

The use of nonpathogenic viruses to target and kill cancer cells is a promising strategy in cancer therapy. However, many types of human cancer are resistant to the oncolytic (cancer-killing) effects of virotherapy. In this study, we identify a host cellular protein, SIRT1, that contributes to the sensitivity of prostate cancer cells to infection by a prototypical oncolytic virus. Knockout of SIRT1 activity increases the sensitivity of prostate cancer cells to virus-mediated killing. At the molecular level, SIRT1 is controlled by a small microRNA termed miR-34a. Altogether, SIRT1 and/or miR-34a levels may serve as predictors of response to oncolytic-virus therapy.
KEYWORDS: SIRT1, VSV, apoptosis, miRNA, oncolytic virus
ABSTRACT
Oncolytic virotherapy represents a promising experimental anticancer strategy, based on the use of genetically modified viruses to selectively infect and kill cancer cells. Vesicular stomatitis virus (VSV) is a prototypic oncolytic virus (OV) that induces cancer cell death through activation of the apoptotic pathway, although intrinsic resistance to oncolysis is found in some cell lines and many primary tumors, as a consequence of residual innate immunity to the virus. In the effort to improve OV therapeutic efficacy, we previously demonstrated that different agents, including histone deacetylase inhibitors (HDIs), functioned as reversible chemical switches to dampen the innate antiviral response and improve the susceptibility of resistant cancer cells to VSV infection. In the present study, we demonstrated that the NAD+-dependent histone deacetylase SIRT1 (silent mating type information regulation 2 homolog 1) plays a key role in the permissivity of prostate cancer PC-3 cells to VSVΔM51 replication and oncolysis. HDI-mediated enhancement of VSVΔM51 infection and cancer cell killing directly correlated with a decrease of SIRT1 expression. Furthermore, pharmacological inhibition as well as silencing of SIRT1 by small interfering RNA (siRNA) was sufficient to sensitize PC-3 cells to VSVΔM51 infection, resulting in augmentation of virus replication and spread. Mechanistically, HDIs such as suberoylanilide hydroxamic acid (SAHA; Vorinostat) and resminostat upregulated the microRNA miR-34a that regulated the level of SIRT1. Taken together, our findings identify SIRT1 as a viral restriction factor that limits VSVΔM51 infection and oncolysis in prostate cancer cells.
IMPORTANCE The use of nonpathogenic viruses to target and kill cancer cells is a promising strategy in cancer therapy. However, many types of human cancer are resistant to the oncolytic (cancer-killing) effects of virotherapy. In this study, we identify a host cellular protein, SIRT1, that contributes to the sensitivity of prostate cancer cells to infection by a prototypical oncolytic virus. Knockout of SIRT1 activity increases the sensitivity of prostate cancer cells to virus-mediated killing. At the molecular level, SIRT1 is controlled by a small microRNA termed miR-34a. Altogether, SIRT1 and/or miR-34a levels may serve as predictors of response to oncolytic-virus therapy.
INTRODUCTION
Oncolytic virotherapy represents a promising approach to experimental cancer therapy, based on the use of different viruses as self-expanding biotherapeutics that selectively infect and kill cancer cells (1, 2). The therapeutic potential of oncolytic viruses (OVs) has been highlighted by the approval of the oncolytic recombinant herpesvirus talimogene laherparepvec (T-VEC) for the treatment of inoperable malignant melanoma (3) as well as by recent preclinical trials combining OV therapies with the use of immune checkpoint inhibitors (4, 5). Despite significant progress in OV immunotherapy, the heterogeneity of the clinical response to OV-based therapies and the engagement of the adaptive immune response against viral rather than tumor antigens represent crucial obstacles to the large-scale clinical implementation of oncolytic virotherapy against multiple types of cancer. In addition, many primary human malignancies and some tumor cell lines maintain a residual innate immune response that renders tumor cells resistant to OV infection and virus-mediated cell killing (6).
To overcome these challenges, numerous efforts have sought to improve OV therapeutic efficacy through viral engineering or the development of combination strategies. For example, a genetic variant of vesicular stomatitis virus (VSV), VSVΔM51, contains a deletion at methionine 51 in the matrix protein that improves its tumor specificity and impairs its replication in normal cells that have functional antiviral defenses (7, 8). Previous studies have demonstrated the synergistic effect of different agents, including histone deacetylase (HDAC) inhibitors (HDIs), as reversible chemical switches to dampen the innate antiviral response and improve the susceptibility of resistant cancer cells to VSVΔM51 infection and spread (9). We demonstrated that suberoylanilide hydroxamic acid (SAHA; Vorinostat) stimulated NF-κΒ activity via modulation of RelA/p65 signaling, leading to induction of autophagy, suppression of the interferon (IFN)-mediated response, and subsequent enhancement of VSV replication and apoptosis (10).
Among the HDAC family members, sirtuins represent a family of seven NAD+-dependent enzymes that belong to class III HDACs, all with distinct structures, functions, and subcellular localizations. Sirtuins have emerged as important regulators of different physiological and pathological processes, including metabolic disease, age-related disorders, neurodegeneration, inflammation, and cancer (11–13). SIRT1 (Silent mating type information regulation 2 homolog 1) is the best characterized among the sirtuins, and its levels are significantly elevated in prostate, ovarian, and colorectal cancer (14–16). A role for SIRT1 in promoting tumor cell growth and chemoresistance in androgen-refractory prostate cancer has been reported previously (17, 18). Furthermore, SIRT1 inhibition has been reported to suppress cell growth and induce cell cycle arrest or apoptosis in cancer cells (19, 20).
SIRT1 expression can be regulated at both transcriptional and posttranscriptional levels, and several mechanisms involved in dysregulation of SIRT1 in cancer cells have been proposed previously (21). In cancer cells, inactivation of the tumor suppressor p53 by genetic or epigenetic mechanisms leads to upregulation of SIRT1 transcription (22). On the other hand, the RNA binding protein HuR, a potential oncoprotein, stabilizes SIRT1 mRNA through 3′-untranslated region (3′-UTR) interactions leading to elevated SIRT1 levels (23), thus suggesting that the posttranscriptional regulation of SIRT1 may also be important in governing SIRT1 expression in tumors.
SIRT1 expression is also regulated by specific microRNAs (miRNA), small (18-to-24-nucleotides) noncoding RNAs that elicit their regulatory effects by base pairing with specific sequences (miRNA response element) in the 3′-UTR of target mRNAs, causing their degradation or inhibition of translation (24). MicroRNAs—miR-34a and miR-132—bind directly to the 3′-UTR of SIRT1 mRNA, leading to suppression of SIRT1 expression in colon and prostate cancer cells (25, 27) and adipocytes (26), respectively. miR-34a is a recognized tumor suppressor gene (25, 28), and inhibition of SIRT1 by miR-34a results in increased acetylation of p53, as well as expression of p21 and PUMA, which regulate the cell cycle and apoptosis, respectively (25).
Sirtuins, and, in particular, SIRT1, have been identified as evolutionarily conserved antiviral defense factors. Small interfering RNA (siRNA)-mediated knockdown of individual sirtuins increased the virus production in human cells infected with human cytomegalovirus (HCMV) or influenza A virus (29). SIRT1 was reported to inhibit different viruses, including VSV (30) and Kaposi’s sarcoma-associated herpesvirus (KSHV) (31).
In the present study, we demonstrated that SIRT1 plays a key role in the permissivity of prostate cancer cells to VSVΔM51 replication and oncolysis. SIRT1 knockdown by siRNA was sufficient to sensitize PC-3 cells to VSVΔM51 infection and replication. Furthermore, HDI-dependent enhancement of VSVΔM51 infection and cancer cell killing was directly linked to the HDI-mediated decrease in SIRT1 expression. Mechanistically, HDIs such as SAHA, resminostat (RESM), and MS-275 upregulated the microRNA miR-34a, which controls in part the levels of SIRT1. Taken together, our findings identify SIRT1 as a viral restriction factor that limits VSVΔM51 infection and oncolysis in prostate cancer cells.
RESULTS
Inhibition of HDAC1 and HDAC3 induces S-phase depletion and G2/M-phase accumulation in PC-3 cells.
As a part of our studies to examine the mechanistic basis of the synergism between HDIs and oncolytic VSV, we examined the effect of SAHA in human prostate cancer PC-3 cells and found that SAHA treatment caused a dose-dependent reduction in the number of PC-3 cells in the S phase and a concomitant accumulation of cells in the G2/M phase (Fig. 1A). The number of PC-3 cells in the S phase decreased from ∼16% to 3% of the total cells with increasing concentrations of SAHA. SAHA-mediated G2/M cell cycle arrest was accompanied by enhanced susceptibility to VSVΔM51 infection, as reflected by the dose-dependent increase in the number of VSVΔM51 green fluorescent protein-positive (VSVΔM51-GFP+) cells, resulting to ∼90% infectivity (Fig. 1B). Furthermore, an increase in the level of VSVΔM51-mediated PC-3 cell killing was also observed in cell cycle-arrested SAHA-treated cells (Fig. 1C), and, consistent with the G2/M cell cycle arrest, addition of SAHA increased expression of cell cycle inhibitor p21WAF1 (cyclin-dependent kinase inhibitor 1A [CDKN1A]) (Fig. 1D).
FIG 1.

Inhibition mediated by HDAC1 and HDAC3 induces a marked depletion of S-phase cells and increase in the levels of G2/M cells in PC-3 cell cycle arrest. (A) The cell cycle distribution of PC-3 cells treated for 24 h with increasing concentrations of SAHA (0 to 5 μM) was analyzed by propidium iodide (PI) staining by flow cytometry. The percentages of cells in the G0/G1, S, and G2/M phases were evaluated by measuring DNA content. Data represent means ± SD of results from three independent experiments. (B and C) PC-3 cells, pretreated as described for panel A, were subsequently infected with VSVΔM51-GFP (MOI of 10−2). Infectivity was quantified at 24 h postinfection (p.i.) by flow cytometry. (B) Cell death was assessed using annexin V (AnnV)/7AAD staining by flow cytometry. (C) Data represent means ± SD of results from three experiments. (D) PC-3 cells treated with SAHA (5 μM) for 24 h (T0), as well as PC-3 cells pretreated with SAHA and then infected with VSVΔM51-GFP (MOI of 10−2) for a subsequent 24 h, were analyzed for CDKN1A gene expression by qPCR. Gene expression levels were calculated using the threshold cycle (ΔΔCT) method. Data represent means ± SD of results from three independent experiments. The same samples were used to obtain total cell extracts and analyzed by immunoblotting for p21 protein levels. β-Actin was used as a loading control. Results are from a representative experiment. F.I., fold increase. (E to G) PC-3 cells were treated with SAHA (5 μM), RESM (5 μM), TBSA (10 μM), or MS-275 (10 μM) for 24 h (T0) or were left untreated (DMSO). (E) The cell cycle distribution was analyzed by PI staining by using flow cytometer. (F) CDKN1A, CDK6, and CCDN1 gene expression levels were evaluated by qPCR analysis. The gene expression levels were calculated using the ΔΔCT method. Data are representative of results from three independent experiments. (G) Total cell extracts were analyzed by immunoblotting for p21, p16INK4A, IκB α, SQSTM1, and LC3B. GAPDH was used as a loading control. Acetyl-α-tubulin and acetyl-histone H3 were used as controls for analysis of the specific activity of the different HDAC inhibitors. Results are from a representative experiment.
Next, the effect of different HDIs on cell proliferation was examined to determine the relative contributions of different HDACs to the enhanced susceptibility of PC-3 cells to VSVΔM51 infection. Tubastatin A (TBSA; an HDAC6 specific inhibitor), MS-275 (a specific HDAC1/HDAC3 inhibitor), and resminostat (RESM; a well-known HDAC1/HDAC3/HDAC6 inhibitor) were compared to the pan-HDAC inhibitor SAHA for the ability to induce cell cycle arrest. As seen with SAHA, RESM treatment caused a reduction in the percentage of S-phase cells (from 13.2% ± 1.0% to 4.2% ± 1.1%) and accumulation of cells in the G2/M phase (from 23.5% ± 2.8% to 36.3% ± 2.3%); MS-275 treatment likewise decreased the number of cells in S phase (from 13.2% ± 1.0% to 2.4% ± 0.8%). Conversely, TBSA treatment did not significantly impact S-phase and G2/M-phase distribution, thus indicating that simultaneous inhibition of HDAC1 and HDAC3 was involved in the PC-3 cell cycle arrest (Fig. 1E). Consistent with these results, SAHA, RESM, MS-275 treatments led to the upregulation of CDKN1A gene expression while downregulating cyclin-dependent kinase 6 (CDK6) and cyclin D1 (CCDN1), which are key regulators of the G1/S transition (Fig. 1F). Further, SAHA, RESM, and MS-275 treatments led to the upregulation of p21 expression and to decreased levels of IκBα as an indication of increased NF-κB activity, as well as to enhanced autophagic flux, detected by augmented levels of p62/SQSTM1 and increased lipidated LC3B II accumulation (Fig. 1G). Moreover, SAHA and RESM treatments led to the upregulation of the cell cycle regulator p16INK4A. In contrast, TBSA treatment did not induce p21 increase and was not sufficient to induce NF-κB and autophagic flux activation.
Inhibition of HDAC1 and HDAC3 sensitizes PC-3 to VSVΔM51 infection and VSV-mediated cell death.
As measured by flow cytometry analysis of VSVΔM51-GFP+ cells, SAHA, RESM, MS-275, and TBSA all synergized with VSV to increase the level of PC-3 infection from 11.9% ± 4.6% (VSVΔM51 alone) to 85.2% ± 3.9%, 82.1% ± 9.7%, 77.8% ± 4.2%, and 48.4% ± 9.9%, respectively, at 24 h postinfection (p.i.) (Fig. 2A and B). Enhanced cell killing was elicited by combinatorial treatment with SAHA, RESM, and MS-275 based on the increase in the proportion of annexin V+ cells (from 2.3% ± 1.2% in VSVΔM51 alone to 26.1% ± 4.3%, 20.0% ± 6.1%, and 16.6% ± 1.7%, respectively), whereas treatment with TBSA did not increase the proportion of annexin V+ cells (2.1% ± 0.5%) (Fig. 2C). Levels of expression of both BH3-only proapoptotic genes (Puma and Noxa) were upregulated in SAHA-, RESM-, and MS-275-treated and VSV-infected cells, whereas antiapoptotic genes Mcl1 and Bcl-xL were downregulated by SAHA, RESM, and MS-275 treatment but not in TBSA-treated infected cells (Fig. 2D). Consistent with the activation of the intrinsic apoptotic pathway, Bcl-xL protein expression was reduced, while downstream caspase-3 was activated upon SAHA, RESM, or MS-275 combinatorial treatment, leading to poly(ADP-ribose) polymerase (PARP) cleavage (Fig. 2E). In addition, the use of either SAHA treatment or RESM treatment induced the production of proinflammatory cytokines interleukin-6 (IL-6) and IL-8 in VSV-infected cells, while MS-275 treatment induced the upregulation of IL-8 (Fig. 2F). Collectively, the features of VSV-positive HDI-treated cells—cell cycle arrest, p21 and p16INK4A upregulation, inflammatory cytokine release—are reminiscent of the senescence-associated secretory phenotype (SASP) identified in many cells undergoing metabolic stress.
FIG 2.

Inhibition of HDAC1 and HDAC3 sensitizes PC-3 cells to VSVΔM51 infection and VSV-mediated cell death. PC-3 cells were pretreated with SAHA (5 μM), RESM (5 μM), TBSA (10 μM), or MS-275 (10 μM) or were left untreated (DMSO) and were subsequently infected with VSVΔM51-GFP (MOI of 10−2). (A and B) Infectivity was determined at 24 h p.i. by fluorescence microscopy (A) or quantified by flow cytometry (B). (C) Cell death was assessed using annexin V/7AAD staining by flow cytometry. Data represent means ± SD of results from three experiments. (D) Expression of proapoptotic (left panel) and antiapoptotic (right panel) genes was evaluated by qPCR analysis. (E) Total cell extracts were analyzed by immunoblotting for PARP, cleaved caspase-3, and Bcl-xL. β-Actin was used as a loading control. Results are from a representative experiment. (F) Gene expression of proinflammatory cytokines (IL-6 and IL-8) was evaluated by qPCR analysis at 24 h p.i. (D and F) Gene expression levels were calculated using the ΔΔCT method. Data represent means ± SD of results from three independent experiments.
Pharmacological and genetic inhibition of SIRT1 expression enhances VSVΔM51 infectivity and cell death in PC-3 cells.
Given that the development of a SASP-like phenotype may have an impact on the susceptibility of PC-3 to VSVΔM51 infection and replication, we next sought to evaluate the role of the antisenescence gene SIRT1 as a potential candidate HDAC involved in regulating SASP and/or VSV infectivity. Interestingly, treatment with either SAHA, RESM, or MS-275 significantly downregulated SIRT1 expression by more than 4-fold compared to untreated cells, whereas TBSA did not inhibit SIRT1 expression (Fig. 3A). A robust decrease in the gene expression levels of SIRT1 was confirmed (Fig. 3B). To determine whether there is a causal link between the changes in the SIRT1 levels and the enhanced sensitivity of PC-3 to VSVΔM51 infection, we used the SIRT1 inhibitor nicotinamide (NAM), which is an active form of nicotinic acid (vitamin B3) and a precursor of NAD+ (32, 33). As shown in Fig. 3C, NAM-mediated inhibition of SIRT1 enzymatic activity increased the percentage of infected cells from ∼10% to 60% to 80% of GFP-positive cells in a dose-dependent manner. NAM treatment was accompanied by a reduction of SIRT1 levels and by upregulation of p21 as well as by modulation of autophagic flux (Fig. 4D).
FIG 3.

Inhibition of SIRT1 expression enhances VSVΔM51 infectivity and cell death in PC-3 cells. (A and B) PC-3 cells were treated with SAHA (5 μM), RESM (5 μM), TBSA (10 μM), or MS-275 (10 μM) for 24 h or were left untreated (DMSO) and were subsequently infected with VSVΔM51-GFP (MOI of 10−2) for 24 h. (A) Total cell extracts were analyzed by immunoblotting for SIRT1, IκB α, and VSV G. β-Actin was used as a loading control. Results are from a representative experiment. (B) Analysis of Sirt1 gene expression by qPCR. Gene expression level was calculated using the ΔΔCT method. Data represent means ± SD of results from three independent experiments. (C and D) PC-3 cells were treated with increasing concentrations of nicotinamide (NAM) for 24 h and subsequently infected with VSVΔM51-GFP (MOI of 10−2) for 24 h. (C) Infectivity was determined at 24 h p.i. by fluorescence microscopy or quantified by flow cytometry. (D) Total cell extracts were analyzed by immunoblotting for SIRT1, p21, p62/SQSTM1, and LC3B. GAPDH was used as a loading control. Results are from a representative experiment. (E to I) PC-3 cells were transfected with control (CTR) or Sirt1 siRNA and were infected 48 h later with VSVΔM51-GFP (MOI of 0.01 or 0.05). (E) Whole-cell extracts were analyzed by immunoblotting for SIRT1 and VSV G. GAPDH was used as a loading control. Results are from a representative experiment. Lipo, Lipofectamine; siCTR, small interfering control; siSIRT1, small interfering SIRT1. (F and G) Viral infectivity was determined at 24 h p.i. by fluorescence microscopy (F) and measured by flow cytometry (G). Data represent means ± SD of results from three independent experiments. (H) Viral RNA levels were measured by qPCR. (I) Cell death was assessed using 7-AAD staining by flow cytometry.
FIG 4.

Resveratrol-mediated SIRT1 activation reverses the synergistic effect of SAHA on VSVΔM51 replication. (A) PC-3 cells were treated with resveratrol (Rsv) at different concentrations for 2 h prior to the addition of SAHA (5 μM) or not (DMSO) for 24 h and were subsequently infected with VSVΔM51-GFP (MOI of 10−2). Infectivity was determined at 24 h p.i. by fluorescence microscopy (A) or quantified by flow cytometry (B). Data represent means ± SD of results from three experiments. (C) Total lysates were analyzed by immunoblotting for SIRT1 expression. GAPDH was used as a loading control. Results are from a representative experiment. (D) DU-145 cells were treated with Rsv 50 μM or not for 24 h, and subsequently infected with VSVΔM51-GFP (MOI of 10−2). Infectivity was determined at 24 h p.i. by fluorescence microscopy or quantified by flow cytometry. Data represent means ± SD of results from three experiments. (E to H) U-2 OS cells were transfected with SIRT1 WT, H363Y mutant or the empty vector (vec) and 24 h later were infected with VSVΔM51-GFP (MOI of 10−3 or 10−2). Viral infectivity was determined at 24 h p.i. by fluorescence microscopy (E) and measured by flow cytometry as the percentage of GFP-positive cells (F) and GFP mean fluorescence intensity (MFI) (G). (H) Whole-cell extracts were analyzed by immunoblotting for FLAG, SIRT1, and VSV G. L.E., long exposure; S.E., short exposure. GAPDH was used as a loading control. Results are from a representative experiment. Data represent means ± SD of results from three independent experiments.
Next, the expression of Sirt1 was inhibited using specific siRNA directed against Sirt1. Knockdown efficiency was estimated at ∼85% in immunoblot analysis comparing control and siRNA-transfected cells (Fig. 3E, lanes 4 to 6 compared to lanes 7 to 9). Strikingly, the silencing of Sirt1 led to a dramatic increase in the number of VSVΔM51-GFP+ cells following infection (Fig. 3F and G), altogether representing a 5-fold to 10-fold increase in the number of GFP+ PC-3 cells from less than 10% to >50% positivity at a multiplicity of infection (MOI) of 0.01. Measurements of the intracellular levels of VSV RNA likewise reflected increased replication of VSV (Fig. 3H); also, VSV G protein expression levels were 4-fold higher in VSV-infected PC3 cells silenced for Sirt1 (Fig. 3E; compare lane 6 and lane 9). Consistently, enhanced cell killing was observed in Sirt1 knocked-down VSVΔM51-infected cells compared to control cells (Fig. 3I). Furthermore, SIRT1 silencing, as well as its inhibition using the specific SIRT1 inhibitor EX-527, caused a G0/G1 cell cycle arrest, resulting in an accumulation of cells in the G0/G1 phase and concomitant reduction of cells in the S phase, accompanied by reductions in the levels of CDK6 and CCDN1 mRNA (data not shown). Taken together, these results establish an inverse relationship between SIRT1 levels and VSV infection and replication.
Activation and upregulation of SIRT1 impair VSVΔM51 infection in PC-3 cells.
To test whether the use of a SIRT1 agonist to promote SIRT1 expression would inhibit VSV replication, PC-3 cells were treated with resveratrol (Rsv), previously characterized as a potent activator of SIRT1 (34). Resveratrol addition induced the expression of SIRT1 (Fig. 4C) and effectively reversed the synergistic effect of SAHA, resulting in a decrease in VSVΔM51 infectivity (Fig. 4A and B). Moreover, in VSV-sensitive DU-145 prostate cancer cells, addition of Rsv strongly inhibited VSVΔM51 infection (Fig. 4D).
Next, we overexpressed the SIRT1 wild-type (WT) vector, the dominant-negative H363Y mutant without deacetylase activity, or empty vector (vec) in VSV-sensitive U-2 OS cells. Overexpression of SIRT1 WT reduced the number of VSVΔM51-GFP+-infected cells from 23.1% to 7.4% at an MOI of 10−3 (Fig. 4E and F) and lowered the VSV G protein expression level 3-fold (Fig. 4H; compare lanes 2 and 5). Expression of the SIRT1 H363Y mutant did not alter VSV infectivity in U-2 OS cells (Fig. 4E and F), since endogenous SIRT1 levels were already low (Fig. 4H). These results further support the inverse relationship between SIRT1 expression and VSV permissivity.
miR-34a upregulation augments the permissivity of PC-3 cells to VSVΔM51 infection.
Regulation of SIRT1 expression is dictated in part by the function of miR-34a, a microRNA that binds to the 3′-UTR of SIRT1 mRNA and controls SIRT1 expression (25). Consistent with the observations described above, expression of miR-34a after SAHA or RESM treatment increased 3-fold (Fig. 5A), concomitant with a reduction in SIRT1 expression (Fig. 3A). Inhibition of miR-34a using a specific antagonist (anti-miR-34a) blocked SAHA-mediated miR-34a induction (Fig. 5B) and counteracted the synergistic effect of SAHA, resulting in a decrease in the proportion of VSVΔM51-GFP+-infected cells, from 77.6% ± 4.7% to 37.4% ± 2.8% (Fig. 5C and D).
FIG 5.

miR-34a upregulation augments sensitivity of PC-3 cells to VSVΔM51 infection. (A) PC-3 cells were treated with SAHA (5 μM) or RESM (5 μM) for 24 h or were left untreated (DMSO). (B) PC-3 cells were transfected with 40 or 80 nM anti-miR-34a or the negative control (anti-miR ctr neg) and treated with SAHA 5 μM for 24 h or were left untreated (DMSO). (A and B) Total RNA containing miRNA was extracted and subjected to real-time RT-PCR for miR-34a. The expression level of miR34a was normalized to U6. Data are expressed as means ± SD (n = 3). (C and D) PC-3 cells treated as described for panel B were infected with VSVΔM51-GFP (MOI of 10−2). Viral infectivity was determined at 24 h p.i. by fluorescence microscopy (C) and measured by flow cytometry (D). (E to I) PC-3 cells were transfected with 30 nM pre-miR-34a or the negative control (pre-miR neg ctr) for 24 h. (E and F) Total RNA containing miRNA was extracted and analyzed by qPCR for miR-34a (E) and Sirt1 (F) expression. The expression level of miR34a was normalized to U6. The Sirt1 gene expression level was calculated using the ΔΔCT method. Data represent means ± SD of results from three independent experiments. (G to I) PC-3 transfected as described for panel E were infected with VSVΔM51-GFP (MOI of 0.01 or 0.05). (G and H) Viral infectivity was determined at 24 h p.i. by fluorescence microscopy (G) and measured by flow cytometry (H). (I) Viral RNA was measured by qPCR.
Ectopic expression of a mature miR-34a mimic (pre-miR-34a) in PC-3 cells (Fig. 5E) resulted in a concomitant reduction of Sirt1 mRNA levels (Fig. 5F), increased the proportion of VSVΔM51-GFP+-infected cells from 8.9% ± 3.8% to 46.0% ± 7.0% at an MOI of 0.01 (Fig. 5G and H), and augmented the levels of VSV mRNA (Fig. 5I), further corroborating the role of miR-34a in regulating permissivity to VSVΔM51.
In addition, the levels of miR-34a RNA and SIRT1 expression were examined in PC-3, DU-145, and LNCaP cells; as shown in Fig. 6A, LNCaP expressed the highest levels of miR-34a, with DU-145 and PC-3 expressing ∼3-fold-lower and ∼10-fold-lower levels of miR-34a, respectively. Conversely, SIRT1 expression was highest in PC-3 cells and was 1.5-fold and 3-fold lower in DU-145 and LNCaP cells, respectively (Fig. 6B); this reciprocal relationship correlated with the differential sensitivities of these prostate cancer cells to VSVΔM51 infection. LNCaP cells (with high miR-34a and low SIRT1 levels) were the most susceptible to infection (∼90% infected cells), compared to DU-145 (∼60% infected cells) and PC-3 (low miR-34a and high SIRT1) (∼10% infected cells) (Fig. 6C). Taken together, these results demonstrate that the enhanced sensitivity of prostate cancer cells to VSVΔM51 infection is attributable to elevated levels of miR-34a accompanied by a strong reduction in the level of SIRT1 expression.
FIG 6.
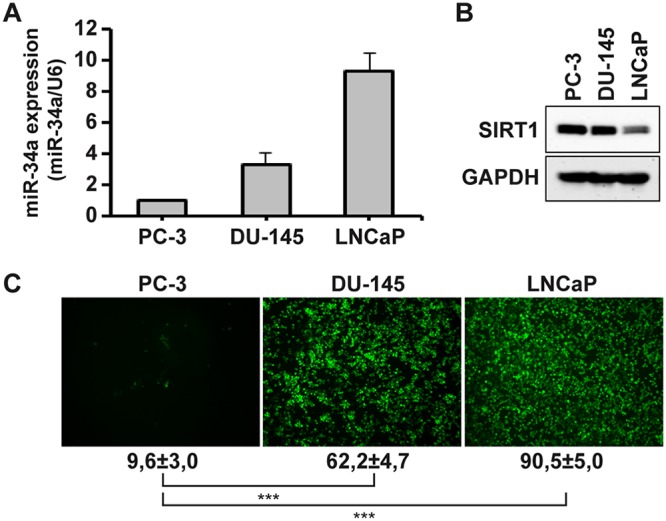
miR-34a and SIRT1 expression levels determine the permissivity of prostate cancer cells to VSVΔM51 infection. (A) Total RNA containing miRNA from PC-3, DU-145, and LNCaP cells was extracted and subjected to real-time RT-PCR for miR-34a. The expression level of miR34a was normalized to U6. Data are expressed as means ± SD (n = 3). (B) Total lysates from PC-3, DU-145, and LNCaP cells were analyzed by immunoblotting for SIRT1 expression. GAPDH was used as a loading control. Results are from a representative experiment. (C) PC-3, DU-145, and LNCaP cells were infected with VSVΔM51-GFP (MOI of 10−2). Infectivity was determined at 24 h p.i. by fluorescence microscopy or quantified by flow cytometry. Data represent means ± SD of results from three experiments.
DISCUSSION
A significant obstacle to the development of oncolytic-virus-based therapies for cancer is the resistance of many cancer cell lines and primary tumors to virus infection, attributable in part to the residual antiviral immunity in the tumor cell population. Another significant roadblock to OV therapy involves the generation of an appropriate adaptive immune response directed against tumor antigens rather than viral antigens. Only recently, with the FDA approval of oncolytic virus T-VEC (talimogene laherparepvec), has it been possible to envision further development of OV immunotherapeutics as a companion therapy to immune checkpoint blockade strategies (3).
Among the many efforts aimed at increasing the efficacy of experimental oncolytic immunotherapies, we and others have explored strategies to potentiate viral oncolysis in OV-resistant cancer cells by combining OVs with small-molecule compounds that enhance virus replication and cell killing (9, 10, 35, 36). In previous studies, we determined that the oncolytic potential of a model oncolytic virus—VSV—was reversibly stimulated in cell lines, animal models, and ex vivo patient samples by the addition of HDIs, a class of therapeutic compounds that alter host gene expression by epigenetic modulation of chromatin structure, via inhibition of HDAC activities (37). We demonstrated that coadministration of HDIs transiently suppressed innate immunity to promote infection and spread of VSV and significantly enhanced antitumor immunity and augmented virus-induced apoptosis (9, 10).
In the present study, we sought to further understand the molecular mechanisms of synergism in HDI and VSVΔM51 combinatorial treatments. We provide evidence that SIRT1 functions as a host restriction factor that limits VSVΔM51 replication in prostate cancer cell lines and that inhibition of SIRT1 by pharmacologic or genetic ablation dramatically increased VSV infection and replication in prostate cancer PC-3 cells. Mechanistically, HDIs such as SAHA, resminostat, and MS-275 upregulated the microRNA miR-34a that controls the levels of SIRT1 expression, as shown schematically in Fig. 7. Consistent with these data, siRNA knockdown of SIRT1 was sufficient to sensitize PC-3 cells to VSVΔM51 infection and replication, whereas prostate cancer cell lines (LNCaP and DU145) expressing low levels of SIRT1 were highly susceptible to VSVΔM51 infection (Fig. 6). The observation that SIRT1 expression was increased in different human cancers such as prostate cancer, colon cancer, and acute myeloid leukemia (14, 16, 38), where it inhibits apoptosis and senescence, suggests that SIRT1 inhibition may be useful in predicting which types of cancer would be amenable to oncolytic-virus treatment.
FIG 7.
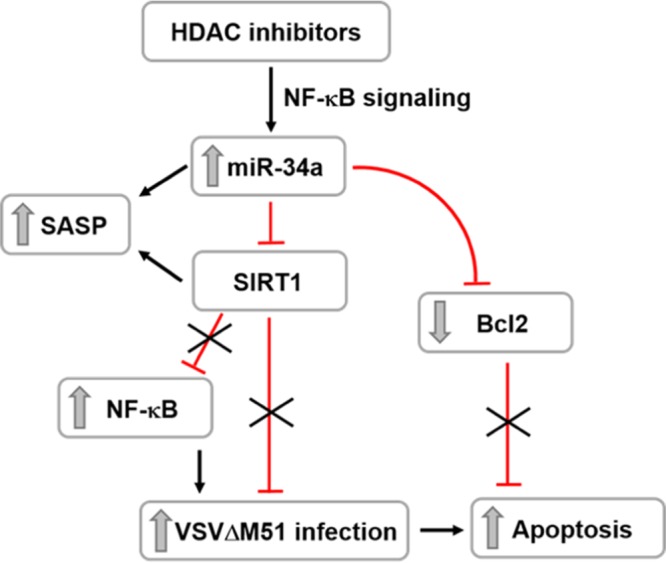
Schematic of SIRT1 and miR-34a-mediated regulatory role in the permissivity of prostate cancer cells to VSVΔM51 replication and oncolysis. HDAC inhibitors upregulate the microRNA miR-34a, which, in turn, negatively regulates the level of SIRT1 and antiapoptotic Bcl2 proteins. SIRT1 inhibition mediates the activation of NF-κB, contributing to the enhancement of VSVΔM51 infection and cancer cell killing.
Cellular replicative senescence has been described as a reprogramming of the cellular phenotype that facilitates increased viral replication, mainly due to impaired viral sensing and dampening of IFN induction by host cells (39). HDAC inhibition elicits a SASP response, due to chromatin remodeling, in the absence of DNA breaks (40). Treatment with several HDIs induced growth arrest and senescence-like phenotypes in different normal and cancer cells (41, 42), through upregulation of cell cycle inhibitors p21WAF1 and p16INK4A. p21, in turn, downregulates a number of genes involved in cell cycle progression. Furthermore, HDAC inhibition causes strong activation of expression of canonical SASP factors, such as interleukin-6 (IL-6) and IL-8, that is dependent upon DNA damage response (DDR) and mediated by ATM and NF-κB (40). Consistently, treatment of PC-3 with HDIs caused significant upregulation of p21 (Fig. 1F and G), together with downregulation of CDK6 and CCDN1 gene expression (Fig. 1F), which halted cell proliferation and induced inflammatory cytokines IL-6 and IL-8 (Fig. 2F).
SIRT1 is the most widely studied among senescence-associated genes and negatively regulates the expression of the SASP component through epigenetic gene regulation. Depletion of SIRT1 augmented the acetylation state of histone 3 (H3) and H4 on the promoter regions of SASP factors even in cells that were not undergoing senescence (43). Interestingly, all tested HDIs, except the specific HDAC6 inhibitor Tubastatin A, elicited a robust decrease in SIRT1 at either the gene expression level or the protein level (Fig. 3A and B).
SIRT1 regulates the function of several important transcription factors, such as p53, FoxO and NF-κB (11). Previous studies have demonstrated that SIRT1 is a potent inhibitor of NF-κB transcription, and acts by directly deacetylating the RelA/p65 subunit in the NF-κB complex. Specific deacetylation of Lys310 inhibited the transactivation function of RelA/p65, increased its export from the nucleus, and consequently decreased transcription of NF-κB-dependent genes (44). Strikingly, we previously demonstrated that HDI treatment resulted in the increased expression of a subset of NF-κB-regulated genes and demonstrated by mass spectrometry that HDI treatment resulted in the maintenance of the acetylated Lys310 in RelA/p65 (10). On the basis of the current studies, it is tempting to speculate that inhibition of SIRT1 activity by SAHA, resminostat, and MS-275 is responsible for the maintenance of the RelA/p65 acetylation at Lys310. Furthermore, several studies have shown that blocking the serine/threonine p38 mitogen-activated protein kinase (p38MAPK) attenuates NF-κB-driven transcriptional activity indirectly, by regulating RelA/p65 acetylation at Lys310 (45). Indeed, pretreatment with the specific p38MAPK inhibitor BIRB-796 reversed SAHA-mediated enhancement of VSVΔM51 infectivity in PC-3 cells, further supporting the key role of p65 acetylation (data not shown).
NF-κB signaling has also been shown to downregulate SIRT1 activity, with NF-κB binding to the miR-34a promoter, leading to the induction of miR-34a expression (46, 47). In turn, miR-34a can regulate cell proliferation, senescence, and apoptosis, through downregulation of multiple targets such as CDK4, CDK6, cyclin D1, E2F3, and BCL2 (28). In this context, miR-34a also targeted Bcl-2 family members in PC-3 cells; levels of Bcl-xL and Mcl1 mRNA were decreased and levels of proapoptotic BH3-only members—Noxa and Puma—were increased following treatment with SAHA, RESM, and MS-275 (Fig. 2D). These findings collectively suggest that activation of the intrinsic mitochondrial death pathway could trigger HDI-induced apoptosis (48).
Reduced levels of miR34a expression have been reported in prostate, colon, and ovarian cancer and in chronic lymphocytic leukemia (CLL), mainly due to aberrant CpG methylation of the miR-34a promoter or altered function of p53, which directly controls miR-34a expression (28, 49–51). In TP53-defective cells such as PC-3 cells, the miR-34a response was mediated by HDAC1 (52), further corroborating our findings on the upregulation of miR-34a expression upon HDAC inhibition (53).
Finally, a more practical use of SIRT1 and/or miR-34a levels may be to exploit these values as predictors of response to OV therapy that could aid in discriminating between cancers that would be amenable to an oncolytic-immunotherapy approach. As yet, we do not know if replication of other oncolytic-virus candidates is also modulated by or is dependent on Sirt1 and/or miR-34a expression. The prognostic value of SIRT1 levels, as well as the prediction of therapeutic efficacy, represent ongoing studies to address the generality of the Sirt1 restriction of oncolytic-virus efficacy.
MATERIALS AND METHODS
Reagent and antibodies (Abs).
SAHA was purchased from Sigma; MS-275 was purchased from AdipoGen; and resminostat, Tubastatin A, nicotinamide, and resveratrol were purchased from Cayman Chemicals and were dissolved in dimethyl sulfoxide (DMSO). Anti-VSV G, anti-SIRT1, anti-p21, anti-p62, anti-Bcl-xL, and anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) were purchased from Santa Cruz Biotechnology. Anti-p16/INK4A was purchased from Invitrogen. Anti-FLAG was purchased from Sigma. Anti-cleaved-caspase-3 (Asp175), anti-PARP, anti-LC3B, anti-IκBα, anti-acetyl-α-tubulin, anti-acetyl-histone H3, and anti-β-actin were purchased from Cell Signaling.
Cell culture.
Human PC-3 cells were provided by Antonio Filippini (Sapienza University), while DU-145 and LNCaP were obtained from Francesca Cutruzzolà (Sapienza University). PC-3, DU-145, and LNCaP cells were cultured in RPMI 1640 medium (Euroclone) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% antibiotics (Euroclone). U-2 OS and BHK-21T7 cells were obtained from ATCC and were grown in Dulbecco’s modified Eagle’s medium (DMEM), 10% FBS, and 1% antibiotics.
VSV production, quantification, and infection.
VSVΔM51-GFP is a recombinant derivative of the VSV Indiana serotype, as previously described (8), and was provided by John Bell (Ottawa Health Research Institute). VSVΔM51 was propagated on Vero cells, and virus titers were quantified by a standard plaque assay method on BHK-21T7 cells as described previously (9).
In vitro treatment with drugs and infection with VSVΔM51.
Cancer cell lines were pretreated for 24 h with the indicated inhibitor and then infected at the indicated MOIs with VSVΔM51 for a subsequent 24 h. Images of GFP-positive cells were obtained by the use of an Olympus fluorescence microscope. The percentage of infected cells was determined based on GFP expression by using a BD FACSCanto II instrument (BD Biosciences). Data were analyzed by using FlowJo software.
Cell cycle, cell viability, and apoptosis analyses.
For cell cycle analyses, after trypsinization, cells were fixed in 70% ethanol at −20°C. Samples were then washed in phosphate-buffered saline (PBS) and incubated for 30 min at 37°C with PI solution (0.1% Triton X-100, 50 μg/ml propidium iodide, 200 μg/ml RNase A). A total of 20,000 events were acquired (BD FACSCanto II, BD Biosciences), and the cell cycle distribution was analyzed by using FlowJo software. Cell viability and apoptosis were assessed by flow cytometry measuring annexin V and 7-aminoactinomycin D (7-AAD)-positive populations.
Protein extraction and immunoblot analysis.
Total protein extracts were obtained by lysing cells for 30 min at 4°C in lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 0.5% NaDoc (sodium deoxycholate), 1% SDS) in the presence of protease and phosphatase inhibitors. Proteins were resolved by SDS-PAGE on 4% to 20% precast Novex Tris-glycine gradient gels (Thermo Fisher Scientific) and blotted onto nitrocellulose membranes (GE Healthcare) (pore size, 0.22 μM). Blots were incubated with the indicated primary antibodies, extensively washed with TBST, and, after incubation with horseradish peroxidase (HRP)-labeled goat anti-rabbit or goat anti-mouse Abs (GE Healthcare), developed with an enhanced chemiluminescence (ECL) detection system per the instructions of the manufacturer (Thermo Fisher Scientific).
mRNA extraction, total RNA extraction, and quantitative PCR (qPCR) assay.
RNA extraction was performed by using an RNeasy minikit (Qiagen), and cDNA was synthesized using a High-Capacity cDNA reverse transcription (RT) kit (Applied Biosystems). Real-time RT-PCR was carried out using TaqMan Fast Advanced Master Mix (Applied Biosystems). The expression level of the analyzed genes was normalized to that of GAPDH. Total RNA containing miRNA was extracted using NucleoSpin miRNA (Macherey-Nagel) for quantitation of miR-34a, and cDNA was synthesized using a TaqMan MicroRNA reverse transcription kit (Applied Biosystems). Real-time RT-PCR was performed using a TaqMan MicroRNA assay kit (Applied Biosystems). The expression level of miR34a was normalized to that of U6.
siRNA, plasmid, and cell transfection.
For siRNA experiments, PC-3 cells were transfected with 40 pmol of human Sirt1 or with control siRNA purchased from Santa Cruz Biotechnology diluted in Opti-MEM (Gibco), using Lipofectamine RNAiMax as indicated by the manufacturer’s instructions. PC-3 cells were incubated for 48 h and then infected with VSVΔM51 at the indicated MOI.
The FLAG-SIRT1 WT vector and the H363Y mutant were gifts from Michael Greenberg (Addgene plasmid catalog no. 1791 and catalog no. 1792 [RRID:Addgene_1791 and RRID:Addgene_1792, respectively]) (54). Two hundred nanograms of SIRT1 WT or the H363Y mutant or empty vector was transfected in U-2 OS cells by using Lipofectamine 3000 as indicated by manufacturer’s instructions. U-2 OS cells were incubated for 24 h and then infected with VSVΔM51 at the indicated MOI.
miRNA overexpression and inhibition.
Modulation of miR-34a expression in PC-3 cells was performed using pre-miR-34a (mimic) or anti-miR-34a (inhibitor) (Ambion). A pre-miR negative control and an anti-miR inhibitor negative control were used as scrambled RNA negative controls for mimic and inhibitor, respectively. Transfections were performed using Lipofectamine RNAiMax according to the instructions of the manufacturer. Medium was changed after 6 h, and PC-3 cells were incubated for 24 h and then infected with VSVΔM51 at the indicated MOI.
Statistical analysis.
Values were expressed as means ± standard deviations (SD). Graphs and statistics were computed using Microsoft Excel or GraphPad Prism 5. An unpaired, two-tailed Student's t test was used to determine the significance of the differences between the control and each experimental condition. P values of less than 0.05 were considered statistically significant (***, P < 0.001; **, P < 0.01; *, P < 0.05).
ACKNOWLEDGMENTS
This research was supported by grants from Fondazione Cenci Bolognetti, NIH grant 7R21CA192185-02, and the Italian Association for Cancer Research (IG16901). D.O. was supported by a Carlsberg Foundation International Research Fellowship.
We declare that we have no potential conflicts of interest.
REFERENCES
- 1.Breitbach CJ, Lichty BD, Bell JC. 2016. Oncolytic viruses: therapeutics with an identity crisis. EBioMedicine 9:31–36. doi: 10.1016/j.ebiom.2016.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. 2014. Going viral with cancer immunotherapy. Nat Rev Cancer 14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 3.Rehman H, Silk AW, Kane MP, Kaufman HL. 2016. Into the clinic: talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer 4:53. doi: 10.1186/s40425-016-0158-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen W, Patnaik MM, Ruiz A, Russell SJ, Peng KW. 2016. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood 127:1449–1458. doi: 10.1182/blood-2015-06-652503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chesney J, Collichio F, Andtbacka RHI, Puzanov I, Glaspy J, Milhem M, Hamid O, Cranmer L, Saenger Y, Ross M, Chen L, Kim JJ, Kaufman HL. 1 October 2016, posting date. Interim safety and efficacy of a randomized (1:1), open-label phase 2 study of talimogene laherparepvec (T) and ipilimumab (I) vs I alone in unresected, stage IIIB-IV melanoma. Ann Oncol doi: 10.1093/annonc/mdw379.04. [DOI] [Google Scholar]
- 6.Russell SJ, Peng KW, Bell JC. 2012. Oncolytic virotherapy. Nat Biotechnol 30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 8.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275. doi: 10.1016/S1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS, Yasmeen A, Bismar TA, Kirn D, Falls T, Snoulten VE, Vanderhyden BC, Werier J, Atkins H, Vaha-Koskela MJ, Stojdl DF, Bell JC, Hiscott J. 2008. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A 105:14981–14986. doi: 10.1073/pnas.0803988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shulak L, Beljanski V, Chiang C, Dutta SM, Van Grevenynghe J, Belgnaoui SM, Nguyen TL, Di Lenardo T, Semmes OJ, Lin R, Hiscott J. 2014. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-kappaB-dependent autophagy. J Virol 88:2927–2940. doi: 10.1128/JVI.03406-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michan S, Sinclair D. 2007. Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haigis MC, Sinclair DA. 2010. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi JE, Mostoslavsky R. 2014. Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev 26:24–32. doi: 10.1016/j.gde.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR. 2007. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res 67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 15.Shuang T, Wang M, Zhou Y, Shi C. 2015. Over-expression of Sirt1 contributes to chemoresistance and indicates poor prognosis in serous epithelial ovarian cancer (EOC). Med Oncol 32:260. doi: 10.1007/s12032-015-0706-8. [DOI] [PubMed] [Google Scholar]
- 16.Jiang K, Lyu L, Shen Z, Zhang J, Zhang H, Dong J, Yan Y, Liu F, Wang S. 2014. Overexpression of SIRT1 is a poor prognostic factor for advanced colorectal cancer. Chin Med J (Engl) 127:2021–2024. [PubMed] [Google Scholar]
- 17.Kojima K, Ohhashi R, Fujita Y, Hamada N, Akao Y, Nozawa Y, Deguchi T, Ito M. 2008. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun 373:423–428. doi: 10.1016/j.bbrc.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 18.Nakane K, Fujita Y, Terazawa R, Atsumi Y, Kato T, Nozawa Y, Deguchi T, Ito M. 2012. Inhibition of cortactin and SIRT1 expression attenuates migration and invasion of prostate cancer DU145 cells. Int J Urol 19:71–79. doi: 10.1111/j.1442-2042.2011.02888.x. [DOI] [PubMed] [Google Scholar]
- 19.Yuan H, Su L, Chen WY. 2013. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther 6:1399–1416. doi: 10.2147/OTT.S37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Z, Fang D. 2013. The roles of SIRT1 in cancer. Genes Cancer 4:97–104. doi: 10.1177/1947601912475079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T, Liu PY, Marshall GM. 2009. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res 69:1702–1705. doi: 10.1158/0008-5472.CAN-08-3365. [DOI] [PubMed] [Google Scholar]
- 22.Yi J, Luo J. 2010. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta 1804:1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdelmohsen K, Pullmann R Jr, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, Furneaux H, Gorospe M. 2007. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell 25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. 2009. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 25.Yamakuchi M, Ferlito M, Lowenstein CJ. 2008. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strum JC, Johnson JH, Ward J, Xie HB, Feild J, Hester A, Alford A, Waters KM. 2009. MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol Endocrinol 23:1876–1884. doi: 10.1210/me.2009-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan K, Ge YC, Zhang XP, Wu SY, Feng JS, Chen SL, Zhang LI, Yuan ZH, Fu CH. 2015. miR-34a inhibits cell proliferation in prostate cancer by downregulation of SIRT1 expression. Oncol Lett 10:3223–3227. doi: 10.3892/ol.2015.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita Y, Kojima K, Hamada N, Ohhashi R, Akao Y, Nozawa Y, Deguchi T, Ito M. 2008. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun 377:114–119. doi: 10.1016/j.bbrc.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 29.Koyuncu E, Budayeva HG, Miteva YV, Ricci DP, Silhavy TJ, Shenk T, Cristea IM. 2014. Sirtuins are evolutionarily conserved viral restriction factors. mBio 5:e02249-14. doi: 10.1128/mBio.02249-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campagna M, Herranz D, Garcia MA, Marcos-Villar L, Gonzalez-Santamaria J, Gallego P, Gutierrez S, Collado M, Serrano M, Esteban M, Rivas C. 2011. SIRT1 stabilizes PML promoting its sumoylation. Cell Death Differ 18:72–79. doi: 10.1038/cdd.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He M, Yuan H, Tan B, Bai R, Kim HS, Bae S, Che L, Kim JS, Gao SJ. 2016. SIRT1-mediated downregulation of p27Kip1 is essential for overcoming contact inhibition of Kaposi's sarcoma-associated herpesvirus transformed cells. Oncotarget 7:75698–75711. doi: 10.18632/oncotarget.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. 2003. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J Biol Chem 278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 33.Sauve AA, Schramm VL. 2003. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry 42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- 34.Tang HM, Gao WW, Chan CP, Cheng Y, Deng JJ, Yuen KS, Iha H, Jin DY. 2015. SIRT1 suppresses human T-cell leukemia virus type 1 transcription. J Virol 89:8623–8631. doi: 10.1128/JVI.01229-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. 2010. Intelligent design: combination therapy with oncolytic viruses. Mol Ther 18:251–263. doi: 10.1038/mt.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olagnier D, Lababidi RR, Hadj SB, Sze A, Liu Y, Naidu SD, Ferrari M, Jiang Y, Chiang C, Beljanski V, Goulet ML, Knatko EV, Dinkova-Kostova AT, Hiscott J, Lin R. 2017. Activation of Nrf2 signaling augments vesicular stomatitis virus oncolysis via autophagy-driven suppression of antiviral immunity. Mol Ther 25:1900–1916. doi: 10.1016/j.ymthe.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen TL, Wilson MG, Hiscott J. 2010. Oncolytic viruses and histone deacetylase inhibitors–a multi-pronged strategy to target tumor cells. Cytokine Growth Factor Rev 21:153–159. doi: 10.1016/j.cytogfr.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM. 2005. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 39.Kim JA, Seong RK, Shin OS. 2016. Enhanced viral replication by cellular replicative senescence. Immune Netw 16:286–295. doi: 10.4110/in.2016.16.5.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. 2012. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res 72:2251–2261. doi: 10.1158/0008-5472.CAN-11-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terao Y, Nishida J, Horiuchi S, Rong F, Ueoka Y, Matsuda T, Kato H, Furugen Y, Yoshida K, Kato K, Wake N. 2001. Sodium butyrate induces growth arrest and senescence-like phenotypes in gynecologic cancer cells. Int J Cancer 94:257–267. doi: 10.1002/ijc.1448. [DOI] [PubMed] [Google Scholar]
- 42.Place RF, Noonan EJ, Giardina C. 2005. HDACs and the senescent phenotype of WI-38 cells. BMC Cell Biol 6:37. doi: 10.1186/1471-2121-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayakawa T, Iwai M, Aoki S, Takimoto K, Maruyama M, Maruyama W, Motoyama N. 30 January 2015, posting date. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One doi: 10.1371/journal.pone.0116480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. 2004. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saha RN, Jana M, Pahan K. 2007. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J Immunol 179:7101–7109. doi: 10.4049/jimmunol.179.10.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Wang K, Chen X, Meng H, Song M, Wang Y, Xu X, Bai Y. 2012. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol Biol 13:4. doi: 10.1186/1471-2199-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forte E, Salinas RE, Chang C, Zhou T, Linnstaedt SD, Gottwein E, Jacobs C, Jima D, Li QJ, Dave SS, Luftig MA. 2012. The Epstein-Barr virus (EBV)-induced tumor suppressor microRNA MiR-34a is growth promoting in EBV-infected B cells. J Virol 86:6889–6898. doi: 10.1128/JVI.07056-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matthews GM, Newbold A, Johnstone RW. 2012. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res 116:165–197. doi: 10.1016/B978-0-12-394387-3.00005-7. [DOI] [PubMed] [Google Scholar]
- 49.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR, Fearon ER. 2007. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 50.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. 2007. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A 104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, Nikitin AY. 2010. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res 16:1119–1128. doi: 10.1158/1078-0432.CCR-09-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao J, Lammers P, Torrance CJ, Bader AG. 2013. TP53-independent function of miR-34a via HDAC1 and p21(CIP1/WAF1.). Mol Ther 21:1678–1686. doi: 10.1038/mt.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S. 2011. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One 6:e24099. doi: 10.1371/journal.pone.0024099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]