

Approximately 2/3 of adults worldwide are latently infected with herpes simplex virus 1. Upon reactivation, the virus has the ability to evade neutralizing antibodies by moving through cell junctions, but the mechanism of direct cell-to-cell spread is poorly understood. The machinery that assembles between cells includes the viral fusion proteins and various accessory proteins that prevent cells from fusing. Alterations in four proteins will dysregulate the machinery, allowing neighboring cells to fuse to make syncytia, but this can be prevented by removing various individual accessory proteins to further disable the machinery. Previously, the accessory protein UL21 was found to be important for the activity of some syncytial variants but not others. In this study, we discovered that UL16, gE, and gI all act differently in how they control the fusion machinery. A better understanding of the mechanism of cell-to-cell spread may enable the development of drugs that block it.
KEYWORDS: cell-to-cell spread, HSV-1, syncytia, tegument, UL16, gE, gI
ABSTRACT
Like all the herpesviruses, herpes simplex virus encodes machinery that enables it to move through cell junctions to avoid neutralizing antibodies. This cell-to-cell spread mechanism requires the viral fusion machinery (gD, gH/gL, and gB) and numerous accessory proteins. Of all of these, minor alterations to only four proteins (gB, gK, UL20, or UL24) will dysregulate the fusion machinery, allowing the formation of syncytia. In contrast, removal of individual accessory proteins will block cell-to-cell spread, forcing the virus to transmit in a cell-free manner. In the context of a Syn variant, removal of a required accessory protein will block cell fusion, again forcing cell-free spread. This has been investigated most thoroughly for gBsyn variants, which lose their syncytial phenotype in the absence of several accessory proteins, including gE, gI, UL16, and UL21, which are known to physically interact. Recently it was found that UL21 is not needed for gKsyn-, UL20syn-, or UL24syn-induced cell fusion, and hence it was of interest to ascertain whether gE, gI, and UL16 are required for Syn variants other than gBsyn. Null mutants of these were each combined with seven syncytial variants distributed among gK, UL20, and UL24. Surprisingly, very different patterns of accessory protein requirements were revealed. Indeed, for the three gKsyn variants tested, two different patterns were found. Also, three mutants were able to replicate without causing cytopathic effects. These findings show that mutations that produce Syn variants dysregulate the cell-to-cell-spread machinery in unique ways and provide clues for elucidating how this virus moves between cells.
IMPORTANCE Approximately 2/3 of adults worldwide are latently infected with herpes simplex virus 1. Upon reactivation, the virus has the ability to evade neutralizing antibodies by moving through cell junctions, but the mechanism of direct cell-to-cell spread is poorly understood. The machinery that assembles between cells includes the viral fusion proteins and various accessory proteins that prevent cells from fusing. Alterations in four proteins will dysregulate the machinery, allowing neighboring cells to fuse to make syncytia, but this can be prevented by removing various individual accessory proteins to further disable the machinery. Previously, the accessory protein UL21 was found to be important for the activity of some syncytial variants but not others. In this study, we discovered that UL16, gE, and gI all act differently in how they control the fusion machinery. A better understanding of the mechanism of cell-to-cell spread may enable the development of drugs that block it.
INTRODUCTION
The membrane fusion machinery of herpes simplex virus 1 (HSV-1) plays two distinct roles for transmission of infections. One is during entry of cell-free virus into the host, which occurs either at the plasma membrane or within endocytic vesicles (1). This process begins when gD (Fig. 1A) binds to one of its cellular receptors—nectin-1, HVEM, or 3-O-sulfated heparan sulfate (2–5)—and then transmits a signal through the heterodimer gH/gL to the viral fusogen, gB (6, 7), which mediates fusion of the viral and host membranes (8). These four proteins are inherently fusogenic, and thus, when they are coexpressed in the absence of other viral proteins, the cells are stimulated to fuse with each other (9). However, wild-type (WT) HSV-1 normally does not result in cell fusion despite the presence of these viral glycoproteins on the plasma membranes of the infected cells (10–12), and it is quite clear that gD, gH/gL, and gB are tightly regulated by other viral proteins (13–15), tyrosine phosphorylation (16), and perhaps other unknown components.
FIG 1.

Syncytial variants display unique properties. (A) Diagram of proteins required for cell-to-cell spread that are relevant to this study. The fusion machinery consists of gB, gH/gL, and gD. Basic residues in gD that are needed for cell-to-cell spread are indicated (+++). Eight substitutions in gB, gK, UL20, and UL24 that dysregulate the fusion machinery are represented with red dots, and the specific changes are listed below the diagram. The accessory proteins investigated are gE, gI, and UL16, which are highlighted in yellow. (B) Vero cells were infected with each Syn variant, cell lysates were harvested at 18 h postinfection (hpi), and immunoblots were probed for VP5, gE, gI, and UL16 expression. (C) Vero cells were infected with each Syn variant at a low MOI and overlaid with 0.5% agarose in DMEM. At 36 hpi, 30 individual syncytia were imaged for each variant: the average areas are shown. (D) Vero cells were infected at a low MOI, incubated for 36 h, fixed, and stained with DAPI and an antibody against the major capsid protein, VP5. Corresponding bright-field and fluorescent images were taken for each variant. The scale bar is set at 50 μm.
The other transmission role for the virus fusion machinery is in cell-to-cell spread. This poorly understood mechanism sends mature virions, contained within cytoplasmic vesicles, to lateral cell junctions so that they can enter neighboring, uninfected cells (17–19). HSV-1 uses this mode of transmission when moving from mucosal epithelial cells into nearby sensory neurons and back again as it establishes latent infections and later reactivates (20). This mechanism enables the virus to avoid neutralizing antibodies (21), and thus, in cell culture, wild-type HSV-1 can still form large plaques even when such antibodies are present in the growth medium. In contrast, mutants defective for cell-to-cell spread make tiny plaques under these conditions because they can only spread via the cell-free mode. For example, viruses lacking gE (Fig. 1A) are severely reduced in their ability to form plaques in the presence of neutralizing antibodies, even though they have no defect in the production of infectious virions (18, 22). Moreover, gE-null viruses cannot spread from epithelial cells into neurons in mouse models (23, 24).
How the viral fusion machinery and its regulatory components mediate the process of cell-to-cell spread is unclear. It has been proposed that this machinery may create new cell junctions since none typically exist between the axons of sensory neurons and nearby epithelial cells; it may also modify existing junctions between epithelial cells to make them compatible for cell-to-cell spread (16). In any case, a large number of HSV-1 mutants have been found that dysregulate the viral machinery and cause infected cells to fuse, forming multinucleated cells known as syncytia (25). Each of the mutations responsible for these Syn variants falls into one of four viral genes, namely those encoding gB, gK, UL20, or UL24 (Fig. 1A) (26). Moreover, cells infected with wild-type HSV-1 can be induced to fuse when treated with salubrinal (27), and although the target of this drug is unknown, inhibitors of PTP1B (a tyrosine phosphatase) block both cell fusion and cell-to-cell spread (16). Thus, there are at least two host factors that regulate the viral fusion machinery: PTP1B and an unidentified tyrosine kinase.
The relevance of the Syn phenotype to cell-to-cell spread can perhaps best be seen by the overlap of viral proteins needed for both. As one example, gE is needed both for cell-to-cell spread (as noted above) and for the gBsyn phenotype. That is, when all of gE or just its cytoplasmic tail is absent, gBsyn viruses are no longer capable of making cells fuse, but instead just make lytic plaques (28–31). Thus, in the case of gBsyn variants, their mutations do not merely destroy the regulatory machinery so that gD, gH/gL, and gB are free to fuse cells indiscriminately like they do when expressed alone (9). Otherwise, accessory proteins like gE would not be required for syncytium formation. Something more subtle is involved in the creation of Syn variants.
There is very little known about the requirements for “accessory” proteins among Syn variants other than gBsyn. One study reported that UL11 and gM are required for both the gBsyn and gKsyn phenotypes (32), but UL20syn and UL24syn were not examined. The first indication that there are differences arose from a study of UL21, which together with UL16 and UL11, forms a complex on the tail of gE (Fig. 1A) that is required for the gBsyn phenotype (30). While passaging a UL21-null mutant, which is defective for cell-to-cell spread, it quickly gave rise to Syn variants, and these mapped to the gene for gK, but the Syn phenotypes of UL20syn and UL24syn do not require UL21, either (33). Thus, UL21 was found to be required only for gBsyn-induced cell fusion, providing evidence that syn mutations may dysregulate the viral machinery in unique ways. Moreover, gBsyn, gKsyn, UL20syn, and UL24syn have been shown to respond in strikingly different ways to salubrinal and PTP1B inhibitors (16). More thorough analyses of the accessory protein requirements among the various Syn variants are needed because these are likely to provide additional clues for the mechanism of cell-to-cell spread.
The experiments described here focus on three proteins in the complex with UL21 and UL11 (Fig. 1A), all of which have been reported to be required for the gBsyn phenotype (30, 32). One is UL16, which makes direct contacts with UL21, UL11, gE, and gD (34–37), and because of its central position in this interaction network, it seemed likely to be required for all the Syn variants, even though UL21 is not. The other proteins are gE and gI, which are well known to form a heterodimer (38, 39). Because the external domain of gE has a discrete function that is essential for cell-to-cell spread (40) and has been hypothesized to perhaps bind a host receptor (22), we expected that gE/gI would exhibit matching requirements and be required for all the Syn variants. As described below, these studies produced several surprising results.
RESULTS
Approach for constructing mutant viruses and confirming their phenotypes.
Since gE, gI, and UL16 have been previously reported to be important for the gBsyn phenotype, our initial goal was to make null mutants of these in the background of a gKsyn variant, a UL20syn variant, and a UL24syn variant, for a total of 9 new viruses. To limit the selection of unintended mutations, all the DNA alterations were made in Escherichia coli via bacterial artificial chromosome (BAC) recombineering rather than by using genetic selections in infected Vero cells. All the clones were screened via restriction endonuclease digestions, and those that had no obvious genome rearrangements were sequenced to confirm that the expected mutations were present. Furthermore, after transfecting the mutant BACs into Vero cells, the resulting viruses were passaged just once to make virus stocks, thereby limiting the selection of suppressor mutations.
Early in this investigation we obtained surprising results, with gE seeming to be dispensable for certain Syn variants. To provide further confidence in our observations, we took the approach of making multiple gE- and gI-null viruses independently and with different ways of preventing expression. In addition, we decided to expand the number of Syn variants to include three in gK, two in UL20, and two in UL24 (Fig. 1A). We also constructed two different gI-null derivatives of a gBsyn variant (A855V) because we had never verified the previous report that this protein is required for that Syn phenotype (28). Because it was very unlikely that a spurious compensating mutation would occur in each independently constructed virus, revertants were not made. Importantly, all of the results described below were consistent, meaning that gE- and gI-null mutants made in different ways had the same phenotypes.
In the case of UL16, the various syn mutations were introduced into a well-characterized null mutant (41), which has been shown to be nonsyncytial in the context of a gBsyn mutant. Confirmation that the phenotypes were due to the absence of UL16 was made possible by the availability of G5 cells, a UL16-complementing cell line derived from Vero cells (42). Thus, the previously constructed Δ16/gBsyn virus (30) regains the Syn phenotype on G5 cells. In the end, the total number of new viruses constructed for this study grew to 36 (Table 1) so as to give us confidence in the results obtained.
TABLE 1.
gE, gI, and UL16 deletion mutants for each Syn varianta
| New virus | Parentb | Engineered mutation |
|---|---|---|
| gE-null | WT | gE signal peptide codons replaced with 3 stops |
| ΔgE(galK) | WT | ED-TM codons of gE replaced with galK cassette |
| ΔgE.stop | WT | ED-TM codons of gE replaced with 2 stops |
| gE-null/gK.A40V | gK.A40V | gE signal peptide codons replaced with 3 stops |
| gE-null/gK.L118Q | gK.L118Q | gE signal peptide codons replaced with 3 stops |
| gE-null/gK.G167D | gK.G167D | gE signal peptide codons replaced with 3 stops |
| ΔgE(galK)/gK.A40V | gK.A40V | ED-TM codons of gE replaced with galK cassette |
| ΔgE(galK)/gK.L118Q | gK.L118Q | ED-TM codons of gE replaced with galK cassette |
| ΔgE.stop/gK.G167D | gK.G167D | ED-TM codons of gE replaced with 2 stop codons |
| gE-null/UL20.F222A | UL20.F222A | gE signal peptide codons replaced with 3 stops |
| gE-null/UL20.R209A | UL20.R209A | gE signal peptide codons replaced with 3 stops |
| ΔgE(galK)/UL20.F222A | UL20.F222A | ED-TM codons of gE replaced with galK cassette |
| gE-null/UL24.G121A | UL24.G121A | gE signal peptide codons replaced with 3 stops |
| gE-null/UL24.E99A/K101A | UL24.E99A/K101A | gE signal peptide codons replaced with 3 stops |
| ΔgE(galK)/UL24.G121A | UL24.G121A | ED-TM codons of gE replaced with galK cassette |
| ΔgI | WT | Deletion of gI codons 2–383 |
| gI-null | WT | Replace gI valine 10 codon with 2 stops |
| ΔgI/gB.A855V | gB.A855V | galK replacement of gI codons 2–383 |
| gI-null/gB.A855V | gB.A855V | Replace gI valine 10 codon with 2 stops |
| ΔgI/gK.A40V | gK.A40V | Deletion of gI codons 2–383 |
| ΔgI/gK.L118Q | gK.A40V | Deletion of gI codons 2–383 |
| ΔgI/gK.G167D | gK.L118Q | Deletion of gI codons 2–383 |
| gI-null/gK.A40V | gK.G167D | Replace gI valine 10 codon with 2 stops |
| ΔgI/UL20.F222A | UL20.F222A | Deletion of gI codons 2–383 |
| ΔgI/UL20.R209A | UL20.F222A | Deletion of gI codons 2–383 |
| gI-null/UL20.F222A | UL20.R209A | Replace gI valine 10 codon with 2 stops |
| ΔgI/UL24.G121A | UL24.G121A | Deletion of gI codons 2–383 |
| ΔgI/UL24.E99A/K101A | UL24.G121A | Deletion of gI codons 2–383 |
| gI-null/UL24.G121A | UL24.E99A/K101A | Replace gI valine 10 codon with 2 stops |
| ΔUL16/gK.A40V | ΔUL16 | A40V substitution inserted into gK |
| ΔUL16/gK.L118Q | ΔUL16 | L118Q substitution inserted into gK |
| ΔUL16/gK.G167D | ΔUL16 | G167D substitution inserted into gK |
| ΔUL16/UL20.F222A | ΔUL16 | F222A substitution inserted into UL20 |
| ΔUL16/UL20.R209A | ΔUL16 | R209A substitution inserted into UL20 |
| ΔUL16/UL24.G121A | ΔUL16 | G121A substitution inserted into UL24 |
| ΔUL16/UL24.E99A/K101A | ΔUL16 | E99A/K101A substitutions inserted into UL24 |
Syncytial variants display unique properties.
The eight Syn variants used in this study (Fig. 1A) were previously inserted into the KOS genome (30, 33), and before removing accessory proteins gE, gI, or UL16, we first wanted a clear view of how each behaved in comparison to the others. Immunoblotting confirmed that all eight strains expressed the three viral proteins of interest (Fig. 1B). In the case of the three gKsyn variants, gE levels seemed to be slightly reduced, but what was most striking was that the level of gI was reduced ∼80% for the UL24.E99A/K101A Syn mutant, even though approximately equal amounts of cell lysate were loaded in each lane, as indicated by the similar levels of VP5 (the major capsid protein). This may be of interest since little is known about how UL24 controls the cell-to-cell spreading machinery to prevent fusion, but we did not pursue the observation any further.
Although cells infected with Syn variants will eventually die to create plaques in a monolayer, there is a long period during which syncytia rapidly form and expand from the site of the initially infected cell. For each of the eight Syn variants, we compared the average expansion sizes of 30 individual syncytia 36 h after infecting cells at a low multiplicity of infection (MOI) (Fig. 1C). No lytic plaques were observed, and most of the variants produced similarly sized syncytia, with the exception of gK.A40V and gK.L118Q, which have substitutions in the large external, N-terminal domain of this protein.
To more closely evaluate the fusogenic phenotype of each variant, monolayers were fixed, and the infected cells were identified by immunostaining for VP5, while nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). The syncytia induced by gB.A855V, gK.A40V, and gK.L118Q were relatively uniform in appearance, with hundreds of nuclei clustering together in the middle of one large syncytium (Fig. 1D, top three rows). However, the syncytia induced by gK.G167D and each of the UL20syn and UL24syn variants did not exhibit uniformity, despite identical starting cell densities. Instead, their nuclei were clustered into several smaller pockets within the syncytium, as seen in the DAPI column (Fig. 1D, bottom five rows). These data emphasize that although all the Syn variants are fully fusogenic, they differ in ability to spread and in visual appearance. Of course, the main question is whether these fusion phenotypes are lost in the absence of gE, gI, or UL16.
gE is not universally required for the Syn phenotype.
Mutants that are incapable of expressing of gE (encoded by the US8 gene) were constructed in three different ways (Fig. 2A). The gE-null mutant was made by inserting three stop codons in the place of the coding sequence for the signal peptide. The ΔgE(galK) mutant was made by replacing the coding sequence for the ecto- and transmembrane domains of gE with a galK cassette. For the ΔgE.stop mutant, the galK cassette was removed and replaced with two stop codons. All three of these mutants leave the overlapping US8A gene and its promoter intact (Fig. 2A), which may not be important since the KOS strain expresses an extended and possibly inactive version of this protein (43). Immunoblotting of infected-cell lysates confirmed that the mutants do not express gE, but they do make wild-type amounts of its binding partner, gI (Fig. 3A, left panel). In high-MOI experiments where cell-to-cell spread is not needed, all three mutants produced virus titers equivalent to the wild type (Fig. 2D), as reported for gE deletion mutants made in other labs (18, 44, 45). In the absence of any syn mutations, all three mutants produced lytic plaques that were small, consistent with a defect in cell-to-cell spread (data not shown), but the question of interest was what happens in the context of the Syn variants (i.e., the viruses listed in the top section of Table 1).
FIG 2.
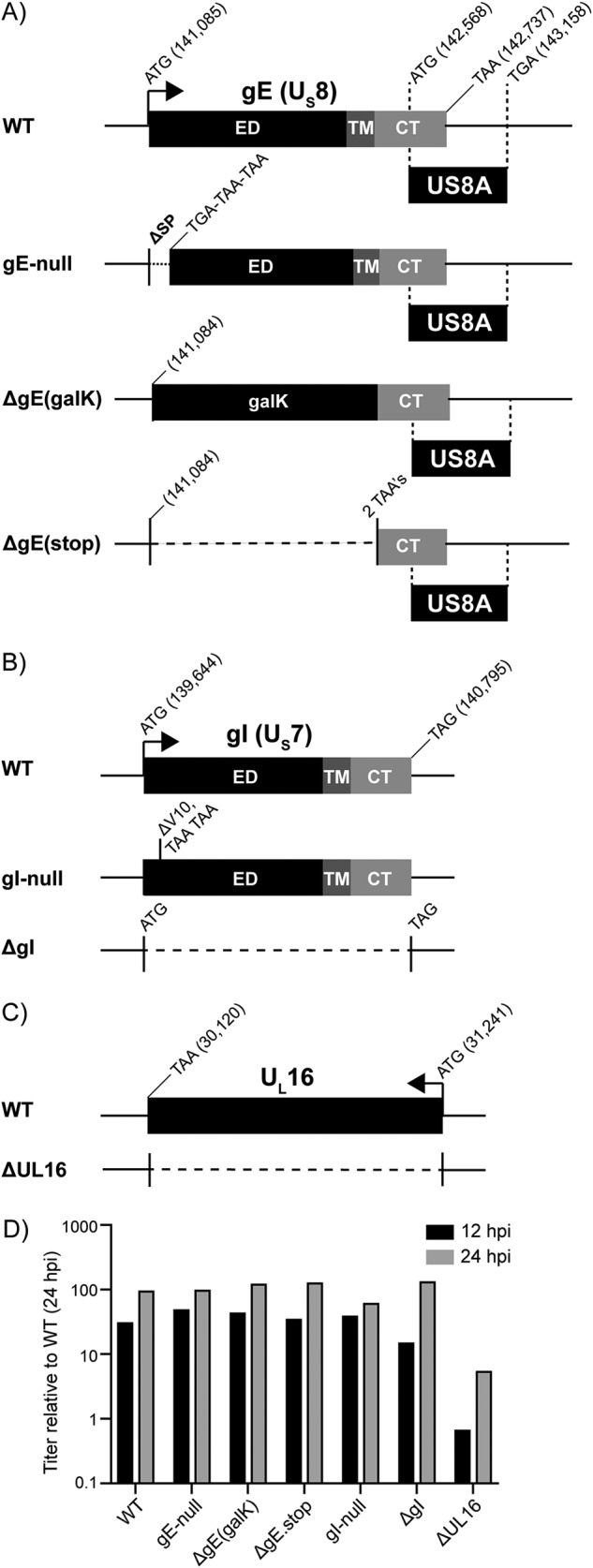
Construction of HSV-1 deletion mutants. (A) An illustration of the US8 (gE) gene and the gE deletion viruses. Sequences encoding the ectodomain (ED), transmembrane domain (TM), and cytoplasmic tail (CT) are indicated. The locations of the start and stop codons within the KOS reference genome are denoted (GenBank accession no. JQ673480.1). Also included is the US8A gene, which overlaps the CT-coding region of US8. The gE-null mutant has three stop codons in place of the signal peptide (SP)-coding sequence. The ΔgE(galK) mutant has a galK cassette in place of the ED-TM-coding sequence. The ΔgE(stop) mutant has two stop codons in place of the ED-TM coding sequence. (B) An illustration of the US7 (gI) gene and the gI deletion viruses. Positions of the ED-, TM-, and CT-coding sequences are indicated. The gI-null mutant has two stop codons in the place of valine codon 10. The ΔgI mutant lacks codons 2 to 383. (C) An illustration of the UL16-coding sequence, which was deleted to make the ΔUL16 mutant. (D) For each deletion mutant, four plates of Vero cells were infected at an MOI of 5. Two plates were harvested at both of the indicated times, and the average titers (cells plus medium) were determined via plaque assays. These were normalized to the wild type at 24 hpi and plotted.
FIG 3.

gE is not universally required for the Syn phenotype. (A) Vero cells were infected with gE deletion viruses with or without syn mutations. Cell lysates were harvested 18 to 20 hpi, and immunoblots were probed for expression of VP5, gE, and gI. (B) Vero cells were infected with gE-null/gKsyn variants at a low MOI. At 48 hpi, the cell were fixed and stained with DAPI and an antibody against VP5. Corresponding bright-field and fluorescent images were taken for each variant. (C) For each of the indicated mutants, the areas of 30 individual syncytia were measured at either 36 hpi (gKsyn parents) or 42 hpi (mutants lacking gE). The reduced average areas of the mutants relative to their gKsyn parent are plotted. ****, P < 0.0001, Student's t test. Additionally, each site of infection was scored as syncytial, mixed, or lytic (as defined in the text), with the linear distribution of these phenotypes shown graphically.
We had predicted that gE would be essential for all the Syn variants, and that hypothesis was true for UL20syn and UL24syn, where all six mutant viruses failed to induce any cell fusion (data not shown because there is nothing to report except the presence of tiny, lytic plaques). Immunoblotting confirmed that gE was absent (Fig. 3A, two panels on the right), and although the levels of gI were unaffected for the UL20syn mutants, we found that this protein was noticeably reduced (∼80%) for UL24.E99A/K101A, as seen earlier (Fig. 1B). Moreover, the two clones of UL24.G121A also exhibited reductions of gI.
On the other hand, our hypothesis was refuted by the gKsyn mutants. In the case of gK.A40V, the two independently constructed mutants, gE-null/gK.A40V (Fig. 3B, top row) and ΔgE(galK)/gK.A40V (not shown), were fully syncytial (i.e., no lytic plaques were observed), even though immunoblotting confirmed their inability to express gE (Fig. 3A, second panel). Fusion was also observed for gK.L118Q when combined with the gE-null (Fig. 3B, middle row) or ΔgE(galK) mutant (not shown). In contrast, gK.G167D became lytic when gE expression was eliminated (example shown in Fig. 3B, bottom). This experiment provides the first example of an accessory protein (gE) being differentially required for fusion in an allele-specific manner. That is, some gKsyn mutants require gE for fusion, but others do not (Table 2).
TABLE 2.
Summary of the accessory protein requirements for each Syn mutanta
| Accessory proteins | Cell-to-cell spread | gBsyn | gKsyn |
UL20syn |
UL24syn |
||||
|---|---|---|---|---|---|---|---|---|---|
| A855V | A40V | L118Q | G167D | R209A | F222A | E99A/K101A | G121A | ||
| gE | + | + | – | – | + | + | + | + | + |
| gI | + | – | – | – | – | – | – | + | + |
| UL16 | + | + | – | – | + | + | + | + | + |
| UL21 | + | + | – | – | – | – | – | – | – |
The results for UL21 were previously reported and are shown for comparison (33).
To quantify our observations for all the gKsyn variants, cells were infected at a low MOI and incubated with an agarose overlay, and images of at least 30 sites of infection were obtained for each virus. gKsyn parent-infected cells were imaged 36 h postinfection because waiting any longer resulted in lysis of the syncytia. Because viruses lacking gE have spreading defects, cells infected with these viruses were imaged at 42 h postinfection so that the sites of infection would be large enough to accurately identify. The area of each site of infection was measured, and each was assigned a score of “syncytial,” “mixed,” or “lytic” (Fig. 3C). “Syncytial” indicates that the entire site of infection consisted of a mutinucleated syncytium, like the Syn parents. “Mixed” was defined as a plaque that had at least 5 nuclei in a syncytium directly juxtaposed to lytically infected cells. Finally, “lytic” plaques contained no fused cells. The results show that both of the gK.A40V viruses lacking gE were fully syncytial. Both gK.L118Q viruses lacking gE exhibited somewhat varied results, with many being fully syncytial and others being mixed (center graph). In contrast, both gK.G167D viruses lacking gE were severely defective for syncytium formation, producing only mixed or lytic plaques (right graph). In addition to revealing an allele-specific requirement for gE, these results show that a small site of infection is not predictive of the ability of a mutant to cause fusion.
gI is only required for UL24syn.
We expected that Syn mutants lacking gI would behave similarly to those lacking gE because these proteins form a heterodimer that is needed for cell-to-cell spread (18, 46) and together create an efficient Fc receptor that binds IgG and aids in viral immune evasion (47, 48). To prevent expression of gI (encoded by the US7 gene), two methods were employed (Fig. 2B). The gI-null mutant was made by replacing the codon for valine at position 10 with two stop codons. However, if an alternative translational start site was present near this position, then a derivative of gI would be produced with a shortened but possibly functional signal peptide. Therefore, the ΔgI mutant was made, in which the coding sequence was removed (codons 2 to 383). Immunoblotting of infected-cell lysates confirmed that both mutants lack expression of gI, with little effect on gE other than a slight alternation in the pattern of glycosylation (Fig. 4A, top). In high-MOI experiments where cell-to-cell spreading is not needed, both viruses produced titers equivalent to those of the wild type (Fig. 2D). Next, the two gI mutants were combined with the Syn mutants, and the cell fusion ability was examined for each virus (Table 1, middle section).
FIG 4.

gI is only required for UL24syn. (A) Cells were infected with gI deletion mutants, harvested 18 hpi, and the lysates analyzed for VP5, gE, and gI expression via immunoblotting. The blots are arranged according to the type of Syn variant. (B) Vero cells were infected with the indicated mutants at a low MOI. At 36 hpi, they were fixed and stained with DAPI and antibodies specific for VP5. Corresponding bright-field and fluorescent images were taken for each variant. (C) For each of the indicated mutants, the areas of 30 individual syncytia were measured at 36 hpi. The reduced average areas of the mutants relative to their Syn parent are plotted. P values were calculated by the Student's t test. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Additionally, each site of infection was also scored as syncytial, mixed, or lytic (as defined in the text), with the linear distribution of these phenotypes shown graphically.
A previous study showed that gE and gI are essential for the gBsyn phenotype (28), and although we previously confirmed the gE requirement (30), the need for gI had not been tested in our laboratory. We were quite surprised to find that in the absence of gI expression (Fig. 4A, top), both of our gBsyn variants exclusively made syncytia (Fig. 4B, top two rows). Although the sites of infection were somewhat smaller than those of the parent, no mixed or lytic plaques were observed (Fig. 4C, leftmost panel). These viruses were passaged only once after transfecting the BACs into Vero cells, making it unlikely that compensating mutations could have arisen. Nevertheless, we extracted viral DNA and sequenced the genes for gK, UL20, and UL24 in the gI-null/gBsyn mutant, but no alterations were found (i.e., no other syn mutations were present). Although we used the same gBsyn mutant as the previous study (A855V), there is a strain difference (KOS versus SC16), which may account for the discrepancy, but that remains to be tested. Since our hypothesis of matching requirements for gE and gI was wrong, it was difficult to predict the phenotypes of the other Syn mutants in the absence of gI. If anything, it seemed likely that this glycoprotein would not be required for any of them.
For gKsyn and UL20syn, gI was indeed found to be dispensable for cell fusion (Fig. 4). The allele-specific requirement seen for gE with the three gKsyn variants was not found for gI, with all mutants exhibiting fusion activity (for example, see Fig. 4B, third row). The absence of gI expression was confirmed by immunoblotting (Fig. 4A, second panel), and quantification of 30 sites of infection for each revealed they were smaller but fully syncytial (Fig. 4C, second panel). Likewise, all the UL20syn variants were confirmed to lack gI (Fig. 4A, third panel) and to be syncytial (example shown in Fig. 4B, fourth row). Quantification revealed that the sites of infection were smaller but essentially all syncytial, with an occasional mixed site (Fig. 4C, third panel).
In contrast to all the other Syn variants, UL24syn was found to require gI. In its absence, which was confirmed by immunoblotting (Fig. 4A, bottom), small, lytic plaques were observed (Fig. 4B, bottom two rows). Quantification of 30 sites of infection for each mutant revealed that some of the plaques were mixed, but no pure syncytia were seen (Fig. 4C, right panel). Thus, it is quite clear that the UL24syn phenotype depends on gI (Table 2). Because UL24syn mutants can reduce the expression of gI (see Fig. 1B and Fig. 3A) and complete deletion of gI eliminates the UL24syn phenotype, it seems likely that some sort of interaction (direct or indirect) takes place between these two proteins, but this possibility was not explored further.
The requirement for UL16 among Syn mutants matches that for gE.
UL16 interacts directly with the cytoplasmic tail of gE and also with UL21 (Fig. 1A). Thus, it was difficult to predict whether Syn mutants lacking this protein would behave more like those lacking gE, where it was required for six of the eight Syn mutants tested, or those lacking UL21, where it is needed only for the gBsyn phenotype (Table 2). It was also possible that UL16 mutants would exhibit a unique profile.
The gBsyn phenotype has already been reported to depend on UL16 (30), and that was confirmed (not shown). To examine the other seven syn alleles, they were each inserted into the ΔUL16 virus (Fig. 2C), a well-described mutant that lacks the entire UL16 coding sequence (41). PCR analyses confirmed that genomes of the resulting viruses (Table 1, bottom section) were smaller than the wild type by the expected 1,100 bp (not shown). We prepared virus stocks on G5 cells, which contain the UL16-through-UL21 segment of the KOS genome (42) and can complement UL16-null mutants (41). On these cells, all the mutants produced sites of infection that were at least as large as the ΔUL16 parent (Fig. 5A, left column). As expected, a closer inspection revealed that all contained syncytia, except for the ΔUL16 parent and the two UL20syn mutants, whose cell fusion phenotypes are suppressed by wild-type UL20 produced by the G5 cells.
FIG 5.

Unexpected spreading defects among ΔUL16/syn variants. (A) Plaque assays for each of the indicated mutant viruses were done on G5 (complementing) or Vero cells. The G5 plaque assays (left column) were fixed at 3 days postinfection (dpi), and the Vero plaque assays (right column) were fixed at 5 dpi. Plaques were visualized by staining with crystal violet. (B) Vero cells or G5 cells were infected with the indicated viruses at an MOI of 3. At 24 hpi, infected cell lysates and the media were harvested separately, and virus titers were measured with plaque assays on G5 cells. The left panel shows the average total virus titers obtained from two measurements for each mutant in each cell type. A Student's t test was used to analyze the reductions in titer of the ΔUL16/syn mutants compared to the ΔUL16 parent in Vero cells. **, P < 0.01. The right panel shows the percentage of infectious virions released into the medium when Vero or G5 cells were infected with the ΔUL16 parent. Release of the ΔUL16/syn variants is shown only for Vero cells because in G5 cells the syncytia that are produced are prone to bursting. (C) Noncomplementing Vero cells were infected at an MOI of 0.05 with the ΔUL16 parent or the three non-plaque-forming mutants, all of which had been produced in Vero cells. At 18 h postinfection, the cells were fixed and stained with DAPI and an antibody against VP5. (D) Vero cells were infected with WT, gE deletion, gI deletion, or ΔUL16 viruses at a low MOI and incubated in medium containing 5 mg/ml pooled human IgG to block cell-free virus spread. At 48 hpi, the cells were fixed and stained with DAPI and an antibody specific for VP5. Representative sites of infection are shown with scale bar set to 50 μm. (E) For each virus, the average area of infection in the presence of neutralizing antibody was measured. For comparisons with the wild type (left panel), 40 sites were averaged. For comparison of ΔUL16 with itself in the presence or absence of neutralizing antibody (right panel), 30 sites were averaged. A Student's t test was used to analyze the area size reductions. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To ascertain whether UL16 is required for the Syn phenotypes, Vero cells were infected with viruses produced on the G5 cells. The ΔUL16 parent produced virus titers that were about 10-fold lower than those of the wild type (Fig. 2D), which was expected based on an earlier study showing that this viral protein is needed for efficient envelopment of cytoplasmic capsids (41). Consequently, sites of infection take longer to expand on Vero cells, and we incubated the cultures for 5 days. When the seven ΔUL16/syn viruses were examined, we were surprised to find that three of them did not produce plaques of any type at all (Fig. 5A, right column), even after incubating the cultures for 7 days (not shown). Even more striking, these viruses have syn mutations in three different genes—the specific alleles encoding gK.G167D, UL20.F222A, and UL24.E99A/K101A.
We expected the non-plaque-forming mutants to be defective for virus production in Vero cells, but this was not the case. To examine this, G5 or Vero cells were infected with the three non-plaque-forming mutants at MOI of 3, so that virus spreading was not required, and at 24 h postinfection, the numbers of infectious viruses were measured via plaque assays on G5 cells (Fig. 5B, left panel). Although the total virus titers (cell lysates plus growth media) were reduced as much as 10-fold compared to those of the ΔUL16 parent, it is clear that all three mutants replicated. We also measured the percentage of each virus population that was released into the growth medium from Vero cells (Fig. 5B, right panel). One mutant seemed to have an egress defect, but the other two behaved essentially like their ΔUL16 parent, and therefore, virion release cannot explain the absence of plaques on Vero cells. Although virus titers were measured on G5 cells, the complementing UL16 gene is under the control of its native promoter, and hence, UL16 should not be present in these cells until the virus enters and reaches the late phase of infection several hours later. To rigorously show that the three non-plaque-forming viruses can progress to the late stage of infection in the complete absence of UL16, Vero cell-produced mutants were used at low MOI on fresh Vero cells, rather than G5 cells. After 18 h, the numbers of cells producing VP5 (another late protein) were the same as for the ΔUL16 parent (Fig. 5C). While it is perplexing as to why these three mutants are unable to produce plaques in Vero cells, it is clear that UL16 is required for their ability to fuse cells (Table 2).
Related to these observations, we found no difference in the ability of the ΔUL16 parent to egress from Vero cells compared to G5 cells (Fig. 5B), even though total virus production is greatly reduced when UL16 is absent (Fig. 2D). The Vero-versus-G5 egress result is interesting because it suggests that UL16 plays no role in getting infectious virions out of cells even though it has a very important role in producing them. Thus, UL16 appears to have another, separate function in the mechanism of cell-to-cell spread. To test this, Vero cells were infected with the ΔUL16 mutant at a low MOI and then incubated in growth media containing neutralizing antibodies to prevent cell-free spread. The four gE- and gI-null viruses were included as controls because the viral proteins they lack are known to be important for cell-to-cell spread (18, 49). After incubation for 48 h, nuclei were stained with DAPI, and infected cells were identified by immunostaining with antibodies against VP5. All the mutants exhibited spreading defects relative to the wild type (Fig. 5D), and the areas of at least 40 sites of infection were measured for each (Fig. 5E, left panel). Viruses lacking gE produced sites of infection that were just 10% the size of the wild type, whereas the viruses lacking gI were reduced only 50%. This suggests a more prominent role for gE in cell-to-cell spread, at least for the KOS strain. In contrast, the UL16-null virus was found to have a much more severe spreading defect, with the average site of infection being reduced by about 2 logs, but this comparison is misleading because all the other viruses (except the ΔUL16 mutant) produced titers equal to that of the wild type (Fig. 2D). Therefore, we also compared the ΔUL16 mutant to itself in the presence or absence of neutralizing antibody, and this revealed a 10-fold reduction in the areas of infection (right panel), which matches that of gE. Thus, UL16 seems to be a critical component of the cell-to-cell spread machinery.
The remaining four ΔUL16/syn variants, which do make visible sites of infection, have their substitutions scattered across gK, UL20, and UL24, and to complete our analysis, they were examined for their abilities to cause cell fusion. Close inspection of the small sites of infection for ΔUL16/UL20.R209A and ΔUL16/UL24.G121A on Vero cells (Fig. 5B) revealed that there were no syncytia present. Taken together with the non-plaque-forming mutants, it is clear that UL16 is required for cell fusion induced by UL20syn and UL24syn. In the case of gK, those mutants with the A40V or L118Q substitutions continued to be fully fusogenic in the absence of UL16 (Fig. 6A), although their syncytia were smaller and took longer to form than their gKsyn parents, which have UL16 (Fig. 6B). Collectively, these data show that the requirement for UL16 among all the Syn variants mirrors that of gE (Table 2).
FIG 6.

The requirement for UL16 among Syn mutants matches that for gE. (A) Vero cells were infected at a low MOI with ΔUL16/gK.A40V or ΔUL16/gK.L118Q. All the other ΔUL16/syn variants produced either no plaques or fully lytic plaques. At 48 hpi, the cells were fixed and stained with DAPI or an antibody for VP5. Representative bright-field and fluorescent images were taken for each variant. (B) For each of the indicated mutants, the areas of 30 individual sites of infection were measured at either 36 hpi (gKsyn parents) or 48 hpi (mutants lacking UL16). The reduced average areas of the mutants relative to their gKsyn parent are plotted. ****, P < 0.0001, Student's t test. Additionally, each site of infection was scored as syncytial, mixed, or lytic (as defined in the text), with the linear distribution of these phenotypes shown graphically.
DISCUSSION
Although syncytia have been observed in the lesions caused by HSV-1 infections (50), the mutants responsible for cell fusion are minor variants that are not thought to be important for the transmission and biology of the disease. While it is easy to dismiss Syn mutants as being irrelevant, the reason for studying them is to seek clues to the poorly understood mechanism of cell-to-cell spread, which is critically important for all the herpesviruses and dysregulated in these variants. As an example, a study of salubrinal-induced fusion of HSV-1-infected cells has revealed that PTP1B, a host tyrosine phosphatase, is critical for cell-to-cell spread (16).
Differential requirements for accessory proteins.
As others first began to imagine long ago (51), we hypothesize that the process of cell-to-cell spread requires machinery located at cell junctions with moving parts that act in a definite but unknown sequence of molecular events. Infected cells have the potential to fuse with adjacent cells because the viral fusion proteins (gD, gH/gL, and gB) are part of the machinery at cell junctions (52), but it is tightly regulated. Of all the components that are involved in cell-to-cell spread, only four proteins can be altered to cause rampant fusion: gB, gK, UL20, and UL24.
There are two ways to turn off the machinery once it is dysregulated, and both have revealed differential effects among Syn mutants. The first method uses certain drugs that can block syncytium formation. For example, melittin blocks fusion of gKsyn but not gBsyn mutants (53), but differential responses have also been observed with cyclosporine (54), heparin-Na+ (51), and, more recently, inhibitors of PTP1B (16). Precisely why the mutants respond differently is unclear, even in the case of PTP1B inhibitors, where the critical tyrosines remain to be identified. In any case, these observations clearly indicate that Syn mutants alter the cell-to-cell-spread machinery in unique ways, which is not surprising since it is known that syn mutations fall within four different genes.
The second way to prevent fusion when the machinery is dysregulated is by removing other viral components, but those pieces depend upon which syn mutation is present. This was discovered first for UL21 (33), which is required for cell-to-cell spread but is only needed for gBsyn (Table 2). Also, removal of the membrane-proximal basic residues in the cytoplasmic tail of gD (Fig. 1A) will block the gBsyn phenotype, but not that of gKsyn. Here, we have shown that gE, gI, and UL16, which are all needed for cell-to-cell spread and reside in the same interaction network with UL21, are differentially required among the Syn mutants but in ways that were difficult to predict. Collectively, these findings suggest that certain regulatory proteins are bypassed or disengaged from the cell-to-cell-spread machinery by some syn mutations but not others. For example, the disrupting substitutions of UL20syn variants lock the fusion machinery in the “on” position, with removal of gI or UL21 having no effect, but removal of gE or UL16 shuts off the fusion activity (Table 2).
At this point, it is difficult to speculate on precise molecular details that would explain our observations because so little is understood about the cell-to-cell-spread machinery, and the differential protein requirements among Syn mutants have only just begun to emerge. In broad strokes, we note that the only Syn mutants that continue to exhibit cell fusion in the absence of gE, gI, UL16, or UL21 (Table 2) are those with changes on the external side of the plasma membrane, namely gK.A40V and gK.L118Q (Fig. 1A). These are also the two mutants that produce the largest syncytia (Fig. 1C). All the other Syn mutants, including the other gKsyn derivative (G167D), have changes on the cytoplasmic side. The N-terminal domain of gK directly binds to the ectodomain of gB and can modulate its fusion activity (13, 55, 56). Moreover, deletions in this domain (e.g., gKΔ31–68) abolish cell fusion in the context of a gBsyn mutant, but syncytium formation can be restored by trans-expression of the first 82 residues of gK with the gKΔ31-68/gBsyn mutant (55). Hence, it would be interesting to know whether trans-expression of the A40V derivative of the same peptide would induce cell fusion in the absence of the gBsyn alteration. In any case, it is clear that mutants like gK.A40V do not completely disassemble the cell-to-cell-spread machinery to allow unregulated fusion by gD-gH/gL-gB, because the gKsyn phenotype is still dependent upon gM and UL11 (32).
The only Syn mutant known to have a substitution in the cytoplasmic portions of gK is G167D (Fig. 1A). It is possible that this change works indirectly to alter the external, N-terminal domain of gK to dysregulate gB activity, but it may induce fusion by another mechanism. This substitution resides in the first of the two cytoplasmic loops (57), and the other loop interacts with UL20 (58), which is important for virion envelopment and transport of the proteins to the cell surface (58). UL20 also interacts with gB (13) and can exert its own influence on the fusion machinery, as shown by UL20syn mutants (59). Thus, it is possible that G167D acts indirectly through wild-type UL20 to subtly alter the UL20-gB interaction and induce cell fusion. Consistent with this, G167D has the same accessory protein requirements as the UL20syn variants (Table 2).
Differing roles for gE and gI.
The heterodimeric relationship of gE and gI is well known, and it has long been thought that these two glycoproteins function as a unit. For instance, gE has a weak IgG binding activity, but together with gI, the complex becomes a much more robust Fc receptor (48). Also, studies of gE- and gI-null viruses have shown that each replicates normally on Vero cells yet forms small plaques, while in vivo studies demonstrate that both viruses exhibit severe spreading defects (18, 49, 60), all of which was thought to be due to gE and gI working together for cell-to-cell spread (45, 46). Thus, we were quite surprised to find that the requirements for gE and gI differed among the Syn mutants (Table 2), suggesting that these proteins influence the cell-to-cell spread machinery in different ways.
Prior to this study, gB.A855V was the only Syn mutant that had been examined with regard to requirements for gE and gI, and consistent with the view that these glycoproteins work as a unit, both were found to be needed for cell fusion (28, 49). Thus, we were skeptical when our experiments showed gI to be dispensable for this Syn mutant, but the result was confirmed with an independently constructed virus. We can imagine two possible explanations for why our result is at odds with the literature. First, because the original mutant was made prior to BAC recombineering methods and required several rounds of plaque purifications, it is possible that other, unintended, mutations were present to block the Syn phenotype. The other, more likely, explanation is that our mutants were constructed in the KOS strain, while the original mutant was constructed in SC16 (28). Consistent with this, gI has been shown to contain a polymorphic, 7-residue, tandem repeat region in its ectodomain (61), with the number of repeats ranging from 2 to 8. These contain similar blocks of serines and threonines (most commonly STPSTTT), which can be utilized for O-glycosylation and hence constitute a mucin domain (62). In the KOS strain, gI contains two repeats, whereas SC16 contains 3. Differing amounts of O-linked glycans might impact the structure of gI and perhaps influence its relationship with gE. The polymorphic nature of gI in different strains and its potential influence on the Syn phenotype merit further investigation.
Although dispensable for gBsyn, gKsyn, and UL20syn, our experiments clearly show that gI is needed for the UL24syn phenotype (Table 2). Moreover, the amount of gI present in infected cells was reduced for UL24syn mutants, and this was most striking in the case of substitutions E99A/K101A (Fig. 1B and Fig. 3A). A reduction in gI expression was also observed for ΔUL16/UL24.E99A/K101A, which was made from a different parent virus (Table 1, data not shown). These observations suggest the possibility that UL24 interacts with and stabilizes gI. This hypothetical interaction could serve in part to regulate the virus fusion machinery indirectly via gI, but, it is possible that UL24 has other interactions with the cell-to-cell-spread machinery, as suggested by studies of a null mutant (63), but this has not yet been examined for point mutants such as the E99A/K101A mutant.
After finding evidence for a potential interaction with gI, it caught our attention that UL24 has recently been shown to antagonize the cGAS-mediated DNA-sensing signaling mechanism to promote virus replication (64). In mice, the E99A/K101A mutant is much less pathogenic than the wild type (65), but why this is the case is not obvious, because this mutant protein alone can still block cGAS signaling (64). The experiments described here suggest a potential explanation, namely that UL24 might normally be moved to adjacent, uninfected cells via the cell-to-cell spread mechanism. Relevant to this hypothesis, UL24 is expressed late during the infection (66) and has not been reported to be a component of the virion (67); hence, it is not delivered via cell-free virus spread. Perhaps UL24 produced during the virion production phase is transferred directly through lateral junctions, perhaps via exosomes, to immediately enhance the infection of neighboring cells.
The role of UL16 in cell-to-cell spread.
UL16 resides on cytoplasmic capsids and participates in a bridging interaction with membrane-bound UL11 (41, 68). Thus, in the absence of UL16, there is a capsid-wrapping defect, which results in virus titers that are 10% of wild type (41). UL16 also binds to the tail of gE (30, 69), but envelopment does not require this interaction because in the absence of gE, wild-type virus titers are produced. As shown here, UL16 is not needed for postenvelopment egress because the percentage of virions released into the culture medium by the ΔUL16 mutant is the same whether this protein is present (G5 cells) or not (Vero cells). However, in the absence of UL16, cell-to-cell spreading is blocked to the same degree as when gE is absent (90%), and the pattern of Syn mutants that require UL16 for cell fusion matches that for gE (Table 2). Thus, it seems likely that UL16 provides its role in cell-to-cell spread as part of the complex on the cytoplasmic tail of gE (30).
One attractive hypothesis is that gE and UL16, along with their various binding partners (Fig. 1A), participate in the assembly and function of modified cell junctions that promote cell-to-cell spread, but another possibility is suggested by the inability of three Syn mutants (one each in gK, UL20, and UL24) to fuse cells, produce plaques, or cause any sort of cytopathic effect when UL16 is absent. These viruses were found to be capable of making virions that can infect fresh monolayers in a cell-free manner, but in plaque assays, the cells looked healthy even after 7 days. Although the explanation for this unexpected phenotype remains to be elucidated, there are two properties of UL16 that raise the possibility that the innate cellular immune response might be enhanced when this viral protein is absent. First, UL16 has been shown to traffic to mitochondria during infection (70), and this is a known site for innate immune signaling (71). Second, in the absence of tegument protein UL37, packaging of UL16 into virions is reduced, suggesting a direct or indirect interaction between these two proteins. This is relevant because UL37 has been shown to be a viral deamidase that targets RIG-I and cGAS during HSV-1 infection, blunting the innate immune response of the cell (72, 73). Thus, it is possible that UL16 and its network of interacting partners (including gK, UL20, and UL24) are structural elements that are needed for defeating cellular defense mechanisms. In this scenario, the three non-plaque-forming mutants infect cells and make progeny viruses, but the infection does not spread because the host defenses are robust. Vero cells do not have a functional interferon pathway (74), and thus, this presumably does not contribute to the spreading defect. However, it has been reported that HSV-1-infected cells produce exosomes that move in a cell-free manner to reduce virus spreading (75, 76). Perhaps cells infected with the non-plaque-forming viruses produce greater numbers or more potent versions of these. In any case, it is intriguing that the UL16 interaction network has putative connections with viral proteins UL24 and UL37, both of which are involved in suppressing innate immune responses.
MATERIALS AND METHODS
Cells and antibodies.
Vero cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 5% fetal calf serum (FCS; HyClone), 5% fetal bovine serum (FBS; HyClone), and 131 μg/ml penicillin-streptomycin (pen/strep; Gibco). G5 cells (42) were grown in DMEM with 5% FCS, 5% FBS, and 1 mg/ml G418 (Gibco). Infected cells were maintained in DMEM supplemented with 2% FBS and pen/strep.
A polyclonal rabbit antibody against VP5 was provided by Richard Courtney (Penn State College of Medicine) and was used at dilutions of 1:1,000 for immunofluorescence and 1:8,000 for immunoblotting. The UL16 antibody was raised in rabbits against GST-UL16 and was used at a dilution of 1:2,000. Rabbit antibodies to gE (UP1725) and gI (UP1928) were a gift from Harvey Friedman (University of Pennsylvania) and were used at dilutions of 1:6,000 and 1:1,000, respectively (77). A horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (eBioscience) was used at 1:10,000.
Viruses and BAC recombineering.
All viruses were constructed in the KOS laboratory strain of HSV-1 contained within a BAC, which was a gift from David Leib (Dartmouth) (78). The two-step BAC recombineering protocol used to generate mutant viruses has been described elsewhere (79, 80). The ΔUL16 virus and the 8 Syn variants were previously constructed and characterized (30, 33, 41). All mutant viruses constructed in this study are listed in Table 1. Each virus was subjected to HindIII digestion to verify there were no obvious genome rearrangements, and all engineered mutations were sequence verified.
Generation of virus stocks.
BAC plasmids containing the genome of HSV-1 were purified from E. coli and transfected into Vero cells with Lipofectamine 2000 (80). At 4 to 5 days posttransfection, when cytopathic effects were evident, the transfected cells and media were harvested and classified as transfection stock. Transfection stock was utilized to infect fresh Vero cells, which were harvested 2 to 3 days postinfection. The cells and media went through three freeze-thaw rounds and were sonicated to create a low-passage-number (P1) viral stock. For ΔUL16 viruses, the transfection and infection stocks were made using G5 complementing cells to minimize the selection of second-site suppressor mutations in the absence of UL16. Plaque assays on Vero cells revealed only tiny plaques with the ΔUL16 viruses, indicating that the recombination frequency in G5 cells (to reinsert the UL16 gene) was too low to be measured in our experiments.
Immunoblotting.
Vero cells were infected at an MOI of 3 with each of the constructed viruses. At 18 to 20 h postinfection, cells were rinsed once with phosphate-buffered saline (PBS) and then harvested in ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl at pH 8, 150 mM NaCl, 1% NP-40, 0.1% SDS, and 0.5% sodium deoxycholate) containing a protease inhibitor cocktail (P8340; Sigma). Nuclei and cellular debris were pelleted out, and 4× sample buffer was added to the cellular lysates. The samples were boiled for 5 min prior to 10% SDS-PAGE, and the separated proteins were transferred to a nitrocellulose membrane, where they were detected with antibodies for VP5, gE, gI, and UL16 in conjunction with the SuperSignal West Pico Plus system (Thermo).
Immunostaining of infected cells.
Vero cells were seeded onto glass coverslips and infected with viruses at an MOI of 0.001 at 37°C. After 1 h, the cells were rinsed and incubated in DMEM containing 2% FBS at 37°C. At the indicated times postinfection, the cells were fixed for 10 min in 4% paraformaldehyde (PFA), rinsed with PBS, permeabilized for 10 min with 0.1% Triton X-100 containing 2% bovine serum albumin (BSA), and blocked for 1 h in PBS containing 2% BSA. Cells were incubated for 1 h with polyclonal rabbit antibody specific for VP5 (1:1,000 dilution) in a humid chamber, rinsed 3× with PBS, and then incubated for 1 h with an Alex 568-conjugated secondary antibody (Life Technologies) at a 1:1,000 dilution. After 3 more PBS rinses, nuclei were stained for 5 min with DAPI (Molecular Probes), and the coverslips were mounted onto slides. Bright-field phase-contrast and fluorescent images were captured with an Olympus IX73 inverted microscope.
For the cell-to-cell spreading assay, Vero cells were infected with the designated viruses at an MOI of 0.001 at 37°C. After 1 h, the cells were rinsed and incubated in DMEM containing 2% FBS and 5 mg/ml pooled human IgG (Equitech Bio). The concentration of IgG used for this assay was previously determined to neutralize all cell-free virus produced during infection (16). After 48 h, the cells were fixed in and stained with VP5 antibodies and DAPI. The plaque areas of VP5-positive cells were measured using the CellSens software.
Fusion assay.
To measure and score syncytia, Vero cells were infected with the viruses at an MOI of 0.001 at 37°C. After 1 h, the cells were rinsed with DMEM and overlaid with a mixture of 0.5% agarose and DMEM. The agarose overlay was composed of a 1:1 ratio of 1% low-melting-temperature agarose (SeaPlaque) and 2× infection medium supplemented with 4% FBS. Cells were incubated at 37°C for 36 h (the Syn variants and gI deletion/syn viruses), 42 h (gE deletion/gKsyn viruses), or 48 h (ΔUL16/gKsyn viruses). At the indicated times postinfection, at least 30 syncytia per virus were imaged using bright-field phase-contrast microscopy with the Olympus IX73 inverted microscope. The area of each syncytium was subsequently measured using the Olympus CellSens software. Each individual syncytium was also assigned a phenotypic score. A score of “syncytial” indicated that the entire area of infection consisted of multinucleated cell. A score of “mixed” meant that the area of infection contained a syncytium with at least 5 nuclei directly juxtaposed to lytically infected cells. Finally, a score of “lytic” meant that no fused cells were apparent.
Virus replication assays.
Six-well plates of Vero cells or G5 cells were infected with the specified viruses at an MOI of 5 at 37°C. After 1 h, the cells were briefly rinsed with a citric acid buffer (135 mM NaCl, 10 mM KCl, 40 mM citric acid at pH 3.0) to inactivate viruses remaining on the cell surface, rinsed one time in DMEM, and then incubated in 1 ml of DMEM plus 2% FBS. For some experiments, the infected cells and media were harvested together at 12 and 24 h postinfection. These samples were subjected to 3 freeze-thaw rounds prior to the titer being determined via plaque assays on Vero cells. In other experiments, only the cell media were collected to measure virus release. For the viruses lacking UL16, titers were measured on G5 cells, except where noted.
Statistical analysis.
A two-tailed Student's t test was utilized to determine statistical significance by using GraphPad Prism (version 8). The indicated significance values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
ACKNOWLEDGMENTS
This study was supported by an NIH grant (AI071286) to J.W.W., which provided support for J.C.C. The NIH was not involved in study design, data collection, interpretation, or the decision to submit this work for publication. The authors declare no conflicts of interest.
We thank Carol Wilson and other lab members for technical support. We also thank Rebecca Craven (Penn State College of Medicine) for help with experimental troubleshooting.
Because this is the final publication from the Wills laboratory, J.W.W. thanks all the scientists who have made his pursuit of virology so much fun for so many years, but especially Richard J. Courtney (deceased), who made it easy to make the transition into herpesvirology during the middle of my career.
REFERENCES
- 1.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 3.Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol 72:7064–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, Peng T, Nicola AV, Montgomery RI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ. 1997. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J Virol 71:6083–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. doi: 10.1016/S0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 6.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subramanian RP, Geraghty RJ. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci U S A 104:2903–2908. doi: 10.1073/pnas.0608374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper RS, Heldwein EE. 2015. Herpesvirus gB: a finely tuned fusion machine. Viruses 7:6552–6569. doi: 10.3390/v7122957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 31:4204–4220. doi: 10.1038/emboj.2012.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albecka A, Laine RF, Janssen AF, Kaminski CF, Crump CM. 2016. HSV-1 glycoproteins are delivered to virus assembly sites through dynamin-dependent endocytosis. Traffic 17:21–39. doi: 10.1111/tra.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau SY, Crump CM. 2015. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 7:915–938. doi: 10.3390/v7030915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J Virol 84:8596–8606. doi: 10.1128/JVI.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avitabile E, Lombardi G, Campadelli-Fiume G. 2003. Herpes simplex virus glycoprotein K, but not its syncytial allele, inhibits cell-cell fusion mediated by the four fusogenic glycoproteins, gD, gB, gH, and gL. J Virol 77:6836–6844. doi: 10.1128/JVI.77.12.6836-6844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster TP, Melancon JM, Baines JD, Kousoulas KG. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J Virol 78:5347–5357. doi: 10.1128/JVI.78.10.5347-5357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmichael JC, Yokota H, Craven RC, Schmitt A, Wills JW. 2018. The HSV-1 mechanisms of cell-to-cell spread and fusion are critically dependent on host PTP1B. PLoS Pathog 14:e1007054. doi: 10.1371/journal.ppat.1007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson DC, Huber MT. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J Virol 76:1–8. doi: 10.1128/JVI.76.1.1-8.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dingwell KS, Brunetti CR, Hendricks RL, Tang Q, Tang M, Rainbow AJ, Johnson DC. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J Virol 68:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol 75:821–833. doi: 10.1128/JVI.75.2.821-833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707. doi: 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dingwell KS, Johnson DC. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J Virol 72:8933–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dingwell KS, Doering LC, Johnson DC. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J Virol 69:7087–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang F, Zumbrun EE, Huang J, Si H, Makaroun L, Friedman HM. 2010. Herpes simplex virus type 2 glycoprotein E is required for efficient virus spread from epithelial cells to neurons and for targeting viral proteins from the neuron cell body into axons. Virology 405:269–279. doi: 10.1016/j.virol.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler C. 1960. Herpes simplex virus. Characteristics of a strain which produces unusually large multinucleated giant cells in tissue culture. Arch Dermatol 82:391–399. doi: 10.1001/archderm.1960.01580030085011. [DOI] [PubMed] [Google Scholar]
- 26.Ambrosini A, Enquist L. 2015. Cell-fusion events induced by alpha-herpesviruses. Future Virol 10:185–200. doi: 10.2217/fvl.14.100. [DOI] [Google Scholar]
- 27.Bryant KF, Macari ER, Malik N, Boyce M, Yuan J, Coen DM. 2008. ICP34.5-dependent and -independent activities of salubrinal in herpes simplex virus-1 infected cells. Virology 379:197–204. doi: 10.1016/j.virol.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis-Poynter N, Bell S, Minson T, Browne H. 1994. Analysis of the contributions of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J Virol 68:7586–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haanes EJ, Nelson CM, Soule CL, Goodman JL. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J Virol 68:5825–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc Natl Acad Sci U S A 109:19798–19803. doi: 10.1073/pnas.1212900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatterjee S, Koga J, Whitley RJ. 1989. A role for herpes simplex virus type 1 glycoprotein E in induction of cell fusion. J Gen Virol 70:2157–2162. doi: 10.1099/0022-1317-70-8-2157. [DOI] [PubMed] [Google Scholar]
- 32.Kim IJ, Chouljenko VN, Walker JD, Kousoulas KG. 2013. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J Virol 87:8029–8037. doi: 10.1128/JVI.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarfo A, Starkey J, Mellinger E, Zhang D, Chadha P, Carmichael J, Wills JW. 2017. The UL21 tegument protein of herpes simplex virus 1 is differentially required for the syncytial phenotype. J Virol 91:e01161-17. doi: 10.1128/JVI.01161-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han J, Chadha P, Meckes DG, Baird NL, Wills JW. 2011. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein E of herpes simplex virus. J Virol 85:9437–9446. doi: 10.1128/JVI.05207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harper AL, Meckes DG, Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol 84:2963–2971. doi: 10.1128/JVI.02015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chadha P, Han J, Starkey JL, Wills JW. 2012. Regulated interaction of tegument proteins UL16 and UL11 from herpes simplex virus. J Virol 86:11886–11898. doi: 10.1128/JVI.01879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmichael JC, Starkey J, Zhang D, Sarfo A, Chadha P, Wills JW, Han J. 2019. Glycoprotein D of HSV-1 is dependent on tegument protein UL16 for packaging and contains a motif that is differentially required for syncytia formation. Virology 527:64–76. doi: 10.1016/j.virol.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson DC, Ligas MW. 1988. Herpes simplex viruses lacking glycoprotein D are unable to inhibit virus penetration: quantitative evidence for virus-specific cell surface receptors. J Virol 62:4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basu S, Dubin G, Basu M, Nguyen V, Friedman HM. 1995. Characterization of regions of herpes simplex virus type 1 glycoprotein E involved in binding the Fc domain of monomeric IgG and in forming a complex with glycoprotein I. J Immunol 154:260–267. [PubMed] [Google Scholar]
- 40.Wisner T, Brunetti C, Dingwell K, Johnson DC. 2000. The extracellular domain of herpes simplex virus gE is sufficient for accumulation at cell junctions but not for cell-to-cell spread. J Virol 74:2278–2287. doi: 10.1128/JVI.74.5.2278-2287.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. 2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88:110–119. doi: 10.1128/JVI.02555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai P, DeLuca NA, Glorioso JC, Person S. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J Virol 67:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Negatsch A, Mettenleiter TC, Fuchs W. 2011. Herpes simplex virus type 1 strain KOS carries a defective US9 and a mutated US8A gene. J Gen Virol 92:167–172. doi: 10.1099/vir.0.026484-0. [DOI] [PubMed] [Google Scholar]
- 44.Polcicova K, Goldsmith K, Rainish BL, Wisner TW, Johnson DC. 2005. The extracellular domain of herpes simplex virus gE is indispensable for efficient cell-to-cell spread: evidence for gE/gI receptors. J Virol 79:11990–12001. doi: 10.1128/JVI.79.18.11990-12001.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farnsworth A, Johnson DC. 2006. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J Virol 80:3167–3179. doi: 10.1128/JVI.80.7.3167-3179.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins WJ, Johnson DC. 2003. Herpes simplex virus gE/gI expressed in epithelial cells interferes with cell-to-cell spread. J Virol 77:2686–2695. doi: 10.1128/JVI.77.4.2686-2695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ndjamen B, Farley AH, Lee T, Fraser SE, Bjorkman PJ. 2014. The herpes virus Fc receptor gE-gI mediates antibody bipolar bridging to clear viral antigens from the cell surface. PLoS Pathog 10:e1003961. doi: 10.1371/journal.ppat.1003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson DC, Frame MC, Ligas MW, Cross AM, Stow ND. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J Virol 62:1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balan P, Davis-Poynter N, Bell S, Atkinson H, Browne H, Minson T. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J Gen Virol 75:1245–1258. doi: 10.1099/0022-1317-75-6-1245. [DOI] [PubMed] [Google Scholar]
- 50.Blank H, Burgoon CF, Baldridge GD, McCarthy PL, Urbach F. 1951. Cytologic smears in diagnosis of herpes simplex, herpes zoster, and varicella. JAMA 146:1410–1412. doi: 10.1001/jama.1951.63670150005012b. [DOI] [PubMed] [Google Scholar]
- 51.Seck T, Lingen M, Weise K, Falke D. 1994. Evidence for a multistep mechanism for cell-cell fusion by herpes simplex virus with mutations in the syn 3 locus using heparin derivatives during fusion from within. Arch Virol 136:173–181. doi: 10.1007/BF01538826. [DOI] [PubMed] [Google Scholar]
- 52.Krummenacher C, Baribaud I, Eisenberg RJ, Cohen GH. 2003. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J Virol 77:8985–8999. doi: 10.1128/JVI.77.16.8985-8999.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baghian A, Kousoulas KG. 1993. Role of the Na+, K+ pump in herpes simplex type 1-induced cell fusion: melittin causes specific reversion of syncytial mutants with the syn1 mutation to Syn+ (wild-type) phenotype. Virology 196:548–556. doi: 10.1006/viro.1993.1510. [DOI] [PubMed] [Google Scholar]
- 54.Walev I, Weise K, Falke D. 1991. Differentiation of herpes simplex virus-induced fusion from without and fusion from within by cyclosporin A and compound 48/80. J Gen Virol 72:1377–1382. doi: 10.1099/0022-1317-72-6-1377. [DOI] [PubMed] [Google Scholar]
- 55.Chouljenko VN, Iyer AV, Chowdhury S, Chouljenko DV, Kousoulas KG. 2009. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J Virol 83:12301–12313. doi: 10.1128/JVI.01329-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Musarrat F, Jambunathan N, Rider PJF, Chouljenko VN, Kousoulas KG. 2018. The amino terminus of herpes simplex virus 1 glycoprotein K (gK) is required for gB binding to Akt, release of intracellular calcium, and fusion of the viral envelope with plasma membranes. J Virol 92:e01842-17. doi: 10.1128/JVI.01842-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foster TP, Alvarez X, Kousoulas KG. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J Virol 77:499–510. doi: 10.1128/JVI.77.1.499-510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J Virol 82:6310–6323. doi: 10.1128/JVI.00147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Charles AS, Chouljenko VN, Jambunathan N, Subramanian R, Mottram P, Kousoulas KG. 2014. Phenylalanine residues at the carboxyl terminus of the herpes simplex virus 1 UL20 membrane protein regulate cytoplasmic virion envelopment and infectious virus production. J Virol 88:7618–7627. doi: 10.1128/JVI.00657-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. 2009. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J Virol 83:8315–8326. doi: 10.1128/JVI.00633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Norberg P, Bergström T, Rekabdar E, Lindh M, Liljeqvist JA. 2004. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J Virol 78:10755–10764. doi: 10.1128/JVI.78.19.10755-10764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Norberg P, Olofsson S, Tarp MA, Clausen H, Bergström T, Liljeqvist JA. 2007. Glycoprotein I of herpes simplex virus type 1 contains a unique polymorphic tandem-repeated mucin region. J Gen Virol 88:1683–1688. doi: 10.1099/vir.0.82500-0. [DOI] [PubMed] [Google Scholar]
- 63.Ben Abdeljelil N, Rochette PA, Pearson A. 2013. The UL24 protein of herpes simplex virus 1 affects the sub-cellular distribution of viral glycoproteins involved in fusion. Virology 444:263–273. doi: 10.1016/j.virol.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 64.Xu H, Su C, Pearson A, Mody CH, Zheng C. 2017. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J Virol 91:e00025-17. doi: 10.1128/JVI.00025-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leiva-Torres GA, Rochette PA, Pearson A. 2010. Differential importance of highly conserved residues in UL24 for herpes simplex virus 1 replication in vivo and reactivation. J Gen Virol 91:1109–1116. doi: 10.1099/vir.0.017921-0. [DOI] [PubMed] [Google Scholar]
- 66.Pearson A, Coen DM. 2002. Identification, localization, and regulation of expression of the UL24 protein of herpes simplex virus type 1. J Virol 76:10821–10828. doi: 10.1128/JVI.76.21.10821-10828.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loret S, Guay G, Lippé R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J Virol 77:11417–11424. doi: 10.1128/JVI.77.21.11417-11424.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeh PC, Han J, Chadha P, Meckes DG, Ward MD, Semmes OJ, Wills JW. 2011. Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J Virol 85:9425–9436. doi: 10.1128/JVI.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chadha P, Sarfo A, Zhang D, Abraham T, Carmichael J, Han J, Wills JW. 2017. Domain interaction studies of herpes simplex virus 1 tegument protein UL16 reveal its interaction with mitochondria. J Virol 91:e01995-16. doi: 10.1128/JVI.01995-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.West AP, Shadel GS, Ghosh S. 2011. Mitochondria in innate immune responses. Nat Rev Immunol 11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao J, Zeng Y, Xu S, Chen J, Shen G, Yu C, Knipe D, Yuan W, Peng J, Xu W, Zhang C, Xia Z, Feng P. 2016. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 20:770–784. doi: 10.1016/j.chom.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, Xie L, Xie N, Liu T, Lee K, Seo GJ, Chen L, Stabell AC, Xia Z, Sawyer SL, Jung J, Huang C, Feng P. 2018. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24:234–248. doi: 10.1016/j.chom.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol 43:247–252. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- 75.Deschamps T, Kalamvoki M. 2018. Extracellular vesicles released by herpes simplex virus 1-infected cells block virus replication in recipient cells in a STING-dependent manner. J Virol 92:e01102-18. doi: 10.1128/JVI.01102-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kalamvoki M, Du T, Roizman B. 2014. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc Natl Acad Sci U S A 111:E4991–E4996. doi: 10.1073/pnas.1419338111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin X, Lubinski JM, Friedman HM. 2004. Immunization strategies to block the herpes simplex virus type 1 immunoglobulin G Fc receptor. J Virol 78:2562–2571. doi: 10.1128/JVI.78.5.2562-2571.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 79.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baird NL, Starkey JL, Hughes DJ, Wills JW. 2010. Myristylation and palmitylation of HSV-1 UL11 are not essential for its function. Virology 397:80–88. doi: 10.1016/j.virol.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]