

Poxviruses are large, double-stranded DNA viruses that replicate entirely in the cytoplasm, an unusual act that activates pathogen sensors and innate antiviral responses. In order to replicate, poxviruses therefore encode a wide range of innate immune antagonists that include F17, a protein that dysregulates the kinase mammalian target of rapamycin (mTOR) to suppress interferon-stimulated gene (ISG) responses. However, the host sensor(s) that detects infection in the absence of F17 and its precise contribution to infection remains unknown. Here, we show that the cytosolic DNA sensor cGAS is primarily responsible for activating ISG responses in biologically relevant cell types infected with a poxvirus that does not express F17. However, in line with their expression of ∼100 proteins that act as immune response and ISG antagonists, while F17 helps suppress cGAS-mediated responses, we find that a critical function of its mTOR dysregulation activity is to enhance poxvirus protein production.
KEYWORDS: F17, cGAS, interferon-stimulated gene, mTOR, poxvirus, protein synthesis, vaccinia virus
ABSTRACT
Despite producing enormous amounts of cytoplasmic DNA, poxviruses continue to replicate efficiently by deploying an armory of proteins that counter host antiviral responses at multiple levels. Among these, poxvirus protein F17 dysregulates the host kinase mammalian target of rapamycin (mTOR) to prevent the activation of stimulator of interferon genes (STING) expression and impair the production of interferon-stimulated genes (ISGs). However, the host DNA sensor(s) involved and their impact on infection in the absence of F17 remain unknown. Here, we show that cyclic-di-GMP-AMP (cGAMP) synthase (cGAS) is the primary sensor that mediates interferon response factor (IRF) activation and ISG responses to vaccinia virus lacking F17 in both macrophages and lung fibroblasts, although additional sensors also operate in the latter cell type. Despite this, ablation of ISG responses through cGAS or STING knockout did not rescue defects in late-viral-protein production, and the experimental data pointed to other functions of mTOR in this regard. mTOR adjusts both autophagic and protein-synthetic processes to cellular demands. No significant differences in autophagic responses to wild-type or F17 mutant viruses could be detected, with autophagic activity differing across cell types or states and exhibiting no correlations with defects in viral-protein accumulation. In contrast, results using transformed cells or altered growth conditions suggested that late-stage defects in protein accumulation reflect failure of the F17 mutant to deregulate mTOR and stimulate protein production. Finally, rescue approaches suggest that phosphorylation may partition F17’s functions as a structural protein and mTOR regulator. Our findings reveal the complex multifunctionality of F17 during infection.
IMPORTANCE Poxviruses are large, double-stranded DNA viruses that replicate entirely in the cytoplasm, an unusual act that activates pathogen sensors and innate antiviral responses. In order to replicate, poxviruses therefore encode a wide range of innate immune antagonists that include F17, a protein that dysregulates the kinase mammalian target of rapamycin (mTOR) to suppress interferon-stimulated gene (ISG) responses. However, the host sensor(s) that detects infection in the absence of F17 and its precise contribution to infection remains unknown. Here, we show that the cytosolic DNA sensor cGAS is primarily responsible for activating ISG responses in biologically relevant cell types infected with a poxvirus that does not express F17. However, in line with their expression of ∼100 proteins that act as immune response and ISG antagonists, while F17 helps suppress cGAS-mediated responses, we find that a critical function of its mTOR dysregulation activity is to enhance poxvirus protein production.
INTRODUCTION
In order to replicate, viruses must evade or counteract a wide range of host nucleotide sensors that detect their presence and mount antiviral responses. Among these, DNA sensors detect the genomes of DNA viruses and DNA produced by retroviruses, as well as broader cellular damage caused by infection (1, 2). Virtually all of these sensors act by phosphorylating the endoplasmic reticulum (ER)-resident protein stimulator of interferon (IFN) genes (STING) (3–6). Activated STING then binds TANK-binding kinase 1 (TBK1) and translocates from the ER to the Golgi complex or lysosomal vesicles (1, 7–9). This in turn activates interferon response factors (IRFs) and nuclear factor κB (NF-κB), resulting in IFN-stimulated gene (ISG) expression (10). Depending on the sensor and pathway activated, distinct ISG responses are mounted to different pathogens or insults. Among DNA sensors, interferon gamma-inducible protein 16 (IFI16) and DNA-dependent protein kinase (DNA-PK) predominantly sense foreign and/or damaged DNA in the nucleus, while in the cytoplasm, vesicle-resident sensors like AIM2 or Toll-like receptor 9 (TLR9) mount inflammatory and antiviral responses in specialized immune cells (1, 2, 11, 12). However, the most potent activator of IFN responses is cyclic-di-GMP-AMP (cGAMP) synthase (cGAS), which is resident in the cytosol and expressed by a wide range of cell types (8, 13, 14). Upon binding DNA, cGAS produces the second messenger cGAMP, which potently activates STING and IFN production (15–25). Most DNA viruses replicate their genomes in the nucleus and are predominantly recognized by nuclear sensors like IFI16 (8, 13, 14). However, because of cellular damage during infection and the formation of defective particles, as well as complex localization dynamics and cross talk between sensors, cGAS can contribute to sensing several nuclear DNA viruses that are primarily recognized by IFI16. This complexity is exemplified by studies of the roles of IFI16 and cGAS during herpes simplex virus 1 (HSV-1) infection (13, 14, 26–28). In contrast, poxviruses are highly unusual in that they replicate entirely in the cytoplasm, where they produce enormous amounts of viral DNA that leaves them particularly sensitive to detection by their hosts (29).
Members of the poxvirus family include variola virus (VarV), which caused smallpox, and vaccinia virus (VacV), a closely related orthopoxvirus that was used as the smallpox vaccine and is now widely considered the prototype for poxvirus research (29). Poxviruses infect a variety of cell types in vivo, including keratinocytes, fibroblasts, and macrophages found in the skin or lungs, the primary sites of exposure, which is followed by systemic spread by immune cells, such as macrophages. Each cell type is capable of mounting distinct responses to infection (25, 30, 31). Further highlighting the enormous complexity in both host responses and viral countermeasures, on average, poxviruses encode ∼200 proteins, of which ∼100 have immunomodulatory functions (29). These include inhibitors of protein kinase R (PKR), JAK-STAT, NF-κB, and IRF signaling pathways that often intervene at several points in the same pathway or block multiple pathways at the same time. In some cases, multiple viral proteins target the same host factor as a fail-safe. Virtually all of the immunomodulatory proteins identified to date are encoded within variable flanking regions of the genome and are expressed early in infection, most likely in preparation for the robust host responses that would otherwise be activated when cytoplasmic DNA replication begins. For more on the broader topic of poxvirus immunomodulation, we refer readers to a recent, very comprehensive review by Smith and colleagues (29). In terms of DNA sensing, it is becoming clear from recent findings that poxviruses have the capacity to counteract this on multiple levels. The host-encoded DNA binding protein barrier-to-autointegration factor (BAF) suppresses VacV infection but is countered by the viral B1 kinase (32), although BAF likely inhibits viral DNA replication directly and operates independently of STING. Among host DNA binding proteins involved in sensing and that signal through STING, DNA-PK can traverse from the nucleus to cytoplasmic viral factories, and at least two viral proteins, C4 and C16, act to prevent DNA-PK-mediated activation of IRF3 (33–35). Studies have also implicated IFI16, although it is not clear whether it plays a direct role in sensing poxvirus infection, given the complexity of its cross talk with cGAS discussed above, or if it operates in all cell types. For example, although IFI16 translocates to viral factories in keratinocytes (36), this only occurs to a limited level in fibroblasts (37) and is not observed in other cell types (38). Furthermore, genetic knockout of IFI16 has no effect on ISG responses to modified vaccinia virus Ankara (MVA) in murine dendritic cells (39), and no poxvirus antagonist of IFI16 has been reported to date, as far as we are aware. Recently, cGAS-STING signaling has emerged as a major effector of host responses to poxvirus infection in several immune cell types, such as dendritic cells and monocytes/macrophages (7, 25, 39–44). Intriguingly, beyond the complexity of in vivo contexts, most studies of responses mounted by the infected cell use MVA, an attenuated strain whose replication is restricted in many cell types and which fails to produce significant levels of late viral proteins. In addition, studies of another VacV mutant (vv811) that lacks 55 genes, including all known inhibitors of NF-κB, along with DNA-PK antagonists like C16, suggest the existence of as-yet-unidentified viral proteins that counteract cGAS-STING activation, some of which may be produced late in infection (40). In line with this, we recently discovered that the late viral protein F17, a component of the lateral bodies of poxvirus particles (45–47), can antagonize ISG production by dysregulating cross talk between mammalian target of rapamycin (mTOR) complex 1 (mTORC1) and mTORC2 (37, 48).
mTORC1 acts as a metabolic rheostat that senses nutrient and energy availability and adjusts a wide range of cellular processes accordingly (49). For example, mTORC1 promotes protein synthesis and represses autophagy, which breaks down proteins and other cellular components, to balance these activities and maintain cellular homeostasis under different conditions (49–54). mTORC1 is also activated by Akt/PKB and enhances protein synthesis and cell growth in response to various mitogenic cues. In addition, mTORC1 regulates broader metabolic functions of the cell, such as lipid metabolism, and responds to a broader range of stimuli, including nucleotide sensing and immune cell activation cues (49, 55–65). This positions mTORC1 as a central regulator of cellular homeostasis, immune cell function, and innate antimicrobial responses.
In contrast, mTORC2 regulates cytoskeletal dynamics and activates Akt (66–68). Given that Akt activates mTORC1, in order to avoid a feedforward activation loop, several mTORC1 substrates repress both phosphoinositide 3-kinase (PI3K) and mTORC2 activity to form a self-balancing regulatory circuit (49, 69, 70). While most viruses identified to date control mTORC1 by targeting upstream signaling (71), F17 directly targets mTORCs by competitively sequestering Raptor and Rictor, key regulatory subunits of mTORC1 and mTORC2, respectively (37, 48, 72). In doing so, F17 disrupts the mTORC1-mTORC2 regulatory circuit. In growth-arrested dermal fibroblasts or macrophages, this has complex outcomes, as is common to perturbations in mTOR signaling in general (49, 51), as this hyperactivates both mTORCs. As such, VacV activates downstream mTORC1 targets that control translation and promotes mTORC2-Akt-mediated degradation of cGAS (37, 73). In the absence of F17, potent ISG responses occur and late-viral-protein production is impaired in both fibroblasts and macrophages. However, these complex phenotypes and mTOR's multifunctionality mean that fundamental questions remain about how mTOR dysregulation contributes to VacV infection. In particular, whether cGAS is required for these host responses and whether it is these responses that suppress viral-protein production in the absence of F17 remain unknown. In addition, the potential contributions of other mTOR-regulated processes to infection remain unclear. Here, we show that cGAS is required for the ISG response in a number of biologically relevant cell types but that this response is not the cause of defects in late-viral-protein production by an F17 mutant. Instead, an independent secondary function of F17-mediated mTOR dysregulation is to maximize viral-protein production in resting cells. Moreover, rescue approaches suggest that F17’s functions as a structural protein and mTOR regulator may be segregated through phosphorylation. These findings provide important insights into the complex multifunctionality of F17 and the varied roles of mTOR dysregulation during late-stage VacV replication.
RESULTS
cGAS mediates ISG responses but does not restrict protein production by an F17 mutant virus.
To begin addressing the question of whether cGAS is required to inhibit late-viral-protein production in the absence of F17, we first examined infection in control or cGAS knockout (KO) monocytic THP1 cells. Cultures were differentiated to macrophages with phorbol 12-myristate 13-acetate (PMA) and infected at a multiplicity of infection (MOI) of 5 with wild-type (WT) VacV or vRR10K, a mutant wherein F17 expression is repressed by a Lac operator system (47, 74). F17 expression is induced by the inclusion of isopropyl-β-d-thiogalactopyranoside (IPTG) in the culture medium (as such, we and others colloquially name the virus iF17) (46, 47, 74). However, F17 is rapidly degraded after entry into the cell (45), enabling the use of this virus in infection studies as a null mutant in the absence of IPTG. We return to the use of IPTG-mediated induction later in this report. Western blotting showed, as we reported previously using unmodified, differentiated THP1s (37), that the expression of early viral proteins, such as E3 or I3, was readily detectable in cells infected with either virus by 24 h postinfection (h.p.i.) and that expression continued through 48 h.p.i. (Fig. 1A). In contrast, postreplicative proteins of the intermediate and late classes, such as G8, D8, and F17 (75), were only detectable at relatively low levels at 24 h.p.i. but were abundantly expressed during later stages, reached by 36 to 48 h. In control THP1s infected with WT VacV, cGAS degradation was detectable at 24 h.p.i., when infection begins to enter the late stage, while more extensive degradation was evident once late-stage replication was fully established, when higher levels of F17 are expressed. Also in line with our prior findings in multiple cell types (37), although cells infected with iF17 expressed E3 and I3 at levels equivalent to the levels in cells infected with WT VacV at both 24 and 48 h.p.i., postreplicative proteins, such as D8 and G8, were only expressed at low levels (Fig. 1A). Most importantly, we found that, at best, only modest increases in viral-protein production occurred in cGAS knockout THP1 cells and that this occurred for both WT and iF17 infections and not specifically for iF17 alone. This suggested that cGAS-mediated ISG responses may not suppress late-viral-protein production in the absence of F17.
FIG 1.

cGAS-STING signaling is required for ISG responses to VacV lacking F17 in human THP1 cells. Control, cGAS KO, STING KO, or IFI16 KO THP1 cells were differentiated to macrophages and mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for the indicated times. (A) Cultures of control or cGAS KO THP1s were infected for 24 h or 48 h and analyzed by Western blotting with antibodies to the indicated proteins. int, intermediate. (B) Cultures were infected for 24 h or 48 h. Whole-cell lysates were subjected to Western blotting with antibodies to the indicated proteins. (C) Top, control or cGAS KO THP1s were infected for 48 h and then analyzed by Western blotting with antibodies to the indicated proteins. Middle and bottom, supernatants were subjected to SEAP (NF-κB) (middle) and Lucia-luciferase (IRF) (bottom) reporter assays. RLU, relative light units. Error bars show standard errors of the means (SEM). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Statistical analysis was performed using analysis of variance (ANOVA) and Tukey’s or Sidak’s test for multiple post hoc comparisons. All data are derived from and representative of at least 3 biological replicates.
To expand upon this initial observation, we next examined whether cGAS or STING was required to mount an ISG response and their respective contributions to infection in the presence or absence of F17. To do this, we first examined infection in commercially available monocytic THP1 cells that have had cGAS, STING, or IFI16 expression genetically knocked out and which carry reporters for IRF and NF-κB activity. Cultures were differentiated and infected as described above. In control THP1s infected with WT VacV, downstream TBK1 activation and ISG induction were not observed at either 24 or 48 h.p.i. (Fig. 1B and C). In addition, ISG56 was degraded, in line with our previous studies in several cell types (37). Notably, the extent of cGAS degradation is somewhat variable at 24 h.p.i. in THP1s, as infection begins to enter late stages, but is always robustly detectable once late-stage infection is established, by 36 to 48 h.p.i. (Fig. 1B and see below). VacV only degrades a subpopulation of cGAS that translocates to initiate host responses, and THP1s express extremely high levels of this protein (37), as is shown again in results described below, making the initial degradation challenging to detect in these cells at times. In control THP1s infected with iF17, cGAS levels remained unchanged or even increased partially, as cGAS is an ISG itself (37), while downstream TBK1 activation and enhanced expression of ISGs (PKR, MxB, and ISG56) was detected (Fig. 1B). Moreover, genetic knockouts demonstrated that both cGAS and STING were essential for the ISG response to iF17 infection at both the 24 h and 48 h time points (Fig. 1B). In contrast, the predominantly nuclear sensor IFI16 was not required to initiate this host response (Fig. 1A), in line with prior studies of murine dendritic cells infected with MVA (39). It must be noted that, over time, uninfected IFI16 knockout cells accumulated higher basal levels of PKR and MxB that made their induction in response to iF17 infection less obvious than in control lines, particularly at later time points. However, lower basal levels of ISG56 made its induction readily detectable, as was the broader suppression of ISG responses by WT VacV in IFI16 knockouts (Fig. 1B). Finally, while our analysis confirmed that cGAS and STING were required to mount an ISG response, we also found that ablation of these responses through knockout of either factor did not restore late viral-protein synthesis by iF17 to any notable extent.
To further analyze the host response, we next infected THP1s as described above and performed secreted embryonic alkaline phosphatase (SEAP) or Lucia luciferase assays to determine NF-κB or IRF activity, respectively. Western blotting confirmed cGAS-dependent phosphorylation of STING and TBK1 during iF17 infection in control but not in cGAS knockout cells (Fig. 1C). Reporter assays showed that neither WT nor iF17 viruses activated NF-κB in control THP1s, while in cGAS knockout cells, NF-κB activity was reduced across either uninfected or infected cells. In contrast, IRF activity was greatly increased specifically during infection by iF17, and this response was absent in cGAS-deficient THP1s (Fig. 1C). This is in line with the notion that F17 is only required to block specific aspects of cGAS-STING signaling to IRFs but continues to express many of the viral factors that prevent NF-κB activation. Indeed, we found previously that iF17 infection progresses through early and intermediate stages normally, and despite activating ISG responses, this virus continues to block IFN release from cells and does not activate the double-stranded RNA (dsRNA) sensor PKR (37), which requires the expression of several early-stage antagonists (76–80; reviewed in reference 48). Overall, these new data demonstrated that, although cGAS is required to mount an antiviral response in the absence of the viral protein that drives its degradation, this response is likely countered by other viral proteins and is not the primary cause of reduced late-viral-protein production during iF17 infection.
To test this in an independent cell type, we next generated cGAS knockouts in lung fibroblast MRC5 cells (Fig. 2A). Similar to our prior findings in primary normal human dermal fibroblasts (NHDFs) (37), VacV caused a notable reduction in cGAS abundance and did not induce host ISG responses in control MRC5 cells. Similar to the results for both NHDF and THP1 cells, the iF17 virus expressed the early proteins E3 and I3 at levels equivalent to the levels in cells infected with WT VacV but failed to produce high levels of postreplicative (intermediate or late) proteins G8, D8, and A14 (Fig. 2A). In addition, iF17 produced lower levels of the late structural protein A10, which was also notably not processed to its faster-migrating form, which occurs during virion maturation (81). This is in line with prior reports of late-stage defects in virion maturation in the absence of F17, which involve failed processing of other structural proteins (46, 47), and suggests that, although this mutant proceeds through early stages of infection, it subsequently encounters both postreplicative-protein production and virion maturation defects. In control cells, iF17 infection also caused robust STING-TBK1 phosphorylation and ISG expression (Fig. 2A). These host responses were inhibited in two independent cGAS knockout MRC5 lines, but this did not restore late-viral-protein production or processing of A10 during iF17 infection. We also noted that, although STING-TBK1 activation and ISG responses were robustly impaired, they were not completely ablated by cGAS knockout in MRC5 cells (Fig. 2A). Examining the expression of some representative DNA sensors across unmodified and reporter THP1 cells alongside MRC5s and NHDFs showed the extent to which cGAS is overexpressed in immune cells compared with its expression in fibroblasts of different kinds (Fig. 2B). However, lung fibroblasts were found to express notably higher levels of DNA-PK, a sensor known to mediate responses to poxvirus infection (33–35). While the precise nature of the sensor(s) in MRC5 cells that mediates these secondary responses to poxvirus infection independently of cGAS remains to be identified, cumulatively, our data revealed that cGAS is an essential sensor during poxvirus infection in immune cells and an important sensor in other biologically relevant cell types. However, the fact that cGAS knockout failed to restore postreplicative-viral-protein levels during iF17 infection in either cell type suggested that, beyond cGAS destabilization and blocking of ISG responses, mTOR dysregulation controlled distinct processes important for maximal viral-protein accumulation.
FIG 2.

cGAS is required for maximal STING activation and ISG responses to VacV lacking F17 in human MRC5 lung fibroblasts. (A) Control or two independent lung fibroblast MRC5 cGAS KO cell lines were mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for 30 h. Whole-cell lysates were subjected to Western blotting. int, intermediate; U>, unprocessed A10; P>, processed A10. (B) Differential abundances of DNA sensors in human cells. Unmodified THP1 cells, commercial THP1 Dual reporter cells used in the experiment whose results are shown in Fig. 1, MRC5 cells, and primary NHDF cells were mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for 40 h (THP1 cells), 48 h (THP1 reporter cells), or 30 h (MRC5 and NHDF cells). Whole-cell lysates were subjected to Western blotting with antibodies to the indicated proteins. Note that cGAS is expressed at far lower levels and degradation of a subpopulation of cGAS is more readily detectable in fibroblasts than in THP1 cells. All data are derived from and representative of at least 3 biological replicates.
F17 is required to maximize viral-protein production in resting cells.
As discussed in the introduction, mTOR activity balances the processes of protein synthesis and the autophagic breakdown of cellular components, including proteins (49). Therefore, we next examined the effects of mTOR inhibition on autophagy and any correlations this might have with viral-protein accumulation. To do this, control or cGAS knockout THP1s were infected as described above in the presence of a dimethyl sulfoxide (DMSO) solvent control or PP242, a catalytic-site inhibitor of mTOR kinase activity. Notably, immune cells have low metabolic and mTOR activity until activated (82–85), and in line with this, the mTOR substrates 4E-BP1 and p70S6 kinase (p70S6K) were predominantly in their hypo- or underphosphorylated state in uninfected THP1s (Fig. 3A) (49). Similar to our prior study with unmodified THP1s, F17 expressed by VacV dysregulated mTOR and stimulated phosphorylation of both substrates, as evidenced by the appearance of slower-migrating species of 4E-BP1 and p70S6K and concomitant decreases in the abundance of faster-migrating underphosphorylated species. This was further confirmed using phosphorylation site-specific (phosphospecific) antibodies against either mTOR substrate. In contrast, intermediate phosphorylation of these substrates was observed during infection with iF17 (Fig. 3A). We showed previously that this reflects continued host control of mTOR in the absence of F17 and facilitates antiviral responses to infection (37). Notably, these patterns of mTOR activity during WT or iF17 infection occurred regardless of the presence or absence of cGAS (Fig. 3A). This suggests that, although these changes in mTOR activity facilitate host responses during iF17 infection, these changes are not caused by the host antiviral response or cGAS activation. As such, intermediate mTOR activation in the absence of F17 most likely reflects cellular responses to the protein-synthetic demands imposed by infection, discussed again below. Examining the autophagy marker LC3 (86), a relatively high basal level of processing of LC3A into LC3B, indicative of autophagy, was observed across all conditions (Fig. 3A and B). This is in line with low levels of mTOR activity and relatively high levels of autophagy in specific immune cell types, which together regulate immune cell activity as discussed previously (56, 59, 64, 87). Also in line with low mTOR activity in THP1s, although treating these cells with PP242 further suppressed mTOR, as determined by dephosphorylation of the mTOR substrates 4E-BP1 and p70S6K, this only modestly increased autophagy in these cells (Fig. 3A and B). Assessing the effects of infection, no significant changes in LC3 processing compared to that in uninfected cells were observed with either WT or iF17 viruses. These observations made using LC3 as an autophagic readout were further confirmed using the autophagy regulator ULK1, whose phosphorylation prevents autophagy (86). While mTOR inhibition suppressed ULK1 phosphorylation, no notable differences were observed in control DMSO-treated cells across uninfected or infected samples. This further demonstrated that infection with either WT or iF17 viruses did not induce significant levels of autophagy in THP1 cells. However, mTOR inhibition caused notable decreases in postreplicative-viral-protein accumulation for both viruses, and this occurred regardless of the presence or absence of cGAS. Notably, early proteins like E3 and I3 were relatively insensitive to mTOR inhibition, which suggests, as discussed again below, that mTOR activation is particularly important during the transition to the later stages of infection where there is a high protein synthesis demand to support virus replication.
FIG 3.

Effects of poxvirus infection and mTOR inhibition on mTOR substrates and autophagy in THP1 cells. (A) Control or cGAS KO THP1s were differentiated to macrophages and then mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for 36 h. Cells were treated with DMSO or the mTOR inhibitor PP242 (2.5 μM) at the time of infection. Whole-cell lysates were subjected to Western blotting with antibodies to the indicated proteins. Fast-migrating nonphosphorylated (0) and slower-migrating species of differently phosphorylated (P) forms of 4E-BP1 or p70S6K are shown. Note that hypo- and underphosphorylated forms of 4E-BP1 accumulate when mTOR is inactive, while p70S6K is completely dephosphorylated. With intermediate signaling in iF17-infected cells, more hypophosphorylated 4E-BP1 (lowest band) is detected than in WT-infected samples. S.E., short exposure; int, intermediate; U>, unprocessed A10; P>, processed A10. Very low basal levels of A10 were produced in THP1 cells infected with iF17. However, detectable levels were present in cGAS KO cells that further demonstrate the lack of A10 processing during iF17 infection. (B) LC3B was quantified as the percentage of total LC3 (LC3A and LC3B) using densitometry measurements of autoradiographs. Error bars show SEM. *, P ≤ 0.05. Statistical analysis was performed using ANOVA and Sidak’s test for multiple post hoc comparisons. All data are derived from and representative of at least 3 biological replicates.
To explore this further, we next examined infection in MRC5 cells under different culture conditions. Unlike the case for differentiated THP1s, mTOR activity was high and basal autophagy levels were much lower in confluent MRC5 cultures maintained in low-serum (0.2% fetal bovine serum [FBS]) medium as determined by measurements of LC3 processing (Fig. 4A and B). In line with this low basal level of autophagy, treating these cells with PP242 induced large increases in LC3 processing in both uninfected and infected cells. This was further confirmed by the large decrease in ULK1 phosphorylation that was caused in all samples upon treatment with PP242 (Fig. 4A). However, despite a low basal level and readily detectable induction of autophagy in these cells, similar to the results for THP1s, there were no significant changes in autophagy in response to infection with either WT or iF17 viruses. Moreover, despite dramatically different levels of autophagy between THP1 and MRC5 cells, mTOR inhibition significantly reduced postreplicative-viral-protein accumulation in both cell types (Fig. 3A and Fig. 4A). This is in line with a broader requirement for mTOR in the case of both viruses, regardless of their use of host- or F17-regulated mTOR to drive late-viral-protein production. In contrast, early proteins E3 and I3 appeared less dependent on mTOR in both cell types. This again emphasizes the particular importance of mTOR activity during the postreplicative phase of infection and the timing of its activation by F17. The lower dependence of early proteins on mTOR activity may reflect a lower protein-synthetic burden on the cell at early stages but is also in line with our broader understanding of mTOR-mediated translational control, wherein cellular proteins also exhibit a range of dependencies, discussed again below (37, 49, 88, 89).
FIG 4.

Effects of poxvirus infection and mTOR inhibition on mTOR substrates and autophagy in MRC5 cells under different growth conditions. (A and B) Confluent cultures of MRC5s cultured in 0.2% FBS medium (low serum) were mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for 30 h. Cells were treated with DMSO or the mTOR inhibitor PP242 (2.5 μM) at 8 h.p.i. (C and D) MRC5s cultured in normal growth medium (normal serum) were infected as described for panels A and B. Cells were treated with DMSO or PP242 (2.5 μM) at 8 h.p.i., and samples were harvested at 30 h.p.i. int, intermediate; U>, unprocessed A10; P>, processed A10. Note that processing of A10 makes the difference in total protein levels between WT and iF17 samples appear smaller due to separation of signals. Fast-migrating nonphosphorylated (0) and slower-migrating species of differently phosphorylated (P) forms of 4E-BP1 or p70S6K are shown. (B and D) LC3B was quantified as the percentage of total LC3 (LC3A and LC3B) using densitometry measurements of autoradiographs. Error bars show SEM. *, P ≤ 0.05; **, P ≤ 0.01. Statistical analysis was performed using ANOVA and Sidak’s test for multiple post hoc comparisons. All data are derived from and representative of at least 3 biological replicates.
In subconfluent MRC5 cells cultured in normal growth medium (5% FBS), basal LC3 processing was higher than in confluent cultures and more in line with the levels in THP1s (Fig. 4C and D). The mTOR inhibitor PP242 increased LC3B processing but only to statistically significant levels in uninfected cells. In line with this, only low levels of ULK1 phosphorylation were detectable, while mTOR inhibition further reduced this across both uninfected and infected cells. However, regardless of varying levels of basal autophagy and autophagic responses across THP1 or MRC5 cells or across MRC5s in different growth states, no significant changes in autophagy could be detected in response to infection with either WT or iF17 viruses. Despite this, mTOR inhibition again suppressed postreplicative-viral-protein synthesis in MRC5s under high-serum conditions, with differential effects on individual viral proteins similar to those observed under low-serum conditions. Overall, from these comparisons, no clear induction of autophagy in response to infection could be detected, nor did correlations between levels of autophagy induction upon mTOR inhibition and effects on viral-protein accumulation emerge. Instead, the extent of defects in late-viral-protein production during iF17 infection compared with the late-viral-protein production during WT infection appeared greatest in THP1 or MRC5 cells cultured under low-serum conditions, despite opposing levels of basal autophagy in each cell type, and the defects were less severe in MRC5s cultured in normal-serum medium despite high levels of LC3B processing. This suggested that the extent of defects in viral-protein production in the absence of F17 was more dependent on the signaling and translational activity of the cell and that there was likely only a limited role for autophagy during infection. This notion is in line with a number of studies showing that autophagy has neither negative nor positive impacts on infection by several poxviruses, including VacV (90–93).
An important point arising from our experiments with MRC5s is the effect that VacV infection has on mTOR substrates in different cell types. In several cell types that we have examined previously (37), as well as THP1s as shown above and NHDFs as shown below, infection with WT VacV but not iF17 causes increases in 4E-BP1 and p70S6K phosphorylation. However, in MRC5 cells, there were very distinct patterns of mTOR activity in response to either WT or iF17 viruses. In iF17-infected cells, host responses appeared to involve a partial activation of mTOR in resting cells but a decrease in activity in cycling cells, which likely reflects responses to different cellular demands imposed by iF17 infection under each condition (Fig. 4A and C). However, across both conditions, mTOR substrate phosphorylation was higher in iF17- than in WT-infected samples. This suggested that mTOR activity was sustained to varying extents in response to iF17 infection, while under both conditions, WT virus suppressed this activity. Beyond comparisons between infected cells, with the exception of modest p70S6K activity under low-serum conditions, infection of MRC5s with WT virus caused decreased mTOR substrate phosphorylation to below the levels observed even in uninfected cells. This opposing and seemingly contradictory behavior is actually in line with what is known about the effects of removing Raptor from mTORC1, which is how F17 targets mTOR (37). Raptor is a substrate gatekeeper for mTOR (49, 94), and depending on cellular metabolic state and the basal level of mTOR activity, removing Raptor has opposing effects: under low-amino acid conditions, Raptor removal activates mTOR, while under amino acid-replete conditions, Raptor removal suppresses mTOR substrate phosphorylation (95, 96). Adding to this complexity, mTOR recognizes distinct motifs and thereby differentially regulates the phosphorylation of substrates like p70S6K and 4E-BP1 (69), which is also evident across our analyses. Our data suggest that MRC5 cells have a high basal level of mTOR activity, perhaps due to the fact that they are telomerase immortalized, and that these cells modulate mTOR differently in response to infection by iF17 depending on culture conditions. However, during WT infection of these cells, dysregulation of mTOR by F17 manifests as suppression of mTOR substrate phosphorylation. This highlights the importance of considering the enormous complexity in how mTOR is used by different cell types in different states for different, multifunctional purposes (49, 51, 97) and the unique nature of F17’s dysregulation of mTORCs.
These observations prompted us to examine the effects of culture conditions and cell state on protein production by iF17 in other cell types. Beginning with primary NHDFs that were either growth arrested in low-serum medium (0.2% FBS) or cycling in normal-serum medium (5% FBS) (98), we again observed no notable autophagic responses to infection under either condition, and yet, clear differences in mTOR substrate phosphorylation became evident (Fig. 5A). In growth-arrested NHDFs, WT VacV caused robust increases in p70S6K and 4E-BP1 phosphorylation, while intermediate mTOR activation in response to iF17 infection was again evident, in line with observations in THP1s described above (Fig. 3A) and as reported previously (37). In cycling NHDFs, however, the basal level of mTOR activity in uninfected cells was higher (Fig. 5A). Under these conditions, the changes in mTOR substrate phosphorylation caused by infection were both subtle and complex. While moderate 4E-BP1 phosphorylation was observed with both viruses, modest suppression of p70S6K phosphorylation was consistently observed only during infection with WT VacV (Fig. 5A). We verified as described below that this is specific to cycling NHDFs only when F17 is expressed in the context of infection, and this observation suggests that cycling NHDFs are in a state that partially resembles that of immortalized MRC5s. This further highlights the importance of considering cell type and state in understanding the effects of F17-mediated mTOR dysregulation. In particular, the clearest phenotypes arise in resting primary NHDFs and THP1 cells, which are arguably the most biologically relevant conditions across cell types examined in this study. However, despite this complexity in how mTOR dysregulation manifests in terms of mTOR substrate phosphorylation in NHDFs cultured under different conditions, the outcomes in terms of the effects on ISG responses and viral-protein production were again in line with those of other cell types examined above. Regardless of culture conditions, similar induction of ISGs occurred in response to iF17 infection, while ISG levels were decreased in the presence of F17 during WT infection (Fig. 5A). Moreover, differences in the accumulation of postreplicative viral proteins during WT versus iF17 infection were notably larger in growth-arrested than in cycling NHDFs (Fig. 5A), similar to correlations that emerged across MRC5 cells earlier (Fig. 4). Cumulatively, these findings suggested that the primary cause of defects in postreplicative-protein accumulation in the absence of F17 was not ISG responses or autophagy but, rather, the growth status of the host cell, which determines the basal levels of mTOR and protein synthesis activity. While the iF17 virus can piggyback off these host activities to produce viral proteins to varying levels, a critical function of F17 is to dysregulate mTOR in order to uncouple its complex regulatory networks while driving protein synthesis. Moreover, these data further suggested that the translationally hyperactivated state of many transformed cell lines may underlie the apparently dispensable nature of F17 for viral-protein synthesis in previous studies (47).
FIG 5.

Cell state and basal mTOR activity determine the degree of defects in late viral-protein accumulation in the absence of F17. (A) Primary NHDFs were maintained as confluent cultures for 72 h in 0.2% FBS medium (Growth-arrested) or maintained in normal growth medium (Cycling). Cultures were then mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for 30 h. Whole-cell lysates were subjected to Western blotting with antibodies to the indicated proteins. Fast-migrating nonphosphorylated (0) and slower-migrating species of differently phosphorylated (P) forms of 4E-BP1 or p70S6K are shown. (B and C) HEK293A (B) or BSC40 (C) cells were mock infected (M) or infected with WT or iF17 virus at an MOI of 5 for 24 h. Cells were treated with DMSO or PP242 (2.5 μM) at the time of infection. Whole-cell lysates were subjected to Western blotting. (D) mTOR activity and responses in normal and transformed cell lines. NHDF, HEK-293A, and BSC40 cells were cultured for 3 days in medium containing 0.2% FBS (St. [serum starved]). Cells were then stimulated with 10% FBS for 1 h. Cultures treated with the mTOR inhibitor PP242 for 1 h are also included. Note that, unlike NHDFs, transformed cell lines do not significantly repress mTOR under low-serum conditions, nor do they significantly activate mTOR in response to FBS stimulation. PP242-treated samples demonstrate that mTOR does regulate 4E-BP1, as well as autophagy regulators, in transformed cells. int, intermediate; U>, unprocessed A10; P>, processed A10. All data are derived from and representative of at least 3 biological replicates.
We recently showed that the iF17 virus produces normal levels of late viral proteins in HEK293A and BSC40 cells (37), in line with the original characterization of this mutant in the commonly used BSC40 cells (47). Our report suggested that this was likely due to the lack of cGAS expression and ISG responses in these cells. However, new data described above revealed that cGAS-mediated ISG responses did not specifically control protein production during iF17 infection and pointed instead to the mTOR activation and protein-synthetic status of the host cell. To test this and the potential contribution of mTOR, we infected HEK293A or BSC40 cells in the presence or absence of mTOR inhibitors. In doing so, we found that in DMSO solvent control-treated HEK293A cells, mTOR activation by WT VacV could be detected similarly to the results for THP1 and NHDF cells, as could intermediate activation during iF17 infection (Fig. 5B). Notably, this was more readily detected using conventional band-shifting approaches that assess all of the phosphorylated species of each protein, while the results for phosphospecific antibodies suggested that mTOR was activated equally by both WT and iF17 infection. This suggests that some aspects of mTOR responses are retained in HEK293A cells, albeit only to a modest degree. Moreover, as HEK293A cells do not mount an ISG response to iF17 infection (37), these changes in mTOR activity further support the notion derived from cGAS knockout THP1s as described above that broader changes in cell state caused by iF17 infection feed into and affect the mTOR pathway. In the case of HEK-293A cells, this iF17-mediated activation is likely to be relatively robust due to the high level of viral-protein production that is supported by transformed cells, which in turn feeds into and influences mTOR activity. In contrast, in BSC40s, basal mTOR substrate phosphorylation was extremely high even in uninfected cells, preventing detection of any changes that might occur during infection by either virus (Fig. 5C). These observations highlight the limited utility of transformed cell lines in understanding how viruses manipulate host signaling pathways in more relevant contexts. Further highlighting their relative hyperactivation, HEK293A and BSC40 cells could not be efficiently serum starved and mTOR responses to serum stimulation were modest at best, in stark contrast to the robust responsiveness of NHDFs (Fig. 5D). In line with the broader translational hyperactivation of transformed cell lines (88), both WT and iF17 viruses produced similar levels of early, intermediate, and late viral proteins in both HEK-293A and BSC40 cells. In addition, both viruses now showed an equal dependence on mTOR activity for maximal viral-protein production (Fig. 5B and C). Notably, mTOR inhibition had only modest effects on early proteins, while all postreplicative proteins tested exhibited notable dependencies on mTOR activity for their accumulation, similar to observations in other cell types described above. Moreover, mTOR inhibition did not affect A10 processing but simply repressed the levels of A10 accumulation; processing was difficult to detect in HEK293A cells infected with WT virus due to the combined effects of reduced protein levels and continued processing of this protein into distinct signals (Fig. 5B), but this was readily seen in BSC40 cells (Fig. 5C). This suggests that virion maturation events requiring F17 are distinct from those involving control of mTOR. Overall, these approaches showed that defects in protein accumulation in the iF17 virus could be rescued to a significant extent in actively cycling cells and completely rescued in translationally hyperactivated transformed cell lines.
Rescue of iF17 suggests regulated partitioning of F17 functions during infection.
As mentioned earlier, iF17 harbors a Lac repressor system that enables IPTG-mediated induction of F17 expression (Fig. 6A) (47, 74). In our hands, IPTG readily induced F17 expression in BSC40 cells infected with the iF17 virus and restored the processing of A10 that is associated with the assembly of virus particles (Fig. 6B). However, IPTG-mediated induction of F17 expression was undetectable in normal cells, such as NHDFs (Fig. 6C). To address this and to rule out the possibility that the absence of thymidine kinase (TK) expression in the iF17 mutant might contribute to some of the phenotypes observed with this virus, we generated an “expression revertant” virus stock (iF17-R). Notably, studies using the iF17 virus report low levels of F17 expression and virus replication (46, 47, 74), and yet, the source of this replication remains unclear. This likely either reflects a low level of leakiness of the repression system across all viral genomes or the emergence of a low percentage of escape mutants. Given the essential nature of F17 for virus replication combined with the nature of the regulatory system that requires a Lac repressor protein to be expressed and a LacO gene repressor binding site to be maintained, we postulated that growing stocks in the absence of IPTG would put high selective pressure on iF17 to generate escape mutants. Remarkably, IPTG-resistant virus emerged which had a minimal lag compared with the growth of normal virus stocks and showed the restoration of significant levels of F17 expression (Fig. 6C). For our experiments, we used the mixed pool of expression revertants to avoid basing any findings on any one particular isolate. However, to get a sense of how these expression revertants may have formed, we PCR amplified and sequenced the TK and F17 regions to determine the predominant mutations present in this stock. Sequencing showed that the F17 promoter, LacO gene repressor binding site, and F17 open reading frame remained unchanged (Fig. 7A). In contrast, we detected sequence variations on either side of the p7.5 promoter and a large truncation of the coding region for the LacO gene repressor, leaving behind only a small portion of the DNA binding domain and lacking essential multimerization and regulatory regions (Fig. 7B and C). This suggested that the predominant mechanism by which expression revertants formed was through mutations in the promoter that likely suppress the expression of the repressor protein and/or mutations resulting in the loss of large portions of the LacI gene repressor open reading frame.
FIG 6.

IPTG-mediated induction of F17 expression is cell-type specific. (A) Schematic of the Lac repressor system in the iF17 virus. The LacI gene repressor under the control of an early/late p7.5 promoter is inserted into the TK gene. The LacO binding site represses F17 expression in the absence of IPTG. ORF, open reading frame. (B) BSC40 cells were infected at an MOI of 5 for 24 h with WT or iF17 viruses in the presence or absence of 5 mM IPTG. Note that restoration of F17 expression in iF17 viruses with IPTG treatment restores processing of A10. (C) Growth-arrested NHDFs were infected at an MOI of 3 for 30 h with the indicated viruses in the presence or absence of 5 mM IPTG. S.E., short exposure; int, intermediate; U>, unprocessed A10; P>, processed A10. Fast-migrating nonphosphorylated (0) and slower-migrating species of differently phosphorylated (P) forms of 4E-BP1 or p70S6K are shown. All data are derived from and representative of at least 3 biological replicates.
FIG 7.

Map of mutations detected in the iF17-R stock. (A) Sequence alignment for LacO gene repressor binding site (red arrow) and F17 open reading frame (yellow arrow). Canonical TAAAT motif in the late promoter is underlined. (B) The partial sequences of the p7.5 early/late promoter (red arrow) and LacI gene repressor (yellow arrow) flanked by left and right TK sequences (orange arrows) at the insertion site. Restriction sites used in the original generation of the vector are shown. Boxed regions highlight sequence divergence detected in the iF17-R stock at the p7.5 promoter or loss of large portions of the LacI gene open reading frame. (C) Sequence alignment and predicted structure of the LacI gene repressor. Red is the N-terminal DNA binding domain, blue is the core regulatory region, green is the C-terminal tetramerization domain, and yellow represents the partial DNA binding domain that remains in the iF17-R virus.
Infection of growth-arrested NHDFs revealed that the iF17-R virus phosphorylated 4E-BP1 and p70S6K, suppressed ISG expression, and enhanced the production of postreplicative viral proteins compared with the levels seen for the parental iF17 virus (Fig. 6C). Notably, suppression of ISG responses and restoration of postreplicative-viral-protein production did not reach the levels observed for WT virus, likely due to lower expression of F17 by the iF17-R virus. While the absence of TK has been shown to reduce virus replication in normal cells in vivo (99), studies suggest there is little requirement for TK in cultured cells, including resting normal cells (100, 101). However, although we cannot completely rule out a contribution from TK to overall virus replication in the iF17 mutant background, our data clearly show that F17 mediates mTOR activation and reveal a dose-dependent correlation between F17 expression and the extent of ISG suppression, as well as viral-protein production across all three viruses, regardless of genetic background. This included the restoration of A10 expression and processing. Similar results were also observed in differentiated THP1 cells, although due to low levels of expression, the processing of A10 was again more difficult to discern in these cells (Fig. 8A). In addition, luciferase assays in these reporter THP1 cells further showed that, although significant IRF activation could be detected in cells infected with iF17-R, the levels of activation were notably lower than with the iF17 virus (Fig. 8B). As such, there was once again a clear dose-dependent correlation between levels of F17 expression and ISG responses in THP1 cells across these three different viruses.
FIG 8.

Restoration of F17 expression and rescue of phenotypes in the iF17 virus using multiple strategies. (A) Differentiated THP1 cells were infected at an MOI of 5 for 48 h with the indicated viruses. S.E., short exposure; int, intermediate; U>, unprocessed A10; P>, processed A10. Note that low-level expression of A10 combined with its processing during iF17-R infection makes A10 detection difficult. Fast-migrating nonphosphorylated (0) and slower-migrating species of differently phosphorylated (P) forms of 4E-BP1 or p70S6K are shown. (B) Differentiated THP1s were infected at an MOI of 5 with the indicated viruses for 48 h. Supernatants were subjected to Lucia luciferase reporter assays to measure IRF activity (n = 3). RLU, relative light units. Error bars show SEM. ****, P ≤ 0.0001; N.S, not significant. Statistical analysis was performed using ANOVA and Tukey’s test for multiple post hoc comparisons. (C) MRC5 cells were infected at an MOI of 5 with the indicated viruses in the presence or absence of 5 mM IPTG for 48 h. L.E., long exposure. All data are derived from and representative of at least 3 biological replicates.
Interestingly, unlike the results for NHDFs or THP1s, we found that IPTG-mediated induction worked to some extent in MRC5 cells infected with iF17 virus (Fig. 8C). In line with the findings shown in Fig. 4, in the absence of IPTG, WT virus suppressed mTOR signaling and ISG responses in MRC5s, while moderate expression of F17 by iF17-R resulted in intermediate phenotypes in this regard. With IPTG treatment, the original iF17 virus was induced to express a lower level of F17 and caused a more moderate reduction in ISG expression than either the WT or iF17-R virus. However, IPTG-treated cells infected with iF17 exhibited more robust mTOR suppression and produced moderately more viral proteins than untreated iF17-R, despite expressing lower levels of F17. Interpreting these two specific phenotypes was complicated by the fact that IPTG increased mTOR signaling in uninfected cells, which may help boost viral-protein production indirectly and confounds our ability to make direct correlations between F17 expression and mTOR activity under conditions with or without IPTG. In addition, IPTG treatment further increased F17 expression by iF17-R, in line with sequencing data suggesting that our revertant pool harbors a mixture of viruses, including some that would likely remain subject to some degree of repression, which would explain why this revertant does not fully restore F17 expression. Again, mTOR activity was suppressed by the iF17-R virus, albeit with the caveat of effects of IPTG itself on mTOR signaling. Beyond mTOR regulation, with increased F17 expression in IPTG-treated cells infected with iF17-R, there was a greater suppression of ISG levels and an increase in postreplicative-viral-protein accumulation. Indeed, despite the confounding effects of IPTG on mTOR signaling and the complexity this generated, this wide range of conditions revealed a striking correlation between F17 expression and the degree of phenotypic rescue, regardless of viral genetic background.
Finally, as an independent approach to rescuing the iF17 virus, we infected NHDFs expressing Flag-tagged forms of F17. Notably, F17 is phosphorylated at Ser53 and Ser62, but this phosphorylation is not required for virion maturation (46). While its function remained mysterious, we recently showed that phosphorylation at these sites serves to enhance F17 binding to Raptor and Rictor to dysregulate mTOR (37). Importantly, previous studies showed that an N-terminal Flag tag, which is negatively charged, mimics F17 phosphorylation (46). As such, Flag tagging enabled us to generate an effectively “constitutively phosphorylated” form of WT F17, alongside a second phosphomimetic S53/62E mutant, both of which could be observed to activate mTOR in uninfected NHDFs (Fig. 9A). It is important to point out that F17 did not express well in growth-arrested NHDFs. This limited us to studying cycling NHDFs, wherein effects on mTOR are relatively subtle and, in the context of infection, F17 expression leads to partial suppression of p70S6K (Fig. 5A). Indeed, low levels of p70S6K suppression were detected in Flag-F17-expressing NHDFs infected with iF17, as well as in Flag control or Flag-F17-expressing cells infected with WT virus (Fig. 9B). As such, outside the context of infection, F17 is sufficient to modestly activate mTOR in cycling NHDFs, but in the context of infection, the effects of F17 result in modest mTOR suppression. This further illustrates the context-specific effects of F17 on mTOR signaling that depend on cell state, with that state being influenced by infection itself. However, despite these limitations to studying mTOR using this approach, the expression of Flag-F17 or Flag-F17-S53/62E enhanced both postreplicative-viral-protein production and the ability to suppress ISG responses during infection with iF17 (Fig. 9B). We also observed that, although viral-protein production was restored in iF17-infected cells by exogenous expression of Flag-F17, processing of A10 was not. This may be due to insufficient Flag-F17 levels to support virion maturation. However, more of the unprocessed form of A10 was observed in both of the Flag-F17-expressing lines infected with WT VacV than in Flag-expressing controls (Fig. 9B). Although preliminary at this point, this observation suggests that the phosphorylated form of F17 does not function in and may even impair virion maturation and that phosphorylation might direct a subpopulation of F17 away from maturing virions specifically to control mTOR.
FIG 9.
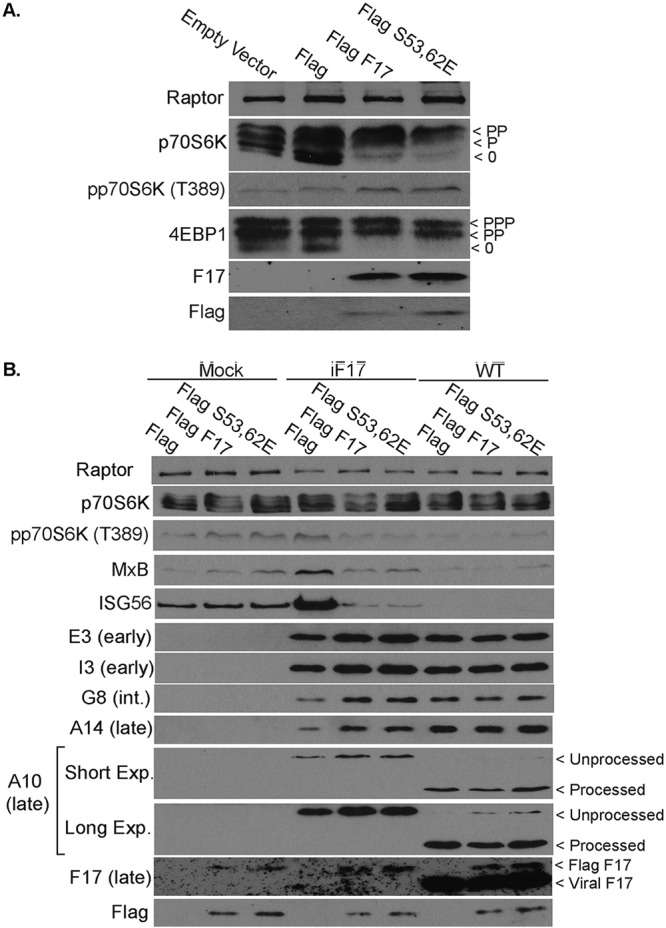
Rescue of phenotypes in the iF17 virus in NHDFs expressing Flag-tagged F17. (A) Stable pools of NHDFs transduced with empty vector or expressing Flag control, Flag-F17, or Flag-F17-S53/62E proteins. Fast-migrating nonphosphorylated (0) and slower-migrating species of differently phosphorylated (P) forms of 4E-BP1 or p70S6K are shown. (B) NHDFs stably expressing Flag control or Flag-tagged forms of F17 or F17-S53/62E were mock infected or infected at an MOI of 5 with iF17 or WT virus for 30 h. The combined effects of exogenous F17 and infection result in complex p70S6K phosphorylation patterns. However, F17 expression rescues iF17 from ISG responses and restores postreplicative-viral-protein production, but not processing of A10. In WT-infected cells, Flag-F17 expression increases the levels of unprocessed A10. Long or Short Exp., long or short exposure. All data are derived from and representative of at least 3 biological replicates.
DISCUSSION
Similar to recent studies of attenuated MVA using either gene knockouts in murine dendritic cells or small hairpin RNA (shRNA)-mediated depletion in human THP1 cells (39, 40), here we find that cGAS or STING knockout in THP1 cells prevents IRF activation and host responses to VacV lacking F17. We further find that lung fibroblast MRC5 cells express far lower levels of cGAS than differentiated THP1s or macrophages, and yet, cGAS remains central to the ability of MRC5s to mount a full response to VacV infection. Thus, our findings extend the importance of cGAS-mediated sensing of poxviruses to a variety of natural target cell types. Intriguingly, residual responses that persist in MRC5 cells lacking cGAS could relate to our observation that these cells express higher levels of sensors like DNA-PK, which also initiates responses to VacV infection (33–35). While the exact sensor(s) involved in this secondary response remains to be determined, our findings highlight the diverse array of sensing mechanisms that poxviruses likely encounter during infection in vivo. There are also some notable differences between the extents of responses induced by MVA and iF17 (vRR10K) viruses. MVA lacks or has mutations in several genes, in addition to being unable to make late viral proteins like F17. Studies have shown that cells infected with MVA not only activate STING, they also secrete high levels of IFNs and chemokines (39, 40, 102). In contrast, the iF17 virus only exhibits defects as it transits to late stages of infection in normal cells, and despite activating the cGAS-STING pathway, it retains the ability to block IFN release (37) and, as we show here, also prevents NF-κB activation. This demonstrates that F17 has a specific role in modulating cGAS-STING signaling to IRFs but that it is not required to control other sensing pathways, downstream effectors, or the broader antiviral response. Furthermore, while the manuscript was under review, a second poxvirus-encoded cGAS antagonist was identified, B2 (also termed “poxin”) (103). B2 is expressed early and degrades cGAMP. Intriguingly, in unpublished experiments, we had observed that the addition of cGAMP to iF17-infected THP1 cells lacking cGAS failed to induce ISG responses to levels observed in uninfected cells, but at high doses, some activation was detected. This supports the notion that B2 is expressed as an early-stage antagonist of cGAS and that this factor is also expressed by the iF17 virus. However, unlike F17, B2 is not a conserved poxvirus gene and is notably absent in viruses like VarV (103). Based on these cumulative findings, a model can be proposed wherein B2 helps to counter initial cGAS activation during the early onset of DNA replication, but with the enormous amounts of DNA that are produced as infection progresses, additional functions, including those of F17, are required to drive cGAS degradation and ensure that ISGs are not eventually activated. Indeed, despite the armory of early genes, potent ISG activation occurs in the absence of F17, suggesting that B2 alone is not sufficient to protect against cGAS for the full duration of infection.
Despite complete ablation or robust impairment of ISG responses in THP1 and MRC5 cells, respectively, cGAS knockout did not rescue defects in late-viral-protein production by the iF17 mutant. It is very common for viral mutants lacking specific immunomodulators to exhibit no phenotype in tissue culture because, as discussed above, poxviruses express an enormous range of antagonists of innate immune responses and ISGs, thereby limiting or completely negating the impact of host responses that occur in the absence of any single immunomodulator (102). Our findings using cGAS or STING knockouts also offer new insights into some of our own recent observations regarding cGAS functionality. We found that the iF17 mutant exhibits no defects in late-viral-protein synthesis in HEK293A or BSC40 cells, and we speculated that this was likely due to the lack of cGAS expression in these cells (37). However, reintroduction of cGAS into HEK293A cells failed to reduce late-viral-protein production during infection with the iF17 mutant. We proposed that this might suggest that additional cGAS cofactors or regulators are absent in HEK293A cells, or it could reflect the fact that cGAS exhibits significant levels of nuclear localization in these cells (37). Our findings described here now show that cGAS-mediated responses do not have a significant impact on late-viral-protein production by iF17 in either THP1 or MRC5 cells and suggest that the ability of iF17 to produce high levels of late viral proteins in transformed cells is not due to their lack of antiviral responses but, rather, to their highly activated mTOR and protein-synthetic activities. As such, although F17-mediated dysregulation of mTOR causes degradation of cGAS and contributes to suppressing ISG activation, this represents just one aspect of the hugely complex and often redundant strategies deployed by poxviruses to counter antiviral responses.
Finally, our findings reveal that, independent of immune evasion strategies, a second function of mTOR dysregulation is to maximize protein production in normal cells, particularly resting cells. Indeed, phenotypes of the iF17 virus were rescued in translationally hyperactivated cell lines in an mTOR-dependent manner. And yet, the timing of F17 expression is intriguing, as it begs the question, why not express this protein early to also maximize early-protein production and at the same time degrade cGAS before DNA replication even begins? The answer may lie in how it functions and its complex cell-specific effects, as F17 does not simply activate mTOR, but rather, it dysregulates mTOR (37). On one level, our data suggest that at least some early viral proteins do not have a high requirement for mTOR, negating the need for such a drastic approach to hyperactivate mTOR at early stages. There may be a low demand on protein synthesis early in infection, or early transcripts themselves may dictate a low requirement for mTOR-mediated translational stimulation. Indeed, many host proteins have a low requirement for mTOR activity, which exerts widely differing stimulatory effects on individual mRNAs (37, 49, 88, 89, 98). Late in infection, mTOR dysregulation may serve not only to counteract host responses but to also sustain a higher demand for protein production critical for virion assembly. On another level, in some cell types, such as MRC5s, mTOR dysregulation manifests as reduced phosphorylation of translational regulators like 4E-BP1 and p70S6K. This inhibitory effect may need to be avoided early in infection for the virus to successfully replicate in specific cell types. In this biological context, mTOR suppression late in infection may contribute to host shutoff late in infection. Interestingly, postreplicative viral mRNAs harbor unusual 5′ leader elements that enable both cap-dependent and -independent translation, and VacV also modifies host ribosomes in unusual ways to maximize translation (48, 104–106). While this unusual mode of initiation may help sustain their translation in specific cell types or under various stress conditions, our data show that maximal accumulation of late viral proteins nonetheless remains dependent on mTOR activity, which controls a wide range of translation factors beyond those that control initiation alone (48). It is also important to consider that mTOR functions locally within the cell in complex manners, and in cells where mTOR is partially repressed, the pool of active translation factors that are not repressed may continue to be used for virus replication. Indeed, poxviruses redistribute critical translation factors and ribosomes to viral factories where late viral proteins are made (37, 105, 107–109). As such, dysregulated mTOR likely has complex and varied roles during poxvirus infection, just as the normal mTOR circuitry has in uninfected cells. What is clear, however, is that regardless of cell type, mTOR dysregulation by F17 is needed to maximize the production of viral proteins late in infection when there is a high protein-synthetic burden on the cell, and F17 expression is perfectly timed to accomplish this. Beyond translation, other aspects of host metabolism are broadly remodeled to facilitate late stages of poxvirus replication (110, 111), and F17 may also impact these processes. How mTOR dysregulation contributes to each of these varied processes during late-stage infection will undoubtedly be both exciting and complex to unravel in future studies.
MATERIALS AND METHODS
Cells and viruses.
THP1 Dual reporter knockout (KO) cells were obtained from Invivogen (WT, catalog identifier thpd-nfis; cGAS KO, catalog identifier thpd-kocgas; STING KO, catalog identifier thpd-kostg; IFI16 KO, catalog identifier thpd-koifi16). Unmodified THP1 cells were obtained from Thomas Hope, Northwestern University, as described previously (37). THP1 cells were cultured in RPMI 1640 (Gibco) containing 5% fetal bovine serum (FBS), penicillin-streptomycin solution (HyClone), and 2 mM l-glutamine (Corning). THP1 monocytes were counted using trypan blue, and 5 × 105 undifferentiated cells were used to seed single wells of 12-well plates for viral infections. Seeding of all THP1 cells used 10% Nu-serum growth medium (product number 355000; Corning) and 2 mM l-glutamine (Corning) in the absence of penicillin-streptomycin, and differentiation into macrophages was performed by incubating cells with 30 ng/ml phorbol 12-myristate 13-acetate (PMA) (product number P8139; Sigma) for 48 h. Nu-serum medium devoid of PMA was then used to maintain THP1s for a further 24 h. Jeremy Luban, UMass School of Medicine, kindly provided human embryonic kidney (HEK) 293A cells. Ian Mohr, NYU, kindly provided BSC40 cells. Primary normal human dermal fibroblasts (NHDFs) were obtained from Lonza (catalog number CC-2509). NHDF, BSC40, and HEK293A cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Fisher Scientific) with 5% FBS, 2 mM l-glutamine, and penicillin-streptomycin at 37°C and 5% CO2. NHDFs were subjected to growth arrest by washing confluent cultures three times in PBS and maintaining cells in DMEM with 0.2% FBS, 2 mM l-glutamine, and penicillin-streptomycin for 3 days (98). Telomerase-expressing MRC5 fibroblasts (created via lentiviral transduction as previously described [112]) were cultured as described for NHDFs. Generation of cGAS knockout MRC5s is described below.
Stewart Shuman, Memorial Sloan Kettering, kindly provided wild-type (WT) VacV (Western Reserve). Bernard Moss, NIH, kindly provided iF17 virus (vRR10K). Virus stocks were grown and titrated using BSC40 cells as described previously. Briefly, BSC40s were infected at an MOI of 0.01 until 90% to 100% cytopathic effect was observed. Infected cells were then harvested and freeze-thawed three times. Cell debris was removed by centrifugation, and virus titer was determined by serial dilution and plaque assays on BSC40 cells (107). iF17 (vRR10K) was grown in the presence of 5 mM IPTG to induce F17 expression to generate virus stocks (37, 47). To generate the revertant (iF17-R), the original iF17 virus was simply grown under the same conditions in the absence of IPTG. In experimental setups, VacV infections were performed for the time indicated at an MOI of 5 unless otherwise indicated.
Sequencing analysis of iF17-R and structure modeling.
Genomic VacV DNA was extracted from BSC40 cells infected with iF17-R at an MOI of 5 for 24 h using a genomic DNA minikit (catalog number IB47201; IBI). Regions spanning the F17 and TK genes were amplified by PCR using the following primers: F17 Flanking, forward (GATCTCCATCAAATGCCAGAC) and reverse (AGGTACATGAAATAAGTGCCAG), and TK Flanking (100), forward (ATGAACGGCGGACATATTCAG) and reverse (TTATGAGTCGATGTAACACTTTC).
PCR products were resolved on agarose gels, purified as described below, and sequenced at the Northwestern University core sequencing facility. Sequencing results were analyzed and aligned using the Molecular Evolutionary Genetics Analysis software (MEGA-X), using the published VacV WR sequence with GenBank accession number AY243312.1, the p7.5 late/early promoter sequence with GenBank accession number X55811.1, and the Escherichia coli LacI sequence with GenBank Reference Sequence accession number NC_000913.3 (113–115). Schematics of gene and protein sequences were produced using SeqBuilder 14 with Lasergene 14 (DNAStar).
The predicted TK monomer tertiary structure was generated using the RaptorX server and approaches for tertiary-structure prediction detailed previously (116). Structure visualization and image rendering were performed using Jmol (Jmol: an open-source Java viewer for chemical structures in a three-dimensional view [http://www.jmol.org/]).
CRISPR-Cas9-mediated targeting of cGAS.
Clustered regularly interspaced short palindromic repeat (CRISPR)-mediated cGAS knockout in MRC5/hTert cells was performed via lentiviral delivery of CRISPR-associated protein 9 (Cas9) and the following guide RNAs (gRNAs), which were cloned into the BsmB1 site of pLentiCRISPRv2 (Addgene) with subsequent sequence confirmation: 5′ CAC CGA GAC TCG GTG GGA TCC ATC G 3′ and 5′ AAA CCG ATG GAT CCC ACC GAG TCT C 3′. For lentiviral production, 293T cells were seeded at 6.8 × 105 cells in a 10-cm dish and grown for 24 h prior to transfection with the following plasmids using Fugene 6 transfection reagent (Promega): 2.6 μg gRNA-containing pLentiCRISPRv2 (Addgene), 0.25 μg pVSVg, and 2.5 μg PAX2. After 24 h, the culture medium was replaced with 4 ml of fresh medium, and after a further 24 h of incubation, the lentivirus-containing medium was collected, passed through a 0.45-μm filter, and applied to MRC5 fibroblasts with the addition of Polybrene (10 μg/ml). The medium was refreshed after 24 h, and the cells were allowed to recover for an additional 48 h prior to selection with 1 μg/ml puromycin (VWR). Following transduction with control or gRNA-expressing lentivirions and subsequent puromycin selection, cells were clonally selected by dilution and seeding of individual cells into the wells of 96-well plates.
For genotyping, after expansion, cells were harvested via scraping, spun down at 1,000 × g for 5 min, washed with phosphate-buffered saline (PBS), and spun down again. Cells were resuspended in 500 μl lysis buffer (100 mM NaCl, 100 mM Tris at pH 8, 25 mM EDTA, 0.5% SDS, 0.1 mg/ml proteinase K, 40 μg/ml RNase A) and incubated at 55°C overnight. An equal volume of phenol-chloroform-isoamyl alcohol (25:24:1) was added to the lysates before centrifuging at 12,000 × g and taking the supernatant. The aqueous phase was chloroform extracted (24:1 chloroform-isoamyl alcohol) prior to precipitation with 0.1 volume of 3 M sodium acetate and 2 volumes of 95% ethanol at −20°C. The genomic DNA was centrifuged for 15 min at 16,000 × g, washed with 70% ethanol, and centrifuged again for 15 min at 16,000 × g. After decanting the 70% ethanol, the DNA pellet was resuspended in 60 μl water. Genomic DNA was amplified via touchdown PCR using the primers F1 (5′ GAC AGG GGC ACG GAT TGC CT 3′) and R1 (5′ GGC GGA GGT CTT GGC TTC GT 3′), and specific genomic mutations were identified via Sanger sequencing. Loss of cGAS protein expression was also confirmed by Western blotting, as shown in the figures.
NF-κB and IRF reporter assays.
Reporter assays were performed using THP1-Dual reporter cells obtained from Invivogen (described above) that stably express reporter genes encoding Lucia luciferase and secreted embryonic alkaline-phosphatase (SEAP). Quanti-Luc (Invivogen) and Quanti-Blue (Invivogen) were used to quantify secreted Lucia luciferase and SEAP proteins, respectively. The Lucia luciferase reporter is under the control of an ISG promoter (ISG54) and five IFN-stimulated response elements (ISREs). SEAP is under the control of an IFN-β promoter fused to NF-κB transcriptional response elements. This enables study of both IRF and NF-κB pathway activity in a single assay. Quanti-Luc and Quanti-Blue protocols were performed as described by Invivogen. Briefly, the luminometer was primed with Quanti-Luc solution and set to parameters of 50-μl injection and end measurements of 4-s start time and 0.1-s reading time. Twenty-microliter amounts of cell supernatant were added to 96-well plates and read on the luminometer. For Quanti-Blue solution, 180 μl of solution was added to each well of a flat-bottom 96-well plate. Twenty-microliter amounts of cell supernatants were then added to the wells containing Quanti-Blue solution and incubated for 1 h at 37°C. The optical density was measured at 655 nm using a 96-well plate microplate reader.
Inhibitor treatment.
PP242 (EMD Millipore, MilliporeSigma, or Calbiochem) was used at a concentration of 2.5 μM. Stocks at 1,000× were diluted in medium and added to cultures at the times indicated in figure legends. DMSO was used as the carrier for inhibitors, and equal amounts of DMSO were used across all drug treatment experiments as controls.
Antibodies.
Antibodies to the following proteins were used: Raptor (catalog number 2280S; Cell Signaling Technology), PKR (catalog number12297S; Cell Signaling Technology), MxB (catalog number 13278-1-AP; Proteintech), ISG56 (catalog number 14769S; Cell Signaling Technology), cGAS (catalog number 26416-1-AP; Proteintech), IFI16 (catalog number 14970S or catalog number ab55328; Cell Signaling Technology or Abcam, respectively), STING (catalog number 13647S; Cell Signaling Technology), pSTING (catalog number 85735S; Cell Signaling Technology), TBK1 (catalog number 3504T; Cell Signaling Technology), pTBK1 (catalog number 5483T; Cell Signaling Technology), VacV A10 (catalog number IT-012-010M1; Immune Tech), VacV E3 (1:5,000; Jingxin Cao), VacV I3 (1:3,000; David Evans), VacV A14 (1:3,000; Yan Xiang), VacV F17 (1:10,000; Paula Traktman), VacV D8 (Paula Traktman; 1:3,000), VacV G8 (1:5,000; Jingxin Cao), DNA-PK (catalog number 4602S; Cell Signaling Technology), p70S6K (catalog number 2708S or catalog number 9202S; Cell Signaling Technology), phospho-p70S6K(T389) (catalog number 9234S; Cell Signaling Technology), 4EBP1 (catalog number 9644S; Cell Signaling Technology), phospho-4EBP1(S65) (catalog number 13443S; Cell Signaling Technology), LC3A/LC3B (catalog number 12741S; Cell Signaling Technology), ULK1 (catalog number 8054T; Cell Signaling Technology), phospho-ULK1(S757) (catalog number 6888T; Cell Signaling Technology), and Flag-M2 (Sigma, catalog number SLBW9109). Primary antibodies were diluted at 1:1,000 unless otherwise stated. Secondary antibodies were horseradish peroxidase (HRP)-conjugated mouse IgG (1:3,000; GE Healthcare UK or MilliporeSigma) and rabbit IgG-HRP (1:3,000; GE Healthcare UK or MilliporeSigma).
Generation of stable NHDF pools.
Flag, Flag-F17, and Flag-F17(S53,62E) genes were subcloned into the pQCXIP retrovirus expression vector. Briefly, F17 and F17(S53,62E) genes were amplified from plasmids provided by Paula Traktman, using primers containing an N-terminal Flag tag and the AgeI and BamHI restriction sites as described previously (37, 46). PCR products were resolved in a 1% agarose–TAE (Tris base, acetic acid, and EDTA buffer) gel at 110 V for 1 h and purified using Qiagen QIAquick gel extraction kits. PCR products were digested with AgeI and BamHI followed by ligation into a shrimp alkaline phosphatase (SAP)-treated pQCXIP plasmid backbone that had been digested with AgeI and BamHI. Competent DH5α bacterial cells were then transformed with the ligation reaction by heat shock treatment at 42°C for 1 min followed by 2 min on ice. A 1-ml volume of LB broth was added to the mixture, and the mixture incubated at 37°C for 1 h. Transformed bacteria were plated on LB-ampicillin agar plates and incubated overnight at 37°C. Single colonies were grown overnight at 37°C in LB-ampicillin broth. Plasmids were isolated using plasmid extraction QIAprep spin miniprep kits (Qiagen). Plasmid sequencing and diagnostic digests were performed to verify inserts.
Phoenix-Ampho cells were grown in a 10-cm dish with antibiotic-free DMEM–5% FBS medium. Ten milligrams of pQCXIP Flag, Flag-F17, or Flag-F17(S53,62E) plasmid was transfected using Lipofectamine 3000 transfection reagent. At 24 h posttransfection, the medium was changed to fresh antibiotic-free DMEM supplemented with 5% FBS. Supernatants were harvested 48 h posttransfection, centrifuged at 4,000 rpm for 4 min to remove debris, and filtered through a 0.45-μM filter. Low-passage-number NHDFs were transduced with 2 ml Flag, Flag-F17, or Flag-F17(S53,62E) retroviral vectors. The viral inoculum was removed after overnight incubation, and the medium was replaced with fresh DMEM containing 5% FBS, penicillin-streptomycin, and 2 mM l-glutamine. After 24 h, the medium was replaced with growth medium containing puromycin (1 mg/ml) to select pools with stable expression. Stable NHDF pools were maintained in DMEM containing 5% FBS, 1% penicillin-streptomycin, 2 mM l-glutamine, and 0.2 μg/ml puromycin.
SDS-PAGE and Western blotting.
Whole-cell lysates were prepared by lysis in Laemmli buffer (62.5 mM Tris-HCl at pH 6.8, 2% SDS, 10% glycerol, 0.7 M β-mercaptoethanol, trace amount of bromophenol blue) and boiled for 3 min. Precision Plus Protein dual color standards (10 to 250 kDa) and protein extracts were resolved on polyacrylamide gels with 1× Tris-glycine-SDS running buffer. The gel percentages varied depending on the resolution requirements for different proteins. After SDS-PAGE, the separated proteins were transferred to nitrocellulose membranes using the Mini-Protean trans-blot module (GE Healthcare Life Sciences) at 57 V for 70 min, using a high-glycine transfer buffer. Membranes were blocked in 5% nonfat milk–Tris-buffered saline (TBS) containing 0.1% Tween (TBS-T), washed 3× for 5 min in TBS-T, and incubated at 4°C overnight with primary antibodies diluted in 3% bovine serum albumin–TBS-T. Membranes were again washed 3× for 5 min in TBS-T and incubated for 1 h at room temperature with the specific HRP-conjugated secondary antibody (1:3,000 dilution; GE Healthcare Life Sciences) in 5% nonfat milk TBS-T. Three final 5-min washes in TBS-T were carried out, followed by a 2-min incubation with Pierce ECL Western blotting substrate (Thermo Fisher Scientific) and exposure to X-ray film.
Densitometric measurements of LC3 processing and statistical analysis.
To determine the levels of LC3 processing, autoradiographs were analyzed using densitometry. All measurements were determined using the FIJI distribution of ImageJ (117). Background was removed, and the areas under density lines for LC3A and LC3B from a single sample were measured and summed. The percentage of processed LC3 (LC3B) was calculated against the summed total of LC3A and LC3B and averaged from the results of 3 independent experiments. All graphical and statistical analyses for densitometry and reporter assays were performed using GraphPad Prism, applying the statistical tests detailed in the figure legends in each case.
ACKNOWLEDGMENTS
We thank Paula Traktman, Bernard Moss, Yan Xiang, Jingxin Cao, David Evans, Nathaniel Gray, and David Sabatini for kindly sharing reagents. We thank Colleen Furey and Helen Astar for technical assistance.
This work was supported by the National Institutes of Health (NIH) through grant numbers R01AI127456 to D.W. and R01AI127370 to J.M., as well as by American Cancer Society (ACS) grant number RSG-15-049-01 to J.M. The authors declare no competing financial interests.
REFERENCES
- 1.Chan YK, Gack MU. 2016. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paludan SR, Bowie AG. 2013. Immune sensing of DNA. Immunity 38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. 2009. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A 106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahama M, Fukuda M, Ohbayashi N, Kozaki T, Misawa T, Okamoto T, Matsuura Y, Akira S, Saitoh T. 2017. The RAB2B-GARIL5 complex promotes cytosolic DNA-induced innate immune responses. Cell Rep 20:2944–2954. doi: 10.1016/j.celrep.2017.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q, Sun L, Chen ZJ. 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 9.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, Kranzusch PJ. 2019. Modular architecture of the STING C-terminal tail allows interferon and NF-kappaB signaling adaptation. Cell Rep 27:1165–1175.e5. doi: 10.1016/j.celrep.2019.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, Wittmann G, Latz E, Subklewe M, Hornung V. 2017. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 171:1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radoshevich L, Dussurget O. 2016. Cytosolic innate immune sensing and signaling upon infection. Front Microbiol 7:313. doi: 10.3389/fmicb.2016.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Z, Damania B. 2016. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19:150–158. doi: 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du M, Chen ZJ. 2018. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361:704–709. doi: 10.1126/science.aat1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, Fan Z. 2016. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol 17:369–378. doi: 10.1038/ni.3356. [DOI] [PubMed] [Google Scholar]
- 17.Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. 2018. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun 9:613. doi: 10.1038/s41467-018-02936-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ. 2013. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith GL, Talbot-Cooper C, Lu Y. 2018. How does vaccinia virus interfere with interferon? Adv Virus Res 100:355–378. doi: 10.1016/bs.aivir.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Xu Z, Fuhlbrigge RC, Pena-Cruz V, Lieberman J, Kupper TS. 2005. Vaccinia virus induces strong immunoregulatory cytokine production in healthy human epidermal keratinocytes: a novel strategy for immune evasion. J Virol 79:7363–7370. doi: 10.1128/JVI.79.12.7363-7370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickman HD, Reynoso GV, Ngudiankama BF, Rubin EJ, Magadan JG, Cush SS, Gibbs J, Molon B, Bronte V, Bennink JR, Yewdell JW. 2013. Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe 13:155–168. doi: 10.1016/j.chom.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiebe MS, Traktman P. 2007. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe 1:187–197. doi: 10.1016/j.chom.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scutts SR, Ember SW, Ren H, Ye C, Lovejoy CA, Mazzon M, Veyer DL, Sumner RP, Smith GL. 2018. DNA-PK is targeted by multiple vaccinia virus proteins to inhibit DNA sensing. Cell Rep 25:1953–1965.e4. doi: 10.1016/j.celrep.2018.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters NE, Ferguson BJ, Mazzon M, Fahy AS, Krysztofinska E, Arribas-Bosacoma R, Pearl LH, Ren H, Smith GL. 2013. A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. PLoS Pathog 9:e1003649. doi: 10.1371/journal.ppat.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. 2012. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Almine JF, O’Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, Unterholzner L. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meade N, Furey C, Li H, Verma R, Chai Q, Rollins MG, DiGiuseppe S, Naghavi MH, Walsh D. 2018. Poxviruses evade cytosolic sensing through disruption of an mTORC1-mTORC2 regulatory circuit. Cell 174:1143–1157.e17. doi: 10.1016/j.cell.2018.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai P, Wang W, Cao H, Avogadri F, Dai L, Drexler I, Joyce JA, Li XD, Chen Z, Merghoub T, Shuman S, Deng L. 2014. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog 10:e1003989. doi: 10.1371/journal.ppat.1003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Georgana I, Sumner RP, Towers GJ, Maluquer de Motes C. 2018. Virulent poxviruses inhibit DNA sensing by preventing STING activation. J Virol 92:e02145-17. doi: 10.1128/JVI.02145-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng WY, He XB, Jia HJ, Chen GH, Jin QW, Long ZL, Jing ZZ. 2018. The cGas-Sting signaling pathway is required for the innate immune response against ectromelia virus. Front Immunol 9:1297. doi: 10.3389/fimmu.2018.01297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu F, Niu Q, Fan X, Liu C, Zhang J, Wei Z, Hou W, Kanneganti TD, Robb ML, Kim JH, Michael NL, Sun J, Soong L, Hu H. 2017. Priming and activation of inflammasome by canarypox virus vector ALVAC via the cGAS/IFI16-STING-type I IFN pathway and AIM2 sensor. J Immunol 199:3293–3305. doi: 10.4049/jimmunol.1700698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh A, Shao L, Sampath P, Zhao B, Patel NV, Zhu J, Behl B, Parise RA, Beumer JH, O’Sullivan RJ, DeLuca NA, Thorne SH, Rathinam VAK, Li P, Sarkar SN. 2018. Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA sensor cGAS during DNA virus infection to limit interferon production. Immunity 50:51–63.e5. doi: 10.1016/j.immuni.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt FI, Bleck CK, Reh L, Novy K, Wollscheid B, Helenius A, Stahlberg H, Mercer J. 2013. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep 4:464–476. doi: 10.1016/j.celrep.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 46.Wickramasekera NT, Traktman P. 2010. Structure/function analysis of the vaccinia virus F18 phosphoprotein, an abundant core component required for virion maturation and infectivity. J Virol 84:6846–6860. doi: 10.1128/JVI.00399-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang YF, Moss B. 1991. Vaccinia virus morphogenesis is interrupted when expression of the gene encoding an 11-kilodalton phosphorylated protein is prevented by the Escherichia coli lac repressor. J Virol 65:6101–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meade N, DiGiuseppe S, Walsh D. 2018. Translational control during poxvirus infection. Wiley Interdiscip Rev RNA 10:e1515. doi: 10.1002/wrna.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saxton RA, Sabatini DM. 2017. mTOR signaling in growth, metabolism, and disease. Cell 168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wan W, You Z, Zhou L, Xu Y, Peng C, Zhou T, Yi C, Shi Y, Liu W. 2018. mTORC1-regulated and HUWE1-mediated WIPI2 degradation controls autophagy flux. Mol Cell 72:303–315.e6. doi: 10.1016/j.molcel.2018.09.017. [DOI] [PubMed] [Google Scholar]
- 51.Hatakeyama R, Peli-Gulli MP, Hu Z, Jaquenoud M, Garcia Osuna GM, Sardu A, Dengjel J, De Virgilio C. 2018. Spatially distinct pools of TORC1 balance protein homeostasis. Mol Cell doi: 10.1016/j.molcel.2018.10.040. [DOI] [PubMed] [Google Scholar]
- 52.Gonzalez A, Hall MN. 2017. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36:397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dibble CC, Cantley LC. 2015. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dibble CC, Manning BD. 2013. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rabanal-Ruiz Y, Otten EG, Korolchuk VI. 2017. mTORC1 as the main gateway to autophagy. Essays Biochem 61:565–584. doi: 10.1042/EBC20170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linke M, Fritsch SD, Sukhbaatar N, Hengstschlager M, Weichhart T. 2017. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett 591:3089–3103. doi: 10.1002/1873-3468.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, Lamming DW, Chen ZJ, Horng T, Yeretssian G, Green DR, Blander JM. 2017. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell 171:809–823.e13. doi: 10.1016/j.cell.2017.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hasan M, Gonugunta VK, Dobbs N, Ali A, Palchik G, Calvaruso MA, DeBerardinis RJ, Yan N. 2017. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci U S A 114:746–751. doi: 10.1073/pnas.1611113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, Laustsen A, Peri S, Hansen AL, Bonefeld L, Thyrsted J, Bruun V, Iversen MB, Lin L, Artegoitia VM, Su C, Yang L, Lin R, Balachandran S, Luo Y, Nyegaard M, Marrero B, Goldbach-Mansky R, Motwani M, Ryan DG, Fitzgerald KA, O’Neill LA, Hollensen AK, Damgaard CK, de Paoli FV, Bertram HC, Jakobsen MR, Poulsen TB, Holm CK. 2018. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun 9:3506. doi: 10.1038/s41467-018-05861-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, Dhungana Y, Rankin S, Chen W, Rosencrance C, Yang K, Fan Y, Cheng Y, Easton J, Neale G, Vogel P, Chi H. 2018. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 565:101–105. doi: 10.1038/s41586-018-0806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang F, Alain T, Szretter KJ, Stephenson K, Pol JG, Atherton MJ, Hoang HD, Fonseca BD, Zakaria C, Chen L, Rangwala Z, Hesch A, Chan ESY, Tuinman C, Suthar MS, Jiang Z, Ashkar AA, Thomas G, Kozma SC, Gale M Jr, Fitzgerald KA, Diamond MS, Mossman K, Sonenberg N, Wan Y, Lichty BD. 2016. S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat Immunol 17:514–522. doi: 10.1038/ni.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kroczynska B, Rafidi RL, Majchrzak-Kita B, Kosciuczuk EM, Blyth GT, Jemielity J, Warminska Z, Saleiro D, Mehrotra S, Arslan AD, Fish EN, Platanias LC. 2016. Interferon gamma (IFNgamma) signaling via mechanistic target of rapamycin complex 2 (mTORC2) and regulatory effects in the generation of type II interferon biological responses. J Biol Chem 291:2389–2396. doi: 10.1074/jbc.M115.664995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Datan E, Shirazian A, Benjamin S, Matassov D, Tinari A, Malorni W, Lockshin RA, Garcia-Sastre A, Zakeri Z. 2014. mTOR/p70S6K signaling distinguishes routine, maintenance-level autophagy from autophagic cell death during influenza A infection. Virology 452-453:175–190. doi: 10.1016/j.virol.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. 2016. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun 7:13683. doi: 10.1038/ncomms13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 67.Hresko RC, Mueckler M. 2005. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem 280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 68.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 69.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. 2011. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. 2011. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jan E, Mohr I, Walsh D. 2016. A cap-to-tail guide to mRNA translation strategies in virus-infected cells. Annu Rev Virol 3:283–307. doi: 10.1146/annurev-virology-100114-055014. [DOI] [PubMed] [Google Scholar]
- 72.Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. 2013. mTOR kinase structure, mechanism and regulation. Nature 497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, Jung JU. 2015. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep 13:440–449. doi: 10.1016/j.celrep.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang YF, Moss B. 1991. Inducer-dependent conditional-lethal mutant animal viruses. Proc Natl Acad Sci U S A 88:1511–1515. doi: 10.1073/pnas.88.4.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 85:9899–9908. doi: 10.1128/JVI.05446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Willis KL, Patel S, Xiang Y, Shisler JL. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2alpha pathway in RK13 and HeLa cells. Virology 394:73–81. doi: 10.1016/j.virol.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meng X, Jiang C, Arsenio J, Dick K, Cao J, Xiang Y. 2009. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J Virol 83:10627–10636. doi: 10.1128/JVI.01260-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BR, Silverman RH. 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol 76:5251–5259. doi: 10.1128/jvi.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A 89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davies MV, Chang HW, Jacobs BL, Kaufman RJ. 1993. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J Virol 67:1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jesus DM, Moussatche N, McFadden BBD, Nielsen CP, D’Costa SM, Condit RC. 2015. Vaccinia virus protein A3 is required for the production of normal immature virions and for the encapsidation of the nucleocapsid protein L4. Virology 481:1–12. doi: 10.1016/j.virol.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salmond RJ. 2018. mTOR regulation of glycolytic metabolism in T cells. Front Cell Dev Biol 6:122. doi: 10.3389/fcell.2018.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiu P, Liu Y, Zhang J. 2018. Review: the role and mechanisms of macrophage autophagy in sepsis. Inflammation 42:6–19. doi: 10.1007/s10753-018-0890-8. [DOI] [PubMed] [Google Scholar]
- 84.Borst K, Schwabenland M, Prinz M. 2018. Microglia metabolism in health and disease. Neurochem Int 2018:S0197-0186(18)30385-1. doi: 10.1016/j.neuint.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 85.Weichhart T, Hengstschlager M, Linke M. 2015. Regulation of innate immune cell function by mTOR. Nat Rev Immunol 15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, Wade-Martins R, McMichael A, Klenerman P, Simon AK. 2012. A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T cells. Autophagy 8:677–689. doi: 10.4161/auto.18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McMahon R, Zaborowska I, Walsh D. 2011. Noncytotoxic inhibition of viral infection through eIF4F-independent suppression of translation by 4EGi-1. J Virol 85:853–864. doi: 10.1128/JVI.01873-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lan Y, Wang G, Song D, He W, Zhang D, Huang H, Bi J, Gao F, Zhao K. 2016. Role of autophagy in cellular response to infection with Orf virus Jilin isolate. Vet Microbiol 193:22–27. doi: 10.1016/j.vetmic.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 91.Martyniszyn L, Szulc-Dąbrowska L, Boratyńska-Jasińska A, Niemiałtowski M. 2013. Increased formation of autophagosomes in ectromelia virus-infected primary culture of murine bone marrow-derived macrophages. Acta Virol 57:467–470. doi: 10.4149/av_2013_04_467. [DOI] [PubMed] [Google Scholar]
- 92.Moloughney JG, Monken CE, Tao H, Zhang H, Thomas JD, Lattime EC, Jin S. 2011. Vaccinia virus leads to ATG12-ATG3 conjugation and deficiency in autophagosome formation. Autophagy 7:1434–1447. doi: 10.4161/auto.7.12.17793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang H, Monken CE, Zhang Y, Lenard J, Mizushima N, Lattime EC, Jin S. 2006. Cellular autophagy machinery is not required for vaccinia virus replication and maturation. Autophagy 2:91–95. doi: 10.4161/auto.2.2.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, Maier T. 2016. Architecture of human mTOR complex 1. Science 351:48–52. doi: 10.1126/science.aaa3870. [DOI] [PubMed] [Google Scholar]
- 95.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 96.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. 2002. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189. doi: 10.1016/S0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 97.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. 2013. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev 18:660–672. doi: 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buller RM, Smith GL, Cremer K, Notkins AL, Moss B. 1985. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature 317:813–815. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- 100.Deng L, Fan J, Ding Y, Zhang J, Zhou B, Zhang Y, Huang B. 2017. Oncolytic efficacy of thymidine kinase-deleted vaccinia virus strain Guang9. Oncotarget 8:40533–40543. doi: 10.18632/oncotarget.17125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, Moss B, Bartlett DL. 2001. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 61:8751–8757. [PubMed] [Google Scholar]
- 102.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 103.Eaglesham JB, Pan Y, Kupper TS, Kranzusch PJ. 2019. Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 566:259–263. doi: 10.1038/s41586-019-0928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DiGiuseppe S, Rollins MG, Bartom ET, Walsh D. 2018. ZNF598 plays distinct roles in interferon-stimulated gene expression and poxvirus protein synthesis. Cell Rep 23:1249–1258. doi: 10.1016/j.celrep.2018.03.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jha S, Rollins MG, Fuchs G, Procter DJ, Hall EA, Cozzolino K, Sarnow P, Savas JN, Walsh D. 2017. Trans-kingdom mimicry underlies ribosome customization by a poxvirus kinase. Nature 546:651–655. doi: 10.1038/nature22814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dhungel P, Cao S, Yang Z. 2017. The 5′-poly(A) leader of poxvirus mRNA confers a translational advantage that can be achieved in cells with impaired cap-dependent translation. PLoS Pathog 13:e1006602. doi: 10.1371/journal.ppat.1006602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Walsh D, Arias C, Perez C, Halladin D, Escandon M, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Mohr I. 2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol Cell Biol 28:2648–2658. doi: 10.1128/MCB.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zaborowska I, Kellner K, Henry M, Meleady P, Walsh D. 2012. Recruitment of host translation initiation factor eIF4G by the vaccinia virus ssDNA-binding protein I3. Virology 425:11–22. doi: 10.1016/j.virol.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 109.Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fontaine KA, Camarda R, Lagunoff M. 2014. Vaccinia virus requires glutamine but not glucose for efficient replication. J Virol 88:4366–4374. doi: 10.1128/JVI.03134-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Greseth MD, Traktman P. 2014. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog 10:e1004021. doi: 10.1371/journal.ppat.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu S, Schafer X, Munger J. 2016. Expression of oncogenic alleles induces multiple blocks to human cytomegalovirus infection. J Virol 90:4346–4356. doi: 10.1128/JVI.00179-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baroudy BM, Venkatesan S, Moss B. 1982. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell 28:315–324. doi: 10.1016/0092-8674(82)90349-X. [DOI] [PubMed] [Google Scholar]
- 114.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol 35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Prideaux CT, Kumar S, Boyle DB. 1990. Comparative analysis of vaccinia virus promoter activity in fowlpox and vaccinia virus recombinants. Virus Res 16:43–57. doi: 10.1016/0168-1702(90)90042-A. [DOI] [PubMed] [Google Scholar]
- 116.Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]