

The nidovirus-specific endoribonuclease (NendoU) encoded by PRRSV nonstructural protein 11 (nsp11) is a unique NendoU of nidoviruses that infect vertebrates; thus, it is an attractive target for the development of antinidovirus drugs. Previous studies have revealed that the NendoU of nidoviruses, including porcine reproductive and respiratory syndrome virus (PRRSV) and human coronavirus 229E (HCoV-229E), acts as a type I interferon (IFN) antagonist. Here, for the first time, we demonstrated that overexpression of PRRSV nsp11 also inhibits IFN signaling by targeting the C-terminal interferon regulatory factor (IRF) association domain of IRF9. This interaction impaired the ability of IRF9 to form the transcription factor complex IFN-stimulated gene factor 3 (ISGF3) and to act as a signaling protein of IFN signaling. Collectively, our data identify IRF9 as a natural target of PRRSV NendoU and reveal a novel mechanism evolved by an arterivirus to counteract innate immune signaling.
KEYWORDS: interferon regulatory factor 9, nidovirus-specific endoribonuclease, porcine reproductive and respiratory syndrome virus, type I IFN signaling
ABSTRACT
Porcine reproductive and respiratory syndrome virus (PRRSV) is an arterivirus from the Nidovirales order that causes reproductive failure and respiratory disease in pigs and poses a constant threat to the global pig industry. The PRRSV-encoded nonstructural protein 11 (nsp11) is a nidovirus-specific endoribonuclease (NendoU) that is conserved throughout the Arteriviridae and Coronaviridae families. Previously, our research and that of others demonstrated that PRRSV nsp11 inhibits type I interferon (IFN) production through NendoU activity-dependent mechanisms. Here, we found that PRRSV nsp11 also inhibited IFN-stimulated response element (ISRE) promoter activity and subsequent transcription of IFN-stimulated genes (ISGs). Detailed analysis showed that nsp11 targeted interferon regulatory factor 9 (IRF9), but not transducer and activator of transcription 1 (STAT1) or STAT2, key molecules in the type I IFN signaling pathway. Furthermore, the nsp11-IRF9 interaction impaired the formation and nuclear translocation of the transcription factor complex IFN-stimulated gene factor 3 (ISGF3) in both nsp11-overexpressed and PRRSV-infected cells. Importantly, nsp11 mutations (H129A, H144A, and K173A) that ablate NendoU activity or its cell cytotoxicity also interacted with IRF9 and retained the ability to block IFN signaling, indicating that the nsp11-IRF9 interaction is independent of NendoU activity or cell cytotoxicity of nsp11. Taking the results together, our study demonstrated that PRRSV nsp11 antagonizes type I IFN signaling by targeting IRF9 via a NendoU activity-independent mechanism, and this report describes a novel strategy evolved by PRRSV to counteract host innate antiviral responses, revealing a potential new function for PRRSV nsp11 in type I IFN signaling.
IMPORTANCE The nidovirus-specific endoribonuclease (NendoU) encoded by PRRSV nonstructural protein 11 (nsp11) is a unique NendoU of nidoviruses that infect vertebrates; thus, it is an attractive target for the development of antinidovirus drugs. Previous studies have revealed that the NendoU of nidoviruses, including porcine reproductive and respiratory syndrome virus (PRRSV) and human coronavirus 229E (HCoV-229E), acts as a type I interferon (IFN) antagonist. Here, for the first time, we demonstrated that overexpression of PRRSV nsp11 also inhibits IFN signaling by targeting the C-terminal interferon regulatory factor (IRF) association domain of IRF9. This interaction impaired the ability of IRF9 to form the transcription factor complex IFN-stimulated gene factor 3 (ISGF3) and to act as a signaling protein of IFN signaling. Collectively, our data identify IRF9 as a natural target of PRRSV NendoU and reveal a novel mechanism evolved by an arterivirus to counteract innate immune signaling.
INTRODUCTION
Porcine reproductive and respiratory syndrome (PRRS) is the most economically important infectious disease in the pig industry, resulting in decreased reproductive performance and increased respiratory problems in pigs (1, 2). The etiological agent, PRRS virus (PRRSV) (family Arteriviridae, order Nidovirales), has a positive-polarity single-stranded RNA genome, approximately 15 kb in length, encoding two nonstructural polyproteins (ORF1ab and ORF1b) and eight structural proteins (GP2, E, GP3, GP4, GP5, ORF5a, M, and N) (3, 4). After being cleaved by four virus-encoded proteases, nonstructural protein 1α (nsp1α), nsp1β, nsp2, and nsp4, the two polyproteins are further cleaved into individual nonstructural proteins that perform different functions during the viral life cycle (5). To reproduce rapidly and establish a persistent infection in pigs, PRRSV has developed the ability to resist host interferon (IFN) signaling, which is the key signaling pathway for innate immune responses against viral invasion or replication (1, 6, 7).
Innate immune responses are activated by host pattern recognition receptors (PRRs), which recognize molecular structures called pathogen-associated molecular patterns (PAMPs) that are structurally conserved within a large number of pathogenic organisms (8). Upon recognition of PAMPs, PRRs initiate signaling pathways that ultimately trigger the production of type I IFNs. Subsequently, the Janus kinase/signal transduction and transcription activator (JAK/STAT) pathway is activated by type I IFNs (9, 10). Briefly, type I IFNs bind to their surface receptors (IFNAR1 and IFNAR2) and then activate and phosphorylate the two intracellular tyrosine kinases Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2). These activated tyrosine kinases mainly phosphorylate two transcription factors, namely, signal transducer and activator of transcription 1 (STAT1) and STAT2, which interact with IFN regulatory factor 9 (IRF9) to form IFN-stimulated gene factor 3 (ISGF3) (11). The transcription factor complex ISGF3 (STAT1/STAT2/IRF9) is transported to the nucleus, where it recognizes IFN-stimulated response elements (ISREs), leading to the induction of hundreds of IFN-stimulating genes (ISGs) (12). Many ISGs function as potent antiviral effectors, directly preventing viral infections by targeting viral processes such as viral entry, viral genome replication, viral protein synthesis, or viral body release (13).
During coevolution with their hosts, many viruses have developed elaborate strategies to circumvent the JAK/STAT pathway in IFN signaling (14). Among several known viral evasion strategies, targeting the transcription factor complex ISGF3 is a particularly powerful means of inactivating IFN signaling, because this strategy effectively inhibits common downstream ISG expression. For example, our recent study showed that porcine deltacoronavirus blocks the JAK/STAT pathway by 3C-like protease-mediated cleavage of STAT2 (15). The nonstructural protein 1 (NSP1) of rotavirus mediates the degradation of IRF9 by targeting the C-proximal IRF-binding domain and inhibits IFN-mediated phosphorylation of STAT1 (16, 17). Sendai virus C protein binds STAT1 to block the formation of STAT1/STAT2 heterodimers or STAT1/STAT1 homodimers, which inhibit IFN signaling (18). Previous studies have shown that PRRSV nsp11 inhibits type I IFN production through nidovirus-specific endoribonuclease (NendoU) activity-dependent mechanisms (19–21). However, the NendoU activity of overexpressed nsp11 unexpectedly exhibited extensive substrate specificity in vitro and is extremely toxic to prokaryotic and eukaryotic cells, indicating that the inhibition of IFN production by wild-type (WT) PRRSV nsp11 may be due to its cytotoxicity (21). Here, we found that PRRSV nsp11 also inhibits ISRE promoter activity and the transcription of ISGs, thereby interfering with the type I IFN signaling pathway. Importantly, mutations that eliminate NendoU activity and its cytotoxicity in nsp11 retain the ability to block IFN signaling. Detailed analysis showed that nsp11 inhibited type I IFN signaling by targeting IRF9, a key molecule in the ISGF3 complex, revealing a potential novel function of PRRSV nsp11 in type I IFN signaling.
RESULTS
Identification of PRRSV nsp11 as an antagonist of type I IFN signaling.
Type I IFN signaling induces a potent antiviral response in cells by inducing the expression of hundreds of ISGs, which is vital for the control of viral infections (22). To assess the potential role of PRRSV nsp11 in type I IFN signaling, the mRNA levels of IFN-stimulated gene 15 (ISG15), ISG54, ISG56, and 2'-5'-oligoadenylate synthetase 1 (OAS1) were analyzed in human embryonic kidney cells (HEK-293T) overexpressing hemagglutinin (HA)-tagged PRRSV nsp11. As shown in Fig. 1A, PRRSV nsp11 significantly inhibited the transcription of ISGs induced by IFN-α compared with the control group results. Because of the presence of ISRE in the ISG promoter regions, various concentrations of PRRSV nsp11 expression plasmid and ISRE-luciferase reporter plasmid were cotransfected into HEK-293T cells or porcine kidney cells (PK-15). The results showed that nsp11 strongly inhibited IFN-α-induced ISRE promoter activity in a dose-dependent manner in HEK-293T cells (Fig. 1B) and PK-15 cells (Fig. 1C). These results confirm the antagonistic nature of PRRSV nsp11 in type I IFN signaling.
FIG 1.

PRRSV nsp11 antagonizes type I IFN signaling. (A) HEK-293T cells cultured in 48-well plates were transfected with PRRSV nsp11 expression plasmid or vector (0.5 μg/well). After 24 h, cells were treated with 1,000 U/ml of IFN-α for 8 h and analyzed by qPCR. (B and C) HEK-293T cells (B) or PK-15 cells (C) cultured in 24-well plates were transfected with various concentrations of PRRSV nsp11 expression plasmid (0.4, 0.2, 0.1, or 0 μg/well) along with ISRE-Luc plasmid (0.04 μg/well) and pRL-TK plasmid (0.01 μg/well). After 24 h, cells were treated with 1,000 U/ml of IFN-α for 8 h, followed by luciferase assays. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
PRRSV nsp11 inhibits type I IFN signaling in an endoribonuclease activity-independent manner.
In arterivirus, the nsp11 endoribonuclease is important for viral replication (19, 21, 23–26). Several previous studies have shown that PRRSV nsp11 inhibits type I IFN production in a NendoU activity-dependent manner (19–21). We considered such a possibility for PRRSV nsp11-mediated inhibition of type I IFN signaling to be associated also with its NendoU activity. On the basis of their chemical properties and known residue requirements, the three catalytic residues of endoribonucleases (His129, His144, and Lys173 [numbering based on PRRSV nsp11]) were shown to be directly involved in catalysis (21, 23, 25). Thus, three endoribonuclease catalytic residue mutations, His129Ala (H129A), His144Ala (H144A), and Lys173Ala (K173A), were introduced into PRRSV nsp11 and the corresponding eukaryotic expression plasmids expressing the N-terminally HA-tagged nsp11 endoribonuclease inactive mutants were constructed. To test whether the HA-tagged nsp11 mutants lose endoribonuclease activity, HA-tagged WT nsp11 and three mutants were expressed in Escherichia coli. The recombinant proteins were purified (Fig. 2A), and fluorescence resonance energy transfer (FRET) assays were performed to detect the endoribonuclease activity. As shown in Fig. 2B, the levels of endoribonuclease activity of the three HA-tagged nsp11 mutants were significantly decreased compared with the WT nsp11 (Fig. 2B). However, none of the endoribonuclease inactive mutants (H129A, H144A, or K173A) showed a loss of the ability of nsp11 to inhibit ISRE promoter activity in cells overexpressing HA-tagged nsp11 mutants (Fig. 2C). We also used PRRSV nsp9 as a negative control and porcine epidemic diarrhea virus (PEDV) nsp15 as a positive control. In agreement with a previous study (27), PEDV WT nsp15 suppressed IFN-α-induced ISRE promoter activity whereas such an inhibitory effect was not observed in cells expressing a PEDV nsp15 endoribonuclease inactive mutant (H226A) or PRRSV nsp9 (Fig. 2C). These data suggest that although the NendoU activity of PEDV nsp15 is important for inhibiting IFN signaling, PRRSV nsp11 does not depend on the NendoU activity. We further tested the ability of nsp11 mutants to inhibit IFN-α-induced ISG expression. As shown in Fig. 2D, each endoribonuclease inactive mutant (H129A, H144A, or K173A) of nsp11 also inhibited the IFN-α-induced transcription of ISGs. Since these three mutants are also devoid of cell cytotoxicity (21), our results indicated that the inhibition of IFN signaling observed in nsp11-expressing cells is independent of its endoribonuclease activity and cell cytotoxicity.
FIG 2.

PRRSV nsp11-mediated inhibition of type I IFN signaling is independent of its endoribonuclease activity. (A) SDS-PAGE analysis of the N-terminally HA-tagged form of PRRSV nsp11 and its mutants (H129A, H144A, K173A). (B) FRET-based enzyme activity. The endoribonuclease activity of HA-tagged PRRSV nsp11 and its mutants (H129A, H144A, K173A) is indicated with different colors. Values representing results of experiments performed in triplicate are presented. (C) HEK-293T cells in 48-well plates were transfected with PRRSV nsp11 expression plasmid or its endoribonuclease activity-defective mutants H129A, H144A, and K173A to analyze ISRE promoter activity, as described in the Fig. 1B legend (negative control, PRRSV nsp9; positive control, PEDV nsp15). (D) HEK-293T cells cultured in 24-well plates were transfected with PRRSV nsp11 expression plasmid or vector. After 24 h, cells were treated with 1,000 U/ml of IFN-α for 8 h and analyzed by qPCR in a parallel experiment, as shown in Fig. 1A. *, P < 0.05; **, P < 0.01; ns, not significant.
PRRSV nsp11 disrupts ISGF3-mediated activation of the ISRE promoter.
ISGs are antiviral effectors induced by IFNs through the formation of a tripartite transcription factor, ISGF3, which is composed of STAT1, STAT2, and IRF9 (11). Given the pivotal role of the transcription factor complex ISGF3 in type I IFN signaling, we further investigated whether overexpression of nsp11 inhibits ISGF3-mediated signaling. As shown in Fig. 3, coexpression of the components of transcription factor complex ISGF3 (STAT1, STAT2, and IRF9) significantly activated the ISRE promoter compared with the results seen with the empty plasmid control. However, activation of the ISRE promoter by ISGF3 was significantly inhibited in the presence of PRRSV nsp11 (Fig. 3). Similarly to the results seen with WT nsp11, each endoribonuclease inactive mutant (H129A, H144A, or K173A) was also able to suppress the ISGF3-mediated IRES promoter (Fig. 3), suggesting that the endoribonuclease activity of nsp11 does not govern the ability of nsp11 to block the activity of the ISGF3-induced IRES promoter. Consequently, we speculated that PRRSV nsp11 might target the ISGF3 complex to inhibit type I IFN signaling.
FIG 3.
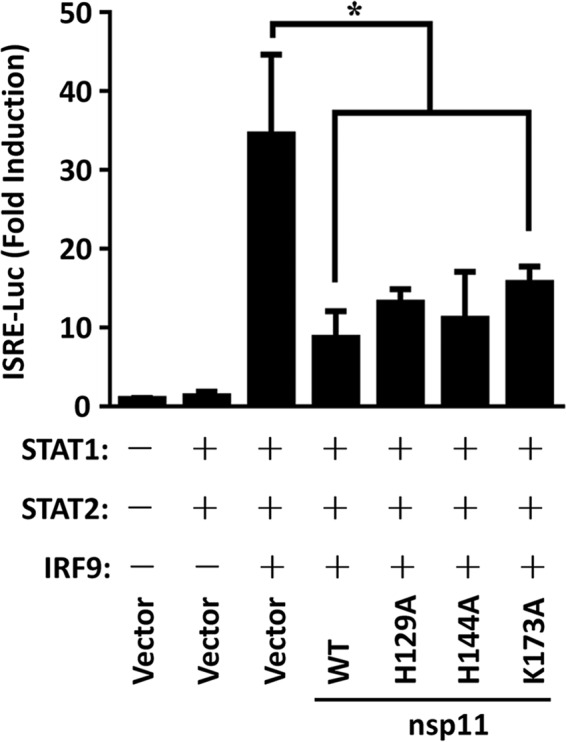
PRRSV nsp11 inhibits ISGF3-induced ISRE promoter activity. HEK-293T cells were cotransfected with PRRSV nsp11 or its endoribonuclease activity-defective mutants (0.2 μg/well), along with porcine ISGF3 complex (STAT1/STAT2/IRF9; 0.3 μg/well), ISRE-Luc plasmid (0.04 μg/well), and pRL-TK plasmid (0.01 μg/well). After 30 h, cells were harvested for luciferase assays. *, P < 0.05.
PRRSV nsp11 does not degrade or block the phosphorylation of STAT1 and STAT2.
Since downregulation of molecules responsible for IFN-activated signal transduction is a mechanism commonly employed by viruses (16, 28–30), we investigated whether nsp11 impairs the endogenous protein levels of STAT1, STAT2, and IRF9. As shown in Fig. 4, no reduction was observed in the protein levels of STAT1, STAT2, and IRF9 as a consequence of the presence of WT nsp11 or expression of its endoribonuclease inactive mutants (H129A, H144A, and K173A [compare lanes 3 and 4 to lane 7]), suggesting that nsp11 does not induce downregulation of the ISGF3 complex.
FIG 4.
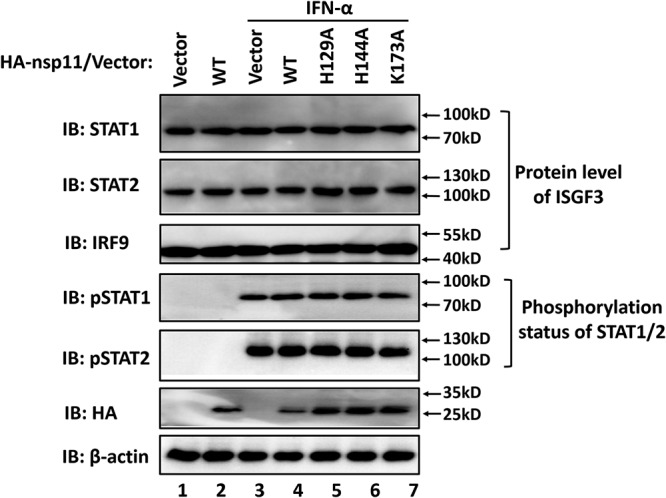
PRRSV nsp11 does not affect the protein level of ISGF3 or the phosphorylation status of STAT1/2. HEK-293T cells were transfected with PRRSV nsp11 expression plasmid or its mutants. After 24 h, cells were treated with IFN-α (1,000 U/ml) for 4 h and collected for Western blot analysis. Antibodies against STAT1, STAT2, IRF9, β-actin, phospho-STAT1-Y701 (pSTAT1), and phospho-STAT2-Y690 (pSTAT2) were utilized to detect each respective endogenous protein. The levels of expression of PRRSV nsp11 and its mutants were assessed with an anti-HA antibody. IB, immunoblotting.
The type I IFN-activated ISGF3 transcription complex containing tyrosine-phosphorylated STAT1 and STAT2 associated with IRF9 is rapidly translocated to the nucleus after IFN treatment, and phosphorylation-dependent activation of STAT1 and STAT2 is critical to mediate IFN-inducible antiviral responses (11, 22). To investigate whether PRRSV nsp11 alters the STAT1 and STAT2 phosphorylation status after IFN-α stimulation, lysates from nsp11-expressing cells were subjected to Western blotting using phospho-STAT1 (STAT1-Y701) and phospho-STAT2 (STAT2-Y690) antibodies (Abs), respectively. The levels of phosphorylated STAT1 and STAT2 were greatly increased after IFN-α treatment, and no difference in the levels was found between nsp11-expressing cells and nsp11-nonexpressing cells (Fig. 4, lanes 3 and 4). Similarly, nsp11 mutants lacking endoribonuclease activity (H129A, H144A, and K173A) had no effect on the phosphorylation of STAT1 and STAT2 (Fig. 4 [compare lane 3 to lanes 5 to 7]). These results indicated that PRRSV nsp11 and its endoribonuclease inactive mutants do not target the ISGF3 complex for degradation or prevent type I IFN-induced STAT1 protein-activating tyrosine phosphorylation.
PRRSV nsp11 interacts with the IRF-association domain (IAD) of IRF9.
Previous studies have shown that many viral proteins can interact with components of the ISGF3 complex to inhibit type I IFN signaling (18, 31, 32). Thus, we next investigated whether nsp11 functions by interacting with the ISGF3 component. To this end, HEK-293T cells were transfected with expression constructs encoding HA-tagged PRRSV nsp11 protein and Flag-tagged porcine STAT1, STAT2, and IRF9. Coimmunoprecipitation (Co-IP) and immunoblotting analyses showed that nsp11 interacted with porcine IRF9, but not STAT1 or STAT2 (Fig. 5A to C). To further confirm the interaction of nsp11 with endogenous IRF9, coprecipitated endogenous IRF9 and PRRSV nsp11 were analyzed by immunoblotting with an anti-IRF9 antibody and an anti-HA antibody, respectively. As shown in Fig. 5D, reciprocal pulldown of both nsp11 and endogenous IRF9 further confirmed the interaction between nsp11 and IRF9.
FIG 5.

PRRSV nsp11 interacts with the IRF-association domain (IAD) of IRF9. (A to C) HEK-293T cells were cotransfected with an expression vector encoding HA-nsp11 and with expression vectors encoding Flag-STAT1 (A), Flag-STAT2 (B), or Flag-IRF9 (C). Immunoblotting analysis was performed to detect the interaction between nsp11 and the ISGF3 complex. The cell lysates were immunoprecipitated (IP) with rabbit polyclonal anti-Flag or anti-HA antibody and immunoblotted with mouse monoclonal anti-HA or anti-Flag antibody, respectively. PRRSV nsp9 served as a negative control in the experiment represented by panel C. (D) HEK-293T cells were transfected with expression vectors encoding HA-nsp11. The cell lysates were immunoprecipitated with mouse monoclonal anti-HA antibody or IgG as a negative control and immunoblotted with anti-IRF9 antibody or anti-HA antibody, respectively (left). Reversed immunoprecipitation was performed with an anti-IRF9 antibody and immunoblotted with an anti-HA antibody or anti-IRF9 antibody, respectively (middle). The horseradish peroxidase-conjugated goat anti-rabbit IgG light chain was used to eliminate the interference between IRF9 and IgG heavy chain. The horseradish peroxidase-conjugated goat anti-mouse IgG heavy chain was used to eliminate the interference between nsp11 and IgG light chain. (E) Schematic representation of porcine IRF9 and its derivatives [IRF9 (1–120), IRF9 (1–200), IRF9 (200–411), and IRF9 (120–411)]. (F) HEK-293T cells were cotransfected with expression construct HA-nsp11 and with the indicated plasmids of porcine IRF9 for 30 h. Cell lysates were immunoprecipitated using anti-Flag antibody and analyzed by immunoblotting using anti-Flag or anti-HA antibodies.
Previous studies demonstrated that IRF9 contains an N-terminal DNA-binding domain (DBD; amino acids [aa] 5 to 118) and a C-terminal IAD (aa 205 to 390) connected by a flexible linker (FL) (33–35). To investigate which domain of porcine IRF9 is involved in nsp11 protein binding, four mutants with deletions of different domains of IRF9, including IRF9 (aa 1 to 120; DBD), IRF9 (aa 1 to 200; DBD-FL), IRF9 (aa 200 to 411; IAD), and IRF9 (aa 120 to 411; FL-IAD), were constructed by mutagenesis (Fig. 5E). HEK-293T cells were cotransfected with various combinations of Flag-tagged full-length or deleted versions of IRF9 and the HA-tagged PRRSV nsp11 protein. As shown in Fig. 5F, Co-IP experiments showed that the C-terminal IRF association domain-deleted mutants of IRF9 [IRF9 (1–120) and IRF9 (1–200)] could not interact with the PRRSV nsp11 protein, indicating that PRRSV nsp11 interacts with the IRF association domain of IRF9 (IRF9-IAD).
PRRSV nsp11-IRF9 interaction impairs the IFN-induced formation and nuclear accumulation of ISGF3.
Since the function of ISGF3 relies on the selective interaction between phosphorylated STAT2 and the IRF-association domain of IRF9 (36, 37), the observed interaction between nsp11 and IRF9-IAD led us to speculate that this interaction may impair the recruitment of phosphorylated STAT2 by IRF9 and the subsequent nuclear accumulation of ISGF3. To test this hypothesis, HEK-293T cells were transfected with the indicated nsp11 expression plasmids and then treated with IFN-α. The PRRSV nsp11 protein and phosphorylated STAT1/STAT2 (STAT1-Y701 and STAT2-Y690) were immunoprecipitated with an anti-IRF9 antibody. As shown in Fig. 6A, IFN-α treatment significantly induced the phosphorylation of STAT1 and STAT2, and IRF9 efficiently pulled down phosphorylated STAT1/STAT2 after IFN-α treatment (compare lanes 1 and 3). However, the amount of phosphorylated STAT1/STAT2 that bound to IRF9 remarkably decreased in the presence of PRRSV nsp11 and its mutants (H129A, H144A, and K173A) (Fig. 6A, lanes 3 and 4 to 7).
FIG 6.

PRRSV nsp11-IRF9 interaction inhibits the IFN-induced formation and nuclear accumulation of ISGF3. (A) HEK-293T cells were transfected with expression vectors encoding HA-nsp11 and its mutants. Endogenous IRF9 was precipitated and immunoblotting analysis was performed with an anti-pSTAT1 antibody (the top panel), anti-pSTAT2 antibody (the second panel), or anti-HA antibody (the fourth panel) to detect the interaction with endogenous IRF9. (B) Phosphorylated STAT1 and STAT2 in nuclear and cytoplasmic fractions. Subcellular fractionation of HEK-293T cells, 4 h after IFN-α treatment, was performed for nuclear and cytoplasmic fractions, followed by Western blotting using STAT1-Y701 antibody, STAT2-Y690 antibody, anti-HSP90 antibody as a cytoplasmic marker, and anti-PARP1 antibody as a nuclear protein marker, as indicated. HA antibody was used to detect the expression of PRRSV nsp11 and its mutants (HA-nsp11).
To further test whether PRRSV nsp11 prevents the IFN-induced nuclear accumulation of ISGF3, we performed nuclear and cytoplasmic fractionation of the cells following IFN-α treatment. Antibodies against STAT1-Y701 and STAT2-Y690 were used to detect the presence of the phosphorylated proteins in the two fractions. As expected, higher levels of STAT1-Y701 and STAT2-Y690 were found in the nuclear fraction than in the cytoplasmic fraction after IFN-α treatment of mock-transfected cells (Fig. 6B, lanes 3 and 10). In contrast, in nsp11-transfected cells after IFN-α stimulation, more ISGF3 complex components (STAT1-Y701, STAT2-Y690, and IRF9) were detected in the cytoplasmic fraction than in the nuclear fraction (Fig. 6B, lanes 4 and 11). Taken together, these findings indicate that PRRSV nsp11 impairs the formation and nuclear translocation of ISGF3.
Because the endoribonuclease inactive mutants of nsp11 retained the ability to block type I IFN signaling, these mutants in particular were still capable of interacting with IRF9. Next, we investigated whether these mutants, lacking endoribonuclease activity, still impaired the formation and nuclear accumulation of ISGF3. Indeed, our data revealed that the PRRSV nsp11 mutants (H129A, H144A, and K173A) still significantly inhibited the IFN-induced formation and nuclear accumulation of ISGF3 and did so to similar extents (Fig. 6B [compare lanes 5 to 7 and lanes 12 to 14]). This provided further support for the notion that the ability of PRRSV nsp11 to block type I IFN signaling is independent of its endoribonuclease activity and cell cytotoxicity, because these three mutants had similar effects with respect to inhibiting the formation and nuclear translocation of ISGF3 compared with WT nsp11.
PRRSV infection inhibits the formation of ISGF3 through the interaction of nsp11 with IRF9.
To exclude the possibility that the observed PRRSV nsp11-IRF9 interaction was an artifact of plasmid overexpression in cell culture, we analyzed this interaction in the context of PRRSV infection. African green monkey kidney cells (Marc-145) were infected with PRRSV for 24 h and then collected at 4 h after IFN-α treatment for immunoprecipitation with anti-nsp11 or anti-IRF9 antibodies to detect the interaction between nsp11 and endogenous IRF9. As shown in Fig. 7A, nsp11 was coimmunoprecipitated with endogenous IRF9 in PRRSV-infected cells. Furthermore, phosphorylated STAT1/STAT2 was also coimmunoprecipitated with endogenous IRF9 upon IFN-α stimulation. However, the formation of STAT1/STAT2/IRF9 heterotrimers was significantly inhibited following PRRSV infection (Fig. 7A [compare lanes 3 and 4]). To further address whether the IFN-induced nuclear accumulation of ISGF3 was also affected by PRRSV infection, nuclear and cytoplasmic fractionation was performed. Compared with uninfected cells, the levels of STAT1-Y701, STAT2-Y690, and IRF9 proteins were reduced in the nuclear fraction of PRRSV-infected cells after IFN-α stimulation (Fig. 7B [compare lanes 3 to 4 and lanes 7 to 8]). Immunofluorescence experiments showed that PRRSV infection inhibited IFN-induced nuclear accumulation of STAT1-Y701 (Fig. 7C), STAT2-Y690 (Fig. 7D), and IRF9 proteins (Fig. 7E) compared to mock-infected cells. In PRRSV-infected cells with IFN stimulation, nsp11 and IRF9 were primarily colocated in the cytoplasm (Fig. 7E). Together, these data indicated that PRRSV infection inhibits the formation and nuclear accumulation of ISGF3 through the nsp11-IRF9 interaction, which was consistent with the results of gene transfection experiments in cells expressing PRRSV nsp11.
FIG 7.

PRRSV infection reduces the formation of ISGF3 through the nsp11-IRF9 interaction. (A) Marc-145 cells were either mock infected or infected with PRRSV at an MOI of 0.5 for 24 h and then treated with IFN-α for 4 h or left untreated. The cells were lysed and immunoprecipitated with an anti-IRF9 antibody and then immunoblotted with an anti-nsp11 antibody (the second panel), an anti-pSTAT1 antibody (the third panel), or an anti-pSTAT2 antibody (the fourth panel) to detect the interaction with endogenous IRF9. (B) Phosphorylated STAT1 and STAT2 in the nuclear and cytoplasmic fractions. An experiment parallel to that outlined for panel A was performed. Subcellular fractionation of Marc-145 cells was performed to obtain nuclear and cytoplasmic fractions, followed by Western blotting using anti-STAT1-Y701, anti-STAT2-Y690, anti-IRF9, and anti-nsp11 antibodies, along with anti-HSP90 antibody as a cytoplasmic marker and anti-PARP1 antibody as a nuclear protein marker, as indicated. (C to E) Marc-145 cells were infected with PRRSV (MOI = 0.5) or uninfected. At 24 h postinfection, the cells were mock-treated or treated with IFN-α (1000 IU/ml) for 4 h. After the cells were fixed and permeabilized, p-STAT1 (C), p-STAT2 (D), and IRF9 (E) was visualized by immunofluorescence staining with rabbit anti-pSTAT1, rabbit anti-pSTAT2, or rabbit anti-IRF9 antibody. The nsp11 protein was detected by the use of a nsp11-specific Mab.
DISCUSSION
During coevolution with their hosts, many viruses have acquired mechanisms to circumvent host innate immune responses. Of note, PRRSV infection has been found to produce abnormally low levels of type I IFNs and to inhibit the ability of type I IFNs to induce antiviral responses (1, 38). The endoribonuclease encoded within the PRRSV nsp11 sequence, which has the uridylate-preferred cleavage site for RNA that is necessary for virus replication, has been found to be a multifunctional protein (19, 21). Previously, it has been demonstrated that PRRSV nsp11 is involved in the inhibition of IFN production through multiple distinct mechanisms, as follows. (i) nsp11 represses the transcription of type I IFNs by inhibiting the activation of transcription factors IRF3 and nuclear factor kappa B (NF-κB), which directly activate the promoters of type I IFNs (19, 20). (ii) nsp11 reduces the levels of transcripts and proteins of mitochondrial antiviral signaling protein (MAVS) and retinoic acid-inducible gene I (RIG-I), two critical factors in the IFN induction pathway (19). (iii) nsp11 removes the ubiquitin chains from IκBα (inhibitor of NF-κB alpha), thereby preventing the proteasomal degradation of IκBα and subsequent liberation of NF-κB (39). (iv) nsp11 recruits the OTU deubiquitinase with linear linkage specificity (OTULIN) to enhance its ability to remove the cellular protein ubiquitin associated with innate immunity, resulting in the additive effect of suppressing type I IFN production (40). However, whether nsp11 also regulates IFN signaling remains unclear. In this report, we present evidence that PRRSV nsp11 suppressed type I IFN signaling by targeting IRF9, a key molecule in the JAK/STAT pathway, revealing a potential new function for the nidovirus endoribonuclease in type I IFN signaling.
PRRSV nsp11, a conserved NendoU within the Arteriviridae and Coronaviridae families, belongs to the Xenopus laevis poly(U)-specific endoribonuclease (XendoU) superfamily and plays an important role in nidovirus replication and pathogenesis (41). The structures of the arterivirus nsp11, coronavirus (CoV) nsp15, and XendoU catalytic domains, essential for endoribonuclease activity, and particularly the active site residues (His129, His144, and Lys173; numbering based on PRRSV nsp11), were found to be highly conserved. Besides PRRSV nsp11, several other studies have reported that CoV nsp15 inhibits IFN production in ectopic expression experiments. In support of this, infection with NendoU activity-deficient CoVs, such as PEDV, murine hepatitis virus, and human CoV 229E (HCoV-229E), produced a remarkably high level of type I IFN in primary cells compared with WT infection, which effectively demonstrated the IFN-antagonistic properties of nsp15 of CoVs (27, 42, 43). These data appear to indicate that endoribonuclease activity is sufficient for NendoU-mediated IFN transcriptional repression; however, the NendoU activity of overexpressed nsp11/nsp15 may unexpectedly mediate nonspecific cleavage, thereby inducing cytotoxicity (21, 41). Unfortunately, it was impossible to determine whether the cell cytotoxicity also contributed to the inhibition of IFN induction by NendoU, as mutations disrupting the ability of NendoU to block IFN production abrogated not only endoribonuclease activity but also its cell cytotoxicity (21). Thus, the observation of IFN transcriptional suppression in previous studies could have been the result of cytotoxicity as a consequence of ectopic overexpression of NendoU. Previous studies have shown that PRRSV WT nsp11 is toxic to E. coli and that the expression levels of WT nsp11 are extremely low and that one of the NendoU mutants (nsp11 K173A) can be expressed at high levels under identical conditions (19, 25). Similar phenomena were observed in our experiments designed to express and purify nsp11. Indeed, in our previous study (21), we found that PRRSV WT nsp11 exhibited cytotoxicity to eukaryotic cells whereas no cytotoxicity associated with the NendoU mutants of nsp11 was detected. Similarly, recombinant nsp11 of equine arteritis virus (EAV) has been reported to be extremely toxic to a variety of hosts (25). Thus, the lower level of expression of PRRSV WT nsp11 may have been due to its cytotoxicity (Fig. 4). Interestingly, our results clearly showed that the PRRSV nsp11 mutants (H129A, H144A, and K173A) devoid of both NendoU activity and cell cytotoxicity retained the ability to block type I IFN signaling by targeting IRF9, as did WT nsp11, indicating that nsp11 has evolved a new mechanism for the impairment of IFN signaling that operates in an endoribonuclease activity-independent and cell cytotoxicity-independent manner, distinct from the inhibition of IFN induction.
As a potent antiviral response, type I IFN signaling can control viral infections by activating the transcription factor complex ISGF3 (STAT1/STAT2/IRF9), resulting in increased transcription of hundreds of ISGs and contributing to the development of an antiviral state (12, 13). It is becoming increasingly apparent that IRF9 is a central factor not only for mediation of but also for regulation and direction of type I IFN responses (44). While many IFN effects, in particular, those associated with type I IFNs, require all three canonical signaling molecules, studies of IRF9 deficiency revealed unique roles for IRF9 that were distinct from those of STAT1 and STAT2 (45, 46). Recently, evidence has emerged that IRF9 is the main viral target of the host's innate immune response. For example, our previous study (31) revealed that porcine bocavirus nonstructural protein 1 (NS1) inhibits the DNA-binding activity of ISGF3 by interacting with IRF9. Another study showed that the E7 oncoprotein of papillomavirus binds to IRF9 to block the formation of the ISGF3 complex (47). Moreover, many virus-encoded proteins, such as varicella-zoster virus ORF63, adenovirus early region 1A protein (E1A), and rotavirus NSP1, mediate IRF9 degradation (16, 48, 49). Human cytomegalovirus also reduces the protein levels of IRF9 in human embryonic lung fibroblasts (50). To survive in the host, PRRSV has also evolved strategies to block IFN signaling. Previous studies have revealed that PRRSV nsp1β, a papain-like proteinase, blocks the nuclear translocation of ISGF3 by inducing degradation of karyopherin-α1 (51, 52). Here, we showed that nsp11 is another PRRSV-encoded antagonist of IFN signaling. PRRSV nsp11 adopts a mechanism distinct from that of nsp1β, i.e., interaction with IRF9, an essential component in the formation of ISGF3. Our data not only highlight the multifaceted control of type I IFN signaling by PRRSV but also uncover a novel mechanism by which PRRSV antagonizes innate immune signaling.
It is well established that IRF9 exhibits several functionally conserved regions among the members of the IRF family, such as an N-terminal DNA-binding domain that recognizes and binds to ISRE motifs in the promoter region of most ISGs and a C-terminal IRF-association domain that is responsible for selective interaction with the coiled-coil domain (CCD) of STAT2 (36, 37). Recently, Rengachari and colleagues reported the crystal structures of IRF9-IAD alone and in a complex with STAT2-CCD (53). The structure of the IRF9-IAD/STAT2-CCD complex revealed that surface features had deviated from their respective paralogs to enable a specific interaction between IRF9-IAD and STAT2-CCD required for ISGF3 function in cells (53). In this study, we found that PRRSV nsp11 specifically interacted with IRF9-IAD. Thus, it may be the case that the targeting of IRF9-IAD by nsp11 sequesters the interaction between IRF9 and STAT2. This may help to explain why the formation and nuclear translocation of ISGF3 were severely impaired in PRRSV nsp11-transfected cells and PRRSV-infected cells. Interestingly, PRRSV nsp11 mutants (H129A, H144A, and K173A) also impaired the formation and nuclear translocation of ISGF3 by interacting with IRF9. Furthermore, the three-dimensional (3D) structures of NendoU activity-defective arterivirus nsp11 (K173A in PRRSV nsp11; H141A and K170A in EAV nsp11) are remarkably similar to those of WT PRRSV nsp11 (21, 23), with an overall root mean square deviation (RMSD) range of 0.408 to 2.000 Å (data not shown), suggesting that the Ala substitution in His129, His144, and Lys173 of PRRSV nsp11 in this study might not prevent the core conformations of nsp11 protein from folding correctly. On the basis of data presented here and those reported by others mentioned above, we speculate that the ability to interact with IRF9 correlated with the correct folding of nsp11 but not with NendoU activity or cell cytotoxicity. Future investigations will be required to identify the structural basis of the nsp11-IRF9 interaction.
Although our data clearly demonstrated that PRRSV nsp11 could be coimmunoprecipitated with endogenous IRF9 in PRRSV-infected cells, thereby inhibiting the formation and nuclear translocation of ISGF3 (Fig. 7), whether PRRSV nsp11 also functions as an antagonist of IFN signaling during PRRSV infection has not been fully understood. One of the obstacles to studying arterivirus with NendoU activity-deficient nsp11 mutants in cell culture is that catalytic residue mutations have been reported to nearly completely abolish viral replication even in IFN-deficient cells (19, 26). Future investigations designed to identify other nonactive site residues of nsp11 that are involved in the nsp11-IRF9 interaction but that do not affect PRRSV replication will be required to fully elucidate the function of nsp11 in IFN signaling. In summary, our data identify PRRSV nsp11 as a newly recognized antagonist in IFN signaling and show that the nsp11-IRF9 interaction is involved in inhibition of the formation and nuclear translocation of ISGF3. This novel function of PRRSV nsp11 offers new insight into the interaction between a viral EndoU and the IFN signaling pathway, potentially aiding the development of novel therapeutic targets and more-effective vaccines against PRRSV.
MATERIALS AND METHODS
Cells and viruses.
HEK-293T, Marc-145, and PK-15 cells, obtained from the China Center for Type Culture Collection, were cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen, USA) supplemented with 10% fetal bovine serum. PRRSV strain WUH3 (GenBank accession number HM853673.2), isolated from the brain of a pig suffering from “high fever” syndrome in China at the end of 2006, was amplified and titrated as described previously (54).
Plasmids.
The luciferase reporter plasmid ISRE-Luc and the cDNA expression constructs encoding porcine STAT1, STAT2, and IRF9 with an N-terminal Flag tag have been described previously (15). The luciferase reporter plasmid ISRE-Luc contained IFN-stimulated response elements in the promoter of most of the ISGs cloned upstream of the firefly luciferase reporter gene. pRL-TK plasmid (Promega) was used as an internal control for normalization of the transfection efficiency. The nsp11 gene of PRRSV strain WUH3 was amplified and cloned into pCAGGS-HA with an N-terminal HA tag. PRRSV nsp11 mutants (H129A, H144A, and K173A) were constructed by overlap extension PCR using specific mutagenic primers (available upon request) in a pCAGGS-HA background. PRRSV nsp11 and its mutants with an N-terminal HA tag were cloned into pGEX-6p-1 with a glutathione S-transferase (GST) tag in the N terminus. IRF9 deletion mutants were also amplified by PCR and constructed in a pCAGGS-Flag background. PEDV nsp15 was amplified from PEDV strain AJ1102 (55) and cloned into vector pCAGGS-HA with a HA tag. PEDV nsp15 mutants (H226A) were constructed using PEDV nsp15 as the template. All constructed plasmids were confirmed by sequencing.
Luciferase reporter gene assay.
HEK-293T or PK-15 cells grown in 48-well plates were cotransfected with reporter plasmid ISRE-Luc (0.04 μg/well), pRL-TK (0.01 μg/well), and the indicated expression plasmids or an empty control plasmid. Where indicated, cells were further treated with IFN-α (PBL Assay Science) (1,000 U/ml) 24 h after the initial cotransfection. Cells were lysed 8 h later, and firefly luciferase and Renilla luciferase activities were measured using a dual-luciferase reporter assay system (Promega) according to the manufacturer's protocol. The relative levels of firefly luciferase activities were standardized as pRL-TK activities. Data are presented as means and standard deviations (SD). P values of <0.05 were considered statistically significant, and P values of <0.01 were considered highly statistically significant.
RNA extraction and quantitative real-time PCR.
In brief, total RNA was extracted from cells using TRIzol reagent (Invitrogen), and RNA (1 μg) was reverse transcribed into cDNA using avian myeloblastosis virus reverse transcriptase (TaKaRa, Japan). The cDNA (1 μl of 25 μl) was then used in a SYBR green real-time PCR assay (Applied Biosystems). The abundance of individual mRNA transcripts in each sample was determined three times and normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. All quantitative PCR (qPCR) primers used in this study have been described previously (15).
Protein expression and purification.
For protein expression, the recombinant prokaryotic expression plasmids encoding the N-terminally GST-HA-tagged WT nsp11 and mutant (H129A, H144A, K173A) proteins were transformed into E. coli strain Trans BL21(DE3) and cultured at 37°C in LB medium containing 50 g/ml ampicillin. When the culture optical density at 600 nm (OD600) reached 0.6 to 0.8, the cells were induced by the use of 0.8 mM IPTG (isopropyl-d-1-thiogalactopyranoside) for 7 h at 27°C. For protein purification, the cultured cells were collected by centrifugation at 4,000 rpm for 10 min and resuspended in phosphate-buffered saline (PBS) followed by disruption with ATS AH-1500. The supernatant was filtered by the use of a filter with a 0.45-mm-pore-size membrane and was then loaded onto a glutathione-Sepharose 4B column (GE Healthcare). To gain GST-free proteins, the harvested fusion proteins with an N-terminally GST tag were incubated with GST-3C rhinovirus protease at a ratio of 40:1 for 8 h at 4°C, and the cleavage products in the cleavage buffer were further separated by reloading onto glutathione-Sepharose 4B column. Finally, the target proteins were eluted by the use of 10 ml cleavage buffer. The purified proteins were stored at −80°C for detection of enzyme activity.
Enzyme activity assay.
The RNA substrate (5′-6-carboxyfluorescein-dA-rU-dA-dA-6-carboxy-N,N,N,N-tetramethylrhodamine-3′) was purchased from Nanjing GenScript Company. The N-terminally HA-tagged PRRSV nsp11/H129A/H144A/K173A proteins and the RNA substrate were placed in a reaction buffer containing 50 mM HEPES (pH 7.5), 50 mM KCl, and 1 mM dithiothreitol (DTT) diluted with 0.1% diethyl pyrocarbonate-treated water. The concentration of proteins used in the assay was 2 μM, and the substrate concentration was 1 μM. The endoribonuclease activity was monitored every 10 min for 60 min in a fluorescence plate reader, with excitation at a wavelength of 492 nm and emission at a wavelength of 518 nm. The results were analyzed by the use of GraphPad Prism software 5.0.
Western blot analyses and subcellular fractionation.
Cells were grown in 60-mm-diameter dishes and harvested with lysis buffer (Beyotime, China) plus 20 nM phenylmethylsulfonyl fluoride (PMSF) and PhosSTOP phosphatase inhibitor (Sigma, USA). Samples were then separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, USA) to determine the protein expression levels. Overexpression of STAT1, STAT2, IRF9, and IRF9 deletion mutants was evaluated using mouse monoclonal anti-Flag antibody (Macgene, China). The expression of PRRSV nsp11 and its mutants was analyzed using mouse monoclonal anti-HA antibody (MBL, Japan). Rabbit polyclonal antibodies against IRF9 (Santa Cruz, USA), STAT1 (Santa Cruz, USA), STAT2 (ABclonal, China), phospho-STAT1 (STAT1-Y701, Cell Signaling Technology, USA), and phospho-STAT2 (STAT2-Y690, ABclonal, China) were used to detect the protein levels and phosphorylated forms of each of the respective endogenous proteins. Monoclonal antibody (MAb) against PRRSV nsp11 was produced from hybridoma cells derived from Sp2/0 myeloma cells and spleen cells of BALB/c mice immunized with recombinant nsp11 protein from PRRSV strain WUH3. The isotype of nsp11-specific MAb is mouse IgG1. The specificity of MAb against PRRSV nsp11 was confirmed by specific reactions performed with pCAGGS-HA-nsp11-transfected cells and PRRSV-infected cells.
To analyze the nuclear translocation of ISGF3, nuclear fractions were extracted from cells using an NE-PER nuclear and cytoplasmic extraction reagents kit (Thermo Fisher Scientific, USA) according to the manufacturer's protocol. The cytoplasmic and nuclear fractions were subjected to Western blotting. Successful isolation was assessed using rabbit polyclonal antibodies against heat shock protein 90 (HSP90; Proteintech, China) and PARP1 (Proteintech, China) with cytoplasmic and nuclear protein markers, respectively.
Coimmunoprecipitation and immunoblotting analyses.
To test the interactions between proteins, HEK-293T cells or Marc-145 cells from each 100-mm-diameter dish were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 20 nM PMSF, and PhosSTOP phosphatase inhibitor (Sigma, USA), and the protein concentration was measured and adjusted. For each immunoprecipitation, 500 μg of cell lysate protein was incubated with 0.5 μg of the indicated antibody and 25 μl of protein A+G agarose (Beyotime, China) overnight at 4°C. The agarose beads were then washed three times with 1 ml of lysis buffer. The precipitates were subjected to 10% SDS-PAGE and subsequent immunoblot analysis using the indicated antibodies. In some immunoblotting analyses, the horseradish peroxidase-conjugated goat anti-rabbit IgG light chain (IPKine, USA) were used to eliminate the interference of the rabbit IgG heavy chain.
Indirect immunofluorescence assay.
Marc-145 cells cultured on coverslips in 24-well plates were infected with PRRSV at a multiplicity of infection (MOI) of 0.5. At 24 h postinfection, the infected cells were mock treated or treated with recombinant human IFN-α (PBL Assay Science) (1,000 U/ml) for 4 h. The cells were then fixed with 4% paraformaldehyde for 15 min and immediately permeabilized using methanol (precooled at −20°C) for 10 min at room temperature (RT). After three washes with PBS, cells were incubated with 5% bovine serum albumin (BSA)–PBS overnight at 4°C and incubated with the primary antibody for 1 h. The antibodies used were as follows: anti-phospho-STAT1 (Cell Signaling Technology, USA), anti-phospho-STAT2 (Cell Signaling Technology, USA), anti-IRF9 (Abclonal, China), and anti-nsp11 MAb. Anti-mouse IgG (H+L) antibody conjugated to Alexa Fluor 594 or anti-rabbit IgG (H+L) antibody conjugated to Alexa Fluor 488 was diluted to 1:500 for use as the secondary antibody, after which the cells were stained with 4’,6-diamidino-2-phenylindole (DAPI; Beyotime, Nantong, China)–PBS for 15 min. Fluorescent images were acquired with a confocal laser scanning microscope (Olympus Fluoview ver. 3.1, Japan).
ACKNOWLEDGMENTS
This work was supported by the Major Project of the National Natural Science Foundation of China (grant 31490602), the National Basic Research Program (973) of China (grant 2014CB542700), the National Natural Science Foundation of China (grants 31372467, 31225027, and 31672566), and the Key Technology R&D Program of China (grant 2015BAD12B02).
REFERENCES
- 1.Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, Renukaradhya GJ. 2016. Porcine Reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci 4:129–154. doi: 10.1146/annurev-animal-022114-111025. [DOI] [PubMed] [Google Scholar]
- 2.Murtaugh MP, Stadejek T, Abrahante JE, Lam TT, Leung FC. 2010. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res 154:18–30. doi: 10.1016/j.virusres.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Fang Y, Snijder EJ. 2010. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res 154:61–76. doi: 10.1016/j.virusres.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kappes MA, Faaberg KS. 2015. PRRSV structure, replication and recombination: origin of phenotype and genotype diversity. Virology 479:475–486. doi: 10.1016/j.virol.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dokland T. 2010. The structural biology of PRRSV. Virus Res 154:86–97. doi: 10.1016/j.virusres.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du TF, Nan YC, Xiao SQ, Zhao Q, Zhou EM. 2017. Antiviral strategies against PRRSV infection. Trends Microbiol 25:968–979. doi: 10.1016/j.tim.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Butler JE, Lager KM, Golde W, Faaberg KS, Sinkora M, Loving C, Zhang YI. 2014. Porcine reproductive and respiratory syndrome (PRRS): an immune dysregulatory pandemic. Immunol Res 59:81–108. doi: 10.1007/s12026-014-8549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 9.Darnell JE Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 10.Ihle JN. 1995. The Janus protein tyrosine kinase family and its role in cytokine signaling. Adv Immunol 60:1–35. doi: 10.1016/S0065-2776(08)60582-9. [DOI] [PubMed] [Google Scholar]
- 11.Qureshi SA, Salditt-Georgieff M, Darnell JE Jr. 1995. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci U S A 92:3829–3833. doi: 10.1073/pnas.92.9.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitini V, Arrigo C, Altavilla G. 2010. How cells respond to interferons. J Clin Oncol 28:E439. doi: 10.1200/JCO.2010.28.9603. [DOI] [PubMed] [Google Scholar]
- 13.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nan YC, Wu CY, Zhang YJ. 11 December 2017, posting date Interplay between Janus kinase/signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front Immunol doi: 10.3389/fimmu.2017.01758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu XY, Wang D, Zhou JW, Pan T, Chen JY, Yang YT, Lv MT, Ye X, Peng GQ, Fang LR, Xiao SB. 28 April 2017, posting date Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J Virol doi: 10.1128/JVI.00003-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnold MM, Barro M, Patton JT. 2013. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol 87:9813–9821. doi: 10.1128/JVI.01146-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen A, Rott L, Phan N, Mukherjee G, Greenberg HB. 2014. Rotavirus NSP1 protein inhibits interferon-mediated STAT1 activation. J Virol 88:41–53. doi: 10.1128/JVI.01501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oda K, Matoba Y, Irie T, Kawabata R, Fukushi M, Sugiyama M, Sakaguchi T. 2015. Structural basis of the inhibition of STAT1 activity by Sendai virus C protein. J Virol 89:11487–11499. doi: 10.1128/JVI.01887-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y, Ke HZ, Han MY, Chen N, Fang WH, Yoo D. 30 Decembe 2016, posting date Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus suppresses both MAVS and RIG-I expression as one of the mechanisms to antagonize type I interferon production. PLoS One doi: 10.1371/journal.pone.0168314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi XB, Wang L, Li XW, Zhang GP, Guo JQ, Zhao D, Chai SJ, Deng RG. 2011. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-beta induction. Mol Immunol 48:1568–1572. doi: 10.1016/j.molimm.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi YJ, Li YW, Lei YY, Ye G, Shen Z, Sun LM, Luo R, Wang D, Fu ZF, Xiao SB, Peng GQ. 2016. A dimerization-dependent mechanism drives the endoribonuclease function of porcine reproductive and respiratory syndrome virus nsp11. J Virol 90:4579–4592. doi: 10.1128/JVI.03065-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takaoka A, Yanai H. 2006. Interferon signalling network in innate defence. Cell Microbiol 8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang MF, Li XR, Deng ZQ, Chen ZH, Liu Y, Gao YN, Wu W, Chen ZZ. 16 December 2017, posting date Structural biology of the arterivirus nsp11 endoribonucleases. J Virol doi: 10.1128/JVI.01309-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi XB, Zhang XZ, Chang YZ, Jiang B, Deng RG, Wang AP, Zhang GP. 6 June 2016, posting date Nonstructural protein 11 (nsp11) of porcine reproductive and respiratory syndrome virus (PRRSV) promotes PRRSV infection in MARC-145 cells. BMC Vet Res doi: 10.1186/s12917-016-0717-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nedialkova DD, Ulferts R, van den Born E, Lauber C, Gorbalenya AE, Ziebuhr J, Snijder EJ. 2009. Biochemical characterization of arterivirus nonstructural protein 11 reveals the Nidovirus-wide conservation of a replicative endoribonuclease. J Virol 83:5671–5682. doi: 10.1128/JVI.00261-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Posthuma CC, Nedialkova DD, Zevenhoven-Dobbe JC, Blokhuis JH, Gorbalenya AE, Snijder EJ. 2006. Site-directed mutagenesis of the Nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J Virol 80:1653–1661. doi: 10.1128/JVI.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng X, van Geelen A, Buckley AC, O'Brien A, Pillatzki A, Lager KM, Faaberg KS, Baker SC. 2019. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type I and type III interferon responses. J Virol 93:e02000-18. doi: 10.1128/JVI.02000-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo L, Luo X, Li R, Xu Y, Zhang J, Ge J, Bu Z, Feng L, Wang Y. 2016. Porcine epidemic diarrhea virus infection inhibits interferon signaling by targeted degradation of STAT1. J Virol 90:8281–8292. doi: 10.1128/JVI.01091-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashour J, Laurent-Rolle M, Shi P-Y, García-Sastre A. 2009. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol 83:5408–5418. doi: 10.1128/JVI.02188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, García-Sastre A. 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang RX, Fang LR, Wang D, Cai KM, Zhang H, Xie LL, Li Y, Chen HC, Xiao SB. 2015. Porcine bocavirus NP1 negatively regulates interferon signaling pathway by targeting the DNA-binding domain of IRF9. Virology 485:414–421. doi: 10.1016/j.virol.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang APP, Bornholdt ZA, Liu T, Abelson DM, Lee DE, Li S, Woods VL, Saphire EO. 23 February 2012, posting date The Ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog doi: 10.1371/journal.ppat.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamura T, Yanai H, Savitsky D, Taniguchi T. 2008. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 34.Banninger G, Reich NC. 2004. STAT2 nuclear trafficking. J Biol Chem 279:39199–39206. doi: 10.1074/jbc.M400815200. [DOI] [PubMed] [Google Scholar]
- 35.Peng YB, Yerle M, Liu B. 2006. Mapping of nine porcine interferon regulatory factor genes. Anim Genet 37:600–601. doi: 10.1111/j.1365-2052.2006.01525.x. [DOI] [PubMed] [Google Scholar]
- 36.Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC. 1997. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J Biol Chem 272:20070–20076. doi: 10.1074/jbc.272.32.20070. [DOI] [PubMed] [Google Scholar]
- 37.Veals SA, Santa Maria T, Levy DE. 1993. Two domains of ISGF3 gamma that mediate protein-DNA and protein-protein interactions during transcription factor assembly contribute to DNA-binding specificity. Mol Cell Biol 13:196–206. doi: 10.1128/mcb.13.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ke H, Yoo D. 2017. The viral innate immune antagonism and an alternative vaccine design for PRRS virus. Vet Microbiol 209:75–89. doi: 10.1016/j.vetmic.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, Fan J, Fang L, Luo R, Ouyang H, Ouyang C, Zhang H, Chen H, Li K, Xiao S. 2015. The nonstructural protein 11 of porcine reproductive and respiratory syndrome virus inhibits NF-kappaB signaling by means of its deubiquitinating activity. Mol Immunol 68:357–366. doi: 10.1016/j.molimm.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su Y, Shi P, Zhang L, Lu D, Zhao C, Li R, Zhang L, Huang J. 2018. The superimposed deubiquitination effect of OTULIN and PRRSV Nsp11 promoted the multiplication of PRRSV. J Virol doi: 10.1128/JVI.00175-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng X, Baker SC. 2018. An “old” protein with a new story: coronavirus endoribonuclease is important for evading host antiviral defenses. Virology doi: 10.1016/j.virol.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng X, Hackbart M, Mettelman RC, O’Brien A, Mielech AM, Yi G, Kao CC, Baker SC. 2017. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc Natl Acad Sci U S A 114:E4251–E4260. doi: 10.1073/pnas.1618310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kindler E, Gil-Cruz C, Spanier J, Li Y, Wilhelm J, Rabouw HH, Züst R, Hwang M, V’kovski P, Stalder H, Marti S, Habjan M, Cervantes-Barragan L, Elliot R, Karl N, Gaughan C, van Kuppeveld FJM, Silverman RH, Keller M, Ludewig B, Bergmann CC, Ziebuhr J, Weiss SR, Kalinke U, Thiel V. 2017. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog 13:e1006195. doi: 10.1371/journal.ppat.1006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suprunenko T, Hofer MJ. 2016. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Factor Rev 29:35–43. doi: 10.1016/j.cytogfr.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Hofer MJ, Li W, Lim SL, Campbell IL. 2010. The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9. J Neurosci 30:1149–1157. doi: 10.1523/JNEUROSCI.3711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofer MJ, Li W, Manders P, Terry R, Lim SL, King NJ, Campbell IL. 2012. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J Virol 86:6932–6946. doi: 10.1128/JVI.07147-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnard P, McMillan NA. 1999. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology 259:305–313. doi: 10.1006/viro.1999.9771. [DOI] [PubMed] [Google Scholar]
- 48.Verweij MC, Wellish M, Whitmer T, Malouli D, Lapel M, Jonjic S, Haas JG, DeFilippis VR, Mahalingam R, Fruh K. 2015. Varicella viruses inhibit interferon-stimulated JAK-STAT signaling through multiple mechanisms. PLoS Pathog 11:e1004901. doi: 10.1371/journal.ppat.1004901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leonard GT, Sen GC. 1996. Effects of adenovirus E1A protein on interferon-signaling. Virology 224:25–33. doi: 10.1006/viro.1996.0503. [DOI] [PubMed] [Google Scholar]
- 50.Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD. 1999. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol 162:6107–6113. [PubMed] [Google Scholar]
- 51.Wang R, Nan Y, Yu Y, Zhang YJ. 2013. Porcine reproductive and respiratory syndrome virus Nsp1beta inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-alpha1 degradation. J Virol 87:5219–5228. doi: 10.1128/JVI.02643-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol 84:11045–11055. doi: 10.1128/JVI.00655-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rengachari S, Groiss S, Devos JM, Caron E, Grandvaux N, Panne D. 2018. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc Natl Acad Sci U S A 115:E601–E609. doi: 10.1073/pnas.1718426115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang D, Cao L, Xu Z, Fang L, Zhong Y, Chen Q, Luo R, Chen H, Li K, Xiao S. 2013. MiR-125b reduces porcine reproductive and respiratory syndrome virus replication by negatively regulating the NF-kappaB pathway. PLoS One 8:e55838. doi: 10.1371/journal.pone.0055838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bi J, Zeng S, Xiao S, Chen H, Fang L. 2012. Complete genome sequence of porcine epidemic diarrhea virus strain AJ1102 isolated from a suckling piglet with acute diarrhea in China. J Virol 86:10910–10911. doi: 10.1128/JVI.01919-12. [DOI] [PMC free article] [PubMed] [Google Scholar]