

HAZV is classified within the Nairoviridae family with CCHFV, which is one of the most lethal human pathogens in existence, requiring the highest biosafety level (BSL) containment (BSL4). In contrast, HAZV is not associated with human disease and thus can be studied using less-restrictive BSL2 protocols. Here, we report a system that is able to rescue HAZV from cDNAs, thus permitting reverse genetic interrogation of the HAZV replication cycle. We used this system to examine the role of a caspase cleavage site, DQVD, within the HAZV nucleocapsid protein that is also conserved in CCHFV. By engineering mutant viruses, we showed caspase cleavage at this site was not required for productive infection and this sequence performs a critical role in the virus life cycle aside from caspase cleavage. This system will accelerate nairovirus research due to its efficiency and utility under amenable BSL2 protocols.
KEYWORDS: Hazara virus, caspases, reverse genetics, virus rescue
ABSTRACT
The Nairoviridae family of the Bunyavirales order comprises tick-borne, trisegmented, negative-strand RNA viruses, with several members being associated with serious or fatal diseases in humans and animals. A notable member is Crimean-Congo hemorrhagic fever virus (CCHFV), which is the most widely distributed tick-borne pathogen and is associated with devastating human disease, with case fatality rates averaging 30%. Hazara virus (HAZV) is closely related to CCHFV, sharing the same serogroup and many structural, biochemical, and cellular properties. To improve understanding of HAZV and nairovirus multiplication cycles, we developed, for the first time, a rescue system permitting efficient recovery of infectious HAZV from cDNA. This system now allows reverse genetic analysis of nairoviruses without the need for high-level biosafety containment, as is required for CCHFV. We used this system to test the importance of a DQVD caspase cleavage site exposed on the apex of the HAZV nucleocapsid protein arm domain that is cleaved during HAZV infection, for which the equivalent DEVD sequence was recently shown to be important for CCHFV growth in tick but not mammalian cells. Infectious HAZV bearing an uncleavable DQVE sequence was rescued and exhibited growth parameters equivalent to those of wild-type virus in both mammalian and tick cells, showing this site was dispensable for virus multiplication. In contrast, substitution of the DQVD motif with the similarly uncleavable AQVA sequence could not be rescued despite repeated efforts. Together, these results highlight the importance of this caspase cleavage site in the HAZV life cycle but reveal the DQVD sequence performs a critical role aside from caspase cleavage.
IMPORTANCE HAZV is classified within the Nairoviridae family with CCHFV, which is one of the most lethal human pathogens in existence, requiring the highest biosafety level (BSL) containment (BSL4). In contrast, HAZV is not associated with human disease and thus can be studied using less-restrictive BSL2 protocols. Here, we report a system that is able to rescue HAZV from cDNAs, thus permitting reverse genetic interrogation of the HAZV replication cycle. We used this system to examine the role of a caspase cleavage site, DQVD, within the HAZV nucleocapsid protein that is also conserved in CCHFV. By engineering mutant viruses, we showed caspase cleavage at this site was not required for productive infection and this sequence performs a critical role in the virus life cycle aside from caspase cleavage. This system will accelerate nairovirus research due to its efficiency and utility under amenable BSL2 protocols.
INTRODUCTION
The Bunyavirales order comprises over 500 RNA viruses, which are the causative agents of infection and disease across a broad range of hosts encompassing insects, plants, animals, and humans. The bunyavirus genome contains between two and eight segments of negative sense RNA, with some species utilizing an ambisense coding strategy for one or more of their segments (1). The recent use of metagenomic techniques (2–4) has led to the discovery of many new diverse bunyaviruses, particularly from within arthropod hosts, and so bunyavirus classification is in a state of flux. Currently, the order is divided into 12 families (1), and viruses associated with human infections are classified within 5 of these, namely, the Arenaviridae, Hantaviridae, Nairoviridae, Peribunyaviridae, and Phenuiviridae families. The Nairoviridae family comprises tick-borne trisegmented RNA viruses, with several members being associated with serious and fatal diseases in both humans and animals. Nairobi sheep disease virus, after which the family was named, causes acute gastroenteritis in susceptible populations of sheep and goats, with an associated mortality rate of 90%, resulting in significant economic impact (5). Crimean-Congo hemorrhagic fever virus (CCHFV) is the most widely distributed tick-borne pathogen on earth (6) and is associated with a devastating human disease known as Crimean-Congo hemorrhagic fever (CCHF), with case fatality rates averaging 30% but rising as high as 80% in specific outbreaks (7, 8). CCHF most commonly results from the bite of a CCHFV-infected tick of the Hyalomma species, which is widespread in Africa, southeast Europe, and Asia, and concerns are growing regarding the widening habitat of permissive tick vectors in the face of global warming (9–12). Due to the associated risks, CCHFV is one of a select group of pathogens classified within hazard group 4, requiring the highest level of biological containment for its propagation (i.e., biosafety level 4 [BSL4]), which has restricted research activity and hindered progress in elucidating the molecular and cellular biology of CCHFV, as well as nairoviruses in general.
Hazara virus (HAZV) is a nairovirus that is closely related to CCHFV, such that it is included within the same serogroup (1); however, it is nonpathogenic in humans. As a consequence, HAZV can be studied in relatively accessible BSL2 facilities, which permits more rapid and cost-effective research.
The genomes of HAZV and CCHFV are trisegmented, with small (S), medium (M), and large (L) segments. The S segment of both viruses encodes the nucleoprotein (N), which encapsidates the RNA segments through its RNA binding ability. In addition, the CCHFV S segment is thought to express a nonstructural protein, using an ambisense transcription strategy, with a role in apoptotic activation (13). The M segment encodes the glycoprotein precursor GPC, which is subsequently cleaved into the structural glycoproteins Gn and Gc and additional nonstructural glycoproteins, whereas the L segment encodes the relatively large RNA-dependent RNA polymerase (RdRp) (14), which is involved in replication and transcription of viral RNA, but also encodes an N-terminal ovarian tumor (OTU) domain with roles in mitigating host cell innate immunity (15).
We and others recently solved the crystal structure of the N proteins of both HAZV and CCHFV, which revealed the location of a conserved caspase-3 cleavage (C3C) site on the exposed apex of the extended arm domain, suggestive of an important role at some stage of the nairovirus multiplication cycle (16–19). One possibility was that the C3C site acted as a caspase-3 decoy, diverting caspase-3 away from its native cellular substrates and thus preventing or prolonging apoptosis activation, as recently found for the related Junin arenavirus (JUNV) (20). In support of this, both CCHFV and HAZV N proteins are extensively cleaved at their C3C sites in mammalian cells, with apoptosis being induced relatively late in infection (21, 22). However, during infection by both HAZV and CCHFV in tick cell lines, the C3C site is subjected to little or no cleavage, revealing a fundamental difference in the tick and mammalian cellular responses to nairovirus infection (22, 23). Recently, a system capable of rescuing infectious CCHFV from cDNA was used to show that the conserved C3C site on the arm apex was dispensable for infection in mammalian cells but, strikingly, mutation of this motif resulted in a significant drop in replication ability during infection of cells of tick origin (23). Taken together, these findings revealed a critical role for the C3C site in CCHFV growth in ticks, although whether the C3C site in HAZV was required remained unknown.
Here, we developed a system for the rescue of infectious HAZV to authentically test the importance of caspase cleavage of HAZV-N protein during infection. By generating a panel of mutant viruses in which the arm domain DQVD was rendered uncleavable, we showed that, while this sequence was critical for virus viability, it was not for allowing caspase cleavage. Instead, our results show that the DQVD site performs a critical role in the virus life cycle aside from acting as a caspase substrate. To the best of our knowledge, this work represents the first recovery of recombinant HAZV (rHAZV) and also the first nairovirus rescue system that permits virus rescue in BSL2 facilities, thus facilitating rapid future gains in the understanding of this important group of viruses.
RESULTS
Characterization of the DQVD caspase cleavage site in HAZV-N.
In previous work, we identified a number of HAZV-N-specific cleavage products during low-multiplicity of infection (MOI) infection of mammalian cells (22). To further characterize these bands, we repeated HAZV infection of human-origin SW13 cells at a higher MOI of 1.0 to increase the presence of these cleaved forms of HAZV-N, thereby facilitating detection via Western blotting. Anti-HAZV-N antiserum detected prominent N-specific cleavage products with apparent molecular masses of approximately 30 and 22 kDa and a temporal expression pattern, being absent during initial stages of infection and most abundant at later time points (Fig. 1A). Previous work by us and others (16, 19) established that the generation of 30- and 22-kDa products resulted from caspase-3 cleavage of HAZV-N at a consensus DQVD motif (Fig. 1B) located at the apex of the HAZV-N arm domain (Fig. 1C). The HAZV-N arm domain also possesses the sequence ENKD, which partially conforms to the caspase-3 cleavage consensus sequence and is immediately upstream of the sequence DQVD, such that these sites overlap, forming the sequence ENKDQVD.
FIG 1.
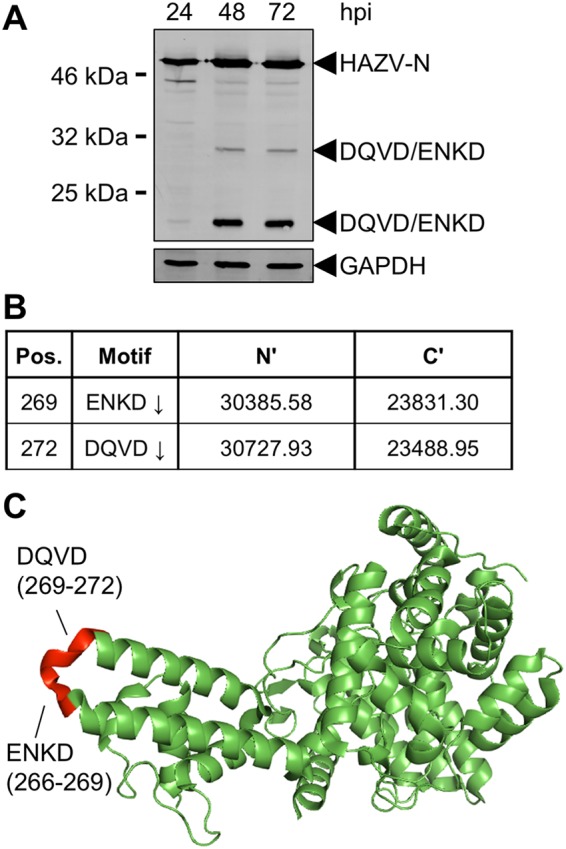
Detection of multiple HAZV-N cleavage products. (A) Western blotting. Monolayers of SW13 cells were infected with HAZV at an MOI of 0.01. At the indicated hours postinfection (hpi), total cell lysates were collected and analyzed for HAZV-N expression by Western blotting with anti-HAZV-N antiserum. (B) Predicted cleavage motifs of HAZV-N and associated predicted molecular weights of N′ and C′ fragments resulting from cleavage. (C) Schematic showing solvent-accessible location of motifs described in panel B.
Recovery of rHAZV.
In order to investigate the role of the ENKD/DQVD caspase cleavage motifs in the authentic context of HAZV infection, we generated a cDNA-based system to allow rescue of infectious HAZV. This system comprised three cDNA plasmids that were designed to transcribe the anti-genomic cRNA strands of the HAZV S, M, and L segments under the control of the bacteriophage T7 RNA polymerase promoter, an approach that has been used by others to rescue bunyaviruses of the Arenaviridae, Peribunyaviridae, and Phenuiviridae families, as well as CCHFV from the Nairoviridae family (24–27). The three corresponding plasmids, pMK-RQ-S, pMK-RQ-M, and pMK-RQ-L, were cotransfected into BSR-T7 cells expressing T7 RNA polymerase, along with plasmid pCAG-T7pol, which allows increased expression of the T7 RNA polymerase and in our hands further increases the efficiency of virus recovery (Fig. 2A). Additional transfections in which the HAZV L segment expression plasmid pMK-RQ-L was omitted were also performed, to act as rescue controls incapable of generating infectious virus.
FIG 2.

Recovery of rHAZV. (A) Schematic showing workflow for recovery of rHAZV. Plasmids were transfected into BSR-T7 cells for the indicated period prior to harvest of supernatant and subsequent 48-h infection of SW13 monolayers. (B) Detection of HAZV-N protein posttransfection (p.Tr) of BSR-T7 cells and 48 h postinfection (p.Inf). Supernatant samples collected from transfected BSR-T7 cells at 72, 96, and 120 h posttransfection (hpt) were used to infect monolayers of SW13 cells. Following a 48-h infection, lysates were collected (p.Inf, lanes 2 to 4) and analyzed for N expression by Western blotting, alongside lysates collected from the initial transfected BSR-T7 cells (p.Tr, lane 1). Recovery of WT rHAZV (Complete) was carried out alongside a control recovery (Control) omitting transfection of the essential pMK-RQ-L plasmid (p.Tr and p.Inf, lanes 5 to 8). Detection of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) abundance was included as a loading control. (C) Titers of infectious rHAZV released into the supernatant at 72, 96, and 120 h posttransfection, as assessed via plaque assay. Error bars represent averages of two repeats.
In cells transfected with all four plasmids (complete transfection), HAZV-N protein was abundantly detected by Western blotting in the primary transfected cultures at 120 h posttransfection (Fig. 2B, p.Tr, lane 1), suggestive of HAZV rescue. As confirmation, supernatants of primary transfected cultures at 72, 96, and 120 h posttransfection were harvested and used to infect further SW13 cells in order to amplify any rescued viruses. At 48 h postinfection, SW13 cell lysates were harvested and tested for the presence of HAZV-N by Western blot analysis, which revealed abundant N production in 96- and 120-h postinfection samples, confirming that virus amplification had occurred (Fig. 2B, p.Inf, lanes 2 to 4). Rescue of wild-type (WT) rHAZV was achieved in all attempts using this optimized protocol, indicating the system was robust and efficient.
In contrast, HAZV-N protein was not detected in control transfected cells that received no pMK-RQ-L (control transfection) in primary transfected cells (Fig. 2B, p.Tr, lane 5). Correspondingly, no N protein was detected in cell lysates harvested at any time point in the postinfection wells (Fig. 2B, p.Inf, lanes 6 to 8).
To examine the efficiency of HAZV rescue, supernatant samples taken from primary transfected BSR-T7 cells at time points of 72, 96, and 120 h posttransfection were used to set up plaque assays in SW13 cells. Analysis of the resulting plaques showed increasing titers of rHAZV in the posttransfection supernatant, increasing through 72-, 96-, and 120-h posttransfection time points (Fig. 2C), with titers of rescued virus reaching over 1.2 × 105 PFU/ml in the 120-h posttransfection supernatants.
Comparison of HAZV and rHAZV growth kinetics.
Although the rescued virus was generated from a cDNA source corresponding precisely to HAZV strain JC280, we wanted to verify that rHAZV displayed growth properties equivalent to those of the parental JC280 isolate, which might have acquired cell culture adaptations through multiple rounds of propagation. To achieve this, we compared multistep growth kinetics of rHAZV alongside the parental virus isolate and mock-infected controls. Following infection at an MOI of 0.001, supernatant samples from each infection scenario were harvested every 24 h for up to 96 h postinfection and were used to titer infectious virus by plaque assay (Fig. 3A). Similar titers for HAZV and rHAZV were observed at all time points, indicating infectious rHAZV displayed growth kinetics equivalent to those of the parental virus. Plaque morphology of rHAZV also resembled that of the parental virus (Fig. 3B), providing further evidence that recombinant and parental viruses possessed indistinguishable growth properties.
FIG 3.
Growth kinetics of rHAZV versus parental isolate. (A) Titers of infectious rHAZV and its parental strain of HAZV harvested at 24-h intervals for 4 days following infection of SW13 cells at an MOI of 0.001. (B) Representative plaque assays used to determine titers of infectious HAZV and rHAZV in panel A, showing plaque morphology to be similar in the two instances. Serial dilutions were generated from virus harvested 24 h postinfection and used to infect SW13 monolayers for 1 h prior to addition of a methylcellulose overlay. Cells were then fixed and stained at 6 days postinfection, and viral plaques were counted.
Confirmation of HAZV rescue from a cDNA source by incorporation of a silent mutation.
To confirm the recombinant source of rHAZV as cDNA, a single nucleotide change of G723T in the N protein open reading frame (ORF) was engineered into pMK-RQ-S to create pMK-RQ-S(G723T). This change was silent at the amino acid level but generated a HindIII recognition site in the corresponding cDNA sequence (Fig. 4A). Plasmid pMK-RQ-S(G723T) replaced the corresponding WT pMK-RQ-S plasmid in a HAZV recovery experiment alongside WT rescue plasmids pMK-RQ-M, pMK-RQ-L, and pCAG-T7pol (described above), and Western blotting of postinfection lysates revealed the abundant presence of HAZV-N, indicating successful mutant virus recovery (Fig. 4B). To verify incorporation of the G723T change, RNA extracted from rHAZV- and rHAZV(G723T)-infected SW13 cell supernatants was used as a template for reverse transcription (RT)-PCR amplification, using primers designed to yield an 802-nucleotide-long cDNA fragment encompassing the HindIII site. While the WT cDNA fragment was not cleaved by HindIII, the corresponding fragment from rHAZV(G723T) was cleaved to generate two products, with lengths of approximately 300 and 500 bp (Fig. 4C and D), corresponding to the fragments expected following HindIII digestion. To confirm that the amplified PCR fragment was templated from a cDNA that originated from an RNA source rather than plasmid carried over from the transfections, we also performed control PCR amplifications without prior RT treatment; these failed to yield a DNA fragment (Fig. 4D). Successful rescue of this rHAZV variant was also confirmed by sequencing of the RT-PCR fragment bearing the introduced HindIII recognition site. Taken together, these findings confirm the utility of the rescue system to efficiently generate both WT and mutant rHAZV variants.
FIG 4.

Confirmation of cDNA origin via recovery of mutant rHAZV. (A) Table outlining the change (underlined) to the cDNA sequence of the HAZV-N ORF and the resulting amino acid sequence. (B) Detection of HAZV-N by Western blotting posttransfection (p.Tr) of BSR-T7 cells and 48 h postinfection (p.Inf). Supernatant samples collected from transfected BSR-T7 cells at 96 h posttransfection were used to infect monolayers of SW13 cells. Following a 48-h infection, lysates were collected and analyzed by Western blotting for N expression. Recovery of rHAZV containing a HindIII restriction site (rHAZV-G723T) was carried out alongside complete control recovery of rHAZV. Detection of GAPDH abundance was included as a loading control. (C) Schematic showing the location of the inserted HindIII restriction site in the S segment ORF cDNA. UTR, untranslated region. (D) Restriction digest of double-stranded DNA fragments following RNA extraction of rHAZV- and rHAZV-G723T-containing supernatants, first-strand synthesis, and PCR amplification of viral genetic material.
Rescue of infectious HAZV variants with alterations to caspase cleavage sites on the N protein arm apex.
Previous work by us and others identified caspase-3 cleavage motifs on both the CCHFV-N (DEVD) and HAZV-N (DQVD) proteins, at the apex of their respective arm domains (16, 17, 19), and mapping of these sites onto the corresponding crystal structures showed these are in precisely superimposable positions (Fig. 5A). Both sites also are preceded by sequences that partially conform to caspase cleavage consensus sequences, suggesting that their cleavage is possible but nevertheless unlikely. These sequences are KHKD in CCHFV and ENKD in HAZV, and both sites overlap the established DEVD/DQVD cleavage sites at a critical aspartic acid reside (D) (Fig. 5A). DEVD and DQVD sites are known to be cleaved during infection of mammalian cells by CCHFV and HAZV, respectively (21, 22), and a mutant CCHFV with the DEVD site changed to a noncleavable AEVA site was shown to be severely growth attenuated in tick cells but phenotypically silent when grown in cells of mammalian origin (23).
FIG 5.

Recovery of mutant rHAZV targeting multiple caspase motifs. (A) Schematic showing locations of caspase cleavage motifs on the apex of the arm domains of HAZV and CCHFV, with amino acid positions indicated numerically. (B) Western blot detection of HAZV-N protein for rHAZV-DQVE, ENKE, and AQVA mutants posttransfection (p.Tr) of BSR-T7 cells and 48 h postinfection (p.Inf) of SW13 cells, using supernatants harvested 72, 96, and 120 h posttransfection. Recovery of all mutants was carried out alongside independent complete control recovery of WT rHAZV. Detection of GAPDH abundance was included as a loading control. (C) Representative plaque assays of supernatants taken 120 h posttransfection, displaying plaque morphology for recovered viruses. (D) Titers of infectious WT rHAZV versus DQVE and ENKE at 24-h intervals following infection of SW13 cells at an MOI of 0.001. (E) Detection of HAZV-N and associated cleavage products at 30 and 32 kDa (*) and 20 kDa (**) following a 48-h infection at an MOI of 0.01.
To investigate the importance of the DQVD and overlapping ENKD motifs in the context of HAZV infection (Fig. 1A and 5A), we created mutant plasmids designed to express HAZV S segments in which the ENKD and DQVD motifs within the N ORF were individually perturbed by mutation. The aspartic acid (D) residues at position 1 within these motifs (underlined above) are known to be critical for caspase recognition and cleavage (28). Therefore, this residue was changed to glutamic acid (E) in both cases, to generate the noncleavable sequences ENKE and DQVE. A double mutant plasmid with the sequence AQVA was also generated, concurrently rendering both ENKD and DQVD motifs uncleavable and allowing direct comparison with the AEVA mutant generated for CCHFV, as described above (23).
As with the rHAZV recoveries described above, the plasmid expressing the WT HAZV S segment, pMK-RQ-S, or a mutant derivative (correspondingly named DQVE, ENKE, or AQVA), was transfected into BSR-T7 cells along with pMK-RQ-M, pMK-RQ-L, and pCAG-T7pol, and supernatants were collected at 72, 96, and 120 h posttransfection and used to reinfect fresh SW13 monolayers for 48 h. At that time point, lysates were collected and examined for HAZV-N expression via Western blotting with anti-HAZV-N antiserum (Fig. 5A), with HAZV-N detection in virus rescue wells indicating successful recovery of both recombinant WT and mutant viruses. Successful rescue was achieved for HAZV-N mutants rHAZV-DQVE and rHAZV-ENKE, with rescue confirmed by sequencing of an RT-PCR fragment bearing the altered cleavage sites (data not shown) amplified from viral RNA harvested from postinfection supernatants. In contrast, the double mutant rHAZV-AQVA could not be rescued, as evidenced by three failed rescue attempts alongside consistent successful rescue of WT rHAZV.
The growth kinetics of rescued rHAZV-ENKE and rHAZV-DQVE alongside WT rHAZV were examined via plaque assay over a 4-day time course (Fig. 5B), and the morphology of resulting plaques was assessed (Fig. 5C). Viral titers at all time points tested showed no significant differences between WT rHAZV and the rHAZV-ENKE or rHAZV-DQVE mutant (Fig. 5D). Taken together, these results show that abrogating caspase cleavage at either of these sites has no detectable effect on virus fitness.
The HAZV-N DQVD caspase cleavage motif is required for rescue of infectious virus but for reasons other than caspase cleavage.
The results of the previous section showed HAZV mutants bearing uncleavable DQVE and ENKE sequences in N could be rescued, whereas an AQVA mutant could not. As expected, abrogation of cleavage at the DQVD site coincided with loss of the customary 30-kDa HAZV-N cleavage product, as measured by Western blot analysis (Fig. 5E). Interestingly, this band was replaced by a band of increased apparent mass, corresponding to approximately 32 kDa. This 32 kDa HAZV-N fragment is too large to correspond to alternative cleavage at the adjacent and overlapping ENKD, so our results show that, during infection with the rHAZV-DQVE mutant, N cleavage occurs at neither the overlapping ENKD site nor the altered DQVE site. The fact that infection with the DQVE mutant does not result in generation of a 30-kDa fragment also shows the ENKD site is not a substrate for caspase cleavage even when the DQVD site can no longer be cleaved, consistent with its only partial similarity to the caspase cleavage consensus sequence. Taken together, these results show that HAZV-N lacking a functional caspase cleavage site at the arm apex can be rescued as infectious virus, and thus a caspase-cleavable site at the arm apex is not a prerequisite for HAZV viability.
Thus, our results show that, individually, the DQVD and ENKD motifs are dispensable for the HAZV life cycle in cultured mammalian cells. However, our observation that the double mutant AQVA could not be rescued despite repeated attempts suggests that simultaneous alteration of both sites critically prevents virus multiplication at some stage of the life cycle. Because we show above that cleavage within the arm domain is not required for virus viability, we suggest the fatal deficiency of the AQVA mutant relates to a function other than caspase cleavability.
This observation is interesting in comparison to the previous findings for CCHFV that the corresponding AEVA mutant, which also acts as a double mutant knocking out both KHKD and DEVD sites, could be rescued as infectious virus and furthermore replicated to WT titers in mammalian cells (23). Taken together, these findings suggest that sequences within the arm apex of CCHFV and HAZV perform different roles, with those of HAZV being highly sensitive to change and those of CCHFV being more tolerant.
The DQVD caspase-3 cleavage motif is also dispensable for HAZV replication in cells of tick origin.
As described above, previous work by others showed that a CCHFV variant with an uncleavable AEVA substitution at the N protein arm apex DEVD site replicates to WT titers in SW13 cells but cannot replicate in tick cells, suggesting a critical role for this motif in the CCHFV life cycle (23). Having shown that mutations at the caspase cleavage site ENKD or DQVD at this location in HAZV-N yield no significant reduction in growth in mammalian cells (Fig. 5C), we next wanted to examine the role of these motifs in replication in tick cells.
To achieve this aim, we examined the ability of the rHAZV-DQVE and rHAZV-ENKE variants to multiply in HAE/CTVM9 tick cells, as determined by their ability to express abundant HAZV-N after 48 h (Fig. 6). Infection with rHAZV carrying individual DQVE and ENKE substitutions resulted in the synthesis of equivalent quantities of N, compared with WT rHAZV, as measured by Western blot analysis with anti-HAZV-N antiserum, categorically showing that the corresponding DQVD and ENKD motifs are nonessential for rHAZV replication in cells of tick origin when individually altered. The equivalent abundance of N proteins expressed by these viruses, compared to WT virus, also suggested these viruses were not growth attenuated in these cells.
FIG 6.
Ability of rHAZV, rHAZV-DQVE, and rHAZV ENKE to replicate in the tick cell line HAE/CTVM9. The ability to replicate was examined via Western blot detection of HAZV-N in lysates taken 48 h postinfection (hpi) at an MOI of 0.01. Actin was included as a loading control.
A more direct comparison of the roles of the arm apex cleavage sites of CCHFV and HAZV would be achieved using equivalent substituting sequences, i.e., AEVA for CCHFV and AQVA for HAZV. This comparison was not possible, however, as the AQVA mutant for HAZV could not be rescued despite repeated efforts. Nevertheless, these results show the ability of the DQVE virus to replicate in tick cells does not require a cleavable caspase site on the apex of the N arm domain.
DISCUSSION
Here we report a rescue system allowing the production of infectious HAZV from cDNA that can be utilized in widespread and amenable BSL2 facilities and that represents an important advance in the study of nairovirus molecular and cellular biology. Previously, a rescue system for the highly pathogenic CCHFV was established, permitting insight into the role of multiple nairovirus-specific features such as the OTU domain within the L protein ORF, as well as characterization of the processing pathway of GPC expressed from the M segment ORF (15, 27). However, due to the extreme pathogenicity of CCHFV, utilization of this rescue system has been restricted to a small number of laboratories that can comply with the highest BSL4 containment protocols. The development of a HAZV rescue system that can be utilized using less-restrictive BSL2 protocols will greatly facilitate and accelerate future nairovirus research. A HAZV minigenome system was also reported recently (29), which allowed the delineation and characterization of the viral mRNA transcription promoters, and, while the utility of this system is restricted to analysis of cis-acting signals involved in RNA synthesis, it will also allow a rapid accumulation of information regarding nairovirus multiplication.
The HAZV rescue system we describe here was capable of generating a titer of WT HAZV of over 1.2 × 105 PFU/ml in primary transfection supernatants. Correspondingly, rescue of WT rHAZV was successful in every attempt and allowed rescue of each of the mutants described here with minimal experimental replicates. Taken together, these findings suggest the HAZV rescue system is highly efficient, a property that will facilitate the rescue of mutant viruses that possess other attenuating lesions.
The N proteins of both HAZV and CCHFV possess caspase-3 cleavage motifs, with the sequences DQVD and DEVD, respectively (16, 30), prominently located at equivalent positions on the apex of their arm domains, suggesting motif conservation is driven by functional importance. In line with this suggestion, previous work has shown both HAZV and CCHFV N proteins are cleaved in vitro at these motifs by purified active caspase-3 (16, 17, 19), as well as by caspase-3 in cultured human cells (22).
To test the importance of cleavage within the HAZV-N arm domain during HAZV infection, we generated mutant viruses for which the overlapping ENKD and DQVD consensus cleavage sites were individually disrupted with glutamic acid substitutions at position 1 (underlined) to render them uncleavable. The fact that both of the resulting viruses, rHAZV-ENKE and rHAZV-DQVE, were viable and replicated with growth kinetics that were indistinguishable from those of WT rHAZV is a critical observation. Because the DQVE mutant is cleaved at neither the DQVE site nor the adjacent ENKD site (Fig. 5C), these findings show cleavage within the arm apex is not a requirement for virus viability.
Our failure to rescue the rHAZV-AQVA mutant despite repeated efforts suggests it is deficient in a critical function. We show that caspase cleavage within the arm apex is not a requirement for virus viability, and indeed viruses that cannot be cleaved at this location show fitness equivalent to that of WT HAZV. Thus, the lack of rescue of rHAZV-AQVD cannot be due to loss of cleavability of the arm apex. This finding is intriguing in light of recent work with CCHFV, for which the equivalent AEVA mutant was found to be phenotypically silent in mammalian cells but significantly reduced in fitness for replication in tick cells. These findings suggest the requirements for HAZV and CCHFV are different, and the molecular basis for these differences likely lies in the subtle differences in arm domain sequences (Fig. 5A). Our results here show the DQVD site is important for rescue but not for caspase cleavage, which provides a starting point for studies to elucidate the critical role of this DQVD motif. What might this role be? One possibility is in N-N multimerization. Previous work has shown the crystallographic N-N interface between adjacent monomers involves the formation of a hydrophobic pocket, comprising six residues from the arm domain of one monomer, that interacts with a single proline reside located at the base of the globular domain of the adjacent monomer (16). Interestingly, neither aspartate (D) residue within the DQVD motif that is altered in the DQVE or AQVA mutant is involved in this interaction, which would thus appear to rule out N-N interactions as the critical role. However, we cannot rule out the possibility that the HAZV N-N interaction that occurs in the assembly of an authentic nairovirus ribonucleoprotein (RNP) is different from that revealed in the HAZV-N crystal structure and that these critical aspartate residues do indeed play important roles in RNP formation. This matter will be resolved by solving the structure of nairovirus RNPs in their native state. An alternative possibility is that the DQVD site is involved in an interaction with a host factor, and the availability of such a component may also explain the differential outcomes of CCHFV infection in mammalian and tick cells.
Previous work by us and others suggested that caspase cleavage of the nairovirus N protein may act as a caspase decoy, as has been shown for JUNV. Our results presented here, showing the HAZV DQVD motif is essential for a role other than cleavage, does not rule this out. In fact, the finding that the rHAZV-DQVE mutant no longer generates the customary 30-kDa band but instead is cleaved to yield a 32-kDa product shows that alternative cleavage sites exist that are amenable to cleavage once the dominant sites are removed. Deciphering the caspase cleavage profiles of nairovirus N proteins is a complex task, as there are a total of 28 aspartate residues that could potentially be cleaved for HAZV; however, we are struck by the high level of accessibility of the HAZV-N protein to caspase digestion. Further work on this and other projects are under way, using the rescue system described here, and we hope that its use will accelerate further understanding of nairovirus molecular and cellular biology.
MATERIALS AND METHODS
Cells and viruses.
BSR-T7 cells (derived from baby hamster kidney, containing a T7 polymerase expression gene) were maintained in Dulbecco’s modified Eagle medium (DMEM) (Sigma-Aldrich) containing 2.5% fetal bovine serum (FBS) (Invitrogen). SW13 cells (derived from human adrenal cortex) were maintained in DMEM containing 10% FBS. All cell culture media were supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin, and cells were grown at 37°C in a 5% CO2 atmosphere. Tick-derived HAE/CTVM9 (Hyalomma anatolicum) cells (31) were maintained at 30°C in L15 minimal essential medium containing 20% FBS, 10% tryptose phosphate broth, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Plasmids.
Full-length cDNAs representing the S, M, and L segments were synthesized (Genewiz) using the HAZV strain JC280 (GenBank accession numbers M86624.1, NC_038710, and DQ076419.1, respectively) as reference and were incorporated into the pMK-RQ plasmid, resulting in the generation of pMK-RQ-S, pMK-RQ-M, and pMK-RQ-L, respectively, able to express S, M, and L segment-specific RNAs. Viral gene segments were flanked by the bacteriophage T7 polymerase promoter and hepatitis delta virus ribozyme to ensure correct formation of 3′ and 5′ terminal sequences, with the cDNAs oriented such that primary T7 polymerase transcripts were positive sense. pCAG-T7pol (Addgene plasmid number 59926) was a gift from Ian Wickersham. Generation of mutant plasmids was achieved using the Q5 site-directed mutagenesis kit (New England Biolabs), according to the manufacturer’s instructions, with all mutant plasmid sequences confirmed via sequencing (Genewiz).
Virus rescue.
Six-well plates were seeded with 2 × 105 BSR-T7 cells/well, 1 day prior to transfection, in 2 ml DMEM supplemented with 2.5% FBS. Sixteen to 24 h later, cells were transfected with 1.2 μg pMK-RQ-S, pMK-RQ-M, and pMK-RQ-L and 0.6 μg pCAG-T7pol, combined with 2.5 μl Mirus TransIT-LT1 transfection reagent (Mirus Bio) per microgram of DNA, in 200 μl Opti-MEM (Life Technologies). For mutant recovery, the WT plasmid was replaced with the corresponding mutant plasmid. A control sample, in which transfection of pMK-RQ-L was omitted, was set up alongside each experiment. Cell supernatants were collected 72, 96, and 120 h posttransfection, and 300 μl supernatant was passaged for 48 h in a 6-well plate of SW13 cells grown in DMEM supplemented with 10% FBS. One hundred microliters of supernatant was also used to titer virus following transfection, using a standard plaque assay protocol.
Virus infections.
SW13 monolayers were infected with HAZV, at the specified MOI, in serum-free DMEM (SFM) at 37°C. After 1 h, the inoculum was removed, cells were washed in phosphate-buffered saline (PBS), and fresh DMEM containing 2.5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin was then applied for the duration of the infection. Infections of HAE/CTVM9 cells of tick origin were carried out at an MOI of 0.01, in a manner similar to that for mammalian cell infections but with an incubation temperature of 30°C.
Inhibition of caspases.
SW13 monolayers were pretreated for 45 min with 20 μM benzyloxycarbonyl-phenylalanyl-alanyl-fluoromethyl ketone (Z-FA-FMK) in SFM at 37°C prior to infection with HAZV at an MOI of 0.1. Following infection, virus and inhibitor were removed, cells were washed three times in PBS, and 2.5% FBS-DMEM containing 20 μM Z-FA-FMK was reapplied. At 24-, 32-, and 48-h time points, total cell lysates were harvested and prepared for Western blotting.
Cell viability assays.
SW13 monolayers were pretreated with a range of Z-FA-FMK concentrations in SFM for 105 min, matching the duration of the pretreatment and the infection stage. At that point, the medium was changed to 2.5% FBS-DMEM containing the same concentration of drug, again matching the infection procedure. Following a 48-h infection period, cell viability was assessed using the CellTiter 96 AQueous One Solution cell proliferation assay (Promega), according to the manufacturer’s instructions.
Western blotting.
For preparation of cell lysates, monolayers were washed in ice-cold PBS, followed by incubation in ice-cold RIPA buffer (150 mM sodium chloride, 1.0% NP-40 alternative, 0.1% SDS, 50 mM Tris [pH 8.0]), and agitated for 120 s. Cells were then harvested via cell scraping and were transferred to prechilled Eppendorf tubes, after which lysates were centrifuged at 20,000 × g for 15 min to pellet insoluble material. SDS-gel loading buffer containing dithiothreitol was added to the supernatant prior to storage at −20°C. Proteins were separated on 12% SDS-polyacrylamide gels by electrophoresis and were transferred to fluorescence-compatible polyvinylidene difluoride (PVDF) membranes. Sheep anti-HAZV-N antiserum, generated as described previously (32), was used to detect HAZV-N, which was subsequently visualized using fluorescently labeled anti-sheep IgG secondary antibodies. Membranes were visualized on the LiCor Odyssey Sa infrared imaging system.
Virus titration.
Determination of virus titers for construction of growth curves was achieved through plaque assays. Briefly, SW13 cells were seeded (2 × 106 cells) into 75-cm2 flasks 24 h prior to infection with HAZV, rHAZV, or mutant rHAZV at an MOI of 0.001. Supernatant was collected at time points of 24, 48, 72, 96, and 120 h postinfection and serially diluted to infect fresh monolayers of SW13 cells in a 6-well plate. Following infection, medium containing virus was removed and replaced (1:1) with 2.5% FBS-DMEM containing 1.6% methylcellulose, and cells were returned to incubate for a further 6 days prior to fixing and staining with crystal violet. Plaques were then counted, and virus titers were determined.
Extraction of viral RNA.
Viral RNA was extracted from cell-free supernatant using the QIamp viral RNA kit (Qiagen) and treated with DNase to remove any contaminating DNA before a cDNA copy was generated using ProtoScript II reverse transcriptase (New England Biolabs), according to the manufacturer’s instructions. PCR amplification of an ∼800-bp fragment using primers specific to the HAZV S segment was achieved using the Q5 high-fidelity polymerase (New England Biolabs). Restriction digest analysis was performed by incubating HindIII (New England Biolabs) with 500 ng PCR product at 37°C for 1 h prior to resolving DNA bands on a 1% agarose gel containing 0.01% SYBR Safe (Thermo Fisher).
ACKNOWLEDGMENTS
We thank L. Bell-Sakyi and the Tick Cell Biobank, University of Liverpool (Liverpool, UK), for kindly providing HAE/CTVM9 cells.
This work was funded by a Public Health England PhD studentship (to J.F.).
J.F., R.A.S., G.S.S., J.M., R.H., and J.N.B. conceptualized the study, J.F., R.A.S., and G.S.S. performed the experimental investigation, J.F. and J.N.B. wrote the original draft manuscript, R.A.S. and G.S.S. reviewed and edited the manuscript, J.M., R.H., and J.N.B. supervised the core team, and J.N.B. provided management and coordination of the research activities and acquired the financial support for the project.
We declare that there are no conflicts of interest.
REFERENCES
- 1.ICTV. 2018. Taxonomy. https://talk.ictvonline.org/taxonomy. Accessed February 2019.
- 2.Makhsous N, Shean RC, Droppers D, Guan J, Jerome KR, Greninger AL. 2017. Genome sequences of three novel bunyaviruses, two novel rhabdoviruses, and one novel nyamivirus from Washington State moths. Genome Announc 5:e01668-16. doi: 10.1128/genomeA.01668-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouquet J, Melgar M, Swei A, Delwart E, Lane RS, Chiu CY. 2017. Metagenomic-based surveillance of Pacific Coast tick Dermacentor occidentalis identifies two novel bunyaviruses and an emerging human ricksettsial pathogen. Sci Rep 7:12234. doi: 10.1038/s41598-017-12047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandler JA, Liu RM, Bennett SN. 2015. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front Microbiol 6:185. doi: 10.3389/fmicb.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies FG. 1997. Nairobi sheep disease. Parassitologia 39:95–98. [PubMed] [Google Scholar]
- 6.Messina JP, Pigott DM, Golding N, Duda KA, Brownstein JS, Weiss DJ, Gibson H, Robinson TP, Gilbert M, William Wint GR, Nuttall PA, Gething PW, Myers MF, George DB, Hay SI. 2015. The global distribution of Crimean-Congo hemorrhagic fever. Trans R Soc Trop Med Hyg 109:503–513. doi: 10.1093/trstmh/trv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ergonul O, Whitehouse CA (ed). 2007. Crimean-Congo hemorrhagic fever: a global perspective. Springer, Dordrecht, The Netherlands. [Google Scholar]
- 8.Schwarz TF, Nsanze H, Ameen AM. 1997. Clinical features of Crimean-Congo haemorrhagic fever in the United Arab Emirates. Infection 25:364–367. doi: 10.1007/BF01740819. [DOI] [PubMed] [Google Scholar]
- 9.Grard G, Drexler JF, Fair J, Muyembe JJ, Wolfe ND, Drosten C, Leroy EM. 2011. Re-emergence of Crimean-Congo hemorrhagic fever virus in central Africa. PLoS Negl Trop Dis 5:e1350. doi: 10.1371/journal.pntd.0001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palomar AM, Portillo A, Santibáñez S, García-Álvarez L, Muñoz-Sanz A, Márquez FJ, Romero L, Eiros JM, Oteo JA. 2017. Molecular (ticks) and serological (humans) study of Crimean-Congo hemorrhagic fever virus in the Iberian Peninsula, 2013–2015. Enferm Infecc Microbiol Clin (Engl Ed) 35:344–347. doi: 10.1016/j.eimce.2017.01.034. [DOI] [PubMed] [Google Scholar]
- 11.Al-Tikriti SK, Al-Ani F, Jurji FJ, Tantawi H, Al-Moslih M, Al-Janabi N, Mahmud MI, Al-Bana A, Habib H, Al-Munthri H, Al-Janabi S, Al-Jawahry K, Yonan M, Hassan F, Simpson DI. 1981. Congo/Crimean haemorrhagic fever in Iraq. Bull World Health Organ 59:85–90. [PMC free article] [PubMed] [Google Scholar]
- 12.Papa A, Christova I, Papadimitriou E, Antoniadis A. 2004. Crimean-Congo hemorrhagic fever in Bulgaria. Emerg Infect Dis 10:1465–1467. doi: 10.3201/eid1008.040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnwal B, Karlberg H, Mirazimi A, Tan Y-J. 2016. The non-structural protein of Crimean-Congo hemorrhagic fever virus disrupts the mitochondrial membrane potential and induces apoptosis. J Biol Chem 291:582–592. doi: 10.1074/jbc.M115.667436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zivcec M, Scholte FEM, Spiropoulou CF, Spengler JR, Bergeron É. 2016. Molecular insights into Crimean-Congo hemorrhagic fever virus. Viruses 8:106. doi: 10.3390/v8040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholte FEM, Zivcec M, Dzimianski JV, Deaton MK, Spengler JR, Welch SR, Nichol ST, Pegan SD, Spiropoulou CF, Bergeron É. 2017. Crimean-Congo hemorrhagic fever virus suppresses innate immune responses via a ubiquitin and ISG15 specific protease. Cell Rep 20:2396–2407. doi: 10.1016/j.celrep.2017.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surtees R, Ariza A, Punch EK, Trinh CH, Dowall SD, Hewson R, Hiscox JA, Barr JN, Edwards TA. 2015. The crystal structure of the Hazara virus nucleocapsid protein. BMC Struct Biol 15:24. doi: 10.1186/s12900-015-0051-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter SD, Surtees R, Walter CT, Ariza A, Bergeron É, Nichol ST, Hiscox JA, Edwards TA, Barr JN. 2012. Structure, function, and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J Virol 86:10914–10923. doi: 10.1128/JVI.01555-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Dutta S, Karlberg H, Devignot S, Weber F, Hao Q, Tan YJ, Mirazimi A, Kotaka M. 2012. Structure of Crimean-Congo hemorrhagic fever virus nucleoprotein: superhelical homo-oligomers and the role of caspase-3 cleavage. J Virol 86:12294–12303. doi: 10.1128/JVI.01627-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, Liu X, Wang X, Dong H, Ma C, Wang J, Liu B, Mao Y, Wang Y, Li T, Yang C, Guo Y. 2015. Structural and functional diversity of nairovirus-encoded nucleoproteins. J Virol 89:11740–11749. doi: 10.1128/JVI.01680-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolff S, Becker S, Groseth A. 2013. Cleavage of the Junin virus nucleoprotein serves a decoy function to inhibit the induction of apoptosis during infection. J Virol 87:224–233. doi: 10.1128/JVI.01929-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlberg H, Tan YJ, Mirazimi A. 2011. Induction of caspase activation and cleavage of the viral nucleocapsid protein in different cell types during Crimean-Congo hemorrhagic fever virus infection. J Biol Chem 286:3227–3234. doi: 10.1074/jbc.M110.149369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuller J, Surtees RA, Shaw AB, Álvarez-Rodríguez B, Slack GS, Bell-Sakyi L, Mankouri J, Edwards TA, Hewson R, Barr JN. 2019. Hazara nairovirus elicits differential induction of apoptosis and nucleocapsid protein cleavage in mammalian and tick cells. J Gen Virol 100:392–402. doi: 10.1099/jgv.0.001211. [DOI] [PubMed] [Google Scholar]
- 23.Salata C, Monteil V, Karlberg H, Celestino M, Devignot S, Leijon M, Bell-Sakyi L, Bergeron É, Weber F, Mirazimi A. 2018. The DEVD motif of Crimean-Congo hemorrhagic fever virus nucleoprotein is essential for viral replication in tick cells. Emerg Microbes Infect 7:190. doi: 10.1038/s41426-018-0192-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. 2000. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J Virol 74:3470–3477. doi: 10.1128/JVI.74.8.3470-3477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowen AC, Noonan C, McLees A, Elliott RM. 2004. Efficient bunyavirus rescue from cloned cDNA. Virology 330:493–500. doi: 10.1016/j.virol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 26.Ikegami T, Won S, Peters CJ, Makino S. 2006. Rescue of infectious Rift Valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J Virol 80:2933–2940. doi: 10.1128/JVI.80.6.2933-2940.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergeron É, Zivcec M, Chakrabarti AK, Nichol ST, Albariño CG, Spiropoulou CF. 2015. Recovery of recombinant Crimean Congo hemorrhagic fever virus reveals a function for non-structural glycoproteins cleavage by furin. PLoS Pathog 11:e1004879. doi: 10.1371/journal.ppat.1004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duclos C, Lavoie C, Denault JB. 2017. Caspases rule the intracellular trafficking cartel. FEBS J 284:1394–1420. doi: 10.1111/febs.14071. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto Y, Ohta K, Kolakofsky D, Nishio M. 2019. A minigenome study of Hazara nairovirus genomic promoters. J Virol 93:e02118-18. doi: 10.1128/JVI.02118-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewson R, Chamberlain J, Mioulet V, Lloyd G, Jamil B, Hasan R, Gmyl A, Gmyl L, Smirnova SE, Lukashev A, Karganova G, Clegg C. 2004. Crimean-Congo haemorrhagic fever virus: sequence analysis of the small RNA segments from a collection of viruses world wide. Virus Res 102:185–189. doi: 10.1016/j.virusres.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 31.Bell-Sakyi L. 1991. Continuous cell lines from the tick Hyalomma anatolicum anatolicum. J Parasitol 77:1006–1008. doi: 10.2307/3282757. [DOI] [PubMed] [Google Scholar]
- 32.Surtees R, Dowall SD, Shaw A, Armstrong S, Hewson R, Carroll MW, Mankouri J, Edwards TA, Hiscox JA, Barr JN. 2016. Heat shock protein 70 family members interact with Crimean-Congo hemorrhagic fever virus and Hazara virus nucleocapsid proteins and perform a functional role in the nairovirus replication cycle. J Virol 90:9305–9316. doi: 10.1128/JVI.00661-16. [DOI] [PMC free article] [PubMed] [Google Scholar]