

Abstract
Binding and uptake of triglyceride-rich lipoproteins (TRLs) in mice depend on heparan sulfate and the hepatic proteoglycan, syndecan-1 (SDC1). Alteration of glucosamine N-sulfation by deletion of glucosamine N-deacetylase-N-sulfotransferase 1 (Ndst1) and 2-O-sulfation of uronic acids by deletion of uronyl 2-O-sulfotransferase (Hs2st) led to diminished lipoprotein metabolism, whereas inactivation of glucosaminyl 6-O-sulfotransferase 1 (Hs6st1), which encodes one of the three 6-O-sulfotransferases, had little effect on lipoprotein binding. However, other studies have suggested that 6-O-sulfation may be important for TRL binding and uptake. In order to explain these discrepant findings, we used CRISPR/Cas9 gene editing to create a library of mutants in the human hepatoma cell line, Hep3B. Inactivation of EXT1 encoding the heparan sulfate copolymerase, NDST1 and HS2ST dramatically reduced binding of TRLs. Inactivation of HS6ST1 had no effect, but deletion of HS6ST2 reduced TRL binding. Compounding mutations in HS6ST1 and HS6ST2 did not exacerbate this effect indicating that HS6ST2 is the dominant 6-O-sulfotransferase and that binding of TRLs indeed depends on 6-O-sulfation of glucosamine residues. Uptake studies showed that TRL internalization was also affected in 6-O-sulfation deficient cells. Interestingly, genetic deletion of SDC1 only marginally impacted binding of TRLs but reduced TRL uptake to the same extent as treating the cells with heparin lyases. These findings confirm that SDC1 is the dominant endocytic proteoglycan receptor for TRLs in human Hep3B cells and that binding and uptake of TRLs depend on SDC1 and N- and 2-O-sulfation as well as 6-O-sulfation of heparan sulfate chains catalyzed by HS6ST2.
Keywords: heparan sulfate 6-O-sulfotransferases, lipoprotein receptors, syndecan-1, triglyceride-rich lipoprotein remnants
Introduction
Hypertriglyceridemia results from aberrant metabolism of triglyceride-rich lipoproteins (TRLs) and constitutes an independent risk factor for developing premature coronary artery disease (Talayero and Sacks 2011; Nordestgaard and Varbo 2014; Peng et al. 2017). TRLs consist of chylomicrons derived from dietary fat, very low-density lipoproteins secreted by hepatocytes and remnant particles that arise from these lipoproteins by the action of several lipases in the vasculature, including lipoprotein lipase, hepatic lipase and endothelial lipase. Clearance of TRLs and their remnants occurs in the liver through three major hepatocyte receptors, low-density lipoprotein receptors (LDLR and the LDLR-related protein 1, LRP1) and syndecan-1 (SDC1), a type of heparan sulfate proteoglycan (HSPG) (Foley and Esko 2010). SDC1 is a transmembrane proteoglycan that contains 2–3 heparan sulfate chains, which make up the binding site for TRLs (Stanford et al. 2009). The absence of SDC1 results in elevated fasting triglycerides and more provocatively delayed postprandial clearance of dietary triglycerides.
Heparan sulfate assembly involves formation of the backbone of the chain catalyzed by EXT1 and EXT2 and a series of processing reactions that modifies sugar residues by variable N-sulfation of glucosamine residues catalyzed by one or more members of the N-acetylglucosamine (GlcNAc) N-deacetylase-N-sulfotransferase (NDST1–4) family of enzymes, epimerization of adjacent d-glucuronic acid (GlcA) residues to l-iduronic acids (IdoA) by C5 epimerase (GLCE), 2-O-sulfation of the IdoA and occasionally GlcA residues by HS2ST, 6-O-sulfation of glucosamine residues catalyzed by HS6ST1–3 and less frequent 3-O-sulfation of glucosamine residues by HS3ST1, 2, 3a, 3b, 4, 5 and 6 (Esko and Selleck 2002; Thacker et al. 2013). The chains assemble while attached to 1 of the 17 HSPG core proteins expressed differentially in various cell types (Lindahl et al. 2015). Two endo-6-O-sulfatases (SULF1–2) can further process the heparan sulfate chains at the plasma membrane or in the extracellular matrix by removal of specific 6-O-sulfate groups in highly sulfated regions of the chains (El Masri et al. 2017).
Studies of mice harboring mutations in Ndst1 and Hs2st established the importance of N-sulfation and 2-O-sulfation in binding and uptake of TRLs in the liver in vivo (MacArthur et al. 2007; Stanford et al. 2010). Previous studies also suggested that TRL binding and uptake did not require functional Hs6st1 in vivo and that 6-O-sulfation in general might not play a role in TRL binding and clearance based on the retention of inhibitory activity of heparin before and after chemical 6-O-desulfation (Stanford et al. 2010). However, heparin is a highly modified form of heparan sulfate, abundantly N-sulfated and 2-O-sulfated. Other evidence indicated that overexpression of the 6-O-sulfatase Sulf2 suppressed uptake of model TRLs (Chen et al. 2010; Hassing et al. 2014). Inhibition of Sulf2 by antisense oligonucleotides doubled HSPG-mediated catabolism in T2DM db/db mice, consistent with the idea that 6-O-sulfation might be important (Hassing et al. 2012).
To study the role of 6-O-sulfation in HSPG-mediated clearance, we undertook the development and characterization of a library of heparan sulfate-deficient mutants in the human hepatoma cell line Hep3B using CRISPR/Cas9 technology. Binding measurements using TRLs demonstrated a requirement for N-sulfation, 2-O-sulfation and 6-O-sulfation, suggesting that the overall charge on the chains is more important than specific subclasses of sulfate groups. Studies of cells lacking one or more of the HS6ST isozymes showed that HS6ST2 is the principle enzyme for generating 6-O-sulfated glucosamine residues in Hep3B cells. We also showed that Hep3B cells express multiple cell surface and secreted HSPGs, which can bind TRLs; however, only SDC1 mediates TRL uptake. Our data demonstrated the importance of fully sulfated heparan sulfate chains on SDC1 for TRL binding and uptake and that this proteoglycan is the dominant TRL proteoglycan receptor in human Hep3B cells.
Results
Developing a library of heparan sulfate-deficient human hepatoma cell lines
To examine the role of heparan sulfate and HSPGs in TRL binding and uptake in human hepatocytes, we first analyzed gene expression profiles in primary human hepatocytes and Hep3B cells, a human hepatoma line often used as a model for human hepatocytes (Figure 1A). Primary human hepatocytes express exostosin family genes (EXT1 and EXT2; the copolymerase responsible for the elongation of heparan sulfate chains); two GlcNAc N-deacetylase-N-sulfotransferases (NDST1 and NDST2); heparan sulfate uronyl 2-O-sulfotransferase (HS2ST); and two isoforms of glucosaminyl 6-O-sulfotransferases (HS6ST1 and HS6ST2). Hep3B had a qualitatively similar expression profile, but the level of expression of HS2ST, HS6ST1 and HS6ST2 was higher (Figure 1A). SULF1 and SULF2 were not expressed in Hep3B (data for primary human hepatocytes are not available). The gene expression pattern of HSPGs was similar in both primary and Hep3B cells, with the notable exception of the glycosylphosphatidylinositol-linked HSPG, glypican-3 (GPC3), which was expressed in Hep3B cells but not in primary hepatocytes (Figure 1B). Several HSPGs showed higher levels of gene expression in Hep3B cells, in particular syndecan-2 (SDC2), GPC6 and the three extracellular matrix HSPGs, collagen 18 (COL18), agrin (AGRN) and perlecan (HSPG2).
Fig. 1.
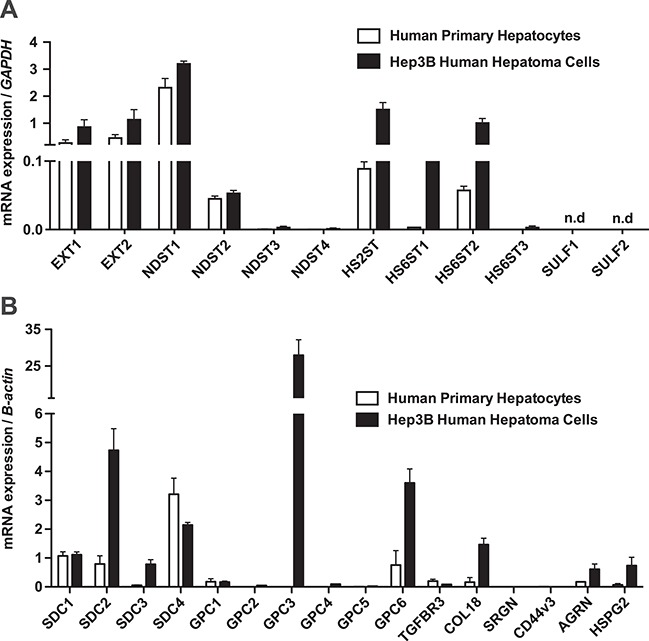
Expression of HSPGs and heparan sulfate biosynthetic genes in primary human hepatocytes and Hep3B cells. mRNA was isolated from human primary hepatocytes (open bars; N = 3 technical replicates) and human hepatoma cell line, Hep3B (filled bars; n = 2 technical replicates). Amplification primers are listed in Supporting Table S1. This figure is available in black and white in print and in color at Glycobiology online.
The application of CRISPR/Cas9 gene targeting technology in Hep3B cells led to successful guided mutational inactivation of EXT1, NDST1, HS2ST, HS6ST1 and HS6ST2; a double mutant bearing mutations in both HS6ST1 and HS6ST2; and SDC1 (Supporting Figure S1). Multiple clonal cell lines were obtained for each targeting experiment. Polymerase chain reaction (PCR) products covering the targeted exons were cloned and sequenced. Lines bearing missense and indels were identified, but only those isolates that bore mutations in both alleles and that resulted in a shift in the reading frame were further characterized. Targeting and sequencing data are provided for mutants in EXT1 (Figure S1A and B), NDST1 (Figure S1C and D), HS2ST (Figure S1E and F), HS6ST1 (Figure S1G and H), HS6ST2 (Figure S1I and J), HS6ST1 in the HS6ST2 mutant (Figure S1K and L) and SDC1 (Figure S1M and N).
Disruption of heparan sulfate biosynthetic genes alters heparan sulfate structure
Each mutant was expanded in culture and processed to obtain a mixed heparan sulfate preparation derived from extracellular matrix, cell surface and intracellular proteoglycans. The material was then treated with a mixture of heparin lyases, which cleaves the chains into disaccharides, each bearing sulfate groups at different positions (N-sulfoglucosamine residues [N-sulfates], 2-O-sulfouronic acids [2-O-sulfates] and 6-O-sulfoglucosamine residues [6-O-sulfates]). After tagging the disaccharides with [12C]aniline, they were mixed with [13C]aniline-tagged disaccharide standards and analyzed by liquid chromatography/mass spectrometry (LC/MS) (Lawrence, Olson, et al. 2008). Quantitative values of the individual disaccharides are summarized in Supporting Table S2 and presented graphically in Figure 2A. This information was then used to calculate the percentage content of N-acetylated (N-acetyl) and N-sulfated glucosamine residues (N-SO3) and 2-O-sulfated uronic acids (2-O-SO3) and 6-O-sulfated glucosamine residues (6-O-SO3; Figure 2B). As expected, EXT1−/− cells did not produce any heparan sulfate due to its key role in chain polymerization (Lin et al. 2000). NDST1−/− cells produced chains that were partially undersulfated, with reductions in all N-sulfated disaccharides (D0S0, D0S6, D2S0 and D2S6) and accumulation of the nonsulfated disaccharide, D0A0. [The disaccharide structure code is used: D0H0, ΔUA-GlcNH2; D0A0, ΔUA-GlcNAc; D0H6, ΔUA-GlcNH26S; D2H0, ΔUA2S-GlcNH2; D0S0, ΔUA-GlcNS; D2H6, ΔUA2S-GlcNH26S; D0A6, ΔUA-GlcNAc6S; D0S6, ΔUA-GlcNS6S; D2A0, ΔUA2S-GlcNAc; D2S0, ΔUA2S-GlcNS; D2A6, ΔUA2S-GlcNAc6S; D2S6, ΔUA2S-GlcNS6S; where ΔUA = 4,5-unsaturated uronic acid and I = iduronic acid. (Lawrence, Lu, et al. 2008)] Undersulfation was incomplete most likely due to the expression of NDST2 (Figure 1), as observed in other cell lines and in various mouse tissues (Ledin et al. 2006; MacArthur et al. 2007). Inactivation of NDST1 also caused a decrease in 6-O-sulfoglucosamine residues because of the preference of HS6STs for N-deacetylated/N-sulfated glucosamine residues (Esko and Selleck 2002). 2-O-sulfouronic acids also decreased due to the preference of the uronyl epimerase to act toward the reducing side of N-sulfated glucosamine units (Hagner-McWhirter et al. 2004). Overall, inactivation of NDST1 reduced N-sulfation by 81 ± 2.6% (abundance of N-sulfation observed in wild-type cells was 27.8 ± 1.4% compared to 5.5 ± 0.3% in mutant, P = 0.001), 2-O-sulfation by 98.3 ± 1.5% (7.1 ± 1.6% in wild type vs. 0.1 ± 0.08% in mutant, P = 0.045) and 6-O-sulfation by 52.8 ± 2.2% (34.8 ± 1.6% in wild type vs. 16.5 ± 1.7% in mutant, P = 0.004).
Fig. 2.
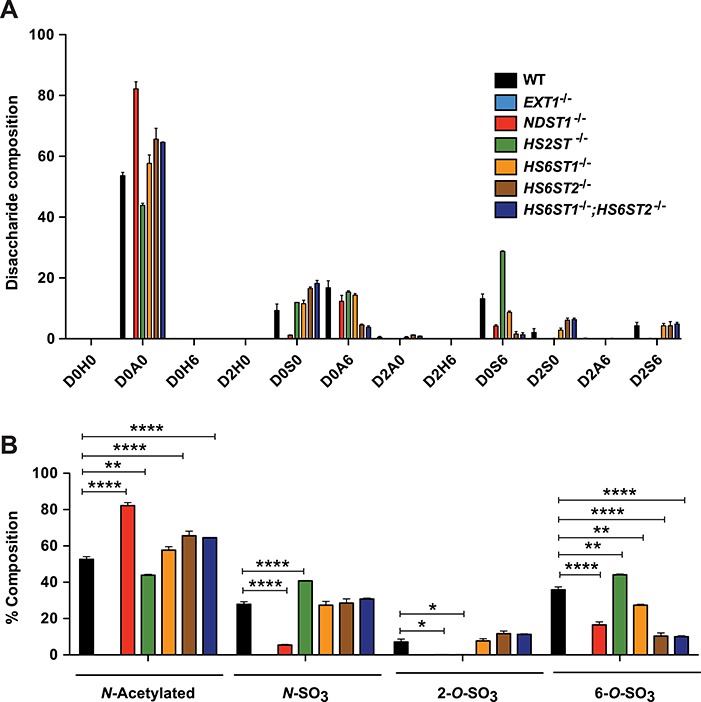
Composition of heparan sulfate in wild-type and mutant Hep3B cells. (A) Heparan sulfate was isolated and purified from wild-type and mutant cell lines, and the disaccharide composition was determined (n = 2 biological replicates). The relative content of individual disaccharides is shown. (B) The number of N-acetylated (N-acetyl), N-sulfated (N-SO3), 6-O-sulfated (6-O-SO3) glucosamine units and 2-O-sulfated uronic acids (2-O-SO3) was calculated. Statistics was calculated by two-way analysis of variance (ANOVA). n.d. means not determined. The disaccharide structure code is used: D0H0, ΔUA-GlcNH2; D0A0, ΔUA-GlcNAc; D0H6, ΔUA-GlcNH26S; D2H0, ΔUA2S-GlcNH2; D0S0, ΔUA-GlcNS; D2H6, ΔUA2S-GlcNH26S; D0A6, ΔUA-GlcNAc6S; D0S6, ΔUA-GlcNS6S; D2A0, ΔUA2S-GlcNAc; D2S0, ΔUA2S-GlcNS; D2A6, ΔUA2S-GlcNAc6S; D2S6, ΔUA2S-GlcNS6S; where ΔUA = 4,5-unsaturated uronic acid and I = iduronic acid (Lawrence, Lu, et al. 2008). This figure is available in black and white in print and in color at Glycobiology online.
HS2ST catalyzes the formation of 2-O-sulfoiduronic acid and less frequently 2-O-sulfoglucuronic acid residues (Rong et al. 2001). As expected, inactivation of HS2ST resulted in loss of 2-O-sulfated disaccharides (D2A0, D2S0 and D2S6) by 98.9 ± 1.1% (P = 0.001). As in other systems, an increase of N-sulfoglucosamine (+40.4 ± 7.6%; P = 0.006) and 6-O-sulfoglucosamine units (+22.4 ± 4.5%; P = 0.02) occurred compared to wild type (Figure 2B) (Bai and Esko 1996; Ledin et al. 2004; Stanford et al. 2010; Dejima et al. 2013; Qiu et al. 2018). D0S6 accounted for most of the increase in N- and 6-O-sulfation (Figure 2A).
Three members of the HS6ST family of sulfotransferases catalyze 6-O-sulfation of N-acetylated and N-sulfoglucosamine residues. All three enzymes catalyze 6-O-sulfation of both glucuronic acid-N-sulfoglucosamine and iduronic acid-N-sulfoglucosamine units, with a preference for iduronic acid-containing disaccharides, with or without 2-O-sulfate substituents (Habuchi et al. 2000; Jemth et al. 2003; Smeds et al. 2003). In mice, inactivation of Hs6st1 and Hs6st2 causes a complete loss of 6-O-sulfation in fetal skin-derived mast cells (Anower et al. 2013). Hep3B cells express all three isozymes as well (Figure 1A). Genetic inactivation of HS6ST1 caused only a slight reduction in D0S6, with an overall decrease of 6-O-sulfation by 21.6 ± 5.5% (P = 0.02; Figure 2A). In contrast, genetic inactivation of HS6ST2 drastically reduced D0A6 and D0S6, resulting in a 70.5 ± 4.1% reduction in 6-O-sulfation (P = 0.001). Compounding mutations in HS6ST1 and HS6ST2 did not alter hepatic heparan sulfate structure to a greater extent than observed in HS6ST2−/− cells, confirming that HS6ST1 does not contribute significantly to 6-O-sulfation in Hep3B cells. The presence of D0A6, D0S6 and D2S6 in the double mutant suggested that HS6ST3 might be active. An analysis of mRNA transcripts by quantitative polymerase chain reaction (qPCR) indicated a mild induction of HS6ST3 mRNA in HS6ST1−/−;HS6ST2−/− cells (Figure 3A). siRNA-mediated knockdown of HS6ST3 reduced mRNA expression by ~50% in both the wild-type Hep3B and HS6ST1−/−;HS6ST2−/− cell lines (Figure 3A). A disaccharide analysis showed that siRNA treatment had no effect in the wild type but caused a further reduction of D0S6 in HS6ST1−/−;HS6ST2−/− cells as compared to cells treated with scrambled siRNA (85.5 ± 13.6% reduction; P = 0.036; Figure 3B). The net effect was reduction of 6-O-sulfate groups and an increase in N-acetylated glucosamine residues (Figure 3C).
Fig. 3.
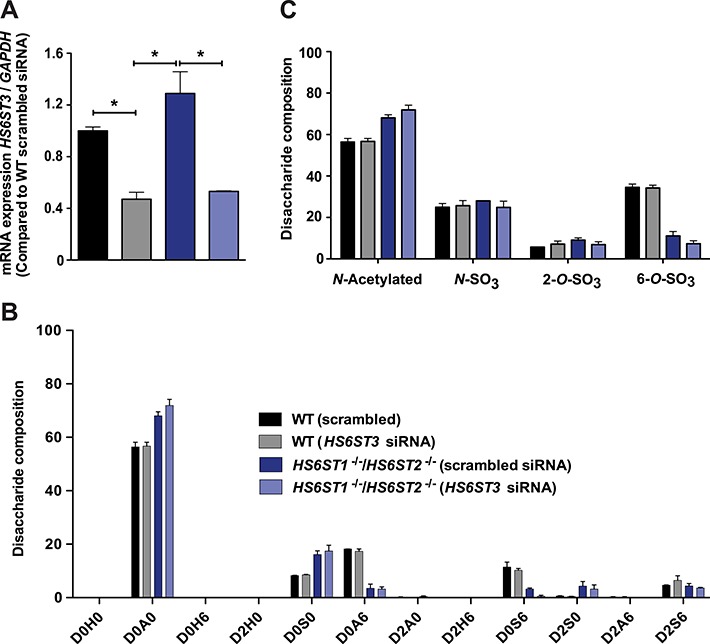
Silencing of HS6ST3 in HS6ST1−/−;HS6ST2−/− cell line. Wild-type Hep3B and HS6ST1−/−;HS6ST2−/− cells were transfected either scrambled or HS6ST3 siRNA (Sigma-Aldrich). (A) HS6ST3 gene expression was analyzed (n = 2 technical replicates). (B) Heparan sulfate was isolated and the disaccharide composition was determined (n = 2 biological replicates). (C) The number of N-acetylated (N-acetyl), N-sulfated (N-SO3), 6-O-sulfated (6-O-SO3) glucosamine units and 2-O-sulfated uronic acids (2-O-SO3) was calculated. Statistics was calculated by two-way ANOVA. This figure is available in black and white in print and in color at Glycobiology online.
Reduction of TRL and FGF2 binding in the mutants
To examine the impact of altering heparan sulfate on TRL uptake, we prepared radioactive TRLs from mouse plasma after feeding the animals [3H] retinol, which is converted to retinol esters and packaged into chylomicrons. The chylomicrons undergo partial lipolysis in the circulation, yielding 3H-labeled remnant particles in the circulation, which can be readily isolated by buoyant density ultracentrifugation (Gordts et al. 2016). The capacity of Hep3B cells to bind these [3H] TRLs was assessed by incubation of wild-type cells and the various mutants with [3H] TRLs at 4°C, followed by solubilization of the cells and counting of samples by liquid scintillation spectrometry. Loss of heparan sulfate in EXT1−/− cells reduced [3H] TRL binding by 5-fold (Figure 4A; P < 0.0001); the residual binding was presumably due to LDLR and LRP1 receptors (Mahley and Huang 2007). Inactivation of NDST1 and HS2ST also reduced [3H] TRL binding by 60.5 ± 1.7% (P < 0.0001) and 62.1 ± 5.5% (P < 0.0001), respectively, consistent with the trend observed in previous studies of hepatocytes derived from mice deficient in these genes (MacArthur et al. 2007; Stanford et al. 2010). Inactivation of HS6ST1 resulted in only a mild reduction in binding (27.3 ± 13%; P = 0.02), whereas inactivation of HS6ST2 had a more pronounced effect (48 ± 15%; P = 0.003). No additional diminution in binding occurred in the compound HS6ST1−/−;HS6ST2−/− cell line. As a control, we challenged Hep3B cells and the mutants with biotinylated basic fibroblast growth factor (FGF2), a well-characterized heparan sulfate-binding protein, and analyzed binding using streptavidin-PE/Cy5 and flow cytometry (Weiss et al. 2015). Abrogating EXT1 expression caused dramatic reduction in binding (96.6 ± 0.1%; P < 0.0001), much like treating cells with heparin lyases (86.3 ± 1.2% reduction; P = 0.0002), as in previous studies of Chinese hamster ovary (CHO) cells and endothelial cells (Yayon et al. 1991; Fuster et al. 2007). Binding was also dramatically reduced in NDST1−/− (78.6 ± 0.1%; P < 0.0001) and HS2ST−/− cells (86.2 ± 0.5%; P < 0.0001), consistent with previous studies demonstrating a preference of FGF2 for N- and 2-O-sulfated heparan sulfate (Turnbull et al. 1992; Kreuger et al. 2001). In contrast, inactivation of HS6ST1 had very little effect on FGF2 binding, whereas inactivation of HS6ST2 actually enhanced binding, an effect that was recapitulated in the double HS6ST1−/−;HS6ST2−/− cell line (Figure 4B). Enhanced binding may reflect the increase in N- and 2-O-sulfation when 6-O-sulfation is reduced (Figure 2C).
Fig. 4.
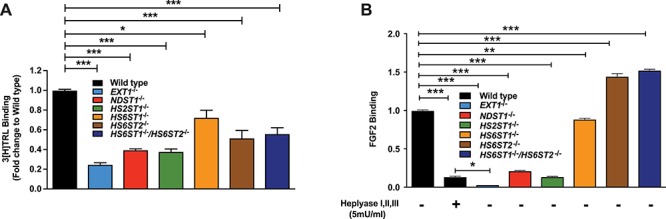
Cell surface heparan sulfate determines TRL and FGF2 binding. (A) Wild-type and mutant Hep3B cells were incubated with [3H] TRLs (100 μg/mL) for 1 h on ice. Bound [3H] TRLs were determined by liquid scintillation counting (n = 3 technical replicates). (B) Cells were incubated with biotinylated FGF2 at 4°C for 1 h. Binding was determined by PE/Cy5-conjugated streptavidin and flow cytometry. Data were analyzed using FlowJo software. A set of Hep3B wild-type cells was treated with heparin lyases I, II and III for 30 min at 37°C prior to incubation with FGF2 (n = 2 technical replicates). Statistics was calculated by one-way ANOVA. This figure is available in black and white in print and in color at Glycobiology online.
Binding of TRLs to clearance receptors results in internalization of the lipoprotein particles and delivery to lysosomes. To evaluate the role of HS in the uptake process, we incubated wild-type HS6ST1−/− and HS6ST2−/− cell lines with DiD-labeled human very low density lipoprotein (VLDL) at 37°C, and at various times the cells were treated with trypsin to remove cell surface bound particles. The extent of uptake was then measured at the indicated time points by flow cytometry, and the rate of TRL uptake was determined from the slope of the uptake curves (Figure 5A). Inactivation of HS6ST1 mildly affected the rate of VLDL internalization (5.5 ± 0.2 in wild type vs. 4.8 ± 0.2 RFU/μg cell protein; P = 0.004), whereas inactivation of HS6ST2 had a dramatic effect (3.8 ± 0.2 RFU/μg cell protein; P < 0.0001). Treatment with heparin lyases reduced the rate of uptake to the same baseline level in all three cell lines. The residual uptake results from internalization mediated by LDLR and LRP1 (Mahley and Huang 2007). Heparin inhibited binding of TRLs to all three receptors.
Fig. 5.
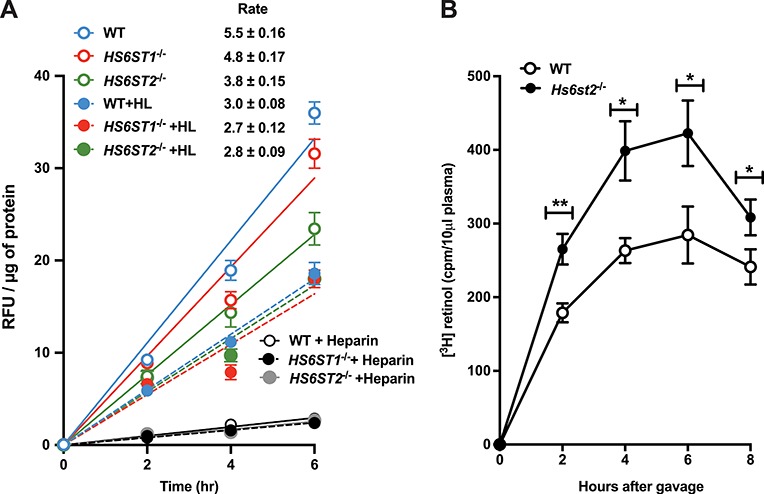
Heparan sulfate 6-O-sulfation is important for TRL uptake. (A) Uptake of TRLs in wild-type and SDC1−/− cells was measured using DiD-VLDL particles. Cells were treated with 100 μg/mL of DiD-VLDL particles in the presence and absence of heparin lyases (HL), or heparin (100 μg/mL). Cell uptake was measured using fluorescence reader, and the values were normalized to total cell protein (n = 2 biological replicates each performed in triplicate). (B) Postprandial clearance was measured by retinol excursion as described (Ishibashi et al. 1996). Briefly, 20 μCi of [11,12-3H] retinol (44.4 Ci/mmol; PerkinElmer) in ethanol was mixed with 1 mL of corn oil. Each mouse received 200 μL of the mixture by oral gavage. Blood was sampled at the times indicated by tail bleed, and radioactivity was measured in triplicate (10 μL serum) by scintillation counting. This figure is available in black and white in print and in color at Glycobiology online.
To determine the significance of this finding in vivo, we also measured TRL clearance in Hs6st2−/− mice. Fasted mice were given a bolus of [3H] retinol in corn oil by oral gavage, and blood was sampled at various time points to determine the plasma level of [3H] counts. In the intestine, [3H] retinol is converted to fatty acid esters and incorporated into newly made chylomicrons, which are metabolized to remnant TRLs in the plasma and cleared through the liver (Ishibashi et al. 1996). As shown in Figure 5B, the area under the curve for Hs6st2−/− mice (filled circles, 2500 ± 210) was 1.5 ± 0.2-fold greater than the wild type (open circles; 1700 ± 150), indicating that the mutant cleared intestinally derived TRLs at a slower rate. The decrease in tracer in both mutant and wild-type animals after 6 h is consistent with previous data showing that LDLR and LRP1 receptors also can clear plasma TRLs (Ishibashi et al. 1996; Horton et al. 1999; MacArthur et al. 2007).
Human hepatic SDC1 mediates TRL clearance in Hep3B cells
Previous studies identified SDC1 as a primary HSPG for TRL metabolism in mice (Stanford et al. 2009). However, in a previous study, we showed that when SDC1 expression was suppressed in Hep3B cells by siRNA, binding and uptake were only partially diminished (~35%), suggesting that either the extent of SDC1 silencing was incomplete or that other heparan sulfate proteoglycans can mediate binding and uptake (Deng et al. 2012). Other investigators have also reported that SDC1 can mediate lipoprotein metabolism in HepG2 cells based on antisense and antibody inhibition experiments (Zeng et al. 1998). In these published studies, TRL binding decreased by about 50–70% by treatment with siRNA or monoclonal antibodies to heparan sulfate.
To analyze the role of SDC1 in greater detail, we generated a SDC1−/− mutant using CRISPR/Cas9 by targeting exon 2. A clonal cell line was identified containing indels that caused frameshift mutations in both alleles (Figure 6A and Figure S2M and N). Western blotting and flow cytometry showed that Hep3B cells express SDC1, whereas SDC1−/− cells do not (Figure 6B and C). Deletion of SDC1 had no significant effect on overall sulfation of heparan sulfate based on disaccharide analysis (Figure 6D) and binding of FGF2 (Figure 6E), consistent with it being a minor proteoglycan. To examine the contribution of SDC1 in TRL binding, we incubated cells with fluorescent DiD-labeled human TRLs at 4°C. Deletion of SDC1 resulted in a modest reduction in binding (24 ± 4%; P = 0.005; Figure 7A). Treatment with heparin lyases decreased binding by 43 ± 6% in the wild type and reduced binding in SDC1−/− cells to the same level observed in the wild type. Given that Hep3B cells express high levels of glypicans (GPC3 and GPC6; Figure 1B), we examined if removal of these proteoglycans by phosphatidylinositol-specific phospholipase C (PIPLC) would affect TRL binding. PIPLC treatment decreased binding to a limited extent in wild-type and SDC1−/− cells (13 ± 3% in wild type; P = 0.04 and 22 ± 5% in SDC1−/− cells; P = 0.012). However, the combination of removing SDC1 by mutation and glypicans by PIPLC treatment reduced binding to the level observed after heparin lyase treatment (Figure 7A). The extent of proteoglycan-independent binding was higher in these experiments than in experiments utilizing [3H] TRLs (Figure 4A), presumably reflecting variable expression of LDLR and members of the LRP family of receptors in different experiments. The addition of heparin, which competes for binding to HSPG, LDLR and LRP1, greatly diminished TRL binding from both the cell lines (89 ± 6% and 83 ± 3%, in wild-type and mutant cells, respectively), consistent with this idea.
Fig. 6.
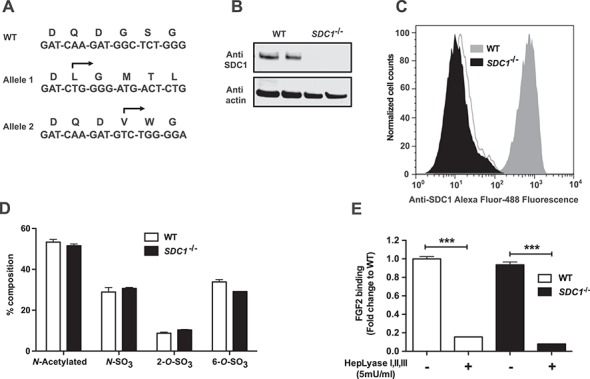
SDC1 null Hep3B cells. (A) Sanger sequencing of a region within exon 2 of SDC1 in wild-type Hep3B cells and in a cloned mutant cell line. The arrows indicate the start site of the altered DNA sequence in the mutant and the predicted amino acid sequence. Each allele resulted in a downstream frameshift mutation. (B) Extracts of wild-type and SDC1−/− cells western blotted with an antibody to human SDC1 after heparin lyase treatment. Anti-actin was used as a loading control. (C) Cell surface SDC1 was detected by flow cytometry. SDC1 null cell line (black filled) does not exhibit cell surface SDC1 as compared to wild type (gray filled). The background is indicated (not filled). (D) Heparan sulfate from wild-type (white bars) and SDC1−/− (black bars) cells was digested with heparin lyases, and the liberated disaccharides were analyzed by liquid chromatography/mass spectrometry (n = 2 biological replicates). (E) FGF2 binding in wild-type and SDC1−/− Hep3B cells before and after treatment of heparin lyases (n = 2 technical replicates). Statistics was calculated by one-way ANOVA. This figure is available in black and white in print and in color at Glycobiology online.
Fig. 7.
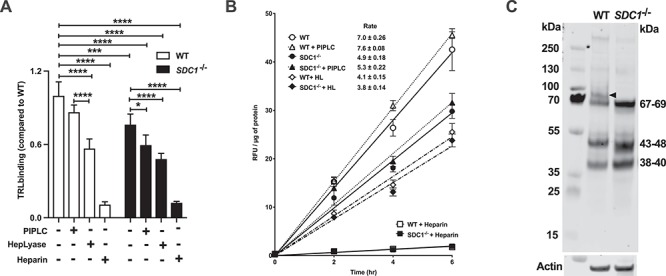
SDC1 regulates TRL uptake. Binding (A) and uptake (B) of TRLs in wild-type and SDC1−/− cells were measured using DiD-VLDL particles. (A) Cells were treated with 100 μg/mL of DiD-VLDL particles in the presence and absence of heparin lyases, PIPLC or heparin for 1 h in the dark on ice. Cell associated particles were measured using fluorescence reader, and the values were normalized to total cell protein (n = 2 biological replicates each performed in triplicate). (B) Wild-type and SDC1−/− cells were incubated at the indicated times at 37°C with 100 μg/mL of DiD-VLDL particles in the presence and absence of heparin lyases, PIPLC and heparin. The cells were treated with trypsin and the fluorescence of internalized particles was measured. The values represent the average fluorescence intensity values normalized to cell protein (n = 2 biological replicates each performed in triplicate). (C) WT and SDC1−/− cell lines were treated with heparin lyase I, II and III (each at 5 mU/mL) and neo-epitopes were detected using 3G10 antibody. SDC1 is indicated by the black arrowhead. Statistics calculated by t-test and two-way ANOVA. This figure is available in black and white in print and in color at Glycobiology online.
The extent of TRL uptake was measured at the indicated time points by flow cytometry, and the rate of TRL uptake was determined from the slope of the uptake curves (Figure 7B). PIPLC treatment did not significantly affect the rate of uptake of VLDL particles in wild-type cells (7.0 ± 0.3 vs. 7.6 ± 0.1 RFU/μg cell protein/min). In contrast, deletion of SDC1 reduced the rate of uptake significantly (4.9 ± 0.2 RFU/μg cell protein/min; P < 0.001; Figure 7B), a surprising finding given that SDC1 is a minor cell surface proteoglycan (Figure 7C, arrowhead). PIPLC treatment of the mutant also did not significantly affect uptake (5.3 ± 0.2 RFU/μg cell protein/min). Treatment of wild-type cells with heparin lyase reduced the rate of uptake almost to the value observed in SDC1−/− cells (4.1 ± 0.2 vs. 4.9 ± 0.2 RFU/μg cell protein/min; P < 0.0001). In contrast, treatment of the mutant with heparin lyases only slightly reduced the rate of uptake in the mutant (to 3.8 ± 0.1 RFU/μg cell protein/min; P < 0.01) compared to untreated cells. Inclusion of heparin dramatically reduced uptake, in parallel to its effect on binding.
Discussion
In this report, we generated a library of mutants of Hep3B cells altered in heparan sulfate assembly and SDC1. We provided genetic evidence that SDC1 and its covalently attached heparan sulfate chains account for at least half of the uptake of TRLs in Hep3B hepatocarcinoma cells, a model for human hepatocytes. All positions of sulfation along the chain, including 6-O-sulfation of glucosamine residues, appear to be important, extending previous findings in mice and murine hepatocytes to human hepatocytes. Importantly, other HSPGs such as the glypicans, SDC2 and SDC4 can bind to TRLs and possibly compete with SDC1, but they do not mediate uptake. Thus, SDC1 is the primary proteoglycan clearance receptor in both mouse and human hepatocytes.
Our original motivation for this study was to determine the role of 6-O-sulfation of heparan sulfate in binding and uptake of TRLs. Williams and colleagues showed early on in cultured cells that binding, uptake and degradation of lipoprotein particles that were modified to contain lipoprotein lipase provided a novel mechanism for cellular uptake dependent on cell surface HSPGs (Williams et al. 1992; Fuki et al. 1997). In vivo evidence supporting this notion was provided by Mahley and coworkers showing that rats injected with heparinase exhibited reduced clearance of plasma TRLs and reduced hepatic uptake (Ji et al. 1993). Subsequently, we showed in a series of studies of genetically altered mice that the heparan sulfate chains specifically attached to SDC1-mediated uptake of remnant TRLs and that these receptors acted independently of other endocytic receptors, LDLR and LRP1 (MacArthur et al. 2007; Stanford et al. 2009; Stanford et al. 2010; Foley et al. 2013). In one set of studies Stanford et al. (2010) showed that N-sulfation and 2-O-sulfation of the chains were important for binding and uptake but not 6-O-sulfation based on the lack of TRL accumulation in mice lacking hepatocyte Hs6st1 and the lack of effect of removal of 6-O-sulfate groups from heparin in competition studies performed in vitro. However, conflicting data emerged from studies of type 2 diabetes (T2D) in mice, which impairs hepatic clearance of atherogenic postprandial remnant lipoproteins (Chen et al. 2010). A microarray transcript analysis of livers from T2D db/db mice fed ad libitum showed that the heparan sulfate glucosamine-6-O-endosulfatase-2 (Sulf2), which catalyzes the removal of 6-O-sulfate groups, was induced. Silencing of Sulf2 in cultured hepatocytes doubled HSPG-mediated catabolism of model remnant TRLs, whereas induction of Sulf2 expression impaired TRL catabolism (Chen et al. 2010). A parallel study in vivo in mice using second-generation antisense oligonucleotides targeting Sulf2 suppressed hepatic Sulf2 mRNA expression and lowered plasma triglyceride levels by 50% (Hassing et al. 2012). These findings implied that 6-O-sulfation indeed plays a role in TRL metabolism. We have now confirmed this finding in Hep3B cells through inactivation of HS6ST2. Thus, all of the classes of sulfate groups on heparan sulfate (N-sulfate, 2-O-sulfate and 6-O-sulfate groups) appear to be important for TRL binding to SDC1 and uptake. It remains to be determined if 3-O-sulfation of glucosamine residues is important for TRL binding and uptake (Thacker et al. 2013).
Lipoproteins carry several apolipoproteins (apo), which help define their identity and metabolism, including their interaction with various clearance receptors. TRLs carry apoE, apoAV and apoB all of which can bind to heparin. Clearance through LDLR and LRP1 is mediated by apoB and apoE, whereas clearance through SDC1 depends on apoE and apoAV (Gonzales et al. 2013). Dong et al. showed that the N-terminal domain of apoE contains the major heparin-binding site and exhibits high affinity for an octasaccharide of the structure D2S6-I2S6-I2S6-I2S6 (Dong et al. 2001; Libeu et al. 2001). ApoAV also binds heparin, but the structural features of heparin required for binding have not been examined (Lookene et al. 2005). Thus, the available data support the idea that apoE and perhaps apoAV bind to heparan sulfate chains that contain 6-O-sulfate as well as N-sulfate and 2-O-sulfate groups. Additional structural studies are needed to determine if binding depends on a specific arrangement of sulfated residues in the heparan sulfate chains on SDC1.
Most cells express multiple HSPGs (Lindahl et al. 2015), including primary human hepatocytes, Hep3B cells and murine primary hepatocytes (Foley et al. 2013) (Figure 1). Binding of TRLs to Hep3B cells was reduced in EXT1-deficient cells to the same extent as observed after heparin lyase treatment, whereas deletion of SDC1 had only a minor impact on binding. Thus, much of the heparan sulfate-dependent binding of TRLs is mediated by HSPGs other than SDC1. In contrast, when TRL internalization was measured, SDC1 accounted for all of the heparan sulfate-dependent uptake, indicating that SDC1 is the dominant HSPG endocytic receptor (Figure 7B). Studies of TRL clearance in SDC1−/− mice support this notion; deletion of other HSPGs had little effect (Stanford et al. 2009). The lack of participation by other HSPGs in clearance may reflect differences in the distribution of the HSPGs (apical vs. basolateral) or their capacity to undergo endocytosis.
Recent studies in humans suggest that heterozygosity of EXT1 or GLCE (GlcA C5 epimerase involved in formation of iduronic acid) has only a minor effect on TRL metabolism. (Hodoğlugil et al. 2011; Mooij et al. 2015). Individuals bearing homozygous loss-of-function mutations in these and other heparan sulfate biosynthetic genes have not been described. This fact and the observation that none of the enzymes involved in heparan sulfate formation have been identified in genome-wide association studies (GWAS) studies of hypertriglyceridemia in humans may reflect the essential requirement of heparan sulfate in human embryonic development and adult physiology (Johansen et al. 2011; Kurano et al. 2016). Systemic homozygous deletion of these genes in mice results in embryonic lethality obviating lipoprotein metabolism studies in adult animals (Bullock et al. 1998; Lin et al. 2000; Grobe et al. 2005; Stickens et al. 2005). Conditional ``flox” alleles inactivated by hepatocyte-specific expression of Cre circumvent this problem (MacArthur et al. 2007; Stanford et al. 2010), but not all genes involved in heparan sulfate formation have been targeted in this way. As an alternative approach, we generated a set of homozygous null mutants in key enzymes involved in heparan sulfate formation. The Hep3B cell lines described here complement recently published libraries of CHO and endothelial cell lines altered in heparan sulfate synthesis (Chen et al. 2018; Qiu et al. 2018). Together, these libraries can be used to study lipoprotein metabolism as well as the many other biological processes affected by HSPGs (Bishop et al. 2007; Iozzo and Sanderson 2011; Xu and Esko 2014; Chung et al. 2016; Afratis et al. 2017; Kjellén and Lindahl 2018).
Materials and methods
Cell culture
Human primary hepatocytes were obtained from a public repository managed by the University of Minnesota under contract from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at the National Institutes of Health (Liver Tissue Cell Distribution System). The analysis of enzyme expression was derived from RNA isolated from cells obtained from a resected liver from a 50-year-old female patient with a colon cancer prior to receiving chemotherapy. HSPG mRNA expression was analyzed in three human hepatocyte preparations, two of which were published previously as Supporting Figure S1 in Deng et al. (2012). Hep3B cells were obtained from the American Type Culture Collection (HB8064, HB 8065; Manassas, VA) and cultured in Minimum Essential Medium [Thermo Fisher Scientific (Carlsbad, CA)] supplemented with 10% fetal bovine serum [Atlanta Biologicals (Flowery Branch, GA)], nonessential amino acids (Thermo Fisher Scientific), penicillin and streptomycin. The cells were maintained at 37°C under an atmosphere of 5% CO2/95% air.
Generation of cell lines
Guide RNAs for targeted genes were designed according to Broad Institute published resources (Ran et al. 2013) and were synthesized by ValueGene. Guide RNAs (100 μM) were annealed using T4 polynucleotide kinase [New England Biolabs (Ipswich, MA)] and integrated into vector pSp-Cas9(BB)-2A-Puro (a gift from Dr. Feng Zhang) using T7 ligase (New England Biolabs). The vectors were sequenced by Sanger sequencing to confirm their correct construction and transfected into Hep3B cells using Lipofectamine 2000 according to the manufacturer’s instructions [Invitrogen (Carlsbad, CA)]. Transfected Hep3B cells were selected with puromycin (3 μg/mL), and single-cell clones were isolated by limiting dilution. Mutations in the targeted region were determined by Sanger sequencing. For siRNA treatment, cells were transfected with scrambled or HS6ST3 siRNA [CCTACAACCTGG, Sigma-Aldrich (St. Louis, MO)] using Lipofectamine 3000 according to manufacturer’s instruction (Invitrogen).
Flow cytometry
Cells were lifted using cell dissociation buffer [Life Technologies (Carlsbad, CA)] and were incubated with biotinylated recombinant FGF2 [R&D Systems (Minneapolis, MN)] on ice (Weiss et al. 2015). Bound FGF2 was measured by reaction of the cells with streptavidin conjugated to phycoerythrin/Cy5 and flow cytometry [BD Biosciences (San Jose, CA)]. SDC1 expression was measured by binding mAb CD-138 [Bio-Rad (Hercules, CA)] followed by Alexa Fluor 488-conjugated secondary antibody. Data were analyzed using the FlowJo software [FlowJo, LLC (Ashland, OR)].
SDS-PAGE and western blotting
Wild-type and SDC1−/− cells were treated with 5 mU each of heparin lyase I, II and III for 30 min at 37°C before extraction with RIPA buffer containing protease inhibitors [cOmplete mini, Roche (Mannheim, Germany)]. Samples (20 μg protein) were subjected to SDS-PAGE (4–12% Bis-Tris, NuPAGE, Invitrogen), blotted on PVDF membranes [Millipore (Temacula, CA)] and probed for SDC1 [anti-CD-138, AbD Serotec (Hercules, CA)] or stub oligosaccharides arising from heparin lyase digestion [3G10, Seikagaku (Tokyo, Japan)].
Liquid chromatography/mass spectrometry
Heparan sulfate was isolated from cells as described previously (Lawrence et al. 2008). Briefly, cells were grown to confluence and digested with 0.4 mg/mL pronase (Sigma-Aldrich) at 37°C for 16 h. Crude glycosaminoglycans (GAGs) were isolated by anion exchange chromatography using DEAE-Sephacel [0.5 mL; GE Healthcare (Chicago, IL)]. The columns were washed with 50 mM sodium acetate buffer (pH 6) containing 0.25 M NaCl, and the GAGs were eluted with buffer containing 2 M NaCl. Heparan sulfate chains were depolymerized with recombinant heparin lyase I, II and III (5 mU/mL of each), and unsaturated disaccharides were analyzed using Glycan Reductive Isotope Labeling-Liquid Chromatography/Mass Spectrometry (Lawrence et al. 2008). The disaccharide structure code is used: D0H0, ΔUA-GlcNH2; D0A0, ΔUA-GlcNAc; D0H6, ΔUA-GlcNH26S; D2H0, ΔUA2S-GlcNH2; D0S0, ΔUA-GlcNS; D2H6, ΔUA2S-GlcNH26S; D0A6, ΔUA-GlcNAc6S; D0S6, ΔUA-GlcNS6S; D2A0, ΔUA2S-GlcNAc; D2S0, ΔUA2S-GlcNS; D2A6, ΔUA2S-GlcNAc6S; D2S6, ΔUA2S-GlcNS6S; where ΔUA = 4,5-unsaturated uronic acid (Lawrence et al. 2008) and I = iduronic acid.
TRL binding and uptake
To prepare radiolabeled TRLs, mice were fasted for 5 h and orally gavaged with corn oil (250 μL) containing 10 μCi of [3H] retinol [PerkinElmer (Waltham, MA), 43 Ci/mmol] (Gordts et al. 2016). Blood was collected through cardiac puncture after 3 h. Plasma samples were centrifuged for 6 h at 38,000 rpm in a Beckman Coulter rotor (42.2Ti), and the top 50 μL fraction was collected and used for TRL-binding studies. Cells were incubated with radioactive lipoproteins at 4°C for 1 h, and counts associated with the cells were measured by liquid scintillation.
Postprandial human TRLs were isolated from 4 h fasted, healthy volunteers by buoyant density ultracentrifugation (δ < 1.006 g/mL). Isolated TRLs (1–2 mg protein) were mixed with 100 μL of 3 mg/mL 1,1-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD; Invitrogen) in dimethylsulfoxide. After 8 h, DiD-labeled TRLs were separated from unincorporated dye by buoyant density ultracentrifugation. Cells were incubated with DiD-TRL particles either at 4°C for 1 h or 37°C for various times. After washing the cells, they were harvested with trypsin and fluorescence intensity was measured using a fluorescence reader (excitation at 644 nm, emission at 665 nm; EnSpire Alpha, PerkinElmer).
Quantitative PCR
RNA was isolated from cells using Qiagen RNeasy kit according to manufacturer’s instruction. mRNA was converted to cDNA using SuperScript III (Invitrogen) according to manufacturer’s instruction. Specific transcripts were amplified using gene specific primers (Supporting Table S1) and quantitated using SYBR Green dye (Bio-Rad).
Statistics. Statistical analyses were performed using Prism Version 5.0 software [GraphPad Software (San Diego, CA)]. Comparisons were analyzed using one-way, two-way ANOVA and t-test as indicated in the figure legends. Data are presented as mean ± SD. *P < 0.01, **P < 0.001, ***P < 0.0001, ****P < 0.00001.
Funding
National Institutes of Health (GM33063 to J.D.E.); Fondation Leducq (01CVD16 to P.L.S.M.G.).
Conflict of interest statement
None declared.
Supplementary Material
References
- Afratis NA, Nikitovic D, Multhaupt HA, Theocharis AD, Couchman JR, Karamanos NK. 2017. Syndecans—key regulators of cell signaling and biological functions. FEBS J. 284:27–41. [DOI] [PubMed] [Google Scholar]
- Anower EKMF, Habuchi H, Nagai N, Habuchi O, Yokochi T, Kimata K. 2013. Heparan sulfate 6-O-sulfotransferase isoform-dependent regulatory effects of heparin on the activities of various proteases in mast cells and the biosynthesis of 6-O-sulfated heparin. J Biol Chem. 288:3705–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XM, Esko JD. 1996. An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J Biol Chem. 271:17711–17717. [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 446:1030–1037. [DOI] [PubMed] [Google Scholar]
- Bullock SL, Fletcher JM, Beddington RS, Wilson VA. 1998. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 12:1894–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Liu ML, Schaffer L, Li M, Boden G, Wu X, Williams KJ. 2010. Type 2 diabetes in mice induces hepatic overexpression of sulfatase 2, a novel factor that suppresses uptake of remnant lipoproteins. Hepatology. 52:1957–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Narimatsu Y, Clausen TM, Gomes C, Karlsson R, Steentoft C, Spliid CB, Gustavsson T, Salanti A, Persson A et al. 2018. The GAGOme: A cell-based library of displayed glycosaminoglycans. Nat Methods. 15:881–888. [DOI] [PubMed] [Google Scholar]
- Chung H, Multhaupt HA, Oh ES, Couchman JR. 2016. Minireview: Syndecans and their crucial roles during tissue regeneration. FEBS Lett. 590:2408–2417. [DOI] [PubMed] [Google Scholar]
- Dejima K, Takemura M, Nakato E, Peterson J, Hayashi Y, Kinoshita-Toyoda A, Toyoda H, Nakato H. 2013. Analysis of Drosophila glucuronyl C5-epimerase: Implications for developmental roles of heparan sulfate sulfation compensation and 2-O-sulfated glucuronic acid. J Biol Chem. 288:34384–34393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Foley EM, Gonzales JC, Gordts PL, Li Y, Esko JD. 2012. Shedding of syndecan-1 from human hepatocytes alters very low density lipoprotein clearance. Hepatology. 55:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Peters-Libeu CA, Weisgraber KH, Segelke BW, Rupp B, Capila I, Hernaiz MJ, LeBrun LA, Linhardt RJ. 2001. Interaction of the N-terminal domain of apolipoprotein E4 with heparin. Biochemistry. 40:2826–2834. [DOI] [PubMed] [Google Scholar]
- El Masri R, Seffouh A, Lortat-Jacob H, Vives RR. 2017. The "in and out" of glucosamine 6-O-sulfation: The 6th sense of heparan sulfate. Glycoconj J. 34:285–298. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. 2002. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 71:435–471. [DOI] [PubMed] [Google Scholar]
- Foley EM, Esko JD. 2010. Hepatic heparan sulfate proteoglycans and endocytic clearance of triglyceride-rich lipoproteins. Prog Mol Biol Transl Sci. 93:213–233. [DOI] [PubMed] [Google Scholar]
- Foley EM, Gordts PL, Stanford KI, Gonzales JC, Lawrence R, Stoddard N, Esko JD. 2013. Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler Thromb Vasc Biol. 33:2065–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuki IV, Kuhn KM, Lomazov IR, Rothman VL, Tuszynski GP, Iozzo RV, Swenson TL, Fisher EA, Williams KJ. 1997. The syndecan family of proteoglycans. Novel receptors mediating internalization of atherogenic lipoproteins in vitro. J Clin Invest. 100:1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster MM, Wang L, Castagnola J, Sikora L, Reddi K, Lee PH, Radek KA, Schuksz M, Bishop JR, Gallo RL et al. 2007. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 177:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales JC, Gordts PL, Foley EM, Esko JD. 2013. Apolipoproteins E and AV mediate lipoprotein clearance by hepatic proteoglycans. J Clin Invest. 123:2742–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE et al. 2016. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. 126:2855–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. 2005. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 132:3777–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi H, Tanaka M, Habuchi O, Yoshida K, Suzuki H, Ban K, Kimata K. 2000. The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J Biol Chem. 275:2859–2868. [DOI] [PubMed] [Google Scholar]
- Hagner-McWhirter A, Li JP, Oscarson S, Lindahl U. 2004. Irreversible glucuronyl C5-epimerization in the biosynthesis of heparan sulfate. J Biol Chem. 279:14631–14638. [DOI] [PubMed] [Google Scholar]
- Hassing HC, Mooij H, Guo S, Monia BP, Chen K, Kulik W, Dallinga-Thie GM, Nieuwdorp M, Stroes ES, Williams KJ. 2012. Inhibition of hepatic sulfatase-2 in vivo: A novel strategy to correct diabetic dyslipidemia. Hepatology. 55:1746–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassing HC, Surendran RP, Derudas B, Verrijken A, Francque SM, Mooij HL, Bernelot Moens SJ, Hart LM, Nijpels G, Dekker JM et al. 2014. SULF2 strongly prediposes to fasting and postprandial triglycerides in patients with obesity and type 2 diabetes mellitus. Obesity (Silver Spring). 22:1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodoğlugil U, Williamson DW, Yu Y, Farrer LA, Mahley RW. 2011. Glucuronic acid epimerase is associated with plasma triglyceride and high-density lipoprotein cholesterol levels in Turks. Ann Hum Genet. 75:398–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shimano H, Hamilton RL, Brown MS, Goldstein JL. 1999. Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J Clin Invest. 103:1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV, Sanderson RD. 2011. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 15:1013–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Perrey S, Chen Z, Osuga J, Shimada M, Ohashi K, Harada K, Yazaki Y, Yamada N. 1996. Role of the low density lipoprotein (LDL) receptor pathway in the metabolism of chylomicron remnants. A quantitative study in knockout mice lacking the LDL receptor, apolipoprotein E, or both. J Biol Chem. 271:22422–22427. [DOI] [PubMed] [Google Scholar]
- Jemth P, Smeds E, Do AT, Habuchi H, Kimata K, Lindahl U, Kusche-Gullberg M. 2003. Oligosaccharide library-based assessment of heparan sulfate 6-O-sulfotransferase substrate specificity. J Biol Chem. 278:24371–24376. [DOI] [PubMed] [Google Scholar]
- Ji ZS, Brecht WJ, Miranda RD, Hussain MM, Innerarity TL, Mahley RW. 1993. Role of heparan sulfate proteoglycans in the binding and uptake of apolipoprotein E-enriched remnant lipoproteins by cultured cells. J Biol Chem. 268:10160–10167. [PubMed] [Google Scholar]
- Johansen CT, Kathiresan S, Hegele RA. 2011. Genetic determinants of plasma triglycerides. J Lipid Res. 52:189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellén L, Lindahl U. 2018. Specificity of glycosaminoglycan-protein interactions. Curr Opin Struct Biol. 50:101–108. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Salmivirta M, Sturiale L, Giménez-Gallego G, Lindahl U. 2001. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 276:30744–30752. [DOI] [PubMed] [Google Scholar]
- Kurano M, Tsukamoto K, Kamitsuji S, Kamatani N, Hara M, Ishikawa T, Kim BJ, Moon S, Jin Kim Y, Teramoto T. 2016. Genome-wide association study of serum lipids confirms previously reported associations as well as new associations of common SNPs within PCSK7 gene with triglyceride. J Hum Genet. 61:427–433. [DOI] [PubMed] [Google Scholar]
- Lawrence R, Lu H, Rosenberg RD, Esko JD, Zhang L. 2008a. Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat Methods. 5:291–292. [DOI] [PubMed] [Google Scholar]
- Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, Cummings RD, Esko JD. 2008b. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem. 283:33674–33684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledin J, Ringvall M, Thuveson M, Eriksson I, Wilén M, Kusche-Gullberg M, Forsberg E, Kjellén L. 2006. Enzymatically active N-deacetylase/N-sulfotransferase-2 is present in liver but does not contribute to heparan sulfate N-sulfation. J Biol Chem. 281:35727–35734. [DOI] [PubMed] [Google Scholar]
- Ledin J, Staatz W, Li JP, Götte M, Selleck S, Kjellén L, Spillmann D. 2004. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem. 279:42732–42741. [DOI] [PubMed] [Google Scholar]
- Libeu CP, Lund-Katz S, Phillips MC, Wehrli S, Hernáiz MJ, Capila I, Linhardt RJ, Raffaï RL, Newhouse YM, Zhou F et al. 2001. New insights into the heparan sulfate proteoglycan-binding activity of apolipoprotein E. J Biol Chem. 276:39138–39144. [DOI] [PubMed] [Google Scholar]
- Lin X, Wei G, Shi ZZ, Dryer L, Esko JD, Wells DE, Matzuk MM. 2000. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev Biol. 224:299–311. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Couchman J, Kimata K, Esko JD. 2015. Proteoglycans and sulfated glycosaminoglycans In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH et al., editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; p. 207–221. [Google Scholar]
- Lookene A, Beckstead JA, Nilsson S, Olivecrona G, Ryan RO. 2005. Apolipoprotein A-V-heparin interactions: Implications for plasma lipoprotein metabolism. J Biol Chem. 280:25383–25387. [DOI] [PubMed] [Google Scholar]
- MacArthur JM, Bishop JR, Wang L, Stanford KI, Bensadoun A, Witztum JL, Esko JD. 2007. Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J Clin Invest. 117:153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. 2007. Atherogenic remnant lipoproteins: Role for proteoglycans in trapping, transferring, and internalizing. J Clin Invest. 117:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooij HL, Bernelot Moens SJ, Gordts PL, Stanford KI, Foley EM, van den Boogert MA, Witjes JJ, Hassing HC, Tanck MW, van de Sande MA et al. 2015. Ext1 heterozygosity causes a modest effect on postprandial lipid clearance in humans. J Lipid Res. 56:665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard BG, Varbo A. 2014. Triglycerides and cardiovascular disease. Lancet. 384:626–635. [DOI] [PubMed] [Google Scholar]
- Peng J, Luo F, Ruan G, Peng R, Li X. 2017. Hypertriglyceridemia and atherosclerosis. Lipids Health Dis. 16:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Shi S, Yue J, Xin M, Nairn AV, Lin L, Liu X, Li G, Archer-Hartmann SA, Dela Rosa M et al. 2018. A mutant-cell library for systematic analysis of heparan sulfate structure-function relationships. Nat Methods. 15:889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong J, Habuchi H, Kimata K, Lindahl U, Kusche-Gullberg M. 2001. Substrate specificity of the heparan sulfate hexuronic acid 2-o-sulfotransferase. Biochemistry. 40:5548–5555. [DOI] [PubMed] [Google Scholar]
- Smeds E, Habuchi H, Do AT, Hjertson E, Grundberg H, Kimata K, Lindahl U, Kusche-Gullberg M. 2003. Substrate specificities of mouse heparan sulphate glucosaminyl 6-O-sulphotransferases. Biochem J. 372:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, Esko JD. 2009. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest. 119:3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Wang L, Castagnola J, Song D, Bishop JR, Brown JR, Lawrence R, Bai X, Habuchi H, Tanaka M et al. 2010. Heparan sulfate 2-O-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J Biol Chem. 285:286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickens D, Zak BM, Rougier N, Esko JD, Werb Z. 2005. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 132:5055–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talayero BG, Sacks FM. 2011. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 13:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker BE, Xu D, Lawrence R, Esko JD. 2013. Heparan sulfate 3-O-sulfation: A rare modification in search of a function. Matrix Biol. 35:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull JE, Fernig DG, Ke Y, Wilkinson MC, Gallagher JT. 1992. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J Biol Chem. 267:10337–10341. [PubMed] [Google Scholar]
- Weiss RJ, Gordts PLSM, Le D, Xu D, Esko JD, Tor Y. 2015. Small molecule antagonists of cell-surface heparan sulfate and heparin-protein interactions. Chem Sci. 6:5984–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Fless GM, Petrie KA, Snyder ML, Brocia RW, Swenson TL. 1992. Mechanisms by which lipoprotein lipase alters cellular metabolism of lipoprotein(a), low density lipoprotein, and nascent lipoproteins. Roles for low density lipoprotein receptors and heparan sulfate proteoglycans. J Biol Chem. 267:13284–13292. [PubMed] [Google Scholar]
- Xu D, Esko JD. 2014. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 83:129–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. 1991. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 64:841–848. [DOI] [PubMed] [Google Scholar]
- Zeng BJ, Mortimer BC, Martins IJ, Seydel U, Redgrave TG. 1998. Chylomicron remnant uptake is regulated by the expression and function of heparan sulfate proteoglycan in hepatocytes. J Lipid Res. 39:845–860. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.