

Abstract
The specificity and action pattern of a β-glucuronidase derived from the pathogenic bacteria Burkholderia pseudomallei and expressed in Escherichia coli as a recombinant protein has been evaluated. While this enzyme shows activity on a number of glycosaminoglycans, our study has focused on its action on heparin, heparan sulfate and their biosynthetic intermediates as well as chemoenzymatically synthesized, structurally defined heparan sulfate oligosaccharides. These heparin/heparan sulfate (HP/HS) substrates examined varied in size and structure, but all contained an uronic acid (UA) residue β-(1→4) linked to a glucosamine residue. On the substrates tested, this enzyme (heparanase Bp) acted only on a glucuronic acid residue β-(1→4) linked to an N-acetylglucosamine, N-sulfoglucosamine or N-acetyl-6-O-sulfoglucosamine residue. A substrate was required to have a length of pentasaccharide or longer and heparanase Bp acted with a random endolytic action pattern on HP/HS. The specificity and glycohydrolase mechanism of action of heparanase Bp resembles mammalian heparanase and is complementary to the bacterial heparin lyases, which act through an eliminase mechanism on a glucosamine residue (1→4) linked to a UA residue, suggesting its utility as a tool for the structural determination of HP/HS as well as representing a possible model for the medically relevant mammalian heparanase. The utility heparanase Bp was demonstrated by the oligosaccharide mapping of heparin, which afforded resistant intact highly sulfated domains ranging from tetrasaccharide to >28-mer with a molecular weight >9000.
Keywords: action pattern, heparan sulfate, heparanase Bp, heparin, specificity
Introduction
Glycosaminoglycans (GAGs) are anionic, polydisperse, linear polysaccharides and are generally located in the cell membrane or in the extracellular matrix (ECM) of all animal tissues (Lindahl et al. 2017). GAGs are classified into four families based on their biosynthetic pathway and their resulting disaccharide repeating units. Heparin/heparan sulfate (HP/HS), chondroitin sulfate/dermatan sulfate, hyaluronic acid and keratan sulfate are the major classes of GAGs. HP/HS are the most structural complex members of GAGs and perform a variety of critical biological functions (Esko and Selleck 2002; Linhardt 2003). HP/HS are biosynthesized in the endoplasmic reticulum and Golgi on serine residues of core proteins through a tetrasaccharide linkage region, β-D-glucuronic acid (GlcA) (1→3) β-D-galactose (Gal) (1→3) β-D-Gal (1→4) β-D-xylose (1→ Serine. A disaccharide repeating unit, →4) GlcA (1→4) N-acetyl-α-D-glucosamine (GlcNAc), called heparosan (HN), extending from this linkage region is biosynthesized in the Golgi through the action of glycosyltransferases. The GlcA residues in the resulting HN chain can be epimerized to α-L-iduronic acid (IdoA) by C5-epimerase and then 2-O-sulfated by a 2-O-sulfotransferase, while the GlcNAc residue can be de-N-acetylated and N-sulfated through the action of a N-deacetylase N-sulfotransferase and subsequently O-sulfated at 6- and/or 3-position by a variety of 6- and/or 3-O-sulfotransferases. HP/HS have enormous structural heterogeneity because the modification of their HN chain backbone, →4 GlcA (1→4) GlcNAc (1→, is incomplete, making the determination of HP/HS sequence extremely difficult. The major disaccharide in HP is →4) IdoA2S (1→4) GlcNS6S (1→ (where S is sulfo) and the major disaccharide in HS is the unsulfated HN disaccharide, →4) GlcA (1→4) GlcNAc (Linhardt et al. 1988; Griffin et al. 1995). Heparin lyases (also known as heparinases), first described in Flavobacterium heparinum in 1956 (Payza and Korn 1956), are a class of enzymes that selectively cleave the glucosamine (1→4) uronic acid (UA) linkage in HP/HS through an β-eliminative mechanism, resulting in non-reducing end unsaturated UA residues (ΔUA, 4-deoxy-α-L-threo-hexenopyranosyluronic acid) having a UV absorbance at 232 nm (Linhardt et al. 1986; Linhardt 1994). There are three well-characterized heparin lyases that act endolytically on HP/HS, heparinase I, heparinase II and heparinase III (Jandik et al. 1994). Heparin lyase I is selective in cleaving highly sulfated polysaccharide chains containing IdoA2S residues, heparin lyase II displays a broad substrate specificity cleaving most of the (1→4) linkages between glucosamine and UA present in HP/HS and heparin lyase III selectively cleaves linkages having a reduced level of sulfation and those principally the glycosidic linkages to GlcNAc residues (Desai et al. 1993a; Desai et al. 1993b). An excellent understanding of heparin lyase I, II and III specificity (Desai et al. 1993a; Desai et al. 1993b) and their ready availability as either native (Lohse and Linhardt 1992) or recombinantly bacterially expressed enzymes (Su et al. 1996) have made these lyases particularly useful in HP/HS sequencing (Liu et al. 1995). Heparin lyases show some size selectively acting better on polysaccharides and oligosaccharides larger than tetrasaccharide in length (Rice and Linhardt 1989).
There is only one known mammalian endo-β-glucuronidase capable of cleaving the HP/HS side chains of HP/HS proteoglycans (Vlodavsky et al. 2018). Mammalian heparanase hydrolyzes the GlcA β-(1→4) GlcNAc/GlcNS linkage and is a β-endoglucuronidase belonging to the glycoside hydrolase 79 [GH79] family. The substrate specificity of mammalian heparanase is only partially understood, currently the known site specificity is that O-sulfo groups are essential for recognition but not N-sulfation, unmodified, N-sulfated and partially epimerized HN are all resistant to mammalian heparanase degradation (Pikas et al. 1998), heparanase cleaves linkages between GlcUA and GlcNS that is 6-O-sulfated or 3, 6-O-disulfated and the target GlcUA residues are in highly sulfated domains (Peterson and Liu 2010; Peterson and Liu 2012, Peterson and Liu 2013). The gene encoding mammalian heparanase results in an inactive pro-enzyme that must be proteolytically cleaved to form the active enzyme (Nardella et al. 2004; Abboud-Jarrous et al. 2005). Mammalian heparanase is involved in multiple biological functions including tumor growth, metastasis, angiogenesis and inflammation, as it cleaves HS (Heyman and Yang 2016). Understanding mammalian heparanase substrate specificity has played a critical role in revealing these biological functions. Although the specificity and action pattern of mammalian heparanase have been well studied (Pikas et al. 1998; Okada et al. 2002; Peterson and Liu 2010; Peterson and Liu 2012), the exact substrate specificity of mammalian heparanase is not completely understood as the enzyme recognizes a fairly long the sequence making a full structural characterization quite difficult (Peterson and Liu 2013).
Enzymes acting on HP/HS are important tools in glycobiology and medicine. A major application for the heparin lyases is for the structural studies of HP-based drugs and HS recovered from tissues. The combined use of heparin lyase I, II and III can nearly completely convert HP/HS to disaccharide products for structural analysis, although some resistant tetrasaccharides with 3-O-sulfoglucosamine residues at their reducing ends are formed (Xiao et al. 2011; Chen et al. 2017). Heparin lyase I has also been utilized both in vitro and in vivo for the elimination of heparin’s anticoagulant activity from blood (Langer et al. 1982). The heparin lyases also have utility in mapping and sequencing of HS/HP and in preparation of potentially useful therapeutic agents (Xiao et al. 2011; Fu et al. 2014). A major limitation of heparin lyases as reagents is that their β-eliminase mechanism results in a loss of information on the C5 chirality of the UA (GlcA vs. IdoA) adjacent to the site of cleavage, making it necessary to deduce UA chirality from an understanding of the specificity of these enzymes. In contrast, the major application of mammalian heparanase is in medicine or basic biology as an important enzyme regulating HS in ECM. Overexpression of mammalian heparanase results in breakdown of HS and release of a variety of HS-bound molecules including growth factors, cytokines and chemokines, thus, making mammalian heparanase for medical applications in cancer diagnosis an attractive target for the development of anticancer drugs (Wu et al. 2015; Pala et al. 2016). Furthermore, mammalian heparanase is involved in other non-cancer related diseases, such as inflammation, wound healing and diabetes (Simeonovic et al. 2013). Furthermore, mammalian heparanase inhibitors have therapeutic potential in the treatment of a number of diseases. Major limitations for using mammalian heparanase as a reagent are that as a pro-enzyme it is difficult to produce the active form and since it is a glycoprotein it often requires preparation in eukaryotic cell culture, making it difficult to obtain large quantities of catalytically active mammalian heparanase.
Recently, Bohlmann and coworkers (Bohlmann et al. 2015) reported a bacterial heparanase, heparanase Bp, an endo-β-glucuronidase, from the pathogenic bacteria Burkholderia pseudomallei, which they cloned and expressed in Escherichia coli to examine its structure and activity. In the current study, we performed a thorough characterization of the specificity and action pattern of heparanase Bp on a variety of substrates, HP, HS, biosynthetic intermediates of HP/HS and chemoenzymatically synthesized, structurally defined HS oligosaccharides. It must be noted that heparanase Bp is an endo-β-glucuronidase that not only acts on HP/HS but also can act on hyaluronan and chondroitin sulfate, acting also as hyaluronidase/chondroitinase cleaving GlcA β(1→3) GlcNAc/N-acetyl-D-galactosamine (Bohlmann et al. 2015). While our studies confirm this observation, we have focused our attention only on the application of this endo-β-glucuronidase only as a heparanase acting on HP/HS, their various biosynthetic intermediates and HS oligosaccharides.
Results
Cloning and production of heparanase Bp
Based on the work of Bohlmann and coworkers, we cloned and expressed heparanase Bp with a glutathione S-transferase (GST) tag. In doing this cloning, we found an error in the Bohlmann paper in that the forward primer used for amplification of the gene encoding heparanase was incorrect as it did not contain a SmaI restriction site for cloning of the amplicon. The polymerase chain reaction (PCR) primers we designed corrected this error and are listed in Table SI. Our construct also had the N-terminal truncation of the putative signal peptide and we used the pGEX-4 T-1 expression vector for cloning, not the pEX vector used by Bohlmann and coworkers. After fermentation to produce the target protein, it was purified on a glutathione column (Figure 1). The molecular weight of purified target protein with TSG tag in lane 4 (Figure 1) was determined to be ~74 kD by SDS–polyacrylamide gel electrophoresis (PAGE). The theoretical size of the protein was calculated (using ExPasy) to be ~74 kD with the GST tag, and ~48 kDa without the GST tag. The cleaved and uncleaved versions of the enzymes were both found to have similar activities, and the cleaved version was used in all of our subsequent studies. Using HS as substrate, we re-examined buffer system and found that Bohlmann and coworkers had correctly identified an acidic pH optimal of 4.5. Thus, we selected 50-mM ammonium acetate (pH 4.5) as a volatile buffer to conduct our studies, which relied on liquid chromatography (LC)–mass spectrometry (MS) analysis.
Fig. 1.
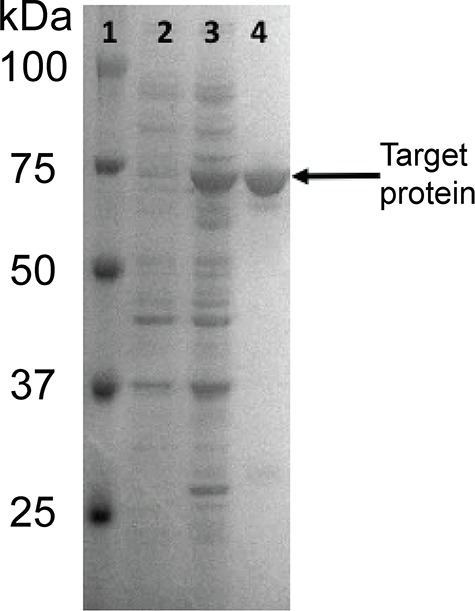
SDS-PAGE analysis of protein fractions. Lanes are 1: molecular weight ladder, 2: uninduced protein fraction, 3: induced protein fraction and 4: purified protein with GST tag. Target protein MW is ~74 kDa.
Action of heparanase Bp on polysaccharide substrates
The enzyme was next tested on multiple substrates HS, HP, HN, N-deacetylated, N-sulfoheparosan (NSHN), completely de-sulfated, N-sulfated heparin (CDNSHP), N-deacetylated, N-sulfated, 6-O-sulfated heparosan (NSHN6S), N-deacetylated, N-sulfated, C5-epimerase treated 2-O-sulfated heparosan (NSHN2S), heparosan partially 6-O-sulfated (HN6S), N-deacetylated, N-sulfated, 6-O-sulfated, C5-epimerase treated 2-O-sulfated heparosan also known as non-anticoagulant heparin (NAHP) (Xiong et al. 2013) (see Table SII for structures). The GST-fused heparanase Bp and the cleaved protein from which the GST tag was recovered both showed similar activity. The specific activity of heparanase Bp acting on HS was 8 μmoles s−1/mg protein. The enzyme acted to completely convert HN and NSHN to disaccharide products, GlcNAc-GlcA and GlcNS-GlcA, respectively. An intermediate level of conversion was observed for HS, HP and NSHN6S; little or no conversion was observed for CDNSHP, NSHN2S and NAHP (Figure 2).
Fig. 2.
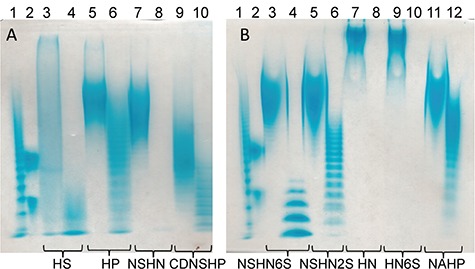
A total of 15% carbohydrate PAGE analysis of before and after heparanase Bp treated heparan sulfate and biosynthetic intermediates substrates. Each substrate has two lanes, left one is before heparanase Bp treated, right one is after heparanase Bp treated. (A) Lane 1, bovine heparin oligosaccharide ladder; lane 2, dp10 and dp20; lanes 3 and 4, HS; lanes 5 and 6, HP; lanes 7 and 8, NSHN; lanes 9 and 10, CDNSHP. (B) Lane 1, bovine heparin oligosaccharide ladder; lane 2, dp10 and dp20; lanes 3 and 4, NSHN6S; lanes 5 and 6, NSHN2S; lanes 7 and 8, HN; lanes 9 and 10, HN6S; lanes 11 and 12, NAHP.
Sites of cleavage in polysaccharide substrates by heparanase Bp
We next focused our attention on HS and HP as these are among the most frequently studied GAGs (Figure 3). We observed a conversion of HS from average molecular weight (Mw) 11,000 to average Mw 3000 and conversion for HP of average Mw 18,000 to average Mw 9000. HS is known to be richer in GlcA than in IdoA/IdoA2S with measured value of ~80% GlcA to ~15% IdoA and ~5% IdoA2S in porcine intestine HS (Griffin et al. 1995). In contrast, HP is composed of ~68% IdoA2S, ~8% IdoA and ~25% GlcA (Linhardt et al. 1988). Thus, these data suggest selectivity for cleavage at GlcA residue. This is consistent with the results of Bohlmann and coworkers, suggesting that heparanase Bp acts selectively on HP/HS cleaving the low sulfated domains affording the resistant highly sulfate domains. The action of heparanase Bp on heparin was next examined by LC-MS mapping of heparin oligosaccharides. Exhaustive treatment of HP with heparanase Bp afforded resistant intact highly sulfated domains ranging from tetrasaccharide to >28-mer with a molecular weight >9000 (Figure S1). LC-MS analysis showed a pattern of highly sulfated domains with a GlcNAc/GlcNAc6S/GlcNS at the non-reducing end, an internal cluster of 1 to >14 disaccharides units containing 1 to 4 sulfo groups and a reducing terminal GlcA residue (Figures S2–S6). The presence of long sequences of highly sulfated domains is consistent with the known structure HP (Turnbull and Gallagher, 1990).
Fig. 3.
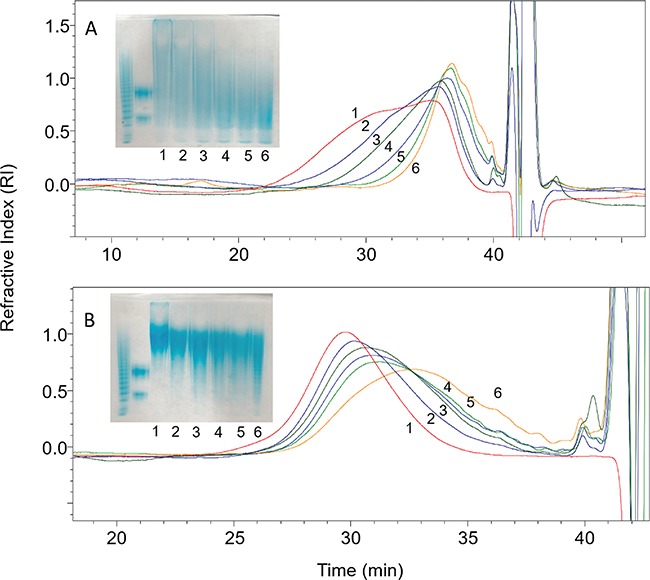
Kinetics analysis of heparanase Bp on HS/HP by PAGE (insert) and by GPC. (A) HS substrate, (B) HP substrate. Enzymatic reaction was terminated at time point 0, 1, 5, 10, 15, 30 min marked as 1, 2, 3, 4, 5, 6.
Action pattern of heparanase Bp on HS
Next, we examined the action pattern of heparanase Bp on HS (Figure 4). Park and Johnson assay (Thompson and Shockman 1968) was used to determine the amounts of reducing ends formed as a function of time (Figure S7) and gel permeation chromatography (GPC) was used to evaluate the change in HS molecular weight as a function of time. Based on a cross-plot of these assays (Figure 4), we conclude that heparanase Bp shows a random endolytic action patterns on HS (Jandik et al. 1994).
Fig. 4.
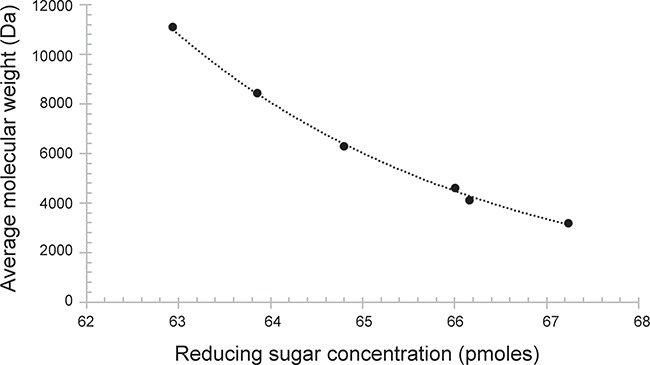
Molecular weight (Da) is plotted as a function of concentration (pmol) of reducing end sugar throughout the time course of heparanase Bp catalyzed depolymerization of HS.
Detailed specificity studies on heparanase Bp acting on defined oligosaccharide substrates
Defined HS oligosaccharides were chemoenzymatically synthesized (Zhang et al. 2017; Xu et al. 2014) to definitively establish the specificity of heparanase Bp. Nine oligosaccharides 1 to 9 (all having p-azidophenyl (Az) groups at their reducing ends) were used in this study and their structures are presented in Table I. The arrows indicated cleavage-site observed in six oligosaccharides (1, 3, 5, 7, 8, 9) of the nine oligosaccharides tested. Four of these oligosaccharides showed multiple cleavage sites (5, 7, 8 and 9) at which heparanase Bp acted. Three oligosaccharides (2, 4 and 6) were not cleaved by heparanase Bp. The cleavage of these defined oligosaccharide substrates was determined using LC-MS. A detailed analysis of nonasaccharide 9 cleavage by heparanase Bp is presented in Figure 5 (see Figures S8–S12 for detailed analyses of the other cleaved oligosaccharides). Three possible sequences of cleavage of nonasaccharide 9 can be visualized (Figure 5B, C and D) based on which of the three possible cleavage sites that is selected first. Each possibility is supported by the four products, a disaccharide (2-3), a trisaccharide (1-2-3) and two tetrasaccharides (2-3-4-5 and 6-7-8-9-Az), observed in LC-MS analysis (Figure 5A). Based on the analysis of the six defined oligosaccharides substrates (Figure 5, Figures S8–S12), we arrive at the specificity of heparanase Bp (Figure 6). Heparanase Bp cuts at the reducing end of a GlcA residue. No cleavage is observed at IdoA, IdoA2S, GlcA2S. The GlcA residue of a heparanase Bp substrate can be linked to β-(1→4) to GlcNAc, GlcNAc6S or GlcNS. No cleavage is observed when the GlcA residue is linked to GlcNS6S or GlcNS6S3S. The minimum substrate size for heparanase Bp appears to be a pentasaccharide or possibly in some cases a tetrasaccharide.
Table I.
Summary of heparanase Bp cleavage site (at arrows) in tested oligosaccharides
| Name | Oligosaccharide structure | Calculated oligosaccharide mass (Da) (Mtheoretical) | Measured oligosaccharide mass (Da) (Mexperimental) |
|---|---|---|---|
| Cleaved substrates | |||
| Hexa-1 | ↓ | 1541.3110 | 1541.3104 |
| GlcNAc-GlcA-GlcNS-IdoA2S-GlcNS-GlcA-Az | |||
| Hexa-3 | ↓ | 1461.3542 | 1461.3498 |
| GlcNAc-GlcA-GlcNS-IdoA-GlcNS-GlcA-Az | |||
| Hepta-5 | ↓ ↓ ↓ | 1637.3863 | 1637.3850 |
| GlcA-GlcNS-GlcA-GlcNS-GlcA-GlcNAc-GlcA-Az | |||
| Octa-7 | ↓ ↓ | 2122.3467 | 2122.3405 |
| GlcNS6S-GlcA-GlcNAc6S-GlcA-GlcNAc6S-GlcA-GlcNAc6S-GlcA-Az | |||
| Octa-8 | ↓ ↓ ↓ | 1802.5194 | 1802.5179 |
| GlcNS-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-Az | |||
| Nona-9 | ↓ ↓ ↓ | 2172.3471 | 2172.3464 |
| GlcA-GlcNS-GlcA-GlcNS-GlcA-GlcNS-IdoA2S-GlcNS-GlcA-Az | |||
| Non-cleaved oligosaccharides | |||
| Hexa-2 | 1701.2247 | 1701.2214 | |
| GlcNAc6S-GlcA-GlcNS6S-IdoA-GlcNS6S-GlcA-Az | |||
| Hexa-4 | 1739.1709 | 1739.1625 | |
| GlcNS6S-GlcA-GlcNS6S-GlcA2S-GlcNS-GlcA-Az | |||
| Hepta-6 | 2075.1166 | 2075.1140 | |
| GlcA-GlcNS6S-GlcA-GlcNS6S3S-IdoA2S-GlcNS6S-GlcA-Az | |||
Fig. 5.
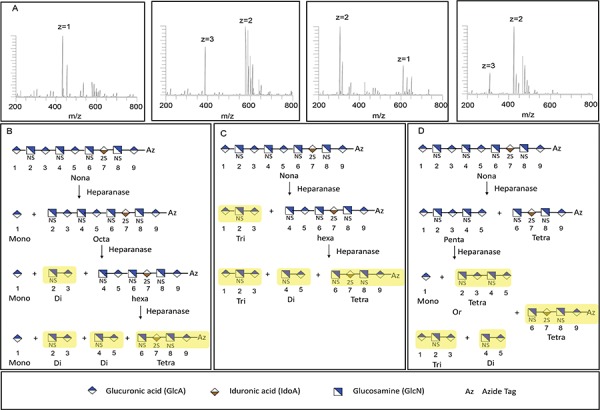
Analysis of Nona-9 (Table I) breakdown by heparanase Bp. Fractions shown in yellow in box (B), (C), (D) were observed in LC-MS analysis A. From left to right in (A), disaccharide 2-3 (GlcNS-GlcA, m/z = 434.0593, z = 1), tetrasaccharide 6-7-8-9-Az (GlcNS-IdoA2S-GlcNS-GlcA-Az, m/z = 386.3915, z = 3; m/z = 580.0922, z = 2), trisaccharide 1-2-3 (GlcA-GlcNS-GlcA, m/z = 304.5419, z = 2; m/z = 610.0905, z = 1), tetrasaccharide 2-3-4-5 (GlcNS-GlcA-GlcNS-GlcA, m/z = 309.6853, z = 3; m/z = 425.0542, z = 2).
Fig. 6.
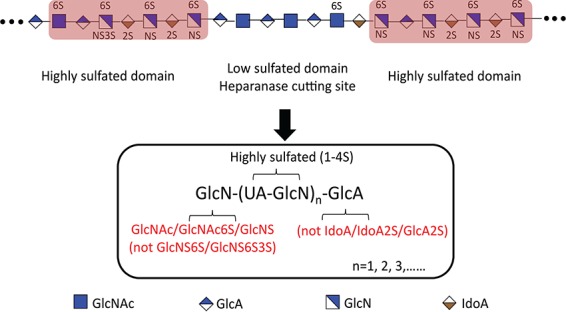
Heparin oligosaccharide products treated with heparanase Bp. Low sulfated domain will be cut by heparanase Bp and form a highly sulfated domain, which has a GlcA residue at the non-reducing end, and it cannot be IdoA/IdoA2S/GlcA2S, and GlcN at the reducing end, which can be modified to GlcNAc/GlcNAc6S/GlcNS, and it cannot be GlcNS6S/GlcNS6S3S.
Discussion
This new bacterially sourced β-endoglucuronidase was first described in 2015 when Bohlmann and coworkers reported a novel heparanase Bp from the invasive pathogenic bacterium B. pseudomallei (Bohlmann et al. 2015). Mammalian heparanase, another β-endoglucuronidase, has been linked to numerous human diseases, including cancer, diabetes and inflammation (Vlodavsky et al. 1999; Vreys and David 2007; McKenzie 2007). The specificity and action pattern of mammalian heparanase have been extensively studied (Pikas et al. 1998; Okada et al. 2002; Peterson and Liu 2010; Peterson and Liu 2012). A comparable understanding of the substrate specificity and action pattern of heparanase Bp is essential for its further application in GAG structural analysis and sequencing, and to potentially provide new insights for the design of potential heparanase inhibitors for related diseases. Although Bohlmann and coworkers tested heparanase Bp activities toward different GAGs, neither the specific substrate recognition properties nor its action pattern were defined. In the present study, we examined heparanase Bp in detail using heparan sulfate biosynthesized intermediates and a series of structurally defined oligosaccharides. The findings of the present study demonstrated heparanase Bp cuts on the reducing end of a GlcA residue and non-reducing end of less sulfated hexosamine residues, GlcNAc/GlcNS/GlcNAc6S.
Biosynthesized heparan sulfate intermediates offer several advantages for analyzing action pattern and specificity of heparanase Bp. First, the sulfation position and level is highly homogeneous in biosynthesized intermediates. Second, multiple sulfation patterns are present in nearly all kinds of heparan sulfate isolated from natural sources. In this study, we used polysaccharides rich in GlcA-GlcNAc, GlcA-GlcNS, GlcA-GlcN, GlcA-GlcN6S, IdoA2S-GlcNAc, GlcA-GlcNAc6S, GlcA2S/IdoA2S-GlcNS6S repeating disaccharide units. We found unmodified HN and NSHN, CDNSHP were susceptible to heparanase Bp cleavage. Highly sulfated polysaccharides were resistant to heparanase Bp. In contrast, it is known that unmodified, N-sulfated and partially epimerized heparosan is resistant to mammalian heparanase degradation (Peterson and Liu 2012). Furthermore, mammalian heparanase is affected by the sulfation pattern around the cleavage site in HS. This suggests that unlike mammalian heparanase, heparanase Bp strongly prefers the low sulfated domain than high sulfated domain.
In this study, we employed a series of oligosaccharides coupled with high-resolution ESI-MS analysis to further determine the specific cutting site. Based on heparanase Bp activities toward biosynthesized HS intermediates, we focused on different sulfated substrates containing a variable array of sites, UA(±2S)-GlcNAc/GlcNS(±6S/3S). The identification of fragments, generated by heparanase Bp acting on the nine representative HS oligosaccharides, and detected by LCMS, provides a better understanding of heparanase Bp specificity. We found that no cleavage takes place at IdoA, IdoA2S, GlcA2S. Furthermore, the GlcA residue of a heparanase Bp substrate can be linked to β-(1→4) to GlcNAc, GlcNAc6S or GlcNS. Notably, no cleavage is observed when the GlcA residue is linked to GlcNS6S or GlcNS6S3S. Moreover, a tetrasaccharide, GlcA-GlcNAc-GlcA-GlcNAc, with a putative cleavage site was observed at the LCMS data, indicating the minimum cutting size for heparanase Bp is a most likely a pentasaccharide. Also, our data indicates that heparanase Bp degrades its substrates in a random endolytic action pattern. While Peterson and Liu indicated that mammalian heparanase degrades its substrates in two modes: consecutive cleavage and gapped cleavage (Peterson and Liu 2012). HP completely treated by heparanase Bp afforded products that were detected by LC-MS. Their mass were consistent with heparin oligosaccharides having a GlcA residue at their reducing end and GlcNAc/GlcNS/GlcNAc6S at their non-reducing end.
In conclusion, a new bacterial enzyme, heparanase Bp, has been prepared as a recombinant E. coli expressed enzyme with high purity and high activity. Mammalian heparanase cleaves the linkage between GlcA and a 6-O-sulfo or a 3,6-O-disulfo GlcNS residue present in a highly sulfated domain and is unable to cleave unmodified HN. In contrast, heparanase Bp cleaves the linkage between GlcA and GlcNAc, GlcNAc6S or GlcNS in a low sulfated domain and is unable to cleave at IdoA and GlcA residues present in highly sulfated domains. Heparanase Bp acts endolytically on both HS and HP making it useful for structural analysis. Indeed, the preservation of the C5 chirality lost when using heparin lyases as structural tools and the cleavage of heparanase Bp at the reducing side of UA (heparin lyase cleavage at the non-reducing site of UA) affords complementary and difficult to obtain information of HS/HP sequence. As an example, our current study provides the first detailed information on the high sulfation domains present in HP. Thus, heparanase Bp represents a new and powerful tool for the study of GAGs.
Materials and methods
Materials
Luria-Bertani (LB) medium with ampicillin (80 μg/mL) was used for the cell growth and transformation screening. Super optimal broth with catabolite repression (SOC) was used for cell recovery after heat shock during the transformation experiments. Additional chemicals were obtained from Sigma Chemical Co. (St. Louis, MO) and GenScript (Piscataway, NJ). Porcine intestinal heparin (18 kDa) and porcine intestinal HS (11 kDa) were from Celsus Laboratories. (Cincinnati, OH). HN, NSHN, CDNSHP, NSHN6S, NSHN2S, HN6S, NAHP were prepared in our own lab (Xiong et al. 2013). Defined structural heparan sulfate oligosaccharides were synthesized in Prof. Jian Liu’s lab (University of North Carolina, Chapel Hill) (Zhang et al. 2017; Xu et al. 2014).
Plasmid construction
Heparanase Bp was expressed as described elsewhere (Bohlmann et al. 2015). The gene encoding heparanase from B. pseudomallei K96243 genomic DNA (Chromosome 1: GenBank CP009538) was synthesized as a g-block fragment (GenScript). After PCR amplification, the gene was cloned into a pGEX-4 T-1 vector at the SmaI and XhoI restriction sites, creating the construct known as pGEX_Heparanase Bp, with a GST tag at the N-terminal for purification. The construct was verified by Sanger DNA sequencing (Genewiz) then transformed into E. coli BL21 Star (DE3) chemically competent cells by heat shock in a 42°C water bath for 45 s. Cells were recovered in SOC medium for 50 min and plated on an LB agar plate supplemented with 80 μg/mL of ampicillin for screening.
The strains, plasmid and primers that were used in this study are listed in Table SI. Plasmid DNA was prepared using an E.Z.N.A plasmid mini kit (OMEGA) and digested DNA fragments were retrieved from agarose gel (Bio-Rad) by an E.Z.N.A. gel extraction kit (OMEGA). FastDigest Restriction endonucleases and a Rapid DNA ligation kit were purchased from Thermo Fisher Scientific. PCRs were carried out using Accuzyme® mix (BIOLINE) according to the manufacturer’s instructions.
Expression and purification of recombinant heparanase Bp
A single colony of E. coli BL21 Star (DE3) containing the plasmid pGEX_Heparanase Bp was picked from streaked agar plates and inoculated into a 20-mL LB medium in 125-mL flasks with 80 μg/mL of ampicillin. The overnight cultures were then inoculated into baffled 2.8-L Erlenmeyer Flasks (Pyrex) containing 1-L LB supplemented with 80-μg/mL ampicillin and incubated in a rotary air shaker (New Brunswick Scientific Innova 44R) at 37°C, 225 rpm until the OD600 was in the range of 0.6–0.8. At this point, the incubation temperature was dropped to 25°C for 1 h, after which 0.1-mM isopropyl-1-thio-β-D-galactopyranoside was added to induce protein expression. After induction, the cells were incubated for an additional 4 h at 25°C. The cell pellet was then collected by centrifugation at 5500 × g for 30 min at 4°C, re-suspended in 30 mL of phosphate buffered saline (PBS) lysis buffer and split into two tubes. The resultant single-cell suspensions were sonicated on ice using a Misonix 3000 sonicator at 70% amplification (10 s on, 10 s off) for 6 min each. The sonicated solutions were centrifuged at 14000 × g for 60 min at 4°C. The supernatant was filtered through a 0.45-μM sterile filter (VWR) to remove any remaining cellular debris and loaded onto a gravity flow column packed with glutathione resin (GenScript) that was pre-equilibrated with column buffer PBS, pH 7.4). The column was kept at 4°C while the filtered supernatant passed through the resin then it was washed with at least four column volumes (CVs) of column buffer to remove any non-specific binding substances. The fusion protein was then eluted with ~3 CV of a reduced glutathione buffer (50 mM Tris-HCl, 10 mM reduced glutathione, pH 8.0). The collected protein was applied to a 10-kDa molecular weight cut-off centrifuge unit (Amicon Ultra-15) for concentrating.
For purification of the cleaved protein without its N-terminal GST tag, the filtered soluble protein fraction was applied to the pre-equilibrated column, after which it was washed with 4 CV of column buffer, followed by 4 CV of PreScission Protease buffer (50 mM Tris HCl, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 8.0). Proteolytic digestion was then initiated by applying PreScission Protease (GenScript) diluted into PreScission buffer according to the size of the column (4.5 mL for a 5-mL column; 2 units enzyme/100 μg of bound GST-tagged protein). The column was capped and incubated at 4°C for 12–16 h to allow efficient binding and even distribution of the PreScission Protease enzyme throughout the column. Once the column was uncapped and flow resumed, the cleaved target protein was eluted with PreScission buffer. Reduced glutathione buffer was then applied to elute GST, unbound GST-target protein and PreScission Protease. The column was washed with 10 CV of regeneration buffer (125 mM Tris-HCl, 150 mM NaCl, pH 8.0) and stored for later use.
The expression and the purity of the target protein in the eluted fractions were assessed by SDS-PAGE. The soluble and insoluble fractions were normalized based on cell density and mixed with loading dye (containing 5% β-mercaptoethanol) in 1:1 ratio. The mixture was then denatured at 100°C for 10 min and loaded into a NuPage 10% Bis-Tris Midi gel (Invitrogen). Electrophoresis was run at 180 volts for 60 min in 1 × MOPS SDS running buffer. The theoretical size of the GST-tagged protein was calculated (using ExPasy) to be ~74 kD, and ~48 kDa without the tag.
Heparanase Bp substrate specificity assay using heparan sulfate and biosynthetic intermediates
Heparan sulfate, heparin (Celsus, Cincinnati, OH) and their biosynthesis family HN, HN6S, NSHN, CDNSHP, NSHN2S, NSHN6S, NAHP (Table SII) were tested as substrate to study the specificity of heparanase Bp. Enzymatic reactions were carried out as follows: 20-μg substrates were treated with 10-μL heparanase Bp (0.7 mg/mL) in 200-μL digestion buffer (50 mM ammonium acetate, pH 4.5) at 37°C overnight.
The products were analyzed by PAGE. Briefly, 10-μg samples were loaded onto a 15% total acrylamide (15% T) resolving gel containing 14.08% (w/v) acrylamide, 0.92% (w/v) N, N′-methylene-bis-acrylamide and 5% (w/v) sucrose. All monomer solutions were prepared in resolving buffer (0.1 M boric acid, 0.1 M Tris, 0.01 M disodium EDTA), 5% stacking gel monomer solution was also prepared in resolving buffer and it contained 4.75% (w/v) acrylamide and 0.25% (w/v) N, N′-methylene-bis-acrylamide. A 10 cm × 7 mm diameter resolving gel column was cast from 4 mL of 15% T monomer solution mixed with 4 μL of TEMED and 12 μL of 10% ammonium persulfate (APS). A stacking gel was cast from 1-mL stacking gel monomer solution mixed with 1 μL of TEMED and 30 μL of 10% APS. Phenol red dye prepared in 50% (w/v) sucrose was added to the sample for visualization of the ion front during electrophoresis. A standard composed of a mixture of heparin oligosaccharides with known molecular weight prepared enzymatically from bovine lung heparin. The gel was visualized with Alcian Blue staining and then digitized with UN-SCAN-it (Silk Scientific).
Heparanase Bp kinetic assay using HP/HS as substrates
Heparanase Bp (10 μL) concentrated at 0.7 mg/mL was added to 100 μg of HP/HS substrates in 200-μL 50-mM ammonium acetate digestion buffer (pH 4.5) and incubated overnight at 37°C. The reactions were terminated at 1, 5, 10, 15, 30 min by boiling for 5 min, and centrifuged at 12,000 × g for 10 min. The supernatant was freeze-dried for PAGE and GPC analysis. The PAGE method was the same as the last section. GPC was performed on the HPLC system consisting of Shimadzu LC-10Ai pump, a Shimadzu CBM-20A controller and a Shimadzu RID-10A refractive index detector (Shimadzu, Kyoto, Japan). A guard column TSK SWXL 6 mm × 4 cm, 7 μL diameter was used to protect two analytical columns: TSK G4000 SWXL 7.8 mm × 30 cm, 8 μm in series with TSK G3000SWXL 7.8 mm × 30 cm, 5 μm (Tosoh Corporation, Tokyo, Japan). The mobile phase was 0.1-M ammonium acetate with 0.02% (w/v) sodium azide. Columns were maintained at 30°C using an Eppendorf column heater (Eppendorf, Hamburg, Germany). A sample injection volume was set at 20 μL (sample concentration was ~5 mg/mL) and a flow rate was set at 0.6 mL/min.
Measurement of glycosidase activity using heparan sulfate as substrate
HS (100 μg) was treated with 10-μL heparanase Bp (0.7 mg/mL) in 200-μL digestion buffer (50 mM ammonium acetate, pH 4.5) at 37°C overnight. The reactions were terminated at 1, 5, 10, 15, 30 min by boiling for 5 min, and centrifuged at 12,000 × g for 10 min. The glycosidase activity of BpHep was determined using a modified Park-Johnson method (35). Briefly, aliquots (20 μL) were heated with 0.05% (w/v) aqueous potassium ferricyanide (20 μL) and 0.53% (w/v) sodium carbonate/0.065% (w/v) potassium cyanide in water (20 μL) in 96 wells plate at 100°C for 15 min. Upon cooling, 100 μL of 0.15% (w/v) ferric ammonium sulfate/0.1% (w/v) SDS in 0.025 M sulfuric acid was added. After 15 min at room temperature, the A690 was measured. Data was converted to glucuronic acid equivalents using a standard curve.
Specificity analysis of heparanase Bp using structural defined oligosaccharides
Structurally defined oligosaccharides (hexa-, hepta-, octa-, nona-) were treated with 10 μL of 0.7-mg/mL heparanase in a total volume of 200 μL of 50-mM ammonium acetate buffer, pH 4.5, at 37°C overnight. The enzymatic reactions were terminated by boiling at 100°C for 5 min. As control experiments, each oligosaccharide was treated with the heat-inactivated heparanase Bp under the same conditions.
Each enzyme digest was analyzed by LC Orbitrap FT MS. A Luna HILIC column (2.0 × 50 mm, 200 Å, Phenomenex, Torrance, CA) was directly connected online to the standard ESI source of LTQ-Orbitrap XL FT MS (Thermo Fisher Scientific, San-Jose, CA). LC parameter: mobile phase A was 5-mM ammonium acetate prepared with HPLC grade water, and mobile phase B was 5-mM ammonium acetate prepared in 98% HPLC grade acetonitrile with 2% of HPLC grade water. The gradient was used from 5% A to 70% A in 7 min then reset to 5% A at a flow rate of 250 μL/min. The source parameters for FTMS were in the negative-ion mode, a spray voltage of 4.2 kV, a capillary voltage of −40 V, a tube lens voltage of −50 V, a capillary temperature of 275°C, a sheath flow rate of 30 L/min and an auxiliary gas flow rate of 6 L/min. All FT mass spectra were acquired at a resolution 60,000 with 200–2000 Da mass range.
Heparanase Bp treatment of heparin
Heparin (20 μg) were treated with 10-μL heparanase Bp (0.7 mg/mL) in 200-μL digestion buffer (50-mM ammonium acetate, pH 4.5) at 37°C overnight. The product was analyzed by HILIC MS on Orbitrap. The method was similar with the method used in oligosaccharide analysis except for a longer column (Luna HILIC column, 2.0 × 50 mm, 200 Å, Phenomenex, Torrance, CA) and separation times.
Funding
National Institutes of Health (grant no. DK111958, CA231074, HL125371); National Science Foundation (CBET grant no. 1604547).
Conflict of interest statement
None declared.
Abbreviations
- APS, ammonium persulfate
-
Az, para-azidophenyl
CDNSHP, completely de-sulfated, N-sulfated heparin
ECM, extracellular matrix
GAGs, glycosaminoglycans
Gal, β-D-galactose
GlcA, β-D-glucuronic acid
GlcNAc, N-acetyl-α-D-glucosamine
GPC, gel permeation chromatography
GST, glutathione S-transferase
NAHP, non-anticoagulant heparin
HN, heparosan
HN6S, heparosan partially 6S
HP, heparin
HS, heparan sulfate
NSHN, N-deacetylated, N-sulfoheparosan
NSHN2S, N-deacetylated, N-sulfated, C5-epimerase treated 2-O-sulfated heparosan
NSHN6S, N-deacetylated, N-sulfated, 6-O-sulfated heparosan
IdoA, α-L-iduronic acid
PAGE, polyacrylamide gel electrophoresis
PBS, phosphate buffered saline
PCR, polymerase chain reaction
UA, uronic acid
References
- Abboud-Jarrous G, Rangini-Guetta R, Aingorn H, Atzmon R, Elgavish S, Peretz T, Vlodavsky I. 2005. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. J Biol Chem. 280:13568–13575. [DOI] [PubMed] [Google Scholar]
- Bohlmann L, Tredwell GD, Yu X, Chang C, Haselhorst T, Winger M, Dyason JC, Thomson RJ, Tiralongo J, Beacham IR et al. . 2015. Functional and structural characterization of a heparanase. Nat Chem Biol. 11:955–957. [DOI] [PubMed] [Google Scholar]
- Chen Y, Lin L, Agyekum I, Zhang X, St. Ange K, Yu Y, Liu J, Amster IJ, Linhardt RJ. 2017. Structural analysis of heparin-derived 3-O-sulfated tetrasaccharides: Antithrombin binding site variants. J Pharm Sci. 106:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai UR, Wang H, Linhardt RJ. 1993a. Specificity studies on the heparin lyases from Flavobacterium heparinum. Biochemistry. 32:8140–8145. [DOI] [PubMed] [Google Scholar]
- Desai UR, Wang H, Linhardt RJ. 1993b. Substrate specificity of the heparin lyases from Flavobacterium heparinum. Arch Biochem Biophys. 306:461–468. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. 2002. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 71:435–471. [DOI] [PubMed] [Google Scholar]
- Fu L, Zhang F, Li G, Onishi A, Bhusker U, Sun P, Linhardt RJ. 2014. Structure and activity of a new low molecular weight heparin produced by enzymatic ultrafiltration. J Pharm Sci. 103:1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CC, Linhardt RJ, VanGorp CL, Toida T, Hileman RE, Schubert RL, Brown SE. 1995. Isolation and characterization of heparan sulfate from crude porcine intestinal mucosa peptidoglycan heparin. Carbohyd Res. 276:183–197. [DOI] [PubMed] [Google Scholar]
- Heyman B, Yang Y. 2016. Mechanisms of heparanase inhibitors in cancer therapy. Exp Hematol. 44:1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandik KA, Gu K, Linhardt RJ. 1994. Action pattern of polysaccharide lyases on glycosaminoglycan. Glycobiology. 4:289–296. [DOI] [PubMed] [Google Scholar]
- Langer R, Linhardt RJ, Cooney CL, Klein M, Tapper D, Hoffberg SM, Larsen A. 1982. An enzymatic system for removing heparin in extracorporeal therapy. Science. 217:261–263. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Couchman J, Kimata K, Esko JD. 2017. Proteoglycans and sulfated glycosaminoglycans. In: Varki, A., Cummings, R. D., Esko, J. D., Stanley, P., Hart, G. W., Aebi, M., Darvill, A. G., Kinoshita, T., Packer, N. H., Prestegard, J. H., Schnaar, R. L., Seeberger, P. H., editors. Essentials of glycobiology. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, Ch. 17.
- Linhardt RJ. 1994. Analysis of glycosaminoglycans with polysaccharide lyases. Curr Protoc Mol Biol. V2:17.13.17–17.13.32. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ. 2003. Heparin: Structure and activity. J Med Chem. 46:2551–2554. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ, Galliher PM, Cooney CL. 1986. Polysaccharide lyases. Appl Biochem Biotech. 12:135–177. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ, Rice KG, Kim YS, Lohse DL, Wang HM, Loganathan D. 1988. Mapping and quantification of the major oligosaccharide components of heparin. Biochem J. 254:781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Desai UR, Han XJ, Toida T, Linhardt RJ. 1995. Strategy for the sequence analysis of heparin. Glycobiology. 5:765–774. [DOI] [PubMed] [Google Scholar]
- Lohse DL, Linhardt RJ. 1992. Purification and characterization of heparin lyases from Flavobacterium heparinum. J Biol Chem. 267:24347–24355. [PubMed] [Google Scholar]
- Nardella C, Lahm A, Pallaoro M, Brunetti M, Vannini A, Steinkühler C. 2004. Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry. 43:1862–1873. [DOI] [PubMed] [Google Scholar]
- Okada Y, Yamada S, Toyoshima M, Dong J, Nakajima M, Sugahara K. 2002. Structural recognition by recombinant human hepranase that plays critical roles in tumor metastasis. J Biol Chem. 277:42488–42495. [DOI] [PubMed] [Google Scholar]
- Pala D, Rivara S, Mor M, Milazzo FM, Roscilli G, Pavoni E, Giannini G. 2016. Kinetics analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology. 26:640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payza AN, Korn ED. 1956. Bacterial degradation of heparin. Nature. 177:88–89. [DOI] [PubMed] [Google Scholar]
- Pikas DS, Li J, Vlodavsky I, Lindahl U. 1998. Substrate specificity of heparanases from human hepatoma and platelets. J Biol Chem. 273:18770–18777. [DOI] [PubMed] [Google Scholar]
- Peterson SB, Liu J. 2010. Unraveling the specificity of heparinase utilizing synthetic substrates. J Biol Chem. 285:14504–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SB, Liu J. 2012. Deciphering mode of action of heparanase using structurally defined oligosaccharides. J Biol Chem. 287:34836–34843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SB, Liu J. 2013. Multi-faceted substrate specificity of heparanase. Matrix Biol. 32:223–227. [DOI] [PubMed] [Google Scholar]
- Rice KG, Linhardt RJ. 1989. Study of defined oligosaccharide substrates of heparin and heparan monosulfate lyases. Carbohyd Res. 190:219–233. [DOI] [PubMed] [Google Scholar]
- Simeonovic CJ, Ziolkowski AF, Wu Z, Choong FJ, Freeman C, Parish CR. 2013. Heparanase and autoimmune diabetes. Front Immunol. 4:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Blain F, Musil RA, Zimmermann JJ, Gu K, Bennett DC. 1996. Isolation and expression in Escherichia coli of hepB and hepC, genes coding for the glycosaminoglycan-degrading enzymes heparinase II and heparinase III, respectively, from Flavobacterium heparinum. Appl Environ Microbiol. 62:2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JS, Shockman GD. 1968. A modification of the Park and Johnson reducing sugar determination suitable for the assay of insoluble material: Its application to bacterial cell walls. Anal Biochem. 22:260–268. [DOI] [PubMed] [Google Scholar]
- Turnbull JE, Gallagher JT. 1990. Molecular organization of heparan sulphate from human skin fibroblasts. Biochem J. 265:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky L, Gross-Cohen M, Weissmann M, IIan N, Sanderson RD. 2018. Opposing functions of heparanase-1 and heparanase-2 in cancer progression. Trends Biochem Sci. 43:18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I et al. . 1999. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat Med. 5:793–802. [DOI] [PubMed] [Google Scholar]
- Vreys V, David G. 2007. Mammalian heparanase: What is the message? J Cell Mol Med. 11:427–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie E. 2007. Heparanase: A target for drug discovery in cancer and inflammation. Br J Pharmacol. 151:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Viola CM, Brzozowski AM, Davies GJ. 2015. Structural characterization of human heparanase reveals insights into substrate recognition. Nat Struct Mol Biol. 22:1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Tappen BR, Ly M, Zhao W, Canova LP, Guan H, Linhardt RJ. 2011. Heparin mapping using heparin lyases and the generation of a novel low molecular weight heparin. J Med Chem. 54:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Bhaskar U, Li G, Fu L, Li L, Zhang F, Dordick JS, Linhardt RJ. 2013. Immobilized enzymes to convert N-sulfo, N-acetyl heparosan to a critical intermediate in the production of bioengineered heparin. J Biotechnol. 167:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Cai C, Chandarajoti K, Hsieh P-H, Li L, Pham TQ, Sparkenbaugh EM, Sheng J, Key NS, Pawlinski R et al. . 2014. Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat Chem Biol. 10:248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Pagadala V, Jester HM, Lim AM, Pham TQ, Goulas AMP, Liu J, Linhardt RJ. 2017. Chemoenzymatic synthesis of heparan sulfate and heparin oligosaccharides and NMR analysis: Paving the way to a diverse library for glycobiologists. Chem Sci. 8:7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]