

Abstract
High‐grade serous ovarian carcinoma (HGSOC) is a major form of ovarian epithelial tumor that is often diagnosed only at an advanced stage when it is already highly aggressive. We performed comprehensive genomic profiling using an analytically validated clinical next‐generation sequencing assay to identify genomic alterations in 450 cancer‐related genes in a cohort of 88 Chinese HGSOC patients. Overall, we detected 547 genomic alterations with an average of 6.2 alterations per tumor. Most of these HGSOC tumors had low tumor mutation burden and were microsatellite stable. Consistent with earlier studies, TP53 mutations were present in the majority (96.6%) of the tumors studied, and mutations in BRCA1/2 that affect DNA repair were also detected frequently in 20.5% of the tumors. However, we observed a 10.2% of mutated genes in the Ras/Raf pathway, all co‐occurring with TP53 mutations in the same tumor, which was unrecognized previously. Our results show that in HGSOC patients, there may be an unrecognized co‐occurrence of TP53 mutations with mutations in Ras/Raf pathway.
Keywords: comprehensive genomic profiling, high‐grade serous ovarian carcinoma, KRAS, TP53
1. INTRODUCTION
Ovarian epithelial tumor is one of the leading causes of cancer‐related deaths in women both in Asia and worldwide.1 Understanding the genomic alterations in HGSOC is critical in improving current therapeutic strategies for HGSOC patients. Several studies had been conducted to better understand the molecular characteristics of HGSOC. Analyses by the Cancer Genome Atlas2 as well as other groups using smaller cohorts3, 4, 5, 6 have repeatedly reported that almost all HGSOC patients harbor mutations in TP53, which are presumed to happen during early tumorigenesis3 and are a defining feature of HGSOC.7 In addition, a smaller proportion of patients also harbor deleterious somatic or germline BRCA1/2 mutations.2 On the contrary, low‐grade serous ovarian cancer is characterized by high prevalence of KRAS and BRAF mutations, and low occurrence of TP53 mutations.8 Thus, TP53 mutation is a necessary condition for HGSOC, while KRAS mutation is generally accepted as a feature of low‐grade serous ovarian cancer.
In this study, we performed comprehensive genomic profiling using an analytically validated clinical next‐generation sequencing (NGS) assay to identify genomic alterations in 450 cancer‐related genes in a cohort of 88 Chinese high‐grade serous ovarian carcinomas, with the aim to better understand the genomic alterations in Chinese HGSOC patients and identify potential opportunities for precision therapy. We show that most of the Chinese HGSOC cases in this cohort also had TP53 mutations, as reported elsewhere.9 To our surprise, we detected that 9 of the 88 (10.2%) Chinese HGSOC tumors had co‐occurring mutations in both TP53 and genes in the Ras/Raf pathway, which was not recognized previously. Preliminary results showed that the co‐occurrence of TP53 and KRAS may more likely to happen in HGSOC patients with endometrial cyst.
2. MATERIALS AND METHODS
2.1. Study samples
Clinical formalin‐fixed, paraffin‐embedded (FFPE) tumor tissue and matched normal tissue (68 blood and 20 paracancerous tissue) were collected from 88 Chinese HGSOC patients. Of the 88 cases, 39 were randomly extracted from the database of the Department of Pathology, Obstetrics & Gynecology Hospital of Fudan University between 2017 and 2018. Of the 88 cases, 18 were collected from Shengjing Hospital of China Medical University, Shenyang, China. These samples were hospital‐based HGSOC patients enrolled during June 2018 to August 2018. The other 31 samples were randomly collected from 22 hospitals in China from 2017 to 2018. All selected cases were informed, and a written informed consent of the patient was received according to the protocols and procedures approved by the Institutional Review Board. All cases were reviewed and confirmed by at least two independent senior pathologists according to the newest edition of WHO classification.10 Immunohistochemistry analysis of p53 protein was performed in all cases. DNA was extracted, and ultra‐deep NGS was performed on hybridization‐captured libraries of 450 cancer genes in a College of American Pathologists (CAP)‐certified laboratory to detect all classes of somatic genomic alterations including substitutions, short and long indels, copy number alterations, and gene rearrangements.
2.2. Next‐generation sequencing
Genomic profiling was performed in the laboratory of OrigiMed (Shanghai, China) using the Yuan Su 450 assay. At least 50 ng of cancer tissue DNA was extracted from each 40‐mm3 FFPE tumor sample using a DNA Extraction Kit (QIAamp DNA FFPE Tissue Kit) according to manufacturer's protocols. All coding exons of 450 key cancer‐related genes and selected introns of 39 genes commonly rearranged in solid tumors were captured by a custom hybridization capture panel. In addition, the probe density was increased to ensure high efficiency of capture in regions with low read depth. Libraries were each diluted to 1.05 nmol L−1 and then sequenced with a mean coverage of 900× for FFPE samples and 300× for matched blood or paracancerous samples on an Illumina NextSeq‐500 Platform.
2.3. Bioinformatics analysis
Reads were aligned to human reference genome hg19 using BWA.11 Single nucleotide variants (SNVs) and short indels were called by MuTect12 following deduplication, base quality recalibration, and local realignment using GATK13 and in‐house pipeline. Short indels were further calibrated using Pindel.14 Copy number variations (CNVs) were called using customized algorithms from the log‐ratio per gene region after normalizing read depths within target regions by EXCAVATOR.15 A customized algorithm was used to estimate tumor cellularity based on allele frequencies of the sequenced single‐nucleotide polymorphisms (SNPs), and detect gene rearrangements, fusions, and long indels. Reliable somatic alterations were detected by comparison with matched normal samples. At least 5 reads were required to support alternative calling. For CNVs, focal CNVs were characterized as genes with ≥ 5 copies for amplification and 0 copies for homozygous deletion. Clinically relevant genomic alterations were further marked as druggable genomic alterations if they match current treatments or clinical trials.
Tumor mutation burden (TMB) was estimated by counting somatic mutations including coding base substitutions and indels per megabase (Mb) of the sequence examined. Known cancer driver mutations and germline alterations in dbSNP were excluded from the TMB calculation. MSI status was inferred based on MANTIS16 score, and microsatellite regions were manually reviewed in Integrated Genomics Viewer (IGV)17 for confirmation.
3. RESULTS
3.1. Patient demographics
Overall, 88 Chinese HGSOC patients were included in this study (Table 1). The morphological features of all cases conformed to typical morphology of HGSOC, composed of solid masses of cells with slit‐like spaces. Papillary, glandular, or cribriform areas can also be seen in some cases. The tumor cells were severely atypical and had an aberrant p53 phenotype, as demonstrated by either diffuse, strong immunostaining or totally negative for p53. Of the 88 cases, 4 were with endometriotic cysts in the same or the opposite ovary. The median age at the time of sequencing was 53 years (range 29‐82 years). Out of 88 samples, 14 harbor germline BRCA1/BRCA2 mutations. The BRCA germline mutation carriers were significantly younger than the non‐carriers (t test P value = 1.8e‐7), indicating strong genetic predisposition. Most patients were at stage III (n = 36, 40.9%) or IV (n = 31, 35.2%) of the disease. Of the 88 (86.4%) samples, 76 were from the original primary tumor, and 11 (12.5%) were from metastatic tumor sites including liver (n = 4, 4.5%), abdominal wall (n = 3, 3.4%), lymph node (n = 2, 2.3%), pelvic wall (n = 1, 1.1%), and intestine (n = 1, 1.1%). The samples taken from tumor metastatic sites were taken at the time of recurrence. Out of 88 patients, 14 received neoadjuvant chemotherapy before surgery.
Table 1.
Patient demographics
| Item | Description |
|---|---|
| Number of patients | 88 |
| Age, median (range) | 53 (range 29‐82) |
| Stage | I: 5 (5.7%) |
| II: 8 (9.1%) | |
| III: 36 (40.9%) | |
| IV: 31 (35.2%) | |
| Unknown: 8 (9.1%) | |
| Site | Primary: 76 (86.4%) |
| Metastatic: 11 (12.5%), including liver (4), abdominal wall (3), lymph node (2), pelvic wall (1), and intestine (1) | |
| Unknown: 1 (1.1%) | |
| Tumor mutation burden, average (range) | 5.2/Mb (0.8‐39/Mb) |
| Microsatellite status | MSI‐high: 3 |
| MSS: 85 | |
| Family history | Of 48 patients with data available, 14 (29.2%) have known family history of cancer |
| Genomic alterations, total and average (range) | 547, average 6.2 per sample (range 1‐15) |
| Actionable genomic alterations | 56 (10.2% of all mutations), in 40 tumors (45.4% of all tumors) |
3.2. Genomic alterations identified in the Chinese HGSOC cohort
A total of 547 genomic alterations (including 529 somatic alterations and 18 germline alterations of BRCA1/2) were detected in these 88 HGSOC tumors, with an average of 6.2 alterations per tumor (range 1‐15; Figure 1). The most frequently mutated gene in this cohort was TP53, which was mutated in 85 of the 88 (96.7%) samples. BRCA1/2 were mutated in 18 of the 88 (20.5%) samples, and more BRCA1 (n = 16 mutations in 15 [17.0%] patients) than BRCA2 (n = 3, 3.4%) mutations were observed. One patient (Pt14) harbored both somatic and germline BRCA1 mutations. Most BRCA1/2 mutations were truncations, and 14 of the 19 (73.7%) BRCA1/2 mutations were identified from the germline samples. Amplifications in PTK2, which encodes focal adhesion kinase (FAK), also had high prevalence in this cohort, as it was detected in 16 of the 88 (18.2%) samples. Other frequently mutated genes in this cohort included NF1 (n = 15, 17.0%), FAM135B (n = 12, 13.6%), RB1 (n = 10, 11.4%), PIK3CA (n = 9, 10.2%), MYC (n = 8, 9.1%), PRKCI (n = 8, 9.1%), and KRAS (n = 6, 6.8%), TERT (n = 6, 6.8%), CCNE1 (n = 6, 6.8%), KMT2C (n = 6, 6.8%), and PTEN (n = 5, 5.7%). This was followed by a long tail of other genes that were altered less frequently in the cohort. The full details of mutations occurred in three or more patients in this cohort were shown in Figure 1.
Figure 1.
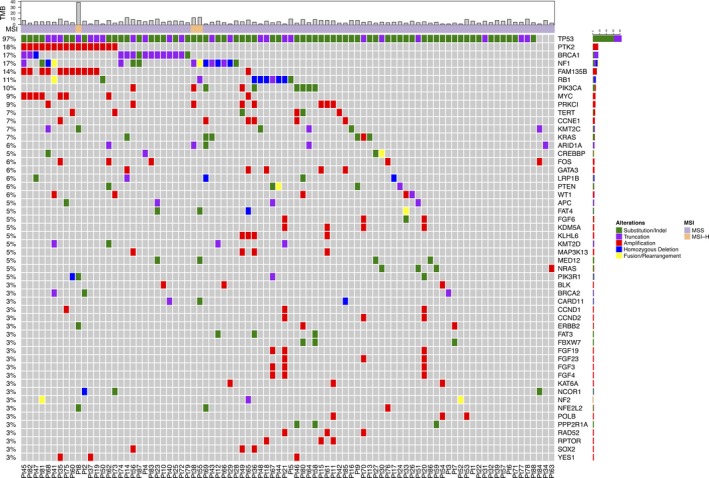
Tile plot of the genomic alterations identified in three or more tumors in this Chinese HGSOC cohort
Tumors in this cohort had an average tumor mutation burden (TMB) of 5.2 mutations per megabase (Mb; range 0‐39 mutations/Mb; see Methods). Most tumors had low tumor mutation burden, and only 4 of the 88 tumors (4.5%) had more than 10 mutations/Mb. Of the 88 (3.4%) tumors, all with TMB higher than 10 mutations/Mb, 3 were tested to be microsatellite instability‐high (MSI‐H), and the remaining 85 tumors (96.6%) were microsatellite stable (MSS; see Methods). Details of the TMB and microsatellite status for each tumor were shown in Figure 1.
3.3. Co‐occurrence of point mutations in the Ras/Raf pathway
Point mutations in KRAS were identified in 5 of the 88 (5.7%) tumors in this cohort, including 4 hotspot KRAS mutations at amino acid 12 (2 G12V, 1 G12A, and 1 G12D), and 1 less common KRAS A59G mutation. Notably, all of these cases had co‐occurring TP53 mutations in the same tumor (Table 2). Pathological review confirmed that all five cases were HGSOC (Figure 2). In addition to KRAS, other notable point mutations in the Ras/Raf pathway in this cohort included three NRAS mutations (1 Q61R, 1 Q61K, and 1 G12C) and one BRAF mutation (D594N). All these tumors also had a co‐occurring TP53 mutation (Table 2). Combined, 9 of the 88 tumors (10.2%) in this Chinese HGSOC cohort had point mutations in the Ras/Raf pathway that co‐occur with TP53 mutations. All these nine co‐occurrences were validated by Sanger sequencing or ddPCR (Supplementary Material Chromas.pdf and ddPCR.pdf). This is the first time that the co‐occurrence of mutated TP53 and KRAS was reported in HGSOC patients.
Table 2.
Co‐occurring point mutations in genes in the Ras/Raf pathway with TP53 mutations
| Individual ID | TP53 mutation | VAF (TP53) | KRAS mutation | VAF (KRAS) | NRAS mutation | VAF (NRAS) | BRAF mutation | VAF (BRAF) |
|---|---|---|---|---|---|---|---|---|
| Pt9 | c.782 + 1G > A | 0.44 | p.G12V | 0.50 | ||||
| Pt13 | p.R248W | 0.06 | p.G12D | 0.01 | ||||
| p.Y205C | 0.01 | |||||||
| Pt14 | p.M243_G244del | 0.43 | p.A59G | 0.17 | ||||
| Pt43 | P.N239* | 0.26 | p.G12V | 0.42 | ||||
| Pt69 | p.C277F | 0.57 | p.G12A | 0.34 | ||||
| Pt30 | p.P64Afs*85 | 0.41 | p.Q61R | 0.42 | ||||
| Pt51 | p.R342* | 0.15 | p.G12C | 0.12 | ||||
| Pt59 | p.R273H | 0.87 | p.Q61K | 0.41 | ||||
| Pt36 | c.920‐2A > G | 0.67 | p.D594N | 0.37 |
Figure 2.
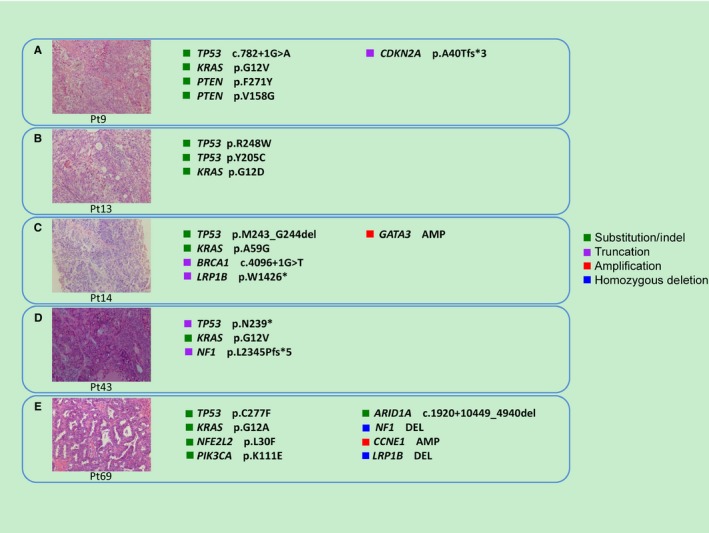
Hemotoxylin and eosin staining of HGSOC tumors in this cohort with co‐occurring KRAS point mutation and TP53 mutations. Total magnification = 10 × 20 = 200×. (A) Pt9, (B) Pt13, (C) Pt14, (D) Pt43, and (E) Pt69. (A‐D) The nuclei of high‐grade serous carcinoma are larger with greater pleomorphism and large nucleoli, diffuse, solid, or nested pattern. (E) Endometrioid carcinoma‐like pattern, irregular serrated luminal contours, and high‐grade nuclei
3.4. Comprehensive genomic profiling revealed many clinically relevant mutations
Of the 547 (10.2%) genomic alterations in this cohort, 56 were associated with targeted therapies that could potentially lead to clinical benefits for the patients. Notable alterations included those occurring in genes involved in DNA repair genes BRCA1/2 or ATM (20 mutations in 19 tumors (21.6%) combined), which sensitize the tumor for poly (ADP‐ribose) polymerase (PARP) inhibitors.18, 19 Of these, somatic BRCA1 mutations were identified in 4 (4.5%) tumors, including 2 truncating mutations (E699* and C1382Sfs*11), 1 homozygous deletion, and 1 single nucleotide variant (SNV) that affected a splice donor site (c.4096 + 1G > T). Germline BRCA1 mutations were identified in 12 (13.6%) tumors, including 10 truncating mutations (5 nonsense mutations including Q126*, F901*, Q1200*, Q1299*, and Q1458*; 5 frameshift deletions including S281Afs*17, Y655Vfs*18, S873Ffs*30, I1824Dfs*3, and E1257Gfs*9) and 2 missense SNVs (R1495K and W1837C). One of the tumors (Pt14) harbored both a somatic BRCA1 mutation (c.4096 + 1G>T) and a germline BRCA1 mutation (S281Afs*17). BRCA2 mutations were identified in 3 (3.4%) tumors (all in germline samples), including 2 truncating mutations (both frameshift deletions, A938Pfs*21 and I2675Nfs*5) and 1 large in‐frame deletion of exons 1‐11. Somatic ATM truncating mutation was identified in 1 tumor (K1057*).
4. DISCUSSION
Comprehensive genomic profiling has become standard practice in cancer cares in recent years with the advancement of NGS technologies and their reduced costs. Here, we studied the mutational profiles of 88 Chinese HGSOC tumors using comprehensive genomic profiling of 450 cancer‐related genes, and were able to reveal a wide range of 547 mutations in all categories (base substitutions, short insertions and deletions, gene rearrangements and fusions, and copy number changes), many of which were clinically relevant. Our results confirmed previous reports about several frequently mutated genes in HGSOC including TP53 and BRCA1/2. Moreover, we also found that mutations in a few important cancer genes, including genes in the Ras/Raf pathway (KRAS, NRAS, and BRAF).
TP53 was mutated in 96.6% tumors in this cohort, which is consistent with many earlier sequencing studies on HGSOC that established TP53 as the dominant mutation in this cancer.2, 3 Previously, the presence of mutations in TP53 had been proposed as a defining feature of HGSOC.7 The three tumors in our cohort which were TP53 mutation negative could possibly be explained by TP53 mutations outside the coding region with unknown function, or TP53 mutations at low frequency below our detection threshold. Hemotoxylin and eosin staining of HGSOC tumors from these three individuals were attached in Supplementary Figures. Although no targeted therapies are yet available for these TP53 mutations, in a recent study, tumor infiltrating T‐cell responses to two TP53 hotspot mutations G245S and Y220C were identified in the context of HLA‐DRB3*02:02 in two separate ovarian cancer patients.20 Among the tumors sequenced in this cohort, five harbored the TP53 Y220C mutation and one had the G245S mutation. Although HLA typing was not performed for this cohort, HLA‐DRB3*02:02 had over 22% population frequency among the Chinese population in the US.21 This suggests that adoptive T‐cell therapy could be a promising direction for some Chinese HGSOC patients, and further studies are needed to better understand their efficacy and identify additional immunogenic TP53 hotspot mutations and relevant HLA‐restriction types.
We observed five co‐occurrences of mutated TP53 and KRAS in our cohort. We then hypothesize that the co‐occurrence was correlated with endometrial cyst, which was more likely to evolve to clear cell carcinoma or endometrioid carcinoma. If this is the case, then the co‐occurrence of mutated TP53 and KRAS could possibly be explained by a mixture of endometrial cyst and high‐grade ovarian carcinoma. By revisiting the clinical data, we were able to identify two endometrial cyst cases out of a total of five patients with mutated TP53 and KRAS. At the same time, we observed 2 endometrial cyst cases in the other 83 patients. To test the independence between TP53 and KRAS co‐occurrence and endometrial cyst status, we applied Fisher's exact test on the contingency table (Table S1) and found that TP53 and KRAS co‐occurrence was significantly dependent on endometrial cyst status (P value = 0.015). Thus, the co‐occurrence of TP53 and KRAS could partly be explained by a mixture of HGSOC and endometrial cyst.
BRCA1/2 germline and somatic mutations were identified in 18 of the 88 (20.5%) patients in this cohort, also at a frequency similar to what had been reported elsewhere.2, 22 BRCA1/2 are key homologous recombination (HR) genes that play crucial roles in DNA double‐strand break repair. Ovarian cancer patients with BRCA1/2 mutations are known to respond better to platinum‐based chemotherapies and have longer overall survivals2, 23 and they also benefit from PARP inhibitor which is currently the most important targeted therapy for HGSOC patients.24 The high prevalence of BRCA1/2 mutations, most of which were from germline samples, highlighted the importance of routine BRCA1/2 testing for Chinese ovarian cancer patients. Several other tumors in the cohort also harbored mutations in other HR genes including ATM, RAC1, and RAD51C.
Mutations in the Ras/Raf pathway and in PIK3CA showed higher prevalence in this Chinese HGSOC cohort. We found that 9 of the 88 (10.2%) tumors in this cohort had point mutations in KRAS (n = 5), NRAS (n = 3) or BRAF (n = 1) in addition to TP53 mutations. KRAS and BRAF had been reported to mutate more frequently in low‐grade serous ovarian carcinoma8, 25 as well as other histological subtypes of ovarian cancer,26 yet their clinical significance in HGSOC is unknown.26 Recently, a study reported that KRAS mutation in ovarian cancer predicts that the tumor may be sensitive to MEK inhibition.27 PIK3CA mutations had been reported previously to occur more frequently in endometrioid and clear cell ovarian cancers28, 29 but rarely in HGSOC.2 Further studies are needed to understand the significance of such co‐occurrences of mutations in the Ras/Raf pathway or PIK3CA (Table 3) with TP53.
Table 3.
Co‐occurring point mutations in PIK3CA with TP53 mutations
| Individual ID | TP53 mutation | VAF (TP53) | PIK3CA mutation | VAF (PIK3CA) |
|---|---|---|---|---|
| Pt35 | R342* | 0.72 | P539R | 0.42 |
| Pt46 | R273H | 0.71 | M1043V | 0.44 |
| Pt58 | R273H | 0.08 | C378W | 0.07 |
| Pt64 | R248W | 0.40 | E542K | 0.26 |
| Pt69 | C277F | 0.57 | K111E | 0.49 |
| Pt80 | R273H | 0.56 | K111N | 0.34 |
PTK2 amplifications were identified in 16 of the 88 (18.1%) tumors in this cohort. Of the 16 tumors with PTK2 amplifications, 11 (68.8%) also had co‐amplifications in FAM135B, a gene located very close to PTK2 on the chromosome 8. PTK2 is located on chromosome 8q24.3 which had been linked to ovarian cancer susceptibility.30 It encodes focal adhesion kinase (FAK), which is a critical component in transmitting signals from extracellular environments into the cell, and its activation in cancer drives tumor progression and metastasis.31 Frequent PTK2 amplification had been reported in ovarian as well as other cancers,32, 33, 34 and was associated with poor overall survival.35 Recently, several FAK inhibitors have been developed and are being tested in clinical trials (NCT01138033, NCT01943292, and NCT00787033). If proved effective, they could bring clinical benefits to these HGSOC patients with PTK2 amplification.
Two tumors in this cohort (2.3%; Pt10 and Pt18) harbored amplifications in both CD274 (PD‐L1) and PDCD1LG2 (PD‐L2). Moreover, 3 of the 88 (3.4%) tumors in this cohort were tested to be MSI‐H (Pt8, Pt38, and Pt55; Figure 1). Together, these 5 (5.7%) tumors might benefit from checkpoint inhibitor treatment. Despite this, several recent early phase clinical trials applying checkpoint inhibitors in ovarian cancer had demonstrated only limited efficacy with response rate between 5% and 20%.36 Further identification of useful biomarkers is needed to better stratify patients and identify those that are more likely to respond. In addition, several ongoing clinical trials are studying the effect of combining checkpoint inhibitors with chemotherapies (NCT02520154), targeted therapies (NCT02484404), or other checkpoint inhibitors (NCT02498600) in ovarian cancers, and results from these trials may provide important insights on how to optimally apply immunotherapy for such patients.
In summary, comprehensive genomic profiling has revealed an unrecognized co‐occurrence of TP53 mutations with mutations in Ras/Raf pathway, for example, KRAS and NRAS, and detected mutations that are informative for choosing personalized treatment regimens in almost half of all the tumors studied in this cohort of Chinese HGSOC patients, demonstrating its values in routine clinical practice.
CONFLICT OF INTEREST
XP, JH, HH, LM, DC, KW, and XD are employees of OrigiMed.
AUTHOR CONTRIBUTIONS
FZ, TZ, XL, and XD wrote the paper. XP, XL, and XD conceived this idea. XZ, XL, and XD reviewed the cases. YZ, HY, XW, and HH manipulated the data. JH, LM, and DC annotated the variants.
Supporting information
ACKNOWLEDGMENT
We thank all the patients who had contributed to tumor samples for this study.
Zhong F, Zhu T, Pan X, et al. Comprehensive genomic profiling of high‐grade serous ovarian carcinoma from Chinese patients identifies co‐occurring mutations in the Ras/Raf pathway with TP53 . Cancer Med. 2019;8:3928–3935. 10.1002/cam4.2243
Fangfang Zhong and Tao Zhu contributed equally to this study.
Data Availability Statement: The data that support the findings of this study are available from the corresponding author upon reasonable request (dongxw@origimed.com).
Contributor Information
Xiuqin Li, Email: lixq@sj-hospital.org.
Xiaowei Dong, Email: dongxw@origimed.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request (dongxw@origimed.com).
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Cancer J Clin. 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2. The Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Labidi‐Galy SI, Papp E, Hallberg D, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun. 2017;8:1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole‐genome characterization of chemoresistant ovarian cancer. Nature. 2015;28:489–494. [DOI] [PubMed] [Google Scholar]
- 6. Cole AJ, Dwight T, Gill AJ, et al. Assessing mutant p53 in primary high‐grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 2016;6:26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vang R, Levine DA, Soslow RA, Zaloudek C, Shih Ie M, Kurman RJ. Molecular alterations of TP53 are a defining feature of ovarian high‐grade serous carcinoma: a rereview of cases lacking TP53 mutations in the cancer genome atlas ovarian study. Int J Gynecol Pathol. 2016;35:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaldawy A, Segev Y, Lavie O, Auslender R, Sopik V, Narod SA. Low‐grade serous ovarian cancer: a review. Gynecol Oncol. 2016;143:433–438. [DOI] [PubMed] [Google Scholar]
- 9. Vang R, Shih I‐M, Kurman RJ. Ovarian low‐grade and high‐grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. 2009;16:267–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurman R, Herrington CS. WHO Classification of Tumours of Female Reproductive Organs. Lyon: IARC Press; 2014. [Google Scholar]
- 11. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England). 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired‐end short reads. Bioinformatics (Oxford, England). 25:2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magi A, Tattini L, Cifola I, et al. EXCAVATOR: detecting copy number variants from whole‐exome sequencing data. Genome Biol. 2013;14:R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kautto EA, Bonneville R, Miya J, et al. Performance evaluation for rapid detection of pan‐cancer microsatellite instability with MANTIS. Oncotarget. 8:7452–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crafton SM, Bixel K, Hays JL. PARP inhibition and gynecologic malignancies: a review of current literature and on‐going trials. Gynecol Oncol. 2016;142:588–596. [DOI] [PubMed] [Google Scholar]
- 19. Mirza MR, Pignata S, Ledermann JA. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol. 2018;29:1366–1376. [DOI] [PubMed] [Google Scholar]
- 20. Deniger DC, Pasetto A, Robbins PF, et al. T‐cell responses to TP53 "Hotspot" mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. 2018;24:5562–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gragert L, Madbouly A, Freeman J, Maiers M. Six‐locus high resolution HLA haplotype frequencies derived from mixed‐resolution DNA typing for the entire US donor registry. Hum Immunol. 2013;74:1313–1320. [DOI] [PubMed] [Google Scholar]
- 22. Pal T, Permuth‐Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104:2807–2816. [DOI] [PubMed] [Google Scholar]
- 23. Vencken P, Kriege M, Hoogwerf D, et al. Chemosensitivity and outcome of BRCA1‐ and BRCA2‐associated ovarian cancer patients after first‐line chemotherapy compared with sporadic ovarian cancer patients. Ann Oncol. 2011;22:1346–1352. [DOI] [PubMed] [Google Scholar]
- 24. Musella A, Bardhi E, Marchetti C, et al. Rucaparib: an emerging parp inhibitor for treatment of recurrent ovarian cancer. Cancer Treat Rev. 2018;66:7–14. [DOI] [PubMed] [Google Scholar]
- 25. Singer G, Oldt R, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low‐grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486. [DOI] [PubMed] [Google Scholar]
- 26. Cuatrecasas M, Villanueva A, Matias‐Guiu X, Prat J. K‐ras mutations in mucinous ovarian tumors: a clinicopathologic and molecular study of 95 cases. Cancer. 1997;79:1581–1586. [DOI] [PubMed] [Google Scholar]
- 27. Nakayama N, Nakayama K, Yeasmin S, et al. KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer. Br J Cancer. 2008;99:2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuo K‐T, Mao T‐L, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–7681. [DOI] [PubMed] [Google Scholar]
- 30. Goode EL, Chenevix‐Trench G, Song H, et al. A genome‐wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sood AK, Coffin JE, Schneider GB, et al. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol. 2004;165:1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ward KK, Tancioni I, Lawson C, et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage‐independent ovarian carcinoma cell growth and tumor progression. Clin Exp Metas. 2013;30:579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoon H, Dehart JP, Murphy JM, Lim ST. Understanding the roles of FAK in cancer: inhibitors, genetic models, and new insights. J Histochem Cytochem. 2015;63:114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sood AK, Armaiz‐Pena GN, Halder J, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest. 2010;120:1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hamanishi J, Mandai M, Konishi I. Immune checkpoint inhibition in ovarian cancer. Int Immunol. 2016;28:339–348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request (dongxw@origimed.com).