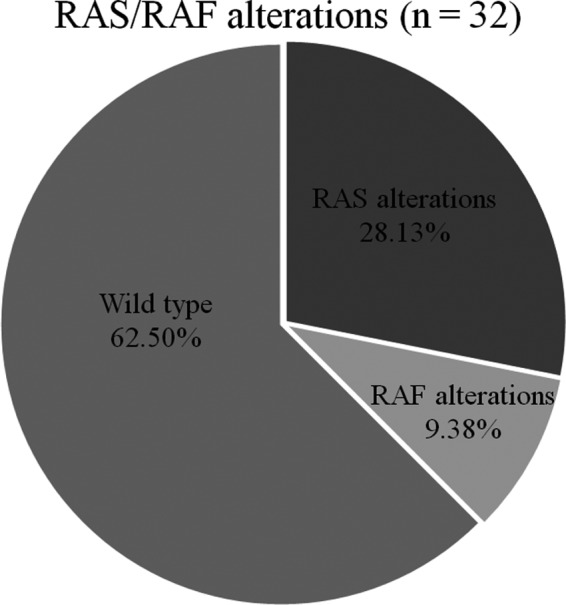
Proportion of RAS, RAF mutations, and RAS/RAF wild‐type status identified by WES. WES, whole‐exome sequencing
Figure 3.
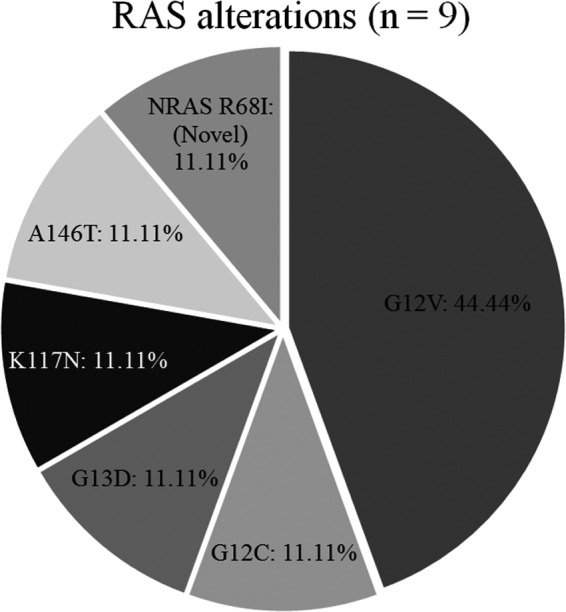
Proportion of RAS alterations identified by WES. WES, whole‐exome sequencing
3.2.2. RAF mutations
Two RAF mutations were found in 9.38% of our patients (Figure 2). Two patients (6.25%) had BRAF V600E mutations. One patient (3.13%) had an ARAF T256fs mutation. None of the CRC patients with RAS mutations harbored a concomitant mutation in RAF. The remaining patients (62.5%) were RAS/RAF wild‐type (Figure 2).
3.2.3. PIK3CA mutations
Three patients (9.38%) had PIK3CA mutation tumors. The mutation variants were R38S, G118D, and D350Y; D350Y was a novel mutation. The non‐synonymous variant at locus 178921566 had a G‐to‐T change mapped in the phosphatidylinositol 3‐kinase, C2 domain, with an allele fraction of 17.53% (total reads 97, variant count 17) (Figure S1B).
3.2.4. TCF7L2 mutations
Two patients (6.25%) had TCF7L2 mutation tumors. The identified variants were R471C, F357L, and G424E, and each patient had two of the three TCF7L2 variants.
3.2.5. SOX9 mutations
Three patients (9.38%) had SOX9 frameshift mutations. One patient had an S431fs mutation, another a G484fs mutation, and the third an S485fs mutation. The G484fs and S485fs mutations were novel variants (Figure S1C).
3.3. CRC‐associated tumor suppressor gene variants
3.3.1. APC mutations
In total, we identified 19 patients (59.38%) with APC alterations. A total of 26 APC mutations were identified in the 19 samples, most of which were nonsense mutations that introduced a premature stop codon (R283*, S320*, Q541*, R564*, R876*, R1114*, Q1294*, E1309*, Q1367*, Q1378*, R1450*, E1544*, Q1916*, and R2204*). Six variants were frameshift deletions (L620fs, D1297fs, E1306fs, G1312fs, E1374fs, and E1397fs), 5 were frameshift insertions (L540fs, L852fs, T1292fs, L1302fs, and E1554fs,), and 1 was a missense mutation (S1400L). Among these mutations, 7 novel mutations were found (L540fs, T1292fs, D1297fs, L1302fs, E1306fs, E1374fs, and Q1916*) (Figure S1D).
3.3.2. TP53 mutations
Overall, TP53 mutations were present in 50% of our CRC patients. Fifteen TP53 mutations were identified in the 16 samples. All variants have been reported (L43fs, K132N, P151S, R175H, C176F, R196*, L206*, M237I, R245C, M246R, E258K, R273H, R273C, R282W, and R306*).
3.3.3. FBXW7 mutations
Six of the 32 samples (18.75%) had a mutation in FBXW7. Four FBXW7 variants were found in the 6 samples. All variants have been reported (G80W, W307C, R347H, and R387C).
3.3.4. SMAD4 mutations
Three patients (9.38%) had SMAD4 mutations. Two variants have been reported previously (G419R and R496H), and the other was novel (Y260_H261delins*). The frameshift variant at locus 48584605 had an A insertion with an allele fraction of 22.18% (total reads 284, variant count 63) (Figure S1E).
3.4. Mismatch repair (MMR) gene variants
3.4.1. MLH1, MSH3, MSH4, PMS1, and PMS2 mutations
Five patients (15.63%) had mismatch repair (MMR) gene mutations. Mutations in the MMR gene included MLH1, MSH3, MSH4, PMS1, and PMS2. The mutation variants were R385C and T117M in MLH1, A61delinsAAPA and E456K in MSH3, E583* in MSH4, R265Q in PMS1, and L633I in PMS2. Among these, MSH3 A61delinsAAPA and E456K, MSH4 E583*, PMS1 R265Q, and PMS2 L633I were novel mutations (Figure S1F‐I). The numbers of variants discovered in the MMR wild‐type and mutation carriers are listed in Tables S1 and S2.
3.5. Altered signaling pathways in CRC
Based on our analytical approach, we identified multiple genes in the RTK‐RAS, PI3K, TGF‐β, WNT, and P53 pathways. The APC gene in the WNT pathway had relatively high levels of somatic mutations compared to genes in the RTK‐RAS, PI3K, TGF‐β, and P53 pathways. We found 10 different altered WNT pathway genes, including LRP5, FZD10, APC, AXIN2, FAM123B, CTNNB1, TCF7L2, SOX9, FBXW7, and ARID1A, confirming the importance of this pathway in CRC. We found that 78.13% of tumors had alterations in the WNT pathway. We also evaluated genetic alterations in the RTK‐RAS, PI3K, TGF‐β, and P53 pathways, with mutation rates of 40.63%, 15.63%, 18.75%, and 56.25%, respectively (Figure 4).
Figure 4.
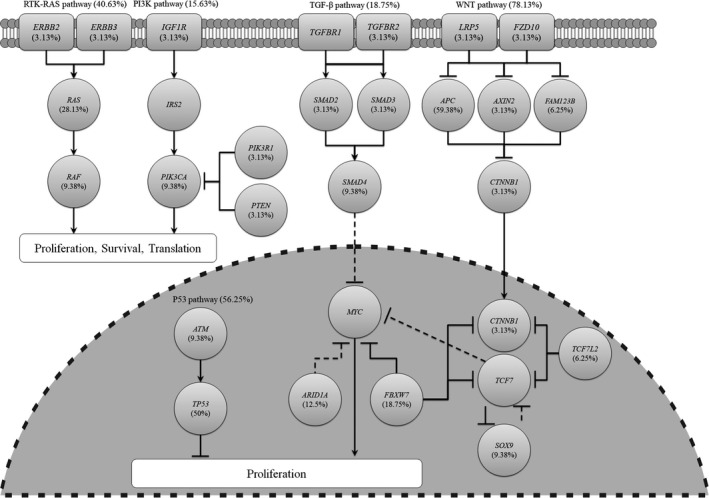
Frequency of genetic changes leading to deregulation of signaling pathways in CRC. CRC, colorectal cancer
3.6. Pathway mutations and associations
We compared the clinicopathological data of CRC patients with mutations in mutation‐related pathways. The RTK‐RAS pathway mutation rate was significantly higher in patients with a tumor size ≤4 cm compared to those with a tumor of >4 cm (57.89% versus 15.38%, P = 0.028). No clinicopathological variables were significantly correlated with WNT, PI3K, TGF‐β, or P53 pathway mutations (Table 3).
Table 3.
Correlation between clinicopathological features and mutational status
| Mutation of WNT pathway | Mutation of RTK‐RAS pathway | Mutation of PI3K pathway | Mutation of TGF‐β pathway | Mutation of P53 pathway | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Yes | Total | P‐Value | No | Yes | Total | P‐Value | No | Yes | Total | P‐Value | No | Yes | Total | P‐Value | No | Yes | Total | P‐Value | ||
| Gender | F | 3 | 9 | 12 | 1.000 | 7 | 5 | 12 | 1.000 | 10 | 2 | 12 | 1.000 | 11 | 1 | 12 | 0.370 | 4 | 8 | 12 | 0.471 |
| M | 4 | 16 | 20 | 12 | 8 | 20 | 17 | 3 | 20 | 15 | 5 | 20 | 10 | 10 | 20 | ||||||
| Age | <62 | 3 | 13 | 16 | 1.000 | 10 | 6 | 16 | 1.000 | 15 | 1 | 16 | 0.333 | 13 | 3 | 16 | 1.000 | 5 | 11 | 16 | 0.285 |
| ≥62 | 4 | 12 | 16 | 9 | 7 | 16 | 12 | 4 | 16 | 13 | 3 | 16 | 9 | 7 | 16 | ||||||
| Tumor Size | ≤4 cm | 3 | 16 | 19 | 0.401 | 8 | 11 | 19 | 0.028 | 17 | 2 | 19 | 0.375 | 15 | 4 | 19 | 1.000 | 10 | 9 | 19 | 0.289 |
| >4 cm | 4 | 9 | 13 | 11 | 2 | 13 | 10 | 3 | 13 | 11 | 2 | 13 | 4 | 9 | 13 | ||||||
| Stage | I, II | 3 | 16 | 19 | 0.384 | 12 | 7 | 19 | 1.000 | 15 | 4 | 19 | 0.624 | 16 | 3 | 19 | 0.653 | 7 | 12 | 19 | 0.710 |
| III, IV | 4 | 8 | 12 | 7 | 5 | 12 | 11 | 1 | 12 | 9 | 3 | 12 | 6 | 6 | 12 | ||||||
| Site | Rectum | 3 | 5 | 8 | 0.327 | 5 | 3 | 8 | 1.000 | 6 | 2 | 8 | 0.578 | 8 | 0 | 8 | 0.296 | 4 | 4 | 8 | 0.704 |
| Colon | 4 | 20 | 24 | 14 | 10 | 24 | 21 | 3 | 24 | 18 | 6 | 24 | 10 | 14 | 24 | ||||||
| LN metastasis | − | 3 | 16 | 19 | 0.401 | 12 | 7 | 19 | 0.720 | 15 | 4 | 19 | 0.625 | 16 | 3 | 19 | 0.666 | 7 | 12 | 19 | 0.473 |
| + | 4 | 9 | 13 | 7 | 6 | 13 | 12 | 1 | 13 | 10 | 3 | 13 | 7 | 6 | 13 | ||||||
P‐Value by Fisher's Exact Test.
4. DISCUSSION
All of the mutated genes discussed in our study have been previously classified as driver genes that confer a selective growth advantage to tumor cells harboring the mutations. CRC is similar to other cancers with only one or multiple driver gene mutations. Tumors with only one driver mutation, always in an oncogene, and with multiple driver mutations contain a combination of oncogene and tumor suppressor gene mutations.15 In our study, of the 4 samples with a single mutation (Table 4), 1 (25%) harbored a mutation in an oncogene (KRAS), and of the 22 samples with 2 or more mutations (Tables 5 and 6), 15 (68.18%) contained a combination of mutations in both oncogenes and tumor suppressor genes.
Table 4.
Single point mutations detected in 32 colorectal cancer samples
| Genes | Mutation | Sex | Age (years) | Differentiation | AJCC stage |
|---|---|---|---|---|---|
| TP53 | p.K132N | F | 57 | Middle | IVB |
| APC | p.Q1294* | M | 57 | Middle | IIIB |
| MSH3 | p.A61delinsAAPA | M | 61 | Middle | IIA |
| KRAS | p.G12C | F | 69 | Middle | IIA |
Table 5.
Double combination mutations detected in 32 colorectal cancer samples
| Gene 1 | Mutation 1 | Gene 2 | Mutation 2 | Sex | Age (years) | Differentiation | AJCC stage |
|---|---|---|---|---|---|---|---|
| ARAF | p.T256fs | FBXW7 | p.W307C | M | 65 | Middle to Low | NA |
| APC | p.Q1916* | MLH1 | p.T117M | M | 72 | Middle | IIA |
| KRAS | p.G12V | TP53 | p.C176F | F | 57 | Middle | IIIB |
| APC | p.Q1367* | TP53 | p.R282W | M | 61 | Middle | IIIB |
| SOX9 | p.G485fs | APC | p.R283* | F | 78 | Middle | IIA |
| APC |
p.L540fs p.R1450* |
TP53 | p.L43fs | M | 35 | Middle | IIA |
| KRAS | p.G12V | APC |
p.R564* p.L1302fs |
M | 68 | Middle | IIA |
| APC | p.Q541* | TP53 | p.M246R | F | 58 | Middle | IIA |
| APC |
p.L620fs p.E1306fs |
TP53 | p.L206* | F | 47 | Middle | I |
| APC |
p.S320* p.E1544* |
FBXW7 | p.R387C | F | 42 | Middle | IIIB |
Table 6.
Three or more combination mutations detected in 32 colorectal cancer samples
| Gene 1 | Mut. 1 | Gene 2 | Mut. 2 | Gene 3 | Mut. 3 | Gene 4 | Mut. 4 | Gene 5 | Mut.5 | Gene 6 | Mut. 6 | Gene 7 | Mut. 7 | Sex | Age | Diff | AJCC stage |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BRAF | p.V600E | TP53 | p.E258K | FBXW7 | p.G80W | M | 55 | Low | IVB | ||||||||
| KRAS | p.G12V | APC | p.E1554fs | TP53 | p.R175H | F | 63 | M | IIA | ||||||||
| SOX9 | p.S431fs | APC |
p.L852fs p.T1292fs |
TP53 | p.R196* | M | 63 | M | |||||||||
| KRAS | p.G13D | APC | p.Q1378* | FBXW7 | p.R347H | M | 63 | M | I | ||||||||
| APC | p.E1374fs | TP53 | p.R273C | SMAD4 | p.R496H | M | 68 | M | IIA | ||||||||
| KRAS | p.A146T | APC | p.G1312fs | TP53 | p.P151S | MLH1 | p.R385C | F | 45 | M | IIA | ||||||
| NRAS | p.R68I | APC | p.E1397fs | TP53 |
p.R245C p.R282W |
FBXW7 | p.R347H | M | 48 | M | I | ||||||
| PIK3CA | P.G118D | APC | p.D1297fs | FBXW7 | p.R387C | TP53 | p.R273H | F | 67 | M | IIA | ||||||
| KRAS | p.G12V | TP53 | p.R273C | SMAD4 | p.G419R | SOX9 | p.S484fs | F | 64 | L | IIA | ||||||
| BRAF | p.V600E | APC |
p.Q1294* p.E1554fs |
TP53 | p.M237I | SMAD4 | p.Y260_H261delins* | M | 44 | M | IVB | ||||||
| KRAS | p.K117N | PIK3CA | p.R38S | TCF7L2 |
p.R471C p.G424E |
APC |
p.R876* p.E1309* p.R2204* |
MSH4 | p.E583* | M | 71 | M to L | IIIC | ||||
| PIK3CA | p.D350Y | TCF7L2 |
p.F357L p.R471C |
APC |
p.R1114* p.Q1378* p.S1400L |
TP53 | p.R306* | MSH3 | p.E456K | PMS1 | p.R265Q | PMS2 | p.L633I | M | 50 | M | IIA |
The integrative analysis of WES data provides insights into pathways that are dysregulated in CRC. The WNT signaling pathway was dysregulated in 78.13% of cases. WNT pathway mutations have been reported in 84.5%% of CRC cases, which is higher than the mutation rate detected in our study.16 In 2012, the TCGA consortium reported that up to 93% of CRC cases involved at least 1 alteration in a known WNT regulator.6 Hyperactivation of the WNT pathway initiates the development of CRC, which predominantly occurs through inactivation of the APC gene.17 Several agents have been investigated to target this pathway, including WNT inhibitors (eg, Rofecoxib, PRI‐724, CWP232291) and a monoclonal antibody against frizzled receptors (e.g., vanituctumab).18 In addition to APC and SOX9, we also identified a novel mutation in AXIN2 (p.R459L) (Figure S1J). The AXIN2 mutation identified in the current study, R459, is located in the region that interacts with β‐catenin.
The frequency of alterations in the RTK‐RAS and PI3K pathways was 40.63% and 15.63%, respectively. RTK‐RAS and PI3K pathway mutations have been found in 60.7% and 30% of CRCs, respectively.16 In a normal cell, RTK‐RAS and PI3K pathways control cell proliferation, differentiation, and survival.19, 20 In a malignant cell, constitutive and aberrant activation of components of these pathways lead to increased cell growth, survival, and metastasis. Small molecule inhibitors, such as Sorafenib and PLX4720, which are currently being used to target BRAF p.V600E, have been developed to target the RTK‐RAS and PI3K pathways. NVP‐BEZ235 and BGT226 are being used to target the PI3K pathway in various cancers.21 In addition to NRAS and PIK3CA, we identified two novel mutations in ERBB2 (p.W9fs) and PIK3R1 (p.S147* and p.L161*) (Figure S1K,L). The PIK3R1 p.S147* and p.L161* mutations were mapped to the Rho GTPase‐activating protein domain.
In our study population, the mutation rate of the TGF‐β and P53 pathways was 18.75% and 56.25%, respectively. TGF‐β and P53 pathway mutations have been described in 28.9% and 69% of CRCs, respectively.16 The TGF‐β signaling pathway has pleiotropic functions, including the regulation of cell growth, apoptosis, cell motility, and invasion. TGF‐β signaling plays a key role in tumor initiation, development, and metastasis. Many TGF‐β pathway inhibitors, such as antisense oligonucleotides, neutralizing antibodies, and receptor kinase inhibitors, have been used in preclinical trials. For example, galunisertib is a TGFβR1 inhibitor that prevents signal transduction.22 Under cellular stress, such as DNA damage, oncogenes, oxidative free radicals, and UV irradiation, the P53 protein is activated. Activation of P53 can induce cell cycle arrest, senescence, and apoptosis. Small molecular inhibitors, such as MIs, nutlins, and RITA, have been tested as therapeutic agents in CRC by activating this pathway.23 In addition to SMAD4, we identified a novel mutation in TGFBR2 (p.D549A) and ATM (p.E650*) (Figure S1M,N). Our relatively low rate of mutations in these 5 critical pathways may reflect our small sample size.
Most CRC samples can be grouped by WNT‐, RTK‐RAS‐, P53‐, TGF‐β‐, and PI3K‐dysregulated pathways. In our study population, 3 samples (3/32, 9.38%) had no mutation in any of these pathways. However, in these 3 samples, 2 had alterations in the Notch signaling pathway (CTBP2, CREBBP, KAT2B, DVL2, and PSEN2). Deregulation of Notch signaling in CRC has been reported.24 The third sample exhibited alterations in cell adhesion molecules (CNTN2, HLA‐DRB1, HLA‐DRB5, and NRXN3). This indicates that it may be necessary to identify other dysregulated pathways to achieve therapeutic benefits.
We also compared the clinicopathological data of CRC patients with the mutational status of important signaling pathways in cancerous tissues. RTK‐RAS pathway mutations were correlated with tumor size (P = 0.028). These results suggest that tumor progression is not linked to increased genetic instability, although this may be due to our small sample size and fact that most cases were stage II (48.39% cases); we need to collect more samples to confirm our results.
In conclusion, we identified recurrent mutations in genes such as APC, TP53, KRAS, and FBXW7, as well as unreported mutations in NRAS, PIK3CA, SOX9, APC, SMAD4, MSH3, MSH4, PMS1 PMS2, AXIN2, ERBB2, PIK3R1, TGFBR2, and ATM in a group of Taiwanese CRC patients. The data presented herein provide more comprehensive characteristics of the top deadly disease and identify a possibility for treating it in a targeted way.
Supporting information
ACKNOWLEDGMENTS
This work has been support by China Medical University Hospital grant (DMR106‐105).
Chang Y‐S, Lee C‐C, Ke T‐W, et al. Molecular characterization of colorectal cancer using whole‐exome sequencing in a Taiwanese population. Cancer Med. 2019;8:3738–3747. 10.1002/cam4.2282
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA: Cancer J Clin. 2015;65(2):87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383(9927):1490‐1502. [DOI] [PubMed] [Google Scholar]
- 3. Strum WB. Colorectal Adenomas. N Engl J Med. 2016;374(11):1065‐1075. [DOI] [PubMed] [Google Scholar]
- 4. Gong J, Cho M, Fakih M. RAS and BRAF in metastatic colorectal cancer management. J Gastrointest Oncol. 2016;7(5):687‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang YS, Chang SJ, Yeh KT, Lin TH, Chang JG. RAS, BRAF, and TP53 gene mutations in Taiwanese colorectal cancer patients. Onkologie. 2013;36(12):719‐724. [DOI] [PubMed] [Google Scholar]
- 6. Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ashktorab H, Mokarram P, Azimi H, et al. Targeted exome sequencing reveals distinct pathogenic variants in Iranians with colorectal cancer. Oncotarget. 2017;8(5):7852‐7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Geißler A‐L, Geißler M, Kottmann D, et al. ATM mutations and E‐cadherin expression define sensitivity to EGFR‐targeted therapy in colorectal cancer. Oncotarget. 2017;8(10):17164‐17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adua D, Di Fabio F, Ercolani G, et al. Heterogeneity in the colorectal primary tumor and the synchronous resected liver metastases prior to and after treatment with an anti‐EGFR monoclonal antibody. Mol Clin Oncol. 2017;7(1):113‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gong J, Cho M, Sy M, Salgia R, Fakih M. Molecular profiling of metastatic colorectal tumors using next‐generation sequencing: a single‐institution experience. Oncotarget. 2017;8(26):42198‐42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee D‐W, Han S‐W, Cha Y, et al. Association between mutations of critical pathway genes and survival outcomes according to the tumor location in colorectal cancer. Cancer. 2017;123(18):3513‐3523. [DOI] [PubMed] [Google Scholar]
- 17. Schatoff EM, Leach BI, Dow LE. WNT signaling and colorectal cancer. Curr Colorectal Cancer Rep. 2017;13(2):101‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bahrami A, Amerizadeh F, ShahidSales S, et al. Therapeutic potential of targeting Wnt/β‐catenin pathway in treatment of colorectal cancer: rational and progress. J Cell Biochem. 2017;118(8):1979‐1983. [DOI] [PubMed] [Google Scholar]
- 19. Nandan MO, Yang VW. An update on the biology of RAS/RAF mutations in colorectal cancer. Curr Colorectal Cancer Rep. 2011;7(2):113‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zenonos K, Kyprianou K. RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol. 2013;5(5):97‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Regad T, Targeting R. Signaling pathways in cancer. Cancers. 2015;7(3):1758‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neuzillet C, Tijeras‐Raballand A, Cohen R, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22‐31. [DOI] [PubMed] [Google Scholar]
- 23. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer—molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015;21(1):84‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vinson KE, George DC, Fender AW, Bertrand FE, Sigounas G. The Notch pathway in colorectal cancer. Int J Cancer. 2016;138(8):1835‐1842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials