

Abstract
Macrophages play an important role in the inflammatory process that contributes to the development of abdominal aortic aneurysm (AAA). Studies of human and mouse AAA tissue reveal expanded populations of macrophages producing an abundance of pro-inflammatory cytokines, including TNF-α, IL-12p40 and high level of metalloprotease 9 (MMP-9) at the late stages of disease. Herein, we show that blockade of IL-12p40 in the early phase of aneurysm development suppresses macrophage expansion, inflammatory cytokine and MMP-9 production and mitigates AAA development. Since IL-12 and IL-23 are related cytokines that share the common p40 subunit, we also evaluate the effect of direct IL-23 blockade on the development of AAA. Specific IL-23p19 blockade prevents AAA progression with the same efficiency as IL-12p40 antagonism, suggesting that the efficacy of anti-IL-12p40 treatment may reflect IL-23 blockade. IL-12p40 and IL-23p19 are also abundantly expressed in human AAA tissue. Our findings have potential translational value since IL-12p40 and IL-23p19 antagonists already exist as FDA-approved therapeutics for various chronic inflammatory conditions.
Subject terms: Mouse, Aneurysm
Introduction
Abdominal aortic aneurysm (AAA) is a common and progressive vascular condition in elderly males that is associated with high risk of mortality due to rupture if left untreated. Although surgical repair is recommended for large aneurysms, medical therapy for small aneurysms is still lacking due to our incomplete understanding of the molecular mechanisms underlying the formation and progression of small AAA1, highlighting the need to further explore and validate candidate therapeutic targets.
Human AAA tissue and animal models suggest that AAA is an inflammatory disease characterized by transmural infiltration of the aortic wall with every type of leukocytes2,3. There is accumulating evidence that macrophages are an important source of matrix-degrading proteases that help to degrade the extracellular matrix (ECM)4,5. The role of metalloproteinases (MMPs) in preclinical models of AAA is well established5,6. And MMPs are expressed in increased amounts in human AAA7,8. More recent studies suggest that genetic polymorphisms of MMPs are associated with vascular dilation or aneurysmal disease9–11. Whether these proteases are directly responsible for the weakening and dilatation of the aortic wall remains a matter of debate12. Moreover, pharmacologic inhibition of MMP activity has not consistently delayed the progression of AAA in clinical trials13.
Macrophages also release a wide range of pro-inflammatory cytokines that may be involved in accelerating the growth of AAA14. IL-12 and IL-23 are related cytokines that share the same p40 subunit; however, the p19 subunit is specific to IL-2315. IL-12 and IL-23 direct the differentiation and expansion of Th1 and Th17 cells, respectively16. Animal studies show that mice deficient in the expression of the common p40 subunit of IL-12 and IL-23 are protected from developing many inflammatory and immune-mediated conditions17–19. IL-12/IL-23 have been indirectly implicated in AAA20–22. Sharma et al. found that genetic disruption of IL-23 attenuated elastase-induced AAA, likely through the suppression of IL-17 expression23. Thus, pharmacological modulation of IL-12/IL-23 may represent an attractive treatment strategy to interrupt the progression of AAA.
Results
Elastase-induced AAA model
We used the well-established elastase-induced AAA model in which transient porcine elastase perfusion (for 5 min) of the infrarenal abdominal aorta on day 0 leads to aneurysmal dilatation at day 1424. AAA is defined as an increase in the aortic diameter (AD) of more than 100% over the pre-elastase perfusion measurements24. Elastase perfusion led to an immediate increase in AD of ~70% (Fig. 1A)25. WT C57BL/6 mice uniformly developed AAA (mean increase in AD of 148% or 0.74 mm) (Fig. 1A,B). AAA development also led to the characteristic elastic fiber fragmentation (Fig. 1C).
Figure 1.

Transient elastase perfusion induces AAA. Mice were transiently perfused with elastase on day 0 and increase in aortic diameter (AD) was measured immediately post-perfusion, and on day 7 or day 14. Elastase perfusion resulted in ∼70% increase in AD on day 0 (post perfusion). AAA was defined as an increase in AD of greater than 100% on day 14 compared with the AD measured prior to elastase perfusion. AAA is expressed as the increase in AD in mm (A) or % (B). AAA was accompanied by elastic fiber degradation on day 14 (C). Scale bar = 250 um.
Elastase-induced AAA leads to macrophage expansion
To examine aortic wall macrophage accumulation during AAA progression, we performed a temporal analysis of macrophage number at different stages of disease post-elastase perfusion (days 3, 7, and 14). We found that macrophages (MOMA-2+ cells) began to accumulate as early as day 3-post elastase perfusion; the number of MOMA-2+ cells in the aortic wall continued to increase from day 3 to day 7 and remained elevated until day 14 (Fig. 2A). In parallel with the temporal expansion in MOMA-2+ cells, we found that the number of cells expressing inflammatory cytokines TNF-α and IL-12p40 peaked at day 7 and remained elevated at day 14 (Fig. 2B,C) while the number of cells expressing MMP-9 rose steadily, peaking on day 14 (Fig. 2D).
Figure 2.

Macrophage expansion during AAA development. Mice were transiently perfused with elastase on day 0 and their aortas were harvested on days 3, 7, and 14. Aortic sections were stained for macrophages (MOMA-2) (A), TNF-α (B), IL12p40) (C), and MMP-9 (D). Scale bar 100 um. Values represent mean ± SEM, n = 6–8 aortas per time point. Values represent mean ± SEM, n = 6–8 aortas time point. ***p < 0.001.
IL-12 drives the inflammatory responses in elastase-induced AAA
IL-12 is mainly known as a pro-inflammatory cytokine produced by activated antigen-presenting cells that induces Th1 differentiation, promotes IFN-γ production and acts as a link between innate and adaptive immunity17. Examination of AAA aortic wall tissue post-elastase perfusion, however, revealed that the majority of IL-12p40 secreting cells were from infiltrating MOMA-2+ cells (~70%) (Fig. S1), suggesting macrophages as the main source of IL-12p40. A recent study shows that IL-12 can “reprogram” macrophage phenotype and activity26, suggesting that modulation of the IL-12 axis may be beneficial to explore as a treatment strategy to interrupt the progression of AAA. To further understand the role of IL-12 in the progression of elastase-induced AAA, we administered a neutralizing monoclonal antibody (Ab) against the p40 subunit of IL-12 on day 3 and day 8-post elastase perfusion. We found that antagonism of IL-12p40 in the early stages of aneurysm development significantly attenuated AAA (increase in AD = 100% or 0.51 mm, n = 8, SD = 21% or 0.11 mm in the IL-12p40 antagonist treatment group versus AD = 155% or 0.750 mm, n = 7, SD = 21% or 0.10 mm in animals administered IgG isotype control, p < 0.001) (Fig. 3A,B). IL-12p40 antagonism significantly suppressed macrophage expansion (Fig. S2), and dampened the production of several macrophage-associated inflammatory mediators, including IL-12p40, IL-6, TNFα, and MMP-9 (Fig. S2) (p < 0.001 compared with isotype control). Moreover, we observed a marked reduction in the number of IL-17A+ cells in aortic wall tissue with IL-12p40 blockade (p < 0.001) (Fig. S2).
Figure 3.

Antagonism of IL-12p40/IL-23p19 attenuates AAA development. Mice were perfused with elastase on day 0 and injected with anti-IL-12p40 or anti-IL-23p19 or isotype control (250ug) on days 3 and 8. AD was assessed on day 14. AAA is expressed as the % increase in AD (A,C) or the change in AD in mm (B,D). Values represent mean ± SEM, n = 7–8 mice per treatment. **p < 0.01, ***p < 0.001.
IL-23 blockade mitigates elastase-induced AAA
IL-17A is an inflammatory cytokine produced by T helper cells in response to IL-23 signaling16. A previous study suggesting that IL-23 deficiency significantly attenuates elastase-induced AAA, perhaps through the modulation of IL-17 producing T helper cells (Th17)23, prompted us to evaluate the effect of direct IL-23 blockade on the development of AAA. Using a blocking Ab specific for the p19 subunit of IL-23, we demonstrated that anti-IL-23p19 treatment significantly dampened the development of AAA (increase in AD = 94% or 0.48 mm, n = 8, SD = 14% or 0.07 mm in the IL-23p19 antagonist treatment group versus AD = 151% or 0.74 mm, n = 10, SD = 16% or 0.08 mm in the isotype control group, p < 0.001) (Fig. 3B,C). Anti-IL-23p19 treatment prevented the onset and expansion of AAA with the same efficiency as IL-12p40 antagonism, suggesting that the efficacy of anti-IL-12p40 treatment may reflect IL-23 inhibition.
Macrophages have distinct phenotypes during AAA development with classically activated macrophages (M1) expressing pro-inflammatory cytokines while alternatively activated macrophages (M2) are involved in extracellular matrix remodeling and repair27. On day 14 post-elastase perfusion we found that aortic wall macrophages expressed both M1 (IL12p40) and M2 (CD206) markers but M2 macrophages were more predominant (Fig. 4). IL-23 blockade inhibited the expansion of macrophages and profoundly reduced the expression of macrophage-associated inflammatory mediators (Fig. S3). Likewise, IL-23 blockade mitigated T cell accumulation (Fig. S4) and IL-17A expression (Fig. S3). Double immunofluorescent staining showed that IL-23p19 blockade also inhibited macrophage polarization, resulting in significantly lower numbers of macrophages expressing either M1 or M2 markers (Fig. 4). IL-23p29 blockade also markedly suppressed the activity of MMPs, the main proteases involved in ECM remodeling (Fig. 5A), apoptotic cell death, as evidenced by decreased TUNEL staining (Fig. 5B), and cell proliferation, as detected by proliferating cell nuclear antigen (PCNA, Fig. 5C). These results suggest that IL-23p19 inhibition dampens the inflammatory responses and MMP expression, preserving the overall architecture of the aortic wall tissue and mitigating aneurysmal dilatation.
Figure 4.
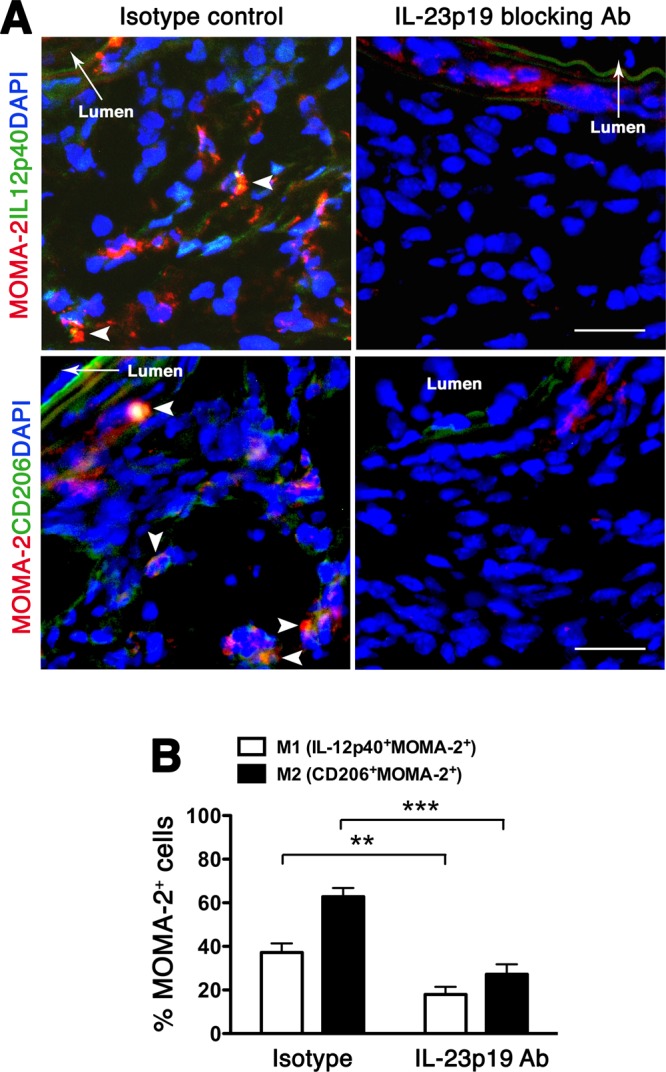
IL-23p19 blockade suppresses macrophage polarization. (A) Aortic sections obtained on day14 were double-stained for MOMA-2 (red) and IL12p40 (green) for M1 and CD206 (green) for M2. DAPI (blue) stained nuclei. Arrows indicate co-localization (yellow). Photographs are representative of 6–8 aortas per treatment. Scale bar = 25 um. (B) Enumeration of M1/M2 on day 14. Values represent mean ± SEM, n = 6–8 mice per treatment. **p < 0.01, ***p < 0.001.
Figure 5.

IL-23p19 blockade suppresses MMP activity and apoptotic cell death. Aortic sections obtained on day 14 were used to detect gelatinolytic activity of MMP by in situ zymography (A) apoptotic cells (TUNEL+, B), cell proliferation (PCNA+, C). Scale bar = 50 um (A, B), 100 um (C). Values represent mean ± SEM, n = 4–6 sections per aorta, 6–8 aortas per treatment. *P < 0.05, **P < 0.001.
IL-12/IL-23 axis is active in human AAA
Next, we sought to determine whether IL-12 and IL-23 could represent relevant therapeutic targets in human AAA. Immunostaining of human AAA tissue (n = 3) for IL12p40 and IL-23p19 confirmed their abundant presence throughout all layers of the aortic wall while non-AAA tissue showed no specific staining (Fig. 6). The IL-23p19 immunostaining corroborates previous studies that examined IL-23 gene expression level in aortic tissue from AAA patients23.
Figure 6.

IL-12/IL-23 in human AAA tissues. Immunohistochemistry of human AAA tissues revealed abundant expression of IL-12p40 and IL-23p19 (orange) throughout all layers of aortic wall tissue. Normal aortic wall tissue showed no specific staining. N = 3 independent AAA samples. L = lumen. Scale bar = 200 um.
Discussion
Careful analysis of aortic wall tissue at different time points post elastase perfusion shows that early stage elastase-induced AAA is associated with the infiltration of cells expressing pro-inflammatory cytokines such as TNF-α, IL-6 and IL-12p40. End-stage experimental aneurysm is associated with a shift towards cells that express high level of MMP-9, which may trigger substantial collateral tissue damage when released, further driving the aneurysmal phenotype. In an oversimplification, macrophages can be divided into two populations, the classically and alternatively activated macrophages. Classically activated macrophages (M1) are generated through IFN-γ and TNF-α stimulation and secrete high levels of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-12) and nitric oxide synthase (NOS). Alternatively activated macrophages (M2) are induced in vitro by IL-4; they promote matrix reorganization and wound healing27. Animal models of AAA suggest that M1 macrophages predominate in early stages of aneurysm development while M2 macrophages are more abundant at late-stage disease28,29. Macrophages are a highly heterogeneous cell population that can rapidly switch their functional phenotype in response to local microenvironmental signals30. And while the definition of M1 and M2 macrophages is well delineated in vitro, their functional distinction and phenotypes in vivo are less clear. Antagonism of IL-23p19 is associated with decreases in both M1 and M2 macrophages, which suggests that the higher M2/M1 macrophage ratio observed in late-stage elastase-induced AAA likely represents an effort to control and repair the aortic wall ECM in response to the pro-inflammatory activities exerted by expansion of M1 macrophages. IL-12 and IL-23 are also important drivers of T cell differentiation and proliferation. While IL-12 (and IFN-γ) promotes Th1 differentiation, IL-23 drives the development of Th17 cells31. Antagonism of IL-12p40 and IL-23p19 not only suppressed the expansion of macrophages but also inhibited T cell proliferation, thus abrogating the Th1/Th17-driven inflammatory responses during elastase-induced AAA development.
The discovery that IL-12 and IL-23 are active participants in the pathogenesis of several immune-mediated diseases have prompted the development of therapeutic agents targeting these cytokines or their receptors. Ustekinumab, the first monoclonal antibody directed against the p40 subunit of IL-12 and IL-23 is approved for the treatment of psoriasis, psoriatic arthritis, and Crohn’s disease16,32,33. Likewise, therapeutic agents targeting IL-23p19 have recently been approved or are in late phase clinical trials for the treatment of psoriasis and Crohn’s disease34–36. Although a previous study finds that genetic disruption of IL-23 attenuated elastase-induced AAA23, our key finding herein is that therapeutic blockade of the IL-12/IL-23 axis effectively suppresses AAA progression when given in the early phase of aneurysm development. On the other hand our results also seemingly contradict findings by Sharma et al. suggesting that deficiency of IL-12p40 promotes angiotensin II (Ang II)-induced AAA37. The discrepancy may be explained by a complete loss of IL-12p40 from birth, which may directly impact the polarization of macrophages in response to various levels of inflammatory modulators at different stages of disease progression.
The role of IL-17 in AAA also remains controversial. A previous study suggests that IL-17 is a critical mediator of elastase-induced AAA and genetic deletion of IL-17 not only protects mice against AAA formation but also leads to significant reduction of several inflammatory cytokines (TNF-α, IFN-γ and MCP-1), indicating that IL-17 is an upstream mediator of the inflammatory cascade23. These results corroborate findings in another study that uses digoxin to attenuate Ang II-induced and elastase-induced AAA, showing that IL-17A related inflammatory responses were suppressed in a dose-dependent manner38. In addition, IL-17 is implicated in the Ang II-induced aortic dissections39. On the other hand, Romain et al. demonstrate that overexpression of suppressor of cytokine signaling 3 (SOCS3) significantly decreases IL-17 expression and markedly increases aneurysm severity in a model of Ang II-induced AAA, leading the investigators to suggest that IL-17 has a protective role in this process40. It is possible that overexpression of SOCS3 modulates the expression of other proinflammatory mediators in Ang II-induced AAA and the indirect decrease in IL-17 is merely coincidental and not causal. Our data herein agree with previous studies suggesting a pro-inflammatory role for IL-17.
In summary, there is currently no drug therapy for AAA. We posit that blockade of IL-12/IL-23 axis as treatment strategy to halt the progression of small aneurysms merits further consideration given the availability of the immunotherapeutics in the clinic combined with the observation that IL-12p40 and IL-23p19 are abundantly expressed in human AAA tissue.
Methods
Animals
WT C57BL/6J mice (Cat# 000664) were obtained from the Jackson Laboratory (Bar Harbor, ME). All mice were kept in a standard pathogen-free environment at Washington University Specialized Research Facility (St. Louis, MO). All animal experiments were performed in accordance to guidelines and protocols approved by the Division of Comparative Medicine at Washington University in St. Louis. The animal protocol is subjected to annual review and approval by The Animal Studies Committee of Washington University.
Human tissues
All research involving human tissues was performed in accordance with relevant guidelines and regulations established by the Washington University in St. Louis Institutional Review Board (IRB) Committee. De-identified human abdominal aorta tissue specimens were provided by Dr. Robert Thompson who obtained them from discarded aortic aneurysm tissues harvested at time of elective surgery through a protocol approved by the Institutional Review Board at Washington University School of Medicine. All participants provided written informed consent. The aortic tissues were snap-frozen, embedded in OCT and stored at −80 °C until use.
Murine elastase-induced AAA
AAA was induced as previously described41. Only male mice were used in these studies, as AAA is a disease with strong male predominance42. Briefly, 8- to 10-week-old male wild-type (WT) C57BL/6J mice were anesthetized with an i.p. injection of ketamine (87 mg kg−1), xylazine (13 mg kg−1), and acepromazine (2 mg kg−1) KXA cocktail. For post-op pain control mice were administered Buprenorphine-SR (0.5 mg kg−1) one hour prior to surgery and the effect was expected to last 72 h. Immediately prior to surgery, lidocaine (0.5%) was injected into the subcutaneous space below the planned incision line. A laparotomy was then performed under sterile conditions. The infrarenal aorta, from the left renal vein to the aortic bifurcation was isolated, ligated and perfused for 5 min and 30 sec with a solution containing 0.145 U/ml type 1 porcine pancreatic elastase (Cat# E-1250, Sigma-Aldrich, St Louis, MO) via infusion pump. The tie was released and the maximal post-perfusion aortic diameter was measured with a calibrated ocular grid and the aortotomy closed. On day 14, unless otherwise indicated, a second laparotomy was performed following anesthesia with KXA cocktail and the aortic diameter measured prior to euthanasia and tissue procurement.
In some studies WT mice were injected i.p. with rat anti mouse-IL-12p40 monoclonal antibody (250 ug on days 3 and 8, Cat# MAB4991, R&D, Minneapolis, MN) or mouse anti mouse-IL-23p19 monoclonal antibody (250 ug on days 3 and 8, Cat# 513806, Biolegend, San Diego, CA). Control groups received either purified rat IgG2a functional grade GOLD (250 ug on days 3 and 8, Cat# I-1177, Leinco Techologies, Fenton, MO) or purified mouse IgG 2b functional grade GOLD (250 ug on days 3 and 8, Cat# I-119, Leinco Techologies, Fenton, MO). The number of mice per treatment is indicated in the text and figure legends. Elastin staining was performed as previously described43.
Immunofluorescence
The mouse aortas were harvested on days 7 or 14, embedded in OCT and sectioned. Cross sections of aortic tissues (9 µm) were fixed in 4% paraformaldehyde, blocked in 8% BSA in PBS and incubated with the following primary antibodies: rat anti-mouse MOMA-2 (1:200 dilution; Cat# ab33451, Abcam, Cambridge, United Kingdom), biotinylated-IL-12p40 (1:100 dilution; Cat# 505302, Biolegend, San Diego, CA), rat anti-mouse CD206 (1:200 dilution; Cat: MCA2235, Bio-Rad [Formerly AbD Serotec], Hercules, CA), or rabbit anti-mouse CD8a (1:200 dilution; Cat# bs-0648R, Bioss antibodies, Woburn, Massachusetts) followed by the appropriate rhodamine red- or FITC-conjugated secondary antibody (1:100–1:200; Jackson ImmunoResearch Laboratories, West Grove, PA) or Streptavidin Alexa Fluor 488 conjugate (1:400 dilution; Cat# S-11223, Molecular Probes, Eugene, OR). Nuclei were counterstained with DAPI. Images were acquired on a Leica DM 2000 microscope fitted with a Leica DMC 4500 color camera and analyzed with Leica Application Suite (LAS) X software. Single- or double-color images were loaded into ImageJ and cell enumeration was performed in a blinded fashion. Data were obtained from 3–4 non-overlapping fields per aortic cross-section, 6–9 sections per aorta, n = 6–8 aortas per time point.
Immunohistochemistry
Immunohistochemical staining was performed on formalin-fixed, OCT-embedded 9 μm sections of mouse or human AAA tissue. Rat anti-mouse MOMA-2 (1:200 dilution; Cat# ab33451, Abcam, Cambridge, United Kingdom), rabbit anti-mouse TNF-α (1:100 dilution; Cat# ab34674, Abcam, Cambridge, United Kingdom), biotinylated anti-mouse IL-12p40(1:100 dilution; Cat# 505302, Biolegend, San Diego, CA), rabbit anti-mouse IL-6 (1:100 dilution; Cat# NB600-1131, Novus biologicals, Littleton, CO), rat anti-mouse MMP-9 (1:200 dilution; Cat# 116103, R&D, Minneapolis, MN), rat anti-mouse IL-17A (1:100 dilution; Cat# 560666, BD Pharmingen, San Jose, CA), biotinylated anti-PCNA (1:100 dilution; Cat# 307904, Biolegend), biotinylated anti-mouse CD3 (1:100 dilution; Cat# 553060, BD Pharmingen) primary antibodies were applied to the frozen cross-sections for 1 hour at RT followed by the appropriate HRP-conjugated secondary antibodies. Data presented was derived from six to nine serial cross-sections that spanned the entire abdominal aorta, with 5–8 aortas per time point. For human tissue, mouse anti-human IL-12p40 (1:100 dilution; Cat# 501801, Biolegend, San Diego, CA), rabbit anti-human IL-23p19 (1:100 dilution; Cat# 3793, ProSci, Poway, CA) antibodies were employed. Staining was performed on at least three independent sections per human AAA tissue samples (n = 3) and the pattern was confirmed on 3–6 independent tissue samples.
In situ zymography
Non-fixed, frozen sections (9 μm) of day 14 aortas were incubated with a fluorogenic gelatin substrate (DQ gelatin at 25 μg/ml, Molecular Probes) for 1 h at room temperature. For negative control, slides were incubated in the presence of 25 mM EDTA. The specific removal of essential divalent cations resulted in no detectable gelatinolytic activity. Images were acquired on a Leica DM 2000 microscope fitted with a Leica DMC 4500 color camera and analyzed with Leica Application Suite (LAS) X software. Images were loaded into ImageJ for analysis. Using the brightness to filter the picture, positively stained areas or the regions of interest (ROIs) were isolated. The ROIs were measured and presented as integrated optical density (IntDen). Data represent 4–6 sections per aorta, 6–8 aortas per treatment.
TUNEL assay
Detection of apoptotic cells was performed using In situ Cell Death Detection Kit, Fluorescein (Cat: 11-684-795-910, Roche) with the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay. Freshly prepared TUNEL reaction mixture were applied to aortic sections for 60 min at 37 °C, rinsed 3–5 times with PBS and mounted with VECTASHIELD mounting medium with DAPI (Cat: H-1200, Vector Laboratories). The TUNEL+ cells were enumerated across the entire aortic section. Data represent 4–6 sections per aorta, 6–8 aortas per treatment.
Statistical analysis
Comparisons between two groups were performed by two-tailed, unpaired t test without correction. Comparisons between multiple groups (≥3) were performed by one-way ANOVA followed by Bonferroni’s posttest to compare all groups of data. F test was used to compare variances within each group of data and the difference in variances was found to be not significant between groups. Data are presented as the mean ± SEM. A p value < 0.05 was considered significant.
Supplementary information
Acknowledgements
The authors would like to thank Dr. Robert Thompson (Washington University) for the de-identified human AAA specimens. This work was supported in part by the VA Merit Review Award 1I01 BX002714 and NIH grants AR067491 and P30 AR073752. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Veterans Affairs or the National Institutes of Health.
Author Contributions
C.T.N.P. and H.Y. conceived the experiments and wrote the manuscript. Y.H., A.A., K.Y. and J.B. performed the experiments the analyzed data. All authors reviewed the manuscript.
Data Availability
Data generated in this current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-46909-y.
References
- 1.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuivaniemi H, Platsoucas CD, Tilson MD., 3rd Aortic aneurysms: an immune disease with a strong genetic component. Circulation. 2008;117:242–252. doi: 10.1161/CIRCULATIONAHA.107.690982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuivaniemi H, Ryer EJ, Elmore JR, Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13:975–987. doi: 10.1586/14779072.2015.1074861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pyo R, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longo GM, et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo GM, et al. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery. 2005;137:457–462. doi: 10.1016/j.surg.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Thompson RW, et al. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96:318–326. doi: 10.1172/JCI118037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson RW, Parks WC. Role of matrix metalloproteinases in abdominal aortic aneurysms. Ann N Y Acad Sci. 1996;800:157–174. doi: 10.1111/j.1749-6632.1996.tb33307.x. [DOI] [PubMed] [Google Scholar]
- 9.Pearce WH, Shively VP. Abdominal aortic aneurysm as a complex multifactorial disease: interactions of polymorphisms of inflammatory genes, features of autoimmunity, and current status of MMPs. Ann N Y Acad Sci. 2006;1085:117–132. doi: 10.1196/annals.1383.025. [DOI] [PubMed] [Google Scholar]
- 10.Jones GT, et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circulation Res. 2017;120:341–353. doi: 10.1161/CIRCRESAHA.116.308765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiotti N, et al. Multiple sites of vascular dilation or aneurysmal disease and matrix metalloproteinase genetic variants in patients with abdominal aortic aneurysm. J Vasc Surg. 2018;67:1727–1735. doi: 10.1016/j.jvs.2017.09.047. [DOI] [PubMed] [Google Scholar]
- 12.Abdul-Hussien H, et al. Collagen degradation in the abdominal aneurysm: a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol. 2007;170:809–817. doi: 10.2353/ajpath.2007.060522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshimura K, et al. Current Status and Perspectives on Pharmacologic Therapy for Abdominal Aortic Aneurysm. Curr Drug Targets. 2018;19:1265–1275. doi: 10.2174/1389450119666171227223331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol. 2015;35:1746–1755. doi: 10.1161/ATVBAHA.115.305269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oppmann B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/S1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 16.Teng MW, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21:719–729. doi: 10.1038/nm.3895. [DOI] [PubMed] [Google Scholar]
- 17.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 18.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elliott M, et al. Ustekinumab: lessons learned from targeting interleukin-12/23p40 in immune-mediated diseases. Ann N Y Acad Sci. 2009;1182:97–110. doi: 10.1111/j.1749-6632.2009.05070.x. [DOI] [PubMed] [Google Scholar]
- 20.Salmon M, et al. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation. 2013;128:S163–174. doi: 10.1161/CIRCULATIONAHA.112.000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng J, Koenig SN, Kuivaniemi HS, Garg V, Hans CP. Pharmacological inhibitor of notch signaling stabilizes the progression of small abdominal aortic aneurysm in a mouse model. J Am Heart Assoc. 2014;3:e001064. doi: 10.1161/JAHA.114.001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marinkovic G, et al. Immunosuppressive drug azathioprine reduces aneurysm progression through inhibition of Rac1 and c-Jun-terminal-N-kinase in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:2380–2388. doi: 10.1161/ATVBAHA.113.301394. [DOI] [PubMed] [Google Scholar]
- 23.Sharma AK, et al. Experimental abdominal aortic aneurysm formation is mediated by IL-17 and attenuated by mesenchymal stem cell treatment. Circulation. 2012;126:S38–45. doi: 10.1161/CIRCULATIONAHA.111.083451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson RW, et al. Pathophysiology of abdominal aortic aneurysms: insights from the elastase-induced model in mice with different genetic backgrounds. Ann N Y Acad Sci. 2006;1085:59–73. doi: 10.1196/annals.1383.029. [DOI] [PubMed] [Google Scholar]
- 25.Zhou HF, et al. CD43-Mediated IFN-gamma Production by CD8+ T Cells Promotes Abdominal Aortic Aneurysm in Mice. J Immunol. 2013;190:5078–5085. doi: 10.4049/jimmunol.1203228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, et al. Interleukin-12 inhibits the hepatocellular carcinoma growth by inducing macrophage polarization to the M1-like phenotype through downregulation of Stat-3. Mol Cell Biochem. 2016;415:157–168. doi: 10.1007/s11010-016-2687-0. [DOI] [PubMed] [Google Scholar]
- 27.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raffort J, et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471. doi: 10.1038/nrcardio.2017.52. [DOI] [PubMed] [Google Scholar]
- 29.Cheng Z, et al. Diverse roles of macrophage polarization in aortic aneurysm: destruction and repair. J Transl Med. 2018;16:354. doi: 10.1186/s12967-018-1731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bettelli E, Kuchroo VK. IL-12- and IL-23-induced T helper cell subsets: birds of the same feather flock together. J Exp Med. 2005;201:169–171. doi: 10.1084/jem.20042279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKeage K. Ustekinumab: a review of its use in psoriatic arthritis. Drugs. 2014;74:1029–1039. doi: 10.1007/s40265-014-0242-4. [DOI] [PubMed] [Google Scholar]
- 33.Dickson I. IBD: Ustekinumab therapy for Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2017;14:4. doi: 10.1038/nrgastro.2016.202. [DOI] [PubMed] [Google Scholar]
- 34.Markham A. Guselkumab: First Global Approval. Drugs. 2017;77:1487–1492. doi: 10.1007/s40265-017-0800-7. [DOI] [PubMed] [Google Scholar]
- 35.Papp KA, et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N Engl J Med. 2017;376:1551–1560. doi: 10.1056/NEJMoa1607017. [DOI] [PubMed] [Google Scholar]
- 36.Feagan BG, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389:1699–1709. doi: 10.1016/S0140-6736(17)30570-6. [DOI] [PubMed] [Google Scholar]
- 37.Sharma N, et al. Deficiency of IL12p40 (Interleukin 12 p40) Promotes Ang II (Angiotensin II)-Induced Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2019;39:212–223. doi: 10.1161/ATVBAHA.118.311969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei Z, et al. Inhibiting the Th17/IL-17A-related inflammatory responses with digoxin confers protection against experimental abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2014;34:2429–2438. doi: 10.1161/ATVBAHA.114.304435. [DOI] [PubMed] [Google Scholar]
- 39.Ju X, et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol. 2013;33:1612–1621. doi: 10.1161/ATVBAHA.112.301049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romain M, et al. Overexpression of SOCS3 in T lymphocytes leads to impaired interleukin-17 production and severe aortic aneurysm formation in mice–brief report. Arterioscler Thromb Vasc Biol. 2013;33:581–584. doi: 10.1161/ATVBAHA.112.300516. [DOI] [PubMed] [Google Scholar]
- 41.Yan H, et al. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler Thromb Vasc Biol. 2016;36:1660–1669. doi: 10.1161/ATVBAHA.116.307786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang CK, et al. Androgen receptor promotes abdominal aortic aneurysm development via modulating inflammatory interleukin-1alpha and transforming growth factor-beta1 expression. Hypertension. 2015;66:881–891. doi: 10.1161/HYPERTENSIONAHA.115.05654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou HF, et al. Fibrinogen-specific antibody induces abdominal aortic aneurysm in mice through complement lectin pathway activation. Proc Natl Acad Sci USA. 2013;110:E4335–4344. doi: 10.1073/pnas.1315512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this current study are available from the corresponding author on reasonable request.