

MAV-1 infection is a useful model to study the pathogenesis of an adenovirus in its natural host. Host factors that control MAV-1 replication and contribute to inflammation and disease are not fully understood. The immunoproteasome is an inducible component of the ubiquitin proteasome system that shapes the repertoire of peptides presented by MHC class I to CD8 T cells, influences other aspects of T cell survival and activation, and promotes production of proinflammatory cytokines. We found that immunoproteasome activity is dispensable in adult mice. However, immunoproteasome deficiency in neonatal mice increased mortality and impaired IFN-γ responses in the lungs. Baseline immunoproteasome subunit expression in lungs of uninfected mice increased with age. Our findings suggest the existence of developmental regulation of the immunoproteasome, like other aspects of host immune function, and indicate that immunoproteasome activity is a critical protective factor early in life.
KEYWORDS: adenovirus, immunoproteasome, interferon gamma, neonatal immunology, respiratory viruses
ABSTRACT
Acute respiratory infection with mouse adenovirus type 1 (MAV-1) induces activity of the immunoproteasome, an inducible form of the proteasome that shapes CD8 T cell responses by enhancing peptide presentation by major histocompatibility complex (MHC) class I. We used mice deficient in all three immunoproteasome subunits (triple-knockout [TKO] mice) to determine whether immunoproteasome activity is essential for control of MAV-1 replication or inflammatory responses to acute infection. Complete immunoproteasome deficiency in adult TKO mice had no effect on MAV-1 replication, virus-induced lung inflammation, or adaptive immunity compared to C57BL/6 (B6) controls. In contrast, immunoproteasome deficiency in neonatal TKO mice was associated with decreased survival and decreased lung gamma interferon (IFN-γ) expression compared to B6 controls, although without substantial effects on viral replication, histological evidence of inflammation, or expression of the proinflammatory cytokines tumor necrosis factor alpha and interleukin-1β in lungs or other organs. T cell recruitment and IFN-γ production was similar in lungs of infected B6 and TKO mice. In lungs of uninfected B6 mice, we detected low levels of immunoproteasome subunit mRNA and protein that increased with age. Immunoproteasome subunit expression was lower in lungs of adult IFN-γ-deficient mice compared to B6 controls. Together, these results demonstrate developmental regulation of the immunoproteasome that is associated with age-dependent differences in MAV-1 pathogenesis.
IMPORTANCE MAV-1 infection is a useful model to study the pathogenesis of an adenovirus in its natural host. Host factors that control MAV-1 replication and contribute to inflammation and disease are not fully understood. The immunoproteasome is an inducible component of the ubiquitin proteasome system that shapes the repertoire of peptides presented by MHC class I to CD8 T cells, influences other aspects of T cell survival and activation, and promotes production of proinflammatory cytokines. We found that immunoproteasome activity is dispensable in adult mice. However, immunoproteasome deficiency in neonatal mice increased mortality and impaired IFN-γ responses in the lungs. Baseline immunoproteasome subunit expression in lungs of uninfected mice increased with age. Our findings suggest the existence of developmental regulation of the immunoproteasome, like other aspects of host immune function, and indicate that immunoproteasome activity is a critical protective factor early in life.
INTRODUCTION
The immunoproteasome is an inducible component of the ubiquitin proteasome system that contributes to the degradation of misfolded and damaged proteins and the processing of proteins to generate peptides that are then presented to CD8 T cells by major histocompatibility complex (MHC) class I (reviewed in references 1 and 2). Stimulation by gamma interferon (IFN-γ) induces the replacement of constitutively active proteasome subunits (β1, β2, and β5) by the immuno subunits β1i (also known as LMP2), β2i (MECL-1), and β5i (LMP7) and the incorporation of the proteasome activator 11S/PA28α/β (3, 4). The resulting immunoproteasome complex is thought to be more efficient than the standard proteasome in generating MHC class I epitopes for recognition by CD8 T cells. Immunoproteasome activity may also contribute to host responses via degradation of IκB and activation of nuclear factor-κB (NF-κB)-mediated pathways (5).
We use mouse adenovirus type 1 (MAV-1) to study the pathogenesis of an adenovirus in its natural host. Host responses that are closely linked to immunoproteasome activity are active during MAV-1 infection. For instance, expression of IFN-γ and proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β), increases in the lungs during MAV-1 respiratory infection (6, 7). CD8 T cells contribute to MAV-1 clearance from the lungs and promote MAV-1-induced pulmonary inflammation (8). We demonstrated that MAV-1 infection upregulates the expression of immunoproteasome subunits and increases immunoproteasome activity in hearts and lungs in an IFN-γ-dependent manner (9). However, deficiency of the β5i subunit or pharmacological inhibition of β5i activity had minimal effect on viral loads, virus-induced inflammation, and cardiac damage. In addition, the establishment of protective immunity was not compromised by β5i deficiency.
Residual activity of β1i and/or β2i may have been sufficient to compensate for β5i deficiency in our previous study. To define effects of immunoproteasome activity on MAV-1 pathogenesis in a more comprehensive manner, we used triple-knockout (TKO) mice that lack β1i, β2i, and β5i and are therefore completely immunoproteasome deficient (10). We hypothesized that IFN-γ-induced immunoproteasome activity mediates virus-induced inflammation during acute infection and optimizes the development of protective antiviral immunity. We found that complete immunoproteasome deficiency affected MAV-1 pathogenesis in an age-dependent manner, impairing IFN-γ responses and increasing mortality in neonatal mice but having minimal effect in adult mice.
RESULTS
Effects of immunoproteasome deficiency on MAV-1 replication in lungs of adult mice.
CD8 T cells contribute to clearance of MAV-1 from the lungs of infected mice (8). Activity of the immunoproteasome, which aids in proteolytic processing of proteins into peptides that are presented by MHC class I to CD8 T cells (2, 11), increases in the lungs of mice during acute infection with MAV-1 (9), suggesting that the immunoproteasome plays an essential role in host defense against MAV-1. Deficiency of a single immunoproteasome subunit β5i does not impair the control of MAV-1 replication (9), potentially due to residual activity of β1i and β2i in β5i deficiency mice. To characterize the effects of complete immunoproteasome deficiency on MAV-1 replication in the lungs, we infected adult C57BL/6 (B6) and TKO mice intranasally (i.n.) with MAV-1. All B6 and TKO mice survived over the course of the experiment (14 days). We detected peak DNA viral loads in the lungs of infected B6 mice at 7 days postinfection (dpi) (Fig. 1A). Lung viral loads in B6 mice were substantially lower at 14 dpi, although still detectable. As a complementary measure of viral replication, we quantified MAV-1 late transcription using reverse transcription-quantitative real-time PCR (RT-qPCR) to assess MAV-1 tripartite leader (TPL) mRNA levels in the lungs. Peak TPL mRNA levels were detected in the lungs of infected B6 mice at 7 dpi; viral gene expression was essentially undetectable in the lungs by 14 dpi. There were no statistically significant differences between lung viral loads or TPL mRNA levels in the lungs of B6 and TKO mice at 7 or 14 dpi (Fig. 1B). These data indicate that immunoproteasome deficiency did not impair control of MAV-1 replication in adult mice.
FIG 1.
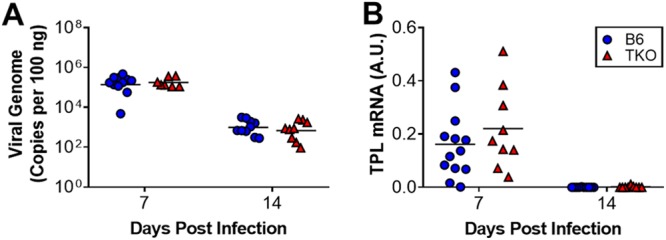
Effects of immunoproteasome deficiency on MAV-1 replication in lungs of adult mice. Adult B6 and TKO mice were infected intranasally with MAV-1. (A) qPCR was used to quantify MAV-1 genome copies in the lungs. DNA viral loads are expressed as copies of MAV-1 genome per 100 ng of input DNA. (B) RT-qPCR was used to quantify MAV-1 TPL mRNA levels in the lungs. mRNA levels are expressed in arbitrary units standardized to GAPDH. Individual circles represent values for individual mice (n = 7 to 13 per group), and horizontal bars represent geometric means (A) or means (B) for each group. Statistical comparisons were made using two-way ANOVA, followed by Bonferroni’s multiple-comparison tests. No statistically significant differences were identified.
Effects of immunoproteasome deficiency on MAV-1-induced pulmonary inflammation in adult mice.
Acute MAV-1 respiratory infection causes pneumonitis that is characterized by a predominantly mononuclear cell infiltrate concentrated around the airways along with scattered areas of hypercellular and thickened alveolar walls (7, 12). We evaluated the lungs of infected adult B6 and TKO mice to assess contributions of the immunoproteasome to virus-induced inflammation. Histological evidence of pulmonary inflammation (Fig. 2A) and total protein concentrations in bronchoalveolar lavage fluid (BALF) (Fig. 2B) were similar in infected adult B6 and TKO mice at 7 and 14 dpi (Fig. 2A). MAV-1 infection was associated with increased BALF protein concentrations (Fig. 2C to E) and whole lung mRNA levels (Fig. 2F to H) of IFN-γ and the proinflammatory cytokines TNF-α and IL-1β, but there were no statistically significant differences between infected B6 and TKO mice at 7 or 14 dpi. Thus, immunoproteasome deficiency did not substantially affect multiple measures of MAV-1-induced pulmonary inflammation during acute respiratory infection.
FIG 2.

Effects of immunoproteasome deficiency on virus-induced pulmonary inflammation. Adult B6 and TKO mice were infected i.n. with MAV-1. Control animals were mock infected with conditioned media. BALF was obtained, and lungs were harvested at the indicated time points. (A) Hematoxylin-and-eosin-stained sections were prepared from paraffin-embedded lungs. Scale bars, 100 μm. (B) Total protein concentration was measured in BALF at 7 and 14 dpi. (C to E) ELISA was used to measure concentrations of the indicated cytokines in BALF. (F to H) RT-qPCR was used to quantify cytokine mRNA levels in the lungs. mRNA levels are expressed in arbitrary units standardized to GAPDH. Individual circles represent values for individual mice (n = 4 to 6 mock-infected mice per group except n = 2 for BALF from mock-infected B6 mice at 14 dpi; n = 7 to 12 infected mice per group), and horizontal bars represent means for each group. Statistical comparisons were made using two-way ANOVA, followed by Bonferroni’s multiple-comparison tests. *, P < 0.05; **, P < 0.01, and ***, P < 0.001 comparing mock-infected and infected mice of the same genotype. No statistically significant differences between B6 and TKO mice were identified.
Effects of immunoproteasome deficiency on adaptive immunity to MAV-1 in adult mice.
Isolated deficiency of a single immunoproteasome subunit, β5i, does not impair the development of protective immunity to MAV-1 following primary infection (9). To determine whether complete immunoproteasome deficiency impaired protective immunity, we infected adult B6 and TKO mice with MAV-1, allowed them to recover, and then rechallenged the mice with MAV-1 at 28 dpi. Controls were rechallenged with conditioned media. As a measure of protective immunity, we assessed viral replication in the lungs 7 days after rechallenge using RT-qPCR to quantify TPL mRNA levels. TPL mRNA was undetectable in the lungs of all rechallenged B6 and TKO mice (Fig. 3A), indicating that protective immunity was intact in immunoproteasome-deficient mice.
FIG 3.

Effects of immunoproteasome deficiency on adaptive immune responses to MAV-1 in adult mice. (A) Adult B6 and TKO mice were infected i.n. with MAV-1. Control animals were mock infected with conditioned media. Mice were reinfected i.n. with MAV-1 at 28 dpi. Lungs were harvested at 7 dpi following reinfection. RT-qPCR was used to quantify TPL mRNA levels in the lungs. mRNA levels are expressed in arbitrary units standardized to GAPDH. Data from mice at 7 dpi. following primary infection (also depicted in Fig. 1B) are presented to the left of the dotted line for reference but are not included in statistical analysis. Individual circles represent values for individual mice (n = 4 to 6 mice per reinfected group), and horizontal bars represent means for each group. Statistical comparisons were made using two-way ANOVA, followed by Bonferroni’s multiple-comparison tests. No statistically significant differences were identified. (B) Neutralization assays were performed in which serum obtained from infected B6 and TKO mice (n = 10 per group) at 21 dpi was used at the indicated dilutions to neutralize in vitro infection of 3T12 mouse fibroblasts by MAV-1.pIXeGFP, a recombinant GFP-expressing MAV-1. Serum from mock-infected mice (n = 4) was used as a control. Accumulating fluorescence expressed as relative fluorescence units (RFU) was used as an indicator of viral replication. The bottom horizontal dashed line represents background fluorescence in wells with uninfected cells. The top horizontal dashed line represents fluorescence in wells with cells infected with virus in the absence of any serum. Statistical comparisons were made using two-way ANOVA followed by Bonferroni’s multiple-comparison tests. ***, P < 0.001, comparing sera from mock-infected and infected mice (both B6 and TKO). No statistically significant differences between sera from infected B6 and TKO mice were identified.
Deficiency of β1i is associated with impaired antiviral antibody production during influenza infection (10). B cell deficiency increases susceptibility to MAV-1 infection following intraperitoneal inoculation, and passive transfer of immune antiserum protects B cell-deficient mice (13). We have not yet defined contributions of virus-specific antibody production to the pathogenesis of MAV-1 respiratory infection. However, the results of our rechallenge experiments suggested that immunoproteasome deficiency was not likely to have substantially altered production of neutralizing antibodies to MAV-1. To verify this, we assessed the ability of serum obtained from B6 and TKO mice at 21 dpi to neutralize in vitro infection of 3T12 mouse fibroblasts by MAV-1.pIXeGFP, a recombinant green fluorescent protein (GFP)-expressing MAV-1 (14, 15). Preincubation of virus with serum from infected B6 or TKO mice inhibited viral replication, assayed by measuring fluorescence in wells of infected cells (Fig. 3B). These data suggest that the production of neutralizing antibody to MAV-1 was not impaired in immunoproteasome-deficient mice.
Effects of immunoproteasome deficiency on susceptibility to MAV-1 infection in neonatal mice.
Neonatal B6 mice are more susceptible than adult B6 mice to MAV-1, with higher lung viral loads, delayed clearance of virus from the lungs, and IFN-γ responses in the lungs that are blunted and delayed in neonatal mice compared to responses in adult mice (7). Isolated deficiency of β5i does not substantially affect MAV-1 pathogenesis in neonatal mice (9), and the data presented above suggest that complete immunoproteasome deficiency in adult TKO mice had minimal effect on MAV-1 pathogenesis. To identify potential age-dependent contributions of the immunoproteasome to MAV-1 pathogenesis, we infected neonatal (7 days old) B6 and TKO mice with MAV-1. All B6 mice survived until at least 11 dpi (Fig. 4A), consistent with our previous reports (7, 9). In contrast, a significant proportion of TKO neonates died or were euthanized because they were moribund between 9 and 11 dpi. To determine whether increased mortality in neonatal TKO mice was related to impaired control of viral replication in TKO mice, we measured viral loads and viral gene expression in the lungs at 7 and 9 dpi. Lung viral loads (Fig. 4B) and TPL mRNA levels (Fig. 4E) were equivalent in neonatal B6 and TKO mice. We also assayed viral replication in the hearts and livers of infected mice to determine whether defects in control of viral replication outside the lung. We detected no statistically significant differences in heart or liver viral loads (Fig. 4C and D) or TPL mRNA levels (Fig. 4F and G) in B6 and TKO mice at either time point. There were no significant differences in brain viral loads or TPL mRNA levels (at 7 dpi; data not shown). Increased mortality in infected neonatal TKO mice was therefore not associated with corresponding increases in viral replication.
FIG 4.

Effects of immunoproteasome deficiency on survival and MAV-1 replication in neonatal mice. B6 and TKO mice were infected intranasally with MAV-1 at 7 days of age. (A) Survival was monitored throughout the course of the experiments (n = 9 B6 mice, n = 19 TKO mice). Statistical comparisons were made using the log-rank (Mantel-Cox) test. (B to G) Lungs, hearts, and livers were harvested from mice at 7 and 9 dpi (n = 8-15 per group). (B to D) qPCR was used to quantify MAV-1 genome copies in lungs, hearts, and livers. DNA viral loads are expressed as copies of MAV 1 genome per 100 ng of input DNA. (E to G) RT-qPCR was used to quantify MAV-1 TPL mRNA levels in lungs, hearts, and livers. The data are shown in arbitrary units standardized to GAPDH. In panels B to G, individual circles represent values for individual mice, and horizontal bars represent geometric means (B to D) or means (E to G) for each group. Statistical comparisons were made using two-way ANOVA, followed by Bonferroni’s multiple-comparison tests. No statistically significant differences between B6 and TKO mice were identified at either time point.
Effects of immunoproteasome deficiency on MAV-1-induced inflammation in neonatal mice.
Multiple cytokines and chemokines are upregulated in the lungs of neonatal mice during acute MAV-1 respiratory infection (7, 16). We used RT-qPCR to quantify the expression of IFN-γ, TNF-α, and IL-1β in the lungs of neonatal B6 and TKO mice (Fig. 5A to C). Expression of all three cytokines was greater in lungs of infected mice than in mock-infected controls. However, IFN-γ mRNA levels were significantly lower in the lungs of infected neonatal TKO mice compared to B6 mice at 7 and 9 dpi (Fig. 5A). There were no significant differences between TNF-α or IL-1β mRNA levels in the lungs of TKO and B6 mice at either time point (Fig. 5B and C). We also quantified cytokine expression in hearts and livers to determine whether immunoproteasome deficiency in neonatal mice affected MAV-1-induced inflammation in other organs. In contrast to our findings in the lungs, there were no statistically significant differences between IFN-γ mRNA levels in the hearts (Fig. 5D) or livers (Fig. 5G) of infected neonatal TKO and B6 mice. TNF-α and IL-1β mRNA levels were also similar in hearts (Fig. 5E and F) and livers (Fig. 5H and I) of infected neonatal TKO and B6 mice. There were no significant differences in IFN-γ, TNF-α, or IL-1β mRNA levels in brains of TKO and B6 mice (at 7 dpi; data not shown). Histological evidence of inflammation was similar in lungs, hearts, and livers of infected neonatal B6 and TKO mice at 9 dpi (Fig. 5J). These data suggest that several aspects of inflammatory responses induced by acute MAV-1 infection were unaffected by immunoproteasome deficiency in neonatal mice. However, in contrast to our findings in adult mice, immunoproteasome deficiency was associated with impaired IFN-γ responses to MAV-1 infection in the lungs.
FIG 5.

Effects of immunoproteasome deficiency on virus-induced inflammation in neonatal mice. B6 and TKO mice were infected intranasally with MAV-1 at 7 days of age. Control animals were mock infected with conditioned media. Lungs, hearts, and livers were harvested from mice at 7 and 9 dpi. RT-qPCR was used to quantify mRNA levels of the indicated cytokines in lungs (A to C), hearts (D to F), and livers (G to I). mRNA levels are expressed in arbitrary units standardized to GAPDH. Individual circles represent values for individual mice (n = 8 to 16 mice per group), and horizontal bars represent means for each group. Statistical comparisons were made using two-way ANOVA, followed by Bonferroni’s multiple-comparison tests. *, P < 0.05; **, P < 0.01, and ***, P < 0.001, comparing mock-infected and infected mice of the same genotype. ††, P < 0.01, and †††, P < 0.001, comparing infected B6 and TKO mice. (J) Hematoxylin-and-eosin-stained sections were prepared from paraffin-embedded lungs, hearts, and livers. Scale bars, 100 μm.
Effects of immunoproteasome deficiency on adaptive immunity to MAV-1 in neonatal mice.
Protective immunity is established following MAV-1 infection of neonatal B6 mice (7). We were unable to assess protective immunity with virus rechallenge following infection of neonatal TKO mice due to the increased mortality compared to B6 mice during acute infection (Fig. 5A). Neutralizing antibody is present in the serum of adult B6 mice as early as 6 dpi after intraperitoneal inoculation (13). We therefore assessed the presence of neutralizing antibody in neonatal B6 and TKO mice at 9 dpi as a separate measure of adaptive immunity. However, we did not detect virus neutralization by serum of infected neonatal B6 or TKO mice (data not shown).
Effects of immunoproteasome deficiency on T cell responses to infection in neonatal mice.
Immunoproteasome activity may affect antiviral CD8 T cell responses by altering the repertoire of virus-derived peptides that are presented by MHC class I and also by promoting T cell development or maturation (reviewed in reference 2). CD8 T cells are recruited to the lungs and hearts of neonatal mice infected with MAV-1 (7, 17). The number and spatial distribution of CD3+ T cells detected by immunohistochemistry in lungs was qualitatively similar in infected neonatal B6 and TKO mice (Fig. 6A). We used flow cytometry to quantify CD4 and CD8 T cells in the lungs and characterize their production of IFN-γ in neonatal B6 and TKO mice at 7 dpi, when we detected less overall IFN-γ expression in the lungs of infected neonatal TKO mice (Fig. 5A). Total leukocyte numbers (Fig. 6B) and numbers of CD4+ (Fig. 6C) and CD8+ (Fig. 6D) cells in the lungs were similar in B6 and TKO mice. Likewise, there were no statistically significant differences in numbers of IFN-γ-producing CD4+ (Fig. 6E) or CD8+ (Fig. 6F) cells, whether or not they were restimulated with anti-CD3 antibody. These data suggest that defects in IFN-γ production in the lungs of neonatal TKO mice were not due to altered numbers of CD4 or CD8 T cells in the lungs or by altered IFN-γ production by CD4 and CD8 T cells.
FIG 6.

Effects of immunoproteasome deficiency on T cell responses to MAV-1 infection in neonatal mice. B6 and TKO mice were infected i.n. with MAV-1 at 7 days of age. (A) Sections from lungs of infected mice harvested at 9 dpi were stained with antibody specific for CD3. CD3+ cells are stained dark brown. Representative images from n = 3 mice per group are presented. Scale bars, 100 μm. (B to F) Lung leukocytes were isolated from lungs of infected mice at 7 dpi. (B) Total cell numbers were quantified with a hemocytometer. Cells were stimulated overnight with anti-CD3 antibody or left unstimulated (PBS controls). Overall numbers of CD4+ (C) and CD8+ (D) T cells in lungs were calculated based on total lung cell counts and the percentage of CD4+ or CD8+ cells in unstimulated fractions, as determined by flow cytometry (n = 6 to 9 per group). Intracellular cytokine staining was used to quantify the percentages of CD4+ (E) and CD8+ (F) cells that were IFN-γ+ (n = 10 to 15 per group). Statistical comparisons were made using the Mann-Whitney test (B to D) or two-way ANOVA (E and F), followed by Bonferroni’s multiple-comparison tests. No statistically significant differences were detected.
Age-dependent expression of immunoproteasome subunits.
Immunoproteasome deficiency affected the pathogenesis of acute MAV-1 infection in an age-dependent manner, with effects on survival and virus-induced IFN-γ expression that were apparent in neonatal mice but not in adult mice. We reasoned that this effect could be related to changes in baseline immunoproteasome activity that developed with age. To assess this possibility, we used RT-qPCR and Western blot to quantify expression of immunoproteasome subunits in the lungs of uninfected mice at multiple ages, including 7 and 11 days old (preweaning), 20 days (shortly before weaning), and 30 days old (postweaning). β1i, β2i, and β5i mRNA levels in lungs increased significantly with age between 7 and 30 days of age (Fig. 7A to C). We detected corresponding increases in the mature form of β1i protein and of β5i protein in whole lung homogenate between 7 and 30 days of age (Fig. 7D).
FIG 7.

Age-based increases in immunoproteasome subunit expression in lungs of uninfected mice. RT-qPCR was used to quantify mRNA levels of the indicated immunoproteasome subunits (A to C) and IFN-γ (E) in lungs harvested from uninfected B6 mice at the indicated ages (n = 3 to 8 mice per group). (D) Immunoproteasome subunit protein was detected by immunoblot in whole lung homogenates from B6 mice at the indicated ages (n = 4 per group). (F to H) RT-qPCR was used to quantify mRNA levels of the indicated immunoproteasome subunits in lungs harvested from uninfected adult (36 to 37 days old) B6 and IFN-γ−/− mice (n = 6 per group). mRNA levels are expressed in arbitrary units standardized to GAPDH. Individual circles represent values for individual mice, and horizontal bars represent means for each group. Statistical comparisons in panels A to C and panel E were made using the Kruskal-Wallis test, followed by Dunn’s multiple-comparison tests. Statistical comparisons in panels F to H were made using Mann-Whitney test (*, P < 0.05; **, P < 0.01).
IFN-γ is the predominant inducer of immunoproteasome activity (18, 19), and immunoproteasome subunit expression is impaired in hearts and lungs of neonatal IFN-γ−/− mice infected with MAV-1 (9). To determine the extent to which age-based changes in IFN-γ expression corresponded to age-dependent increases in immunoproteasome expression, we quantified IFN-γ expression in the lungs of uninfected mice at the ages described above. IFN-γ mRNA levels (Fig. 7E) increased with age similar to increases in immunoproteasome subunit expression with age. Next, we measured immunoproteasome subunit expression in lungs of adult (36 to 37 days old) B6 and IFN-γ−/− mice (Fig. 7F to H). β1i and β5i mRNA levels were significantly lower in IFN-γ−/− mice than in B6 mice, although both β1i and β5i mRNA were readily detectable in IFN-γ−/− mice. There was no statistically significant difference between lung β2i mRNA levels in B6 and IFN-γ−/− mice.
Our data indicated that baseline immunoproteasome subunit expression was present at very low levels in uninfected neonatal mice and then increased with age in a manner that was partially dependent on IFN-γ. However, it remained possible that inducible immunoproteasome activity was intact in neonatal mice to the same extent that it is in adult mice. For instance, we previously demonstrated inducible immunoproteasome subunit expression in hearts and lungs of neonatal mice (9). To determine whether the capacity for inducing immunoproteasome subunit expression was similar in neonatal and adult mice despite differences in baseline expression, we compared induction of β5i expression in lungs of infected neonatal and adult B6 mice during acute MAV-1 infection, characterizing β5i expression near the peak of viral replication and IFN-γ expression that occurs at 9 dpi in neonatal mice (Fig. 4) and 7 dpi in adult mice (Fig. 2). Relative increases in β5i mRNA levels in the lungs of infected neonatal mice compared to mock-infected controls were significantly less than corresponding increases in the lungs of adult mice (Fig. 8A). We observed a similar pattern with IFN-γ mRNA levels in the lungs of neonatal and adult mice (Fig. 8B). Together, our data indicate that baseline levels of immunoproteasome and IFN-γ expression are lower in neonatal mice than in adult mice, and the capacity for induction of immunoproteasome subunit expression is less in neonatal mice than adult mice during MAV-1 infection.
FIG 8.
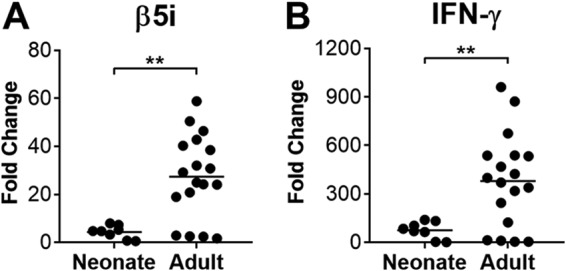
Lung β5i and IFN-γ expression in infected neonatal and adult mice. Neonatal (7 days old) and adult (6 to 8 weeks old) B6 mice were infected i.n. with MAV-1 or mock infected with conditioned media. RT-qPCR was used to quantify expression of β5i (A) and IFN-γ (B) mRNA levels in lungs harvested at 9 dpi (neonatal mice) or 7 dpi (adult mice). Data for are presented as the fold change in infected mice (n = 8 to 18 per group) compared to expression levels measured in mock-infected control mice of the same age (n = 4 per age). Statistical comparisons were made using Mann-Whitney test (**, P < 0.01).
DISCUSSION
Proteasome and immunoproteasome activity contribute to the processing of proteins to generate peptides presented to CD8 T cells by MHC class I (2). CD8 T cells are essential for efficient clearance of MAV-1 from the lungs (8). We have therefore determined the extent to which immunoproteasome activity is required for CD8 T cell-mediated virus clearance. Isolated deficiency of one immunoproteasome subunit, β5i, has no effect (9). Residual β1i and β2i activity in β5i-deficient mice may be sufficient to compensate for absent β5i activity, even though those subunits are likely to be incorporated into proteasome complexes less efficiently in the absence of β5i (20). We addressed this possibility by using TKO mice, which lack all three immunoproteasome subunits and are completely deficient in immunoproteasome activity. Our data indicate that complete immunoproteasome deficiency does not substantially impair host control of viral replication or clearance of virus from the lungs of adult or neonatal mice.
Those results are in accord with other studies examining the effects of immunoproteasome deficiency on viral replication in vivo. For instance, β5i deficiency does not affect control of coxsackievirus B3 (CVB3) replication in the heart (20), and the combined deficiency of β1i and β2i does not affect lymphocytic choriomeningitis virus (LCMV) viral replication in the brain (21). LCMV epitope presentation and T cell responses are impaired in TKO mice, but effects on LCMV replication or other aspects of LCMV pathogenesis in TKO mice were not reported (22). Immunoproteasome deficiency is associated with increased pathogen burden and dissemination in mouse models of Candida albicans (23) and Streptococcus pneumoniae (24) infection, indicating important pathogen-dependent differences in the contributions of the immunoproteasome to host defense. It is possible that immunoproteasome deficiency had an effect on the overall profile of MAV-1-specific epitopes presented by MHC class I to CD8 T cells, as has been demonstrated for LCMV (22), influenza virus (25, 26), and murine cytomegalovirus (27). Because MAV-1-specific MHC class I epitopes have not yet been defined, we are currently unable to address this possibility in a detailed manner. However, our results with TKO mice suggest that any such difference would have minimal implications for CD8 T cell-mediated control of MAV-1 replication. Proteasome activity, which is intact in TKO mice, may be sufficient to compensate for relevant contributions of the immunoproteasome during MAV-1 infection of adult TKO mice. Redundancy in innate and adaptive immune functions, including those that are not influenced by proteasome or immunoproteasome activity, may be sufficient for adequate control of viral replication.
The immunoproteasome may promote inflammation via mechanisms such as degradation of IκB and subsequent activation of NF-κB-mediated signaling (5). In addition, CD8 T cells exert a proinflammatory effect during MAV-1 respiratory infection (8) and myocarditis (our unpublished data). Thus, we hypothesized that MAV-1-induced inflammation would be less in TKO mice than in B6 mice. However, similar to our previous work with neonatal (9) and adult (our unpublished data) β5i-deficient mice, we observed very few differences in virus-induced inflammatory responses in adult and neonatal B6 and TKO mice. Our results contrast with those in studies that show enhanced manifestations of CVB3 myocarditis in β5i-deficient mice (20) or decreased evidence of central nervous system inflammation induced by LCMV in β1i/β2i-deficient mice (21). Even though some manifestations of CVB3 myocarditis are more apparent in β5i-deficient mice than in wild-type mice, CVB3 myocarditis in β5i-deficient mice is not associated with altered expression of proinflammatory cytokines (20). Instead, β5i deficiency is linked to impaired clearance of polyubiquitinated proteins, increased oxidative damage, and enhanced apoptosis in hearts following CVB3 infection. Acute MAV-1 respiratory infection induces apoptosis in the lungs of adult mice (6), but the extent to which turnover of polyubiquitinated proteins or generation of oxidative damage occurs during MAV-1 infection is unknown. If those are not prominent aspects of MAV-1 pathogenesis, then correspondingly minimal effects of immunoproteasome deficiency would be expected.
Immunoproteasome activity contributes to CD8 T cell function via its effects on antigen presentation (2). The immunoproteasome also affects lymphocyte function in ways that are independent of antigen presentation, including intrinsic effects on T cell survival, expansion, differentiation, and activation (28–31). We did not detect substantial differences in BALF IFN-γ concentrations in adult B6 and TKO mice. However, IFN-γ expression was impaired in the lungs of TKO mice, similar to our previous observations in lungs of neonatal β5i-deficient mice (9). A separate study demonstrated decreased IFN-γ production by CD8 T cells isolated from TKO mice infected with LCMV (22). Our data indicate that the effect of immunoproteasome deficiency on IFN-γ expression was not due to decreased numbers of T cells in the lungs or decreased IFN-γ production by CD4 or CD8 T cells in the lungs of neonatal TKO mice infected with MAV-1. It is possible that we would have detected subtler effects of immunoproteasome deficiency on T cell IFN-γ production using different nonspecific or virus-specific stimulation ex vivo.
It is also possible that immunoproteasome deficiency affected IFN-γ production by other types of cells that we did not specifically analyze, such as NK cells and γδ T cells. NK cells are not essential for control of MAV-1 replication in the brains of adult mice (32), but their contribution to other aspects of MAV-1 pathogenesis in adult or neonatal mice has not been defined. Likewise, although γδ T cells are present in the lungs of infected adult mice (33), it is unclear whether they are essential for control of viral replication or contribute to virus-induced inflammation in adult or neonatal mice. In addition, we are aware of no reports that detail requirements of the immunoproteasome for appropriate function of NK cells or γδ T cells. The immunoproteasome is also likely to influence functions of other immune cells, including B cell number, differentiation, and antibody production and macrophage and dendritic cell maturation and cytokine production (10, 34). Our results indicate that immunoproteasome deficiency is unlikely to affect those aspects of immune function during MAV-1 infection. A more detailed analysis of specific cell types may reveal effects that were not addressed in this study.
Immunoproteasome deficiency had a more pronounced effect on outcomes of MAV-1 infection in neonatal mice compared to adult mice. Both adult and neonatal B6 mice typically survive infection, making the mortality in neonatal TKO mice a notable finding. Both the magnitude and timing of the increased mortality in TKO mice resembles effects that we have observed in neonatal IFN-γ-deficient mice (9) and neonatal mice treated with antibody to neutralize IFN-γ (17), raising the possibility that effects of IFN-γ deficiency and neutralization are mediated by downstream effects on immunoproteasome activity. We have previously shown that IFN-γ is not required for control of MAV-1 replication in mice (9, 16, 17), so it is not surprising that impaired IFN-γ production in neonatal TKO mice was not associated with increased viral replication. IFN-γ drives much of the virus-induced inflammation that we have demonstrated in the hearts of neonatal mice (17), differentiating the decreases in inflammation that we detected in IFN-γ-neutralized neonatal B6 mice from the lack of an effect on various measures of inflammation in neonatal TKO mice in this study. Just as immunoproteasome deficiency affected virus-induced IFN-γ expression only in the lungs of neonatal TKO mice, it may be that other organ-specific effects on viral replication, inflammation, or other aspects of pathogenesis not measured in our study accounted for the increased mortality in neonatal TKO mice.
The identification of age-based effects of immunoproteasome deficiency prompted us to investigate potential developmental changes in baseline immunoproteasome subunit expression. We detected low-level expression of immunoproteasome subunits in the lungs of mice early in life that increased with age. Immunoproteasome subunit expression levels generally correlate with activity (9), suggesting that age-based increases in subunit mRNA and protein levels are likely to correspond with increasing levels of immunoproteasome activity. Our results in the lung are similar to results demonstrating age-based increases in immunoproteasome activity in rat lung, spleen, and liver (35–37). In addition, our data suggest that developmental changes in immunoproteasome activity are mediated at least in part by corresponding changes in IFN-γ expression or signaling. These effects may be related to intrinsic changes in immunoproteasome subunit, such as accumulation of histone methylation markers or other types of epigenetic regulation that have been described for developmental regulation of other aspects of immunity such as monocyte function (38). It is also possible that extrinsic factors, such as effects related to the establishment of lung and gut microbiome in early life, contribute to age-based increases in immunoproteasome activity.
Baseline immunoproteasome expression was lower in lungs of uninfected neonatal mice than in adult mice. We also demonstrated that there is less induction of immunoproteasome activity during infection in neonatal mice than in adult mice. This may be related to age-based differences in baseline immunoproteasome expression. However, it is likely that impaired IFN-γ responses in neonatal mice, which we previously demonstrated during MAV-1 respiratory infection (7) and others have demonstrated at baseline and in the context of other infections (reviewed in references 39 and 40), also contributed to age-based differences in virus-induced immunoproteasome subunit expression. Despite these age-based differences, acute MAV-1 infection was still associated with increased immunoproteasome subunit expression in the lungs of neonatal mice, confirming our previous findings in lungs and hearts of neonatal mice (9). Immunoproteasome activity therefore seems likely to be relevant early in life for MAV-1 infection. Less abundant immunoproteasome activity, coupled with relative immaturity of other aspects of immune function in neonatal mice, may provide an explanation for why we observed effects of immunoproteasome deficiency in neonatal TKO mice but not in adult TKO mice during MAV-1 infection. Our results did not allow us to determine whether baseline immunoproteasome activity in hematopoietic cells, inducible activity in hematopoietic cells or other cell types, or both were essential for survival and IFN-γ production in neonatal mice during infection.
In summary, our data indicate that the immunoproteasome makes age-dependent contributions to MAV-1 pathogenesis that are important for survival and IFN-γ production in neonatal mice but are largely dispensable in adult mice. Our findings in uninfected mice of various ages are consistent with a mechanism of developmental regulation of immunoproteasome activity in the lung, which is likely to influence the pathogenesis of MAV-1 infection in neonatal mice. The use of immunoproteasome inhibitors is being explored for treatment of hematologic malignancies and noninfectious inflammatory conditions (41, 42). Inhibition of an inducible host function such as the immunoproteasome would be an appealing strategy to decrease virus-induced inflammation and ensuing disease in an infected host. However, the results of our study suggest that complete inhibition of all immunoproteasome subunit activity during acute adenovirus infection may not be an optimal strategy early in life.
MATERIALS AND METHODS
Mice.
C57BL/6J (B6) mice were obtained from The Jackson Laboratory and bred at the University of Michigan. Mice deficient in β1i, β2i, and β5i (TKO mice; VelociGene modified allele ID VG#1230+Psmb10) (10) were obtained from Kenneth Rock (University of Massachusetts) with the permission of Regeneron Pharmaceuticals, Inc., and bred at the University of Michigan. Adult IFN-γ deficient mice (IFN-γ−/−, B6.129S7-Ifngtm1Ts/J) and B6 controls were purchased from The Jackson Laboratory. All mice were maintained under specific-pathogen-free conditions and provided with food and water ad libitum. All experiments were approved by the University of Michigan Institutional Animal Care and Use Committee.
Virus and cells.
MAV-1 was grown and passaged in mouse 3T6 fibroblasts, and titers of viral stocks were determined by plaque assay on 3T6 cells as previous described (43). MAV-1.pIXeGFP, a recombinant GFP-expressing MAV-1 (14, 15), was kindly provided by Kathy Spindler (University of Michigan). MAV-1.pIXeGFP was also grown and passaged in 3T6 fibroblasts.
Mouse infections.
Adult mice were infected when they were 6 to 8 weeks old. Neonatal mice were infected when they were 7 days old. In a subset of experiments, mice were used without infection or mock infection. Male and female mice were included in all experiments. Adult mice were anesthetized with ketamine and xylazine and then infected intranasally (i.n.) with 105 PFU of MAV-1 in 40 μl of sterile phosphate-buffered saline (PBS). Neonatal mice were manually restrained without anesthesia and were infected i.n. with 105 PFU in 10 μl of sterile PBS. Control adult and neonatal mice were mock-infected i.n. with conditioned medium at an equivalent dilution in sterile PBS. In some experiments, mice were weighed on the day of infection and then intermittently throughout the course of the experiment. All mice were euthanized by ketamine/xylazine overdose at the indicated time points. Organs were harvested, snap-frozen in dry ice, and stored at –80°C.
Assessment of protective immunity using virus rechallenge.
Protective immunity to MAV-1 was assessed as previously described (9, 33). In brief, adult B6 and TKO mice were infected i.n. with 105 PFU of MAV-1. Mice were rechallenged i.n. with 105 PFU of MAV-1 or with conditioned media 28 days after primary infection. Mice were euthanized by a ketamine/xylazine overdose, and lungs were harvested 7 days following rechallenge.
Isolation of DNA and RNA.
Portions of organs were homogenized using sterile glass beads in a Mini-Beadbeater (Biospec Products) for 30 s in 1 ml of TRIzol (Invitrogen). RNA and DNA were then isolated from homogenates according to the manufacturer’s protocol.
Analysis of viral and host gene expression.
Host gene expression was quantified using RT-qPCR. RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Then, 5-μl portions of cDNA were added to reaction mixtures containing Power SYBR green PCR Mix (Applied Biosystems) and forward and reverse primers, each at 200 nM final concentrations in a 25-μl reaction volume. Primers used to detect the MAV-1 tripartite leader (TPL), IFN-γ, IL-1β, TNF-α, β1i, β5i, and GAPDH have been previously described (8, 9, 44). Additional primers were used to detect β2i (forward, 5′-GCTTGTGTTCCGAGATGGAGTC-3′; reverse, 5′-TCCGTTCAAATCAACCCCG-3′). Separate reactions were prepared with primers for mouse GAPDH (glyceraldehyde-3-phosphate dehydrogenase). RT-qPCR analysis consisted of 40 cycles of 15 s at 90°C and 60 s at 60°C. Quantification of target gene mRNA was normalized to GAPDH and expressed in arbitrary units as 2–ΔCT, where CT is the threshold cycle, and ΔCT = CT(target) – CT(GAPDH). In some experiments, relative expression (fold change) was calculated using the comparative CT (ΔΔCT) method.
Analysis of viral loads.
MAV-1 viral loads were measured in organs using quantitative real-time PCR (qPCR) as previously described (7, 17).
Measurement of total protein in bronchoalveolar lavage fluid.
Bronchoalveolar lavage fluid (BALF) was obtained by lavaging lungs three times with the same aliquot of 1 ml of sterile PBS containing protease inhibitor (complete, Mini, EDTA-free tablets; Roche Applied Science). Total protein concentrations in BALF were determined by using a QuickStart Bradford protein assay (Bio-Rad) according to the manufacturer’s instructions.
Measurement of cytokine concentrations in bronchoalveolar lavage fluid.
Cytokine concentrations were determined by enzyme-linked immunosorbent assay (ELISA; Duoset kits; R&D Systems) according to the manufacturer’s directions. ELISA was performed by the University of Michigan Cancer Center Immunology Core.
BALF total and differential cell counts.
BALF cells were counted using a hemocytometer. Differential cell counting was performed as previously described (33).
Measurement of neutralizing antibody in serum.
3T6 cells were plated in clear-bottom, opaque-walled 96-well plates (Greiner Bio-One) at 5 × 104 cells/well and incubated overnight. Serum was heat-inactivated for 60 min at 50°C. Serum dilutions were incubated with separate aliquots of MAV-1.pIXeGFP for 60 min at 37°C. 3T6 cells were then infected with the preincubated MAV-1.pIXeGFP at a multiplicity of infection (MOI) of 1 for 60 min at 37°C. Negative-control wells included uninfected cells. Positive-control wells included cells infected with MAV-1.pIXeGFP that had not been preincubated with serum. Triplicate wells were included for each condition. Infected cells were incubated for 48 h at 37°C in indicator-free Dulbecco modified Eagle medium (Gibco) containing 5% heat-inactivated calf serum and 1% penicillin and streptomycin (Gibco). Fluorescence was measured at 0, 24, and 48 hpi using a Synergy HTX Multi-Mode plate reader with Gen5 software (BioTek Instruments, Inc.).
Detection of immunoproteasome subunit proteins by Western blotting.
Tissue was homogenized in Cell Lytic MT (Sigma) containing protease inhibitor (P8340; Sigma) and phosphatase inhibitor (P2850; Sigma) cocktails (using sterile glass beads in a minibeadbeater. Protein concentrations were determined using a QuickStart Bradford protein assay (Bio-Rad). Equivalent microgram amounts of total protein extracted from tissues were separated by SDS-polyacrylamide gel electrophoresis and transferred to an Immun-Blot polyvinylidene difluoride membrane (Bio-Rad). Western blotting was performed using primary antibodies to β1i (LMP2; A-1, sc-271354), β5i (LMP7; D-2, sc-374089), and β-actin (C4; sc-47778 [all from Santa Cruz Biotechnology]). Mouse IgG kappa binding protein conjugated to horseradish peroxidase (m-IgGκ BP-HRP; sc-516102; Santa Cruz Biotechnology) was used as the secondary reagent to detect all primary antibodies. Membranes were developed using SuperSignal West Femto maximum sensitivity substrate (Thermo Fisher Scientific) and imaged using the FluorChem M system (ProteinSimple).
Intracellular cytokine staining.
Cells isolated from lungs were seeded at 105 cells/well in 96-well plates coated with anti-CD3 antibody (BioLegend, 5 μg/ml) and incubated overnight. Control cells were incubated overnight with PBS. Monensin (Sigma) was added at 3 μM during the last 4 h of culture. After overnight stimulation, cells were preincubated with anti-FcγR monoclonal antibody 2.4G2 to block nonspecific binding before they were stained with antibodies to detect CD4 (L3T4) and CD8 (53-6.7; BD Biosciences). For subsequent intracellular cytokine staining, a fixation/permeabilization solution kit (BD Cytofix/Cytoperm) was used according to the manufacturer’s instructions. Intracellular IFN-γ was stained with phycoerythrin-conjugated rat anti-mouse IFN-γ (XMG1.2; BD Biosciences). Events were acquired on a ZE5 cell analyzer (Bio-Rad), and data were analyzed with FlowJo software (Tree Star).
Histology.
Organs were harvested from a subset of mice and fixed in 10% formalin. Prior to fixation, lungs were inflated with PBS via the trachea to maintain lung architecture. After fixation, organs were embedded in paraffin, and 5-μm sections were obtained for histopathology. Sections were stained with hematoxylin and eosin to evaluate cellular infiltration. Separate sections used for immunohistochemistry were stained with anti-CD3 antibody (Thermo Scientific) to identify CD3+ cells. Sectioning and staining were performed by the University of Michigan Unit for Laboratory Animal Medicine Pathology Cores for Animal Research. Digital images were obtained with an EC3 digital imaging system (Leica Microsystems) using Leica acquisition software (Leica Microsystems). Any adjustments to brightness and contrast in digital images were applied equally to all experimental and control images.
Statistics.
Statistical analysis was performed using Prism 7 (GraphPad Software, Inc.). Viral loads were log transformed for statistical analysis. Differences between two groups at a single time point were analyzed using the Mann-Whitney rank sum test. Differences between groups at multiple time points were analyzed using two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple-comparison tests. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Kathy Spindler and Sharlene Day for helpful reviews of the manuscript and Suzy Dawid for the use of her laboratory’s plate reader. Expert technical assistance from Joel Whitfield in the University of Michigan Cancer Center Immunology Core is greatly appreciated.
This research was supported by the American Heart Association (16GRNT30250013) and by a Charles Woodson Accelerator Award from the University of Michigan Department of Pediatrics.
REFERENCES
- 1.Beling A, Kespohl M. 2018. Proteasomal protein degradation: adaptation of cellular proteolysis with impact on virus-and cytokine-mediated damage of heart tissue during myocarditis. Front Immunol 9:2620. doi: 10.3389/fimmu.2018.02620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy MK, Weinberg JB. 2015. The immunoproteasome and viral infection: a complex regulator of inflammation. Front Microbiol 6:21. doi: 10.3389/fmicb.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubiel W, Pratt G, Ferrell K, Rechsteiner M. 1992. Purification of an 11 S regulator of the multicatalytic protease. J Biol Chem 267:22369–22377. [PubMed] [Google Scholar]
- 4.Ma CP, Slaughter CA, DeMartino GN. 1992. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J Biol Chem 267:10515–10523. [PubMed] [Google Scholar]
- 5.Ebstein F, Kloetzel PM, Kruger E, Seifert U. 2012. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol Life Sci 69:2543–2558. doi: 10.1007/s00018-012-0938-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adkins LJ, Molloy CT, Weinberg JB. 2018. Fas activity mediates airway inflammation during mouse adenovirus type 1 respiratory infection. Virology 521:129–137. doi: 10.1016/j.virol.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Procario MC, Levine RE, McCarthy MK, Kim E, Zhu L, Chang CH, Hershenson MB, Weinberg JB. 2012. Susceptibility to acute mouse adenovirus type 1 respiratory infection and establishment of protective immunity in neonatal mice. J Virol 86:4194–4203. doi: 10.1128/JVI.06967-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molloy CT, Andonian JS, Seltzer HM, Procario MC, Watson ME Jr, Weinberg JB. 2017. Contributions of CD8 T cells to the pathogenesis of mouse adenovirus type 1 respiratory infection. Virology 507:64–74. doi: 10.1016/j.virol.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy MK, Malitz DH, Molloy CT, Procario MC, Greiner KE, Zhang L, Wang P, day SM, Powell SR, Weinberg JB. 2016. Interferon-dependent immunoproteasome activity during mouse adenovirus type 1 infection. Virology 498:57–68. doi: 10.1016/j.virol.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hensley SE, Zanker D, Dolan BP, David A, Hickman HD, Embry AC, Skon CN, Grebe KM, Griffin TA, Chen W, Bennink JR, Yewdell JW. 2010. Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J Immunol 184:4115–4122. doi: 10.4049/jimmunol.0903003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murata S, Takahama Y, Kasahara M, Tanaka K. 2018. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat Immunol 19:923–931. doi: 10.1038/s41590-018-0186-z. [DOI] [PubMed] [Google Scholar]
- 12.Weinberg JB, Stempfle GS, Wilkinson JE, Younger JG, Spindler KR. 2005. Acute respiratory infection with mouse adenovirus type 1. Virology 340:245–254. doi: 10.1016/j.virol.2005.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore ML, McKissic EL, Brown CC, Wilkinson JE, Spindler KR. 2004. Fatal disseminated mouse adenovirus type 1 infection in mice lacking B cells or Bruton’s tyrosine kinase. J Virol 78:5584–5590. doi: 10.1128/JVI.78.11.5584-5590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashley SL, Pretto CD, Stier MT, Kadiyala P, Castro-Jorge L, Hsu TH, Doherty R, Carnahan KE, Castro MG, Lowenstein PR, Spindler KR. 2017. Matrix metalloproteinase activity in infections by an encephalitic virus, mouse adenovirus type 1. J Virol 91:e01412-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tirumuru N, Pretto CD, Castro Jorge LA, Spindler KR. 2016. Mouse Adenovirus Type 1 Early Region 1A Effects on the Blood-Brain Barrier. mSphere 1:e00079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarthy MK, Procario MC, Wilke CA, Moore BB, Weinberg JB. 2015. Prostaglandin E2 production and T cell function in mouse adenovirus type 1 infection following allogeneic bone marrow transplantation. PLoS One 10:e0139235. doi: 10.1371/journal.pone.0139235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy MK, Procario MC, Twisselmann N, Wilkinson JE, Archambeau AJ, Michele DE, day SM, Weinberg JB. 2015. Proinflammatory effects of interferon gamma in mouse adenovirus 1 myocarditis. J Virol 89:468–479. doi: 10.1128/JVI.02077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T, Tanahashi N, Yoshimura T, Tanaka K, Ichihara A. 1994. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J Biochem 115:257–269. doi: 10.1093/oxfordjournals.jbchem.a124327. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka K. 1994. Role of proteasomes modified by interferon-gamma in antigen processing. J Leukoc Biol 56:571–575. doi: 10.1002/jlb.56.5.571. [DOI] [PubMed] [Google Scholar]
- 20.Opitz E, Koch A, Klingel K, Schmidt F, Prokop S, Rahnefeld A, Sauter M, Heppner FL, Volker U, Kandolf R, Kuckelkorn U, Stangl K, Kruger E, Kloetzel PM, Voigt A. 2011. Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLoS Pathog 7:e1002233. doi: 10.1371/journal.ppat.1002233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basler M, Beck U, Kirk CJ, Groettrup M. 2011. The antiviral immune response in mice devoid of immunoproteasome activity. J Immunol 187:5548–5557. doi: 10.4049/jimmunol.1101064. [DOI] [PubMed] [Google Scholar]
- 22.Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, Welsh RM, Karow ML, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rock KL. 2012. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol 13:129–135. doi: 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mundt S, Basler M, Buerger S, Engler H, Groettrup M. 2016. Inhibiting the immunoproteasome exacerbates the pathogenesis of systemic Candida albicans infection in mice. Sci Rep 6:19434. doi: 10.1038/srep19434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirschner F, Reppe K, Andresen N, Witzenrath M, Ebstein F, Kloetzel PM. 2016. Proteasome β5i subunit deficiency affects opsonin synthesis and aggravates pneumococcal pneumonia. PLoS One 11:e0153847. doi: 10.1371/journal.pone.0153847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Norbury CC, Cho Y, Yewdell JW, Bennink JR. 2001. Immunoproteasomes shape immunodominance hierarchies of antiviral CD8+ T cells at the levels of T cell repertoire and presentation of viral antigens. J Exp Med 193:1319–1326. doi: 10.1084/jem.193.11.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pang KC, Sanders MT, Monaco JJ, Doherty PC, Turner SJ, Chen W. 2006. Immunoproteasome subunit deficiencies impact differentially on two immunodominant influenza virus-specific CD8+ T cell responses. J Immunol 177:7680–7688. doi: 10.4049/jimmunol.177.11.7680. [DOI] [PubMed] [Google Scholar]
- 27.Hutchinson S, Sims S, O’Hara G, Silk J, Gileadi U, Cerundolo V, Klenerman P. 2011. A dominant role for the immunoproteasome in CD8+ T cell responses to murine cytomegalovirus. PLoS One 6 doi: 10.1371/journal.pone.0014646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basler M, Lindstrom MM, LaStant JJ, Bradshaw JM, Owens TD, Schmidt C, Maurits E, Tsu C, Overkleeft HS, Kirk CJ, Langrish CL, Groettrup M. 2018. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep 19:e46512. doi: 10.15252/embr.201846512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalim KW, Basler M, Kirk CJ, Groettrup M. 2012. Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J Immunol 189:4182–4193. doi: 10.4049/jimmunol.1201183. [DOI] [PubMed] [Google Scholar]
- 30.Moebius J, van den Broek M, Groettrup M, Basler M. 2010. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur J Immunol 40:3439–3449. doi: 10.1002/eji.201040620. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt C, Berger T, Groettrup M, Basler M. 2018. Immunoproteasome inhibition impairs T and B cell activation by restraining ERK signaling and proteostasis. Front Immunol 9:2386. doi: 10.3389/fimmu.2018.02386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welton AR, Gralinski LE, Spindler KR. 2008. Mouse adenovirus type 1 infection of natural killer cell-deficient mice. Virology 373:163–170. doi: 10.1016/j.virol.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarthy MK, Zhu L, Procario MC, Weinberg JB. 2014. IL-17 contributes to neutrophil recruitment but not to control of viral replication during acute mouse adenovirus type 1 respiratory infection. Virology 456-457:259–267. doi: 10.1016/j.virol.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reis J, Hassan F, Guan XQ, Shen J, Monaco JJ, Papasian CJ, Qureshi AA, Way CW, Vogel SN, Morrison DC, Qureshi N. 2011. The immunoproteasomes regulate LPS-induced TRIF/TRAM signaling pathway in murine macrophages. Cell Biochem Biophys 60:119–126. doi: 10.1007/s12013-011-9183-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abramova EB, Astakhova TM, Sharova NP. 2005. Changes in proteasome activity and subunit composition during postnatal development of rat. Ontogenez 36:205–210. (In Russian.) [PubMed] [Google Scholar]
- 36.Claud EC, McDonald JA, He SM, Yu Y, Duong L, Sun J, Petrof EO. 2014. Differential expression of 26S proteasome subunits and functional activity during neonatal development. Biomolecules 4:812–826. doi: 10.3390/biom4030812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharova NP, Zakharova LA, Astakhova TM, Karpova YD, Melnikova VI, Dmitrieva SB, Lyupina YV, Erokhov PA. 2009. New approach to study of T cellular immunity development: parallel investigation of lymphoid organ formation and changes in immune proteasome amount in rat early ontogenesis. Cell Immunol 256:47–55. doi: 10.1016/j.cellimm.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Bermick JR, Lambrecht NJ, denDekker AD, Kunkel SL, Lukacs NW, Hogaboam CM, Schaller MA. 2016. Neonatal monocytes exhibit a unique histone modification landscape. Clin Epigenetics 8:99. doi: 10.1186/s13148-016-0265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fonseca W, Lukacs NW, Ptaschinski C. 2018. Factors affecting the immunity to respiratory syncytial virus: from epigenetics to microbiome. Front Immunol 9:226. doi: 10.3389/fimmu.2018.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Restori KH, Srinivasa BT, Ward BJ, Fixman ED. 2018. Neonatal immunity, respiratory virus infections, and the development of asthma. Front Immunol 9:1249. doi: 10.3389/fimmu.2018.01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cloos J, Roeten MS, Franke NE, van Meerloo J, Zweegman S, Kaspers GJ, Jansen G. 2017. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev 36:599–615. doi: 10.1007/s10555-017-9699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller Z, Lee W, Kim KB. 2014. The immunoproteasome as a therapeutic target for hematological malignancies. Curr Cancer Drug Targets 14:537–548. doi: 10.2174/1568009614666140723113139. [DOI] [PubMed] [Google Scholar]
- 43.Cauthen AN, Welton AR, Spindler KR. 2007. Construction of mouse adenovirus type 1 mutants. Methods Mol Med 130:41–59. [DOI] [PubMed] [Google Scholar]
- 44.Molloy CT, Adkins LJ, Griffin C, Singer K, Weinberg JB. 2018. Mouse adenovirus type 1 infection of adipose tissue. Virus Res 244:90–98. doi: 10.1016/j.virusres.2017.11.014. [DOI] [PubMed] [Google Scholar]