

Abstract
The antidepressant-sensitive serotonin (5-HT) transporter (SERT) dictates rapid, high-affinity clearance of the neurotransmitter in both the brain and periphery. In a study of families with multiple individuals diagnosed with autism spectrum disorder (ASD), we previously identified several, rare, missense coding variants that impart elevated 5-HT transport activity, relative to wildtype SERT, upon heterologous expression as well as in ASD subject lymphoblasts. The most common of these variants, SERT Ala56 located in the transporter’s cytosolic N-terminus, has been found to confer in transgenic mice hyperserotonemia, an ASD-associated biochemical trait, an elevated brain 5-HT clearance rate, and ASD-aligned behavioral changes. Hyperfunction of SERT Ala56 has been ascribed to a change in 5-HT KM, though the physical basis of this change has yet to be elucidated. Through assessments of fluorescence resonance energy transfer (FRET) between cytosolic N- and C-termini, sensitivity to methanethiosulfonates, and capacity for N-terminal tryptic digestion, we obtain evidence for mutation-induced conformational changes that support an open-outward, 5-HT binding conformation in vitro and in vivo. Aspects of these findings were also evident with another naturally-occurring, C-terminal SERT coding variant identified in our ASD study, Asn605. We conclude that biased conformations of surface resident transporters that can impact transporter function and regulation are an unappreciated consequence of heritable and disease-associated SERT coding variation.
Keywords: serotonin transporter, serotonin, protein conformation, autism spectrum disorder
Graphical Abstract
INTRODUCTION
Autism spectrum disorder (ASD), a neurobehavioral condition with high prevalence and significant, but heterogeneous genetic contributions,1 has been linked to disruptions in serotonin (5-HT) signaling.2 More than 50 years ago, Schain and Freedman3 reported elevated whole blood 5-HT levels (hyperserotonemia) in subjects with ASD. Multiple studies since this time have demonstrated hyperserotonemia in a subset (25-30%) of ASD subjects, primarily derived from elevated 5-HT stores in platelets.3,4 These stores of 5-HT arise from the accumulation by platelet SERT of enterochromaffin cell-derived 5-HT.5 Linkage studies of multiplex ASD families revealed a chromosomal locus at 17q11.2 that harbors SLC6A4, the SERT gene.6-8 As the coding sequence of human SERT is identical in the brain and platelets,5 we hypothesized that the families might harbor ASD-associated SERT coding variants. Indeed, our efforts revealed multiple, rare functional SERT coding variants.6 Remarkably, a common feature of these SERT variants in both subject lymphoblasts and transfected cells was their elevated 5-HT transport activity compared to 5-HT transport conferred by the reference (WT) transporter.6,9,10 Further studies revealed that the most common of these variants, SERT Ala56, induces elevated 5-HT transport6,10 in the absence of changes in either total or surface SERT protein.9,10 Rather, SERT Ala56 demonstrates a reduction in 5-HT KM compared to SERT Gly56, the WT allele,10 suggesting an increased efficiency for 5-HT uptake at sub-saturating 5-HT concentrations. Importantly, the uptake stimulatory impact of the SERT Ala56 variant has been demonstrated in vivo, through studies of SERT Ala56 mice.11,12 Moreover, these mice exhibited hyperserotonemia, increased hippocampal 5-HT clearance, and behavioral features consistent with both core and comorbid medical features of ASD.11,13,14
Mechanistic studies of SERT Ala56 hyperfunction suggest that the variant may exist constitutively in a conformation normally induced transiently by activation through protein kinase G (PKG)15 or p38α MAPK.16,17 Consistent with this model, SERT Ala56 demonstrates no further increase in 5-HT uptake activity when transfected10 or native cells9 are treated with activators of PKG or p38 MAPK. SERT Ala56 is also hyperphosphorylated under basal conditions in these preparations in a p38 MAPK-dependent manner.11,12 Finally, the elevated 5-HT clearance observed in SERT Ala56 mice, as well as multiple physiological and behavioral phenotypes, can be reversed by treatment of these animals with the p38α MAPK inhibitors, MW108 and MW150.13 Another SERT missense variant, the C-terminal substitution Lys605Asn, also displays reduced PKG/p38 MAPK sensitivity.10 A structural basis however, for the kinetic and regulatory changes of SERT Ala56 or SERT Asn605 has not been established.
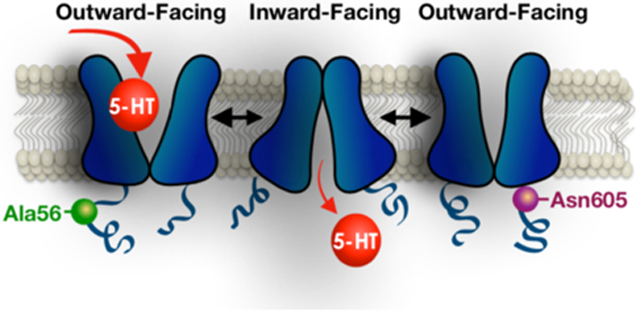
SERT, like other SLC members belonging to the Na+/Cl− dependent neurotransmitter transporter family,18 translocates substrates across the membrane via an alternating access mechanism.19,20 In this model, the binding site for 5-HT and co-transported ions is exposed to the extracellular space via an outward-facing conformation, and upon the binding of substrates, the transporter moves through a substrate-occluded state to an open-inward conformation where substrates are released to the cytoplasm.19 Following 5-HT release, the substrate-free carrier reorients to prepare for the next transport cycle. High resolution structure and modeling studies19,21-23 suggest that during the transport cycle, the transporter undergoes a conformational shift, referred to as a “rocking bundle” mechanism.19 In this model, transmembrane (TM) segments 1-5 and 6-10, which form two inverted repeat bundles, move in relation to each other to open and close internal and external gates that grant or occlude access to the substrate binding site. Whereas residues within the transmembrane domains comprise sites of substrate binding, residues within the transporter’s intracellular N- and C-termini, besides their well-documented contribution to transporter regulation, have become increasingly recognized as determinants of substrate binding affinity, directionality of transport and the rate of substrate translocation.24-29 Herein, we report functional and structural evidence that the N-terminal SERT Ala56 coding variant can impose altered conformations, consistent with a bias toward an outward-facing state. Somewhat similar findings are evident for the C-terminal Asn605 variant, indicating that modest structural changes in either cytoplasmic tails can result in both local and global changes in conformation that for some individuals may impact disease risk.
RESULTS
Conformational Bias of SERT Ala56 Evaluated by Studies of Fenfluramine-Mediated 5-HT Efflux In Vitro and In Vivo.
The alternation of conformations that drives neurotransmitter uptake from the extracellular space participates,30,31 with qualifications,28,32 in the ability of transporters to support movement of elevated intracellular substrate (efflux) into the extracellular space.30 The mutually sequential nature of the alternating access model predicts that alterations in conformational stability that enhance outward-facing conformations will reduce the population of inward-facing conformations, and vice-versa. Thus, if SERT Ala56 more readily acquires outward-facing conformations, efflux of intracellular substrate would concomitantly decrease. Moreover, amphetamines are well known to enhance efflux of substrates by monoamine transporters, including SERT,30 with D-fenfluramine being relatively SERT-selective33 and commonly used to drive SERT-mediated 5-HT efflux in vitro32,34,35 and in vivo36,37 Importantly, the SERT Ala56 variant lies in the SERT N-terminus, which has been shown to dictate the capacity for SERT-mediated 5-HT efflux induced by amphetamine derivatives.28
To examine whether SERT Ala56 displays altered 5-HT efflux rates, we first loaded CHO cells stably expressing WT or SERT Ala56 to equilibrium with [3H]5-HT (20 nM, 1 hr at 37°C). Both WT and SERT Ala56 cells loaded to equivalent levels (Figure S1A), in keeping with the equilibrium nature of the incubations and the equivalent surface expression of WT and SERT Ala56. We then rapidly removed external substrate prior to incubation of cells with 10 μM D-fenfluramine for 1, 3, or 5 min, collecting and quantifying radiolabel in the extracellular media, as described in Experimental Procedures. When cells were exposed to D-fenfluramine, WT cells supported significant (~80%) 5-HT efflux that was consistent from 1-5 min (Figure 1A). However, at both 1 and 3 min, SERT Ala56 cells supported significantly less 5-HT efflux in response to the D-fenfluramine challenge, amounting to ~60-70% 5-HT efflux, which is 10-20% less than the efflux generated by WT cells (Figure 1A). A longer, 5 min incubation with D-fenfluramine, however, resulted in SERT Ala56 efflux reaching WT levels, consistent with reduced kinetic efficiency loading or transporting 5-HT, versus a difference in intrinsic efflux capacity.
Figure 1. Fenfluramine mediated 5-HT efflux of SERT Ala56.

(A) CHO cells stably expressing hSERT (WT) or hSERT Ala56 were loaded for 1 hour with 20 nM [3H] 5-HT and then subjected to 10 μM D-fenfluramine for 1,3, or 5 minutes. Cells expressing SERT Ala56 efflux less [3H]5-HT at 1- and 3-min incubation with D-fenfluramine compared to WT expressing cells (two-way repeated-measures ANOVA; Bonferroni post-hoc test of genotype differences, **P ≤ 0.01; n=4). (B) Hippocampal slices were assessed for D-fenfluramine mediated [3H]5-HT efflux loaded with 400 nM [3H] 5-HT for 30 minutes and then perfused with KRB buffer for 15 min to establish baseline followed by 15 min 3 μM D-fenfluramine pulse and then a 15 min wash out with KRB. Slices prepared from SERT Ala56 mice showed a blunted D-fenfluramine mediated [3H]5-HT efflux compared to WT (two-way repeated-measures ANOVA; Bonferroni post-hoc test of genotype differences, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; n=5). (C) In vivo microdialysis data also show SERT Ala56 mice have a significant decrease in extracellular 5-HT levels in response to fenfluramine infusion (10 μM for 20 min at 1mL/min flow rate) compared to WT in CA3 region of the hippocampus. (D) SERT Ala56 shows decreased extracellular 5-HT levels measured at baseline (average from 0-80 minutes, in nM) compared to WT (unpaired t-test; **P ≤ 0.01; n=7-8). (E) Area under the microdialysis peak from 80-200 min also shows blunted 5-HT efflux in response to D-fenfluramine in SERT Ala56 mice compared to WT (unpaired t-test; *P ≤ 0.05; n=7-8).
To determine whether diminished 5-HT efflux activity is evident in native preparations, we examined the ability of hippocampal slices prepared from WT (SERT Gly56) and SERT Ala56 knock-in (KI) littermate mice to efflux preloaded [3H] 5-HT in response to D-fenfluramine. No differences were obtained in total amount of [3H]5-HT accumulated in the cell following our equilibrium loading protocol (Figure S1B), consistent with a lack of effect of the SERT Ala56 mutation on transporter protein levels in vitro and in vivo.10,11 At low D-fenfluramine concentrations (3 μM, Figure 1B), [3H]5-HT efflux was significantly blunted in slices from SERT Ala56 mice compared to efflux obtained with WT preparations (Figure 1B). However, at higher concentrations of D-fenfluramine (10 and 20 μM), equivalent efflux of [3H]5-HT was observed between the WT and variant (Figure S1C-D). These findings are consistent with a biased conformation of SERT Ala56 in the mouse brain slice preparation that is evident at lower substrate concentrations, but negated at higher substrate concentrations. Another explanation for the observed reduced efflux capacity of SERT Ala56 is an altered affinity for fenfluramine, however in a [3H]citalopram competition binding assay from hippocampal membranes, we found no difference between genotypes in fenfluramine affinity (Figure S2).
Finally, to test whether genotype differences are evident for D-fenfluramine-induced 5-HT efflux in vivo, we assessed extracellular 5-HT levels by microdialysis in the CA3 region of the hippocampus following reverse dialysis of D-fenfluramine in situ (Figure 1C). Across the first 80 minutes of collection, we found baseline extracellular 5-HT levels to be decreased in the SERT Ala56 mice, relative to levels in WT mice (Figure 1D), consistent with the enhanced basal 5-HT clearance observed in the SERT Ala56 mice in the same brain region.11,13 It is important to note that past studies have found no difference in total 5-HT levels between genotypes in various brain regions, including the hippocampus.11,13 At 80 min, we infused 10 μM D-fenfluramine through the dialysis probe, a concentration chosen based on an expectation of ~10% exchange across the dialysis membrane and an effort, established by our ex vivo studies, to deliver a subsaturating D-fenfluramine concentration. As expected,38 we observed a significant time-dependent rise in extracellular 5-HT levels in WT mice. Parallel experiments with SERT Ala56 littermates demonstrated a significantly blunted response to D-fenfluramine compared to WT, analyzed as the area under the peak from 80-200 min, relative to the average baseline calculated for each genotype (Figure 1E). These data provide in vivo evidence supporting the hypothesis that SERT Ala56 biases transporter conformation, most likely outward-facing, that can promote an enhanced rate of extracellular 5-HT clearance while yielding a reduced rate of D-fenfluramine stimulated 5-HT efflux. These findings are also consistent with prior studies with larger SERT N-terminal deletions revealing that this region is critical in modulating efflux potential.28 This observation is further strengthened by studies of the dopamine (DA) and norepinephrine (NE) transporters (DAT and NET, respectively) where findings indicate that amphetamine-induced substrate efflux is influenced by N-terminal residues that can support transporter phosphorylation and or protein interactions.27,39-41
SERT Ala56 Conformation Assessed through Noribogaine Uptake Inhibition.
Ibogaine, a psychoactive molecule derived from the plant Tabemathe iboga, and its active metabolite noribogaine, have been reported to stabilize SERT in an inward-facing conformation.42-44 If SERT Ala56 more frequently assumes an outward-facing conformation, the potency of noribogaine could be observed to be reduced. To examine this issue, we determined the potency of noribogaine for inhibition of [3H]5-HT uptake in transporter-transfected CHO cells as described in Experimental Procedures. Since noribogaine is a non-competitive inhibitor of SERT,45 work by Cheng and Prusoff 46 indicates that the IC50 equals the Ki and is not dependent on the KM of the transporter, which is important considering that SERT Ala56 demonstrates a decreased 5-HT KM compared to WT SERT.9 We found that the IC50 for noribogaine to block 5-HT uptake from hSERT transfected CHO cells was modestly, but significantly, increased for SERT Ala56 as compared to WT SERT (SERT Ala56 = 0.507 nM; R2 = 0.91; 95% CI [0.37, 0.68]; WT SERT = 0.315 nM; R2 = 0.87; 95% CI [0.22, 0.45]; P ≤ 0.05), consistent with the mutant transporter having an increased probability to rest in an outward-facing conformation during noribogaine binding.
SERT Ala56 Impact on Cytoplasmic Domain Apposition Assessed by Fluorescence Resonance Energy Transfer.
Fluorescence resonance energy transfer (FRET) approaches allow for a measurement of the relative distance between proteins or their domains,47 with caveats relating to the orientation of fluorophores potentially appearing as changes in distance.48 Biochemical and modeling studies indicate that as biogenic amine transporters cycle from inward to outward-facing conformations, the N- and C-termini move closer to each other.23,49 We hypothesized that if SERT Ala56 biases the transporter toward an outward-facing conformation, the N- and C-termini of SERT should more often lie closer together than with WT SERT, which might be evident with FRET studies of dually-tagged transporters. We therefore transfected CHO cells individually with either WT or SERT Ala56 constructs that were tagged with cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP), on the N- and C-termini, respectively. The dual addition of CFP and YFP (C-SERT-Y) did not compromise the ability of Ala56 to elevate 5-HT uptake (Figure 2A), nor alter protein expression levels, as assessed by western blot analysis (Figure S3A-B). As predicted, cells transfected with C-Ala56-Y demonstrated a significant increase in FRET efficiency compared to cells transfected with WT C-SERT-Y (Figure 2B-C). Moreover, a population analysis of the distribution of FRET efficiency from C-Ala56-Y transfected cells revealed a much tighter distribution than observed for C-SERT-Y (Figure 2D).
Figure 2: FRET efficiency of SERT N- and C-terminal proximity.

(A) The addition of CFP and YPF tag to the N-&-C terminus respectively, does not impact the ability of SERT Ala56 to enhance [3H] 5-HT uptake (20 nM for 10 min in transfected CHO cells transfected at 37°C), one-way ANOVA followed by Tukey’s post-hoc test, *P ≤ 0.05, n= 6-9; Error bars represent ± SEM. (B) Representative FRET images of transfected CHO cells with indicated CFP-YFP dual tagged constructs with CFP excitation/emission, YFP excitation/emission, calculated FRET efficiency (CFP excitation/YFP emission). (C) FRET efficiency, calculated as described in Methods. (Error Bars ± SEM, One-way ANOVA followed by Tukey’s post hoc test, *P ≤ 0.05, n=22). (D) Histogram of FRET efficiency show C-Ala56-Y distribution is significantly shifted to the right compared to WT (Kruskal-Wallis ANOVA followed by Dunn’s post hoc analysis, P ≤ 0.05, n=22) and C-Asn605-Y split into two Gaussian’s curves.
The naturally-occurring, C-terminal SERT coding substitution Lys605Asn (SERT Asn605) was also found to be transmitted to affected probands in our prior ASD study.6 As shown in our previous work, this variant demonstrates normal 5-HT uptake activity in transfected cells, but like the Ala56 variant, exhibits a complete insensitivity to PKG and p38 MAPK stimulation.10 When the C-SERT-Y construct bearing the Lys605Asn substitution was transfected into CHO cells, we detected no difference from C-SERT-Y in resulting 5-HT uptake (Figure 2A) nor did we observe an increase in the average FRET signal (Figure 2B-C). However, the sum of two Gaussians better fit the distribution of C-Asn605-Y FRET efficiencies (Figure 2D) than a single distribution (two-tailed unpaired t-test; P ≤ 0.001), with the distribution associated with greater FRET efficiencies underlying the distribution that fits the C-Ala56-Y data. These findings suggest that SERT Asn605 may populate one of two distinct conformations, either outward-facing or inward facing, with a limited population of intermediate states, unlike WT SERT. We do not know whether the conformations that these ASD associated variants confer are mimicked in FRET assays using WT SERT treated with compounds that alter SERT conformation and activity, such as fenfluramine or noribogaine, but consider this an important future study.
Sensitivity of 5-HT Transport to Methanethionsulfonates Supports an Increased Propensity for both SERT Ala56 and SERT Asn605 to Reside in an Outward-facing Conformation.
The Substituted Cysteine Accessibility Method (SCAM),50 where the aqueous exposure of cysteine (Cys) residues of functional relevance are queried using inactivating methanethiosulfonate (MTS) reagents, has been commonly used to infer conformation of transporter domains, including those belonging to SERT.51-55 In SERT, the rate of transport inactivation achieved by a membrane permeant MTS reagent such as MTSEA, at an engineered Cys (Ser277Cys) in intracellular loop 2 (ILC2) (substituted on an otherwise reactive Cys depleted transporter (SERT X5C)), provides a quantitative estimate of transporters present in inward-facing conformations.54,56 When we compared the sensitivity of SERT X5C to SERT X5C/Cys277 to MTSEA in transfected HEK-MSR cells, we found the expected increase in transport inactivation for the latter construct due to the addition of solvent exposed Cys277 in a loop critical for conformational cycling (Figure 3A and D). On the X5C/Cys277 background, substitution of either Ala56 or Asn605 for their WT counterparts rendered the Cys277 residue on these transporters less sensitive to MTSEA than SERT X5C/Cys277 (Figure 3A and D), consistent with the terminal variants having a reduction in the population of inward-facing conformations.
Figure 3: Sensitivity of N and C termini ASD SERT variants to MST inactivation of uptake supports outward-facing conformations.

(A) To determine accessibility of the cytoplasmic permeation pathway HEK-MSR cells expressing terminal ASD-associated mutants (SERT Ala56 and Asn605) in the S277C/X5C background were treated with 2 mM MTSEA for the indicated time. Following treatment, cells were assayed for [3H] 5-HT uptake (50 nM for 10 min at 37°C). Remaining activity is plotted as a percent of untreated cells and the values represent the mean ± SEM from four or more independent experiments. SERT Ala56 and Asn605 were less sensitive to MTSEA inactivation, suggesting that Cys277 was less accessible. (B&C) Cys109, located on the extracellular end of TM1, is an extracellular probe for the membrane impermeant thiol-reactive reagent, MTSES (B) and MTSET (C). Both the terminal ASVs (SERT Ala56 and Asn605) are more sensitive to MTSES and MTSET inactivation, suggesting that Cys109 is more accessible in these mutants compared to control cells. (D) Table with time for MTS reagent to decrease total 5-HT uptake in each mutant by 50% (t1/2). One-way ANOVA, Dunnett’s, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n=8.
Inactivation of 5-HT uptake at the endogenous residue Cys109 by a membrane impermeant MTS reagent, such as MTSET or MTSES, has been used successfully to indicate the fraction of transporters in an outward-facing state.57 When the sensitivity to MTSES or MTSET of WT SERT (containing Cys109) was compared to SERT Ala56 (Figure 3B-D), we observed an increased rate of inactivation of the latter construct by both MTS reagents. These findings support an increased propensity for Cys109 in either the Ala56 or the Asn605 constructs to reside in an outward-facing conformation across the duration of the assay. Together, the SCAM analysis assessed the exposure of conformationally-sensitive Cys residues and provided evidence that the conformational equilibrium of both naturally-occurring SERT coding variants shifts to favor an outward-facing state.
Evidence from Limited Proteolysis Studies that SERT Ala56 and SERT Asn605 Favor Outward-Facing Conformations.
The data presented thus far suggest that the Ala56 and the Asn605 variant SERT proteins exhibit a global change in structure that favors an open-outward conformation. SERT is a large, 630 amino acid protein, comprised of 12 transmembrane domains and cytoplasmic N- and C-termini.58 Given the location of SERT Ala56 in the N-terminus, it seemed unlikely to us that, without a local structural alteration, the single methyl group that distinguishes Ala56 from Gly56 (WT) would be able to exert global, conformational effects. Gly56 lies in a region of the N-terminus predicted to be unfolded, possibly allowing conformational alterations of this domain during and throughout the transport cycle.23 Alternatively, N-terminal interacting proteins could stabilize conformations locally and globally by differentially interacting with the Ala56 substituted domain. To examine mutation-induced changes in local N-terminal conformation, we implemented a trypsin accessibility protocol59 using membranes from CHO cells transfected with either WT or mutant SERTs. An N-terminal HA tag was present in WT and mutant constructs to allow us to monitor exposure and trypsin digestion of the N-terminus as a function of time. Importantly, the N-terminus of SERT has been reported to demonstrate differential sensitivity to trypsin cleavage as a result of ionic, substrate or antagonist interactions,28 with the N-termini of outwardly-oriented transporters being relatively protected against trypsin digestion. As shown in Figure 4A, HA-tagged SERT Ala56 demonstrated a greater degree of trypsin resistance as compared to HA-tagged WT SERT. Interestingly, the HA-tagged SERT Asn605 variant also displayed significantly reduced N-terminal trypsin sensitivity (Figure 4B).
Figure 4: Tryptic digestion of the N-terminus of terminal ASD SERT variants.

Membranes (50 μg total) isolated from CHO cells expressing (A) HA-SERT or HA-SERT Ala56 or (B) HA-SERT Asn605 were incubated in buffer containing 150 mM NaCl. Trypsin (4 μg) was added at room temperature for 5, 10, or 15 minutes. Samples were subjected to western blot analysis of HA tag. Percentage of SERT cleaved by trypsin was calculated by dividing the band density of the trypsin sample by the total (labeled as “T” on blot). (Error bars indicate ± SEM Paired t-test. *P ≤ 0.05, **P ≤ 0.01, n=6-9). (C) Ionic substitution of NaCl with 150 mM ChCl did not affect the increased protection of SERT Ala56 N-terminal tryptic digestion at 5 min. Paired t-test. *P ≤ 0.05, n=6-9. (D) Addition of 10 μM D-fenfluramine to membranes normalized SERT Ala56 digestion to WT levels at 5 min (Paired t-test. *P ≤ 0.05, n =10).
Whether a global change in conformation impacts the structure of the N-terminus, or vice-versa, cannot be assessed from the trypsin sensitivity data noted above. However, previous studies28 have demonstrated that the presence or absence of NaCl alters N-terminus trypsin sensitivity in a manner that might be useful to distinguish whether the SERT Ala56 exerts conformational effects independent of changes in global structure. In support of this idea, the N-terminus of WT SERT exhibits reduced trypsin sensitivity when Na+ in the buffer is substituted with impermeant cations, presumably due to a requirement for Na+ interactions at the intramembrane substrate binding site to stabilize transporters in an open-outward conformation.28 We therefore evaluated the trypsin sensitivity of WT HA-SERT and HA-SERT Ala56 in media where Na+ was substituted by choline chloride (ChCl). In these studies, we found that the N-terminus of HA-SERT Ala56 remained less trypsin-sensitive than WT HA-SERT, despite the absence of Na+ (Figure 4C). These findings indicate that the Ala56 substitution imposes local changes in conformation of the SERT N-terminus and/or the differential binding of associated proteins to this domain, in addition to the more global conformational changes revealed by measures of 5-HT uptake/efflux, FRET or transporter functional inactivation by MTS reagents.
Finally, we explored the effects of D-fenfluramine at saturating concentrations on the protection of the SERT N-terminus to tryptic digestion (in the presence of NaCl). We found that the addition of 10 μM D-fenfluramine to membrane preparations had no effect on proteolysis of WT SERT (Figure 4D), consistent with findings of Kern and colleagues using the SERT-targeted amphetamine, p-chloroamphetamine.28 However, D-fenfluramine incubation with HA-SERT Ala56 expressing membranes increased N-terminal trypsin sensitivity to that seen with WT HA-SERT (Figure 4D). These findings indicate that the binding of substrates to SERT is sufficient to relieve N-terminal conformational changes induced by SERT Ala56, suggesting bi-directional communication between the N-terminus and residues forming the substrate binding pocket.
DISCUSSION
Altogether, the studies in our report reveal a remarkable ability of small changes in the sequence of cytoplasmic N- and C-termini to affect both local and global SERT conformations. SERT Ala56 confers elevated 5-HT uptake at sub-saturating substrate concentrations relative to WT SERT, mimicking transient 5-HT transport changes following PKG and p38 MAPK activation.16,17 Consistent with this idea, SERT Ala56 (as well as SERT Asn605) interferes with the ability of PKG and p38 MAPK activators to elevate transporter activity further.9,10 Ala56 lies in the middle of the N-terminus, which has the potential, based on models, to interact with intracellular loop 2 (ICL2) that lies between transmembrane domains 4 and 5. ICL2 has been shown to be conformationally dynamic and bears a site, Thr276, that becomes phosphorylated following PKG activation.15,60,61 Thr276 has been shown to be phosphorylated in the inward-facing conformation upon the activation of PKG which leads to enhanced uptake kinetics.15,62,63 Because SERT Ala56 is insensitive to increased 5-HT uptake upon PKG activation, we speculate that the Ala56 substitution alters an N-terminus:ICL2 interaction to bias conformation in a manner similar to that arising from PKG-dependent Thr276 phosphorylation. Such a model can explain the reduction in 5-HT KM observed with SERT Ala56.9 The ability of the Ala56 substitution to induce N-terminal conformational changes, along with the ability of p38α MAPK inhibition to reduce SERT Ala56 hyperphosphorylation and hyperactivity,10,11,13 suggests that the phosphorylation of one or more sites, such as Thr276, arises following changes in either N-terminal conformation or kinase activation. Further studies are needed to identify these sites and examine their responsiveness to changes in N-terminus structure.
Surprisingly, the Asn605 substitution of the SERT C-terminus impacts both the sensitivity of the N-terminus to trypsin and biases the transporter globally to an outward-facing conformation as seen with the Ala56 variant, without increasing 5-HT uptake. Our FRET studies indicate that the C-terminal mutation may stabilize SERT in either outward-facing or inward-facing conformations, though this results in no net gain in 5-HT uptake. Nonetheless, the Asn605 mutation imparts, like Ala56, an insensitivity of SERT to activators of both PKG and p38 MAPK. A putative p38 MAPK site has been identified in the SERT C-terminus at Thr616.64 Interestingly, Asn605 lies in a critical alpha helical loop which interact with ICL1 to promote proper folding of SERT in an inward-facing conformation ready to traffic to the surface.65
A model worth exploring further is that SERT Ala56 facilitates transporter phosphorylation, such as at Thr276, that stabilizes an outward-facing conformation whereas the Asn605 substitution has bimodal conformational effects, one that enhances N- and C-terminal interactions to stabilize an outward-facing conformation, and another that disrupts p38 MAPK phosphorylation of the C-terminus, stabilizing an inward-facing conformation. Clearly, additional biochemical, structural and modeling studies are needed to clarify these possibilities. Regardless, our findings reveal a capacity for relatively small changes in SERT cytoplasmic domains to drive alterations in transporter conformation, underlying functional and regulatory changes.
EXPERIMENTAL PROCEDURES
Animals and Materials
All experiments using animal subjects were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments involving animal subjects were conducted as preapproved by the Vanderbilt University and Florida Atlantic University Institutional Animal Care and Use Committees. SERT Ala56 and SERT Gly56 (WT) littermate males (129/sv background) 8-12 weeks old were generated from heterozygous breeding. Mouse genotyping was conducted as previously described.11 All reagents, salts and buffers, unless otherwise specified, were obtained from Sigma-Aldrich (St. Louis, MO, USA).
CHO Cell Culture and Transfection
Chinese hamster ovary (CHO) cells used for transfection of human SERT (hSERT) constructs were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Carlsbad, CA, USA), 10% fetal bovine serum (FBS; Thermo Fisher, Waltham, MA, USA) 2 mM L-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C in a 5% CO2 humidified incubator.
Culture of the stably expressing Flp-In™-Cho hSERT and hSERT Ala56 were used as described in Prasad et al. 2009.9 However, to overcome the lower total and surface expression of SERT Ala56 compared to WT, which is believe to be due to 5-HT levels in the FBS in the media (although not shown directly), cells were grown as described above except the DMEM was supplemented with dialyzed FBS (Cat # F0392, Millipore Sigma, Burlington, MA, USA).
D-fenfluramine-Mediated [3H]5-HT Efflux
In vitro D-fenfluramine release, Flp-In™-CHO-stable cell lines expressing SERT Ala56 or WT SERT, previously described,9 were plated at 100,000 cells per well on a 24 well poly-D lysine coated plate. At 24-48 hrs after plating, cells were washed 2X with warmed Krebs-Ringers-HEPES (KRH) assay buffer (130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 10 mM HEPES, 10 μM glucose, 100 μM pargyline, 100 μM ascorbic acid, pH 7.4) and then loaded with 20 nM [3H]5-HT (Hydroxytryptamine Creatinine Sulfate; Specific activity 28 Ci·mmol−1; Perkin-Elmer, NET498, Waltham, MA, USA) for 1 hr at 37°C followed by 2X wash with warmed KRH to remove excess [3H]5-HT. Cells were then subjected to 500 μL total volume of 10 μM D-fenfluramine or vehicle for the indicated time. The supernatant was then collected and 0.1% SDS was added to the wells to lyse the cells to account for unreleased [3H]5-HT. 250 μL of EcoScint XR scintillation fluid (National Diagnostics, Atlanta, GA, USA) was added to the supernatant and the cell lysates and counted using a TriCarb 2900TR scintillation counter (Perkin-Elmer, Waltham, MA, USA). Efflux was calculated by the counts in supernatant divided by the total number of counts (supernatant + cell lysis) in both the presence and absence of D-fenfluramine. To obtain D-fenfluramine mediated efflux, efflux in the absence of D-fenfluramine was subtracted from efflux in the presence D-fenfluramine. Data was analyzed by an ordinary one-way ANOVA followed by Sidak’s multiple comparison test of genotype differences.
Ex vivo D-fenfluramine induced [3H]5-HT efflux was conducted as previously described.66 Briefly, the hippocampus of a mouse was dissected after rapid decapitation and chopped on a cold plate into ~300 μm slices with a razor blade. Slices were placed in 300 μL of oxygenated Krebs’ Ringer Bicarbonate (KRB) buffer (126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2 1.2 mM MgCl2, 1.2 mM NaH2PO4, 10 mM D-glucose, and 21.4 mM NaHCO3, pH 7.4) supplemented with 50 μM pargyline, 50 μM ascorbic acid, and 10 nM nisoxetine and 100nM nomifensine to block NET and DAT, respectively. The slices were incubated with 400 nM [3H]5-HT for 30 minutes at 37°C. The slices were then loaded into the perfusion chamber of the Brandel SF-12 Suprafusion system (Brandel, Gaithersburg, MD, USA), sandwiched between GF/B glass fiber filter discs (Whatman, Maidstone, UK). Chambers containing filter-immobilized brain slices were perfused at a flow rate of 0.7 mL/min with oxygenated KRB buffer for 45 min to remove unloaded [3H]5-HT. Then samples were collected every 2 minutes, with the first 16 min used to establish baseline release, followed by a 16 min perfusion with the indicated concentration of D-fenfluramine, and then an 8 min wash out with KRB buffer. At the end of the experiment, the filters and tissue were collected and lysed in 2 mL of 20% SDS. Five mL of EcoScint XR scintillation fluid (National Diagnostics, Atlanta, GA, USA) was added to each fraction and radioactivity was counted using a TriCarb 2900TR scintillation counter (Perkin-Elmer, Waltham, MA, USA). Data for release is presented as a fraction of the total [3H]5-HT loaded into each sample (amount released + amount remaining in the tissue), normalized as a percent of baseline. Two-way repeated measure ANOVA followed by Bonferroni post-hoc test of genotype differences was used to analyze statistical significance.
D-fenfluramine Competition Binding Assay with [3H]Citalopram
Hippocampal membrane samples from SERT Ala56 mice and their WT littermate controls were prepared as previously described.11 Briefly, after rapid decapitation, the hippocampus was extracted and homogenized utilizing a Teflon-glass homogenizer in 3 mL of 50 mM Tris, pH 7.4 followed by a 20-minute centrifugation at 15,000 X g. The resulting pellet was washed again in 50 mM Tris, pH 7.4 and recentrifuged for 20 minutes at 15,000 X g. The supernatant was discarded and the pellet was resuspended in 50 mM Tris with 120 mM NaCl, pH 7.4. 200 μg of membranes were preincubated with various concentrations of fenfluramine (0.1 nM, 1 nM, 10 nM, 100 nM, 500 nM, 1 μM, 5 μM, 10 μM, 100 μM, 1 mM) for 10 min at 37°C followed by the addition of 5 nM [3H]citalopram (Perkin Elmer, Specific activity 74.5 Ci·mmol−1, NET1039, Waltham, MA, USA) for 1 hr at room temp. Samples were than harvested using a Brandel 48-sample Harvester (Brandel, Gaithersburg, MD, USA) onto GF/B Whatman filters (Whatman, Maidstone, UK). The filters were washed three times with ice-cold PBS buffer and then placed into 7 mL of EcoScint H scintillation fluid (National Diagnostics, Atlanta, GA, USA) overnight. Radioactivity was counted using a TriCarb 2900TR scintillation counter (Perkin-Elmer, Waltham, MA, USA). Binding was calculated and the non-linear fit of log IC50 inhibition was calculated using Prism 7.
In Vivo Microdialysis Assessment of D-fenfluramine Mediated 5-HT Efflux
Mice were anesthetized with isoflurane and placed in a stereotaxic frame using mouse-specific ear bars (Kopf Instruments, Tujunga, CA, USA). A guide cannula (Synaptech, Marquette, MI, USA) was placed 1 mm above the dorsal hippocampus (−1.94 AP from Bregma, ± 2.0 ML and −1.0 DV from dura and secured to the skull with glass ionomer cement (Instech Solomon, Plymouth Meeting, PA, USA). After recovery from surgery, animals were placed in individual dialysis chambers. A microdialysis probe (Synaptech, Marquette, MI, USA) with the active length of 1 mm was inserted into the guide cannula. One end of the tether was attached to the headpiece and the other end attached to a liquid swivel (Instech Solomon, Plymouth Meeting, PA, USA) that was mounted on a counterbalanced arm above the dialysis chamber. The probe was perfused at a flow rate of 1.0 μL/min with artificial cerebral spinal fluid (aCSF) containing 149 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, pH 7.2 at a flow rate of 1.0 μL/min overnight. On the day of the experiment, four baseline dialysis fractions (20 min each) were collected. After the 4th baseline sample the aCSF was switched to aCSF containing 10 μM D-fenfluramine for 20 min. Dialysate samples were stored at −80°C and analyzed by HPLC-EC for 5-HT levels. After the dialysis session, animals were overdosed with sodium pentobarbital, brains removed and post-fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, sectioned, stained, and examined for acceptable probe placement. Extracellular 5-HT levels from 0-80 min at the beginning of the experiment are reported as “Baseline Levels” and reported in nM concentration. Area under the peak was calculated utilizing Prism 7 “Area Under the Curve” function taking the mean from 0-80 min as the baseline for each genotype.
5-HT Transport Assay in SERT Transiently-Transfected CHO Cells
CHO cells were plated on clear bottom 96 well poly-D lysine coated plates at 20,000 cells per well. The next day, cells were transfected with 0.1 μg of DNA per well utilizing TransIT®-LT1 (Mirus, Madison, WI, USA). Forty-eight hours later, cell culture media was removed and plates were washed 3X with 300 μL warmed phosphate buffered saline (PBS) with an ELX microplate washer (Biotek, Winooski, VT, USA). [3H]5-HT uptake assays were performed in 200 μL final volume of KRH assay buffer. Inhibitors were added and incubated at 37°C for 10 min followed by addition of 20 nM (final concentration) [3H]5-HT. For the uptake inhibition studies, noribogaine HCL was custom manufacture under cGMP conditions performed by Ajimoto Omnichem (Lot number: 606950001). Eight serial dilutions of noribogaine from 10 mM to 1 nM were used. After 10 min at 37°C, transport assays were stopped by rapid wash 3X with ice-cold PBS using the ELX microplate washer. MicroScint scintillation cocktail (200 μL) was added to each well and agitated for 1 hr. Counts representing total and non-specific 5-HT uptake were measured using a MicroBeta2 Microplate counter (Perkin-Elmer, Waltham, MA, USA). Specific uptake was determined by subtracting the counts from wells incubated with 10 μM paroxetine for 10 min at 37°C prior to the addition of [3H]5-HT. The non-linear fit of log IC50 inhibition was calculated using Prism 7 to determine statistical significance.
Assessment of SERT N- and C-termini Apposition using Florescence Resonance Energy Transfer (FRET)
CFP, YFP, CFP-YFP, and CFP-SERT-YFP constructs were generously provided by Sonja Sucic and Harald Sittee, Medical University of Vienna. CFP-Ala56-YFP and CFP-Asn605-YFP constructs were generated from these vectors using the Q5 site-directed mutagenesis kit (New England Biolabs, Ipswich, MA, USA). Each construct was individually transfected into CHO cells plated (20,000 cells per dish) onto 35 mm MatTek poly-d-lysine coated dishes (No. 1.5 cover glass, Ashland, MA, USA). Cells were washed 2X with warmed PBS and then imaged in KRH buffer over 5 min in a Tokai Hit Stage top incubator set to 37°C and 5% CO2. We used a 60X oil immersion objective (Plan Apo Lambda, NA 1.4) on an A1R confocal Ti inverted microscope at the Florida Atlantic University Cell Imaging Core. All FRET images were captured utilizing the FRET plugin of NIS Elements Imaging Software (Nikon, Tokyo, Japan). The scan head dichroic mirror optical configurations were set up to acquire the donor alone (Dd: excite CFP at 440nm and dichromatic mirror set to capture CFP 485/35 nm), acceptor alone (Aa; excite YFP at 514 and dichromatic mirror set to capture YFP at 538/33 nm), FRET (Da; excite CFP at 440 nm and dichromatic mirror set to capture YFP at 538/33nm) and bleed through (Ad; excite YPF at 514 nm and dichromatic mirror set to capture CFP 485/35 nm). Negative controls of CFP and YFP constructs were imaged alone to acquire CoA (Acceptor in FRET or DaACCEPTOR) and CoB (Donor in FRET or DaDONER). As a positive FRET control, a fused CFP-YFP construct was also imaged. Images were thresholded manually to regions of interest that were defined by the membrane of the cells and the corrected FRET (Corr FRET) and FRET efficiency (FRET eff) for each image was averaged over the 5 min by the FRET plugin in NIS Elements as followed:
Total expression of C-SERT-Y and variant constructs were assessed by western blot analysis. Briefly, CHO cells were lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) for 1 hr at 4° C and then centrifuged for 20 min at 17,000 X g. 50 μg of the resulting supernatant was incubated with 4X Laemmle buffer (Bio-Rad Laboratories, Hercules, CA, USA) at room temp for 30 min and then separated on a NuPAGE 10% Bis-Tris protein gel (Invitrogen, Carlsbad, CA, USA) and transferred to Immobilon-FL PVDF membrane (Millipore Sigma, Burlington, MA, USA). Protein was visualized by immunoblotting with a rabbit-CFP antibody (BioRad, Hercules, CA, USA) then probed with IRDye 680RD secondary antibody (LI-COR Biosciences, Lincoln, NE, USA). Immunoreactive bands were imaged using the Odyssey Fc (LI-COR Biosciences, Lincoln, NE, USA). Actin levels were measured as a loading control using an HRP-labeled β-actin antibody (Sigma-Aldrich (St. Louis, MO, USA). Densitometry was performed using Image Studio software (LI-COR Biosciences, Lincoln, NE USA).
Sensitivity to Transport Inactivation by Methanethiosulfonate Reagents
ASD variants were introduced into native or cysteine-reduced SERT backgrounds of pcDNA3-hSERT using the Stratagene QuikChange® kit (Agilent Technologies, Santa Clara, CA, USA) and verified by sequencing (Eurofins MWG Operon, Huntsville, AL, USA). Analysis of the intracellular Cys277 probe required introduction of the ASDs into the X5C background (C15A, C21A, C109A, C357I, and C622A)55 prior to transient expression in HEK-MSR cells (Thermo Fisher, Waltham, MA, USA) using TransIT®-LT1 (1 μl per 200 ng of DNA; Mirus, Madison, WI, USA). Cells were maintained in a humidified chamber with 5% CO2 at 37°C in DMEM supplemented with 10% FBS and 600 μg/ml G418. HEK-MSR cells were plated at a density of 10,000 or 50,000 cells/well in 24-well culture plates, allowed to settle and attach for 24 hrs, and then transfected with the mutant plasmids. After 24 hr, cells were washed with 37°C PBS/CM buffer (137 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 1.8 mM KH2PO4, 0.1 mM CaCl2, 1.0 mM MgCl2, pH 7.4) or NMDG-Cl buffer (120 mM NMDG-Cl, 5.4 mM KCl, 1.2 mM CaCl2, 10 mM glucose, 7.5 mM HEPES, pH 7.4) and incubated with 1 mM MTSET, 10 mM MTSES, or 2 mM MTSEA (Biotium, Fremont CA, USA) for 1-10 min at room temp. Post-treatment, cells were washed with PBS/CM or NMDG-Cl buffer and assayed for transport activity by incubation with 50 nM [3H]5-HT for 5 min at 37°C in MKRHG buffer (5 mM Tris, 7.5 mM HEPES, 120 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose, pH 7.4). Cells were dissolved in MicroScint™-20 (Perkin-Elmer, Waltham, MA, USA) scintillation fluid and counts/min of radioactivity were determined using a TopCount NXT scintillation counter (Perkin-Elmer, Waltham, MA, USA). Specific uptake was determined by subtracting uptake observed in non-transfected cells. All experiments were repeated in three or more separate assays and data fit to a one phase decay curve. Half-life values estimated from inactivation were analyzed using a one-way ANOVA followed by a post-hoc Dunnett’s test (GraphPad Software Prism 5).
Limited Proteolysis of SERT N-terminus
CHO cells transfected with HA-tagged SERT constructs were washed with PBS to remove media and then scraped in 1 mL of Trypsin Homogenization Buffer (20 mM HEPES, 2 mM MgCl2, 0.5 M EDTA, pH 7.4). Cells were incubated on ice for 30 min followed by five, 1 sec sonication pulses using a Q125 Sonicator (Fisher Scientific, Waltham, MA, USA). Samples were centrifuged for 10 minutes at 17,000 X g and the resulting pellet was resuspended in trypsin homogenization buffer substituted with 150 mM NaCl or ChCl. For digestion assays, 50 μg of protein was incubated with 4 μg of trypsin (Promega, Madison, WI, USA) for 5, 10 or 15 min on ice. The reaction was stopped by the addition of 8 μg of soybean inhibitor (Sigma-Aldrich, St. Louis, MO, USA). Then, 2X Laemmle buffer (Bio-Rad Laboratories, Hercules, CA, USA) was added to the sample and boiled at 95°C for 3 min. Samples were separated using a NuPAGE 10% Bis-Tris protein gel (Invitrogen, Carlsbad, CA, USA) and transferred to Immobilon-FL PVDF membrane (Millipore Sigma, Burlington, MA, USA). HA-tagged SERT protein was visualized using a mouse anti-HA antibody (BioLegend, San Diego, CA, USA) probed with IRDye 680RD secondary antibody (LI-COR Biosciences, Lincoln, NE, USA). Immunoreactive bands were imaged using the Odyssey Fc (LI-COR Biosciences, Lincoln, NE, USA) and densitometry was performed using Image Studio software (LI-COR Biosciences, Lincoln, NE USA). Tryptic digestion efficiency was determined by calculating the percent uncleaved as the band intensity of tryptic digestion divided by the total band intensity. If there was a 2-fold difference in total SERT expression across genotypes or less than 90% of the protein remained after digestion, the blot was not analyzed. To account for variation in band intensity across blots, paired Student’s t-tests at each time point were used for statistical analysis.
Statistical and Graphical Analyses
Data from experiments was analyzed and graphed using Prism 7.0 (GraphPad Software, Inc., La Jolla, CA, USA). For all analyses, a P ≤ 0.05 was used to infer statistical significance. Specific details of statistical tests are given in each figure legend.
Supplementary Material
ACKNOWLEDGEMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We wish to acknowledge the expert lab support at Vanderbilt University of Christina Svitek, Jane Wright, Tracey Moore-Jarrett, Sarah Sturgeon, Qiao Han, and Angela Steele, and at Florida Atlantic University of Matthew Gross, Sean Mellish, Samantha McGovern, Isabel Stillman and Jana Borner.
FUNDING SOURCE
Financial support for the studies derives from Vanderbilt Pharmacology T32 (GM07628), an Elaine Sanders-Bush Scholar’s Award from the Vanderbilt Silvio O. Conte Center for Neuroscience Research (MAQ). NARSAD YI Award 25230 (MJR) and NIH grants MH096972 (RDB) and MH094527 (RDB).
Footnotes
SUPPORTING INFORMATION
Figure S1. High concentrations of D-fenfluramine mediated [3H] 5-HT efflux; Figure S2. D-fenfluramine competition binding assay with [3H]citalopram; Figure S3. Western blot of total C-SERT-Y and variant constructs.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
- (1).Waye MMY; Cheng HY Genetics and Epigenetics of Autism: A Review. Psychiatry Clin. Neurosci. 2018, 72 (4), 228–244. 10.1111/pcn.12606. [DOI] [PubMed] [Google Scholar]
- (2).Muller CL; Anacker AMJ; Veenstra-VanderWeele J The Serotonin System in Autism Spectrum Disorder: From Biomarker to Animal Models. Neuroscience 2016, 321, 24–41. 10.1016/j.neuroscience.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schain RJ; Freedman DX Studies on 5-Hydroxyindole Metabolism in Autistic and Other Mentally Retarded Children. J. Pediatr. 1961, 58 (3), 315–320. 10.1016/S0022-3476(61)80261-8. [DOI] [PubMed] [Google Scholar]
- (4).Cook EH; Arora RC; Anderson GM; Berry-Kravis EM; Yan S. ya; Yeoh HC; Sklena PJ; Charak DA; Leventhal BL Platelet Serotonin Studies in Hyperserotonemic Relatives of Children with Autistic Disorder. Life Sci. 1993, 52 (25), 2005–2015. 10.1016/0024-3205(93)90685-V. [DOI] [PubMed] [Google Scholar]
- (5).Lesch K; Wolozin BL; Murphy DL; Riederer P Primary Structure of the Human Platelet Serotonin Uptake Site : Identity with the Brain Serotonin Transporter. J. Neurochem 1993, 60, 2319–2322. [DOI] [PubMed] [Google Scholar]
- (6).Sutcliffe JS; Delahanty RJ; Prasad HC; McCauley JL; Han Q; Jiang L; Li C; Folstein SE; Blakely RD Allelic Heterogeneity at the Serotonin Transporter Locus (SLC6A4) Confers Susceptibility to Autism and Rigid-Compulsive Behaviors. Am. J. Hum. Genet. 2005, 77 (2), 265–279. 10.1086/432648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stone JL; Merriman B; Cantor RM; Yonan AL; Gilliam TC; Geschwind DH; Nelson SF Evidence for Sex-Specific Risk Alleles in Autism Spectrum Disorder. Am. J. Hum. Genet. 2004, 75 (6), 1117–1123. 10.1086/426034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cantor RM; Kono N; Duvall JA; Alvarez-Retuerto A; Stone JL; Alarcón M; Nelson SF; Geschwind DH Replication of Autism Linkage: Fine-Mapping Peak at 17q21. Am. J. Hum. Genet. 2005, 76 (6), 1050–1056. 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Prasad HC; Steiner JA; Sutcliffe JS; Blakely RD Enhanced Activity of Human Serotonin Transporter Variants Associated with Autism. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364 (1514), 163–173. 10.1098/rstb.2008.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Prasad HC; Zhu C-B; McCauley JL; Samuvel DJ; Ramamoorthy S; Shelton RC; Hewlett WA; Sutcliffe JS; Blakely RD Human Serotonin Transporter Variants Display Altered Sensitivity to Protein Kinase G and P38 Mitogen-Activated Protein Kinase. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11545–11550. 10.1073/pnas.0501432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Veenstra-VanderWeele J; Muller CL; Iwamoto H; Sauer JE; Owens WA; Shah CR; Cohen J; Mannangatti P; Jessen T; Thompson BJ; et al. Autism Gene Variant Causes Hyperserotonemia, Serotonin Receptor Hypersensitivity, Social Impairment and Repetitive Behavior. Proc. Natl. Acad. Sci. 2012, 109 (14), 5469–5474. 10.1073/pnas.1112345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Veenstra-VanderWeele J; Jessen TN; Thompson BJ; Carter M; Prasad HC; Steiner JA; Sutcliffe JS; Blakely RD Modeling Rare Gene Variation to Gain Insight into the Oldest Biomarker in Autism: Construction of the Serotonin Transporter Gly56Ala Knock-in Mouse. J. Neurodev. Disord. 2009, 1, 158–171. 10.1007/s11689-009-9020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Robson MJ; Quinlan MA; Margolis KG; Gajewski-Kurdziel PA; Veenstra-VanderWeele J; Gershon MD; Watterson DM; Blakely RD P38α MAPK Signaling Drives Pharmacologically Reversible Brain and Gastrointestinal Phenotypes in the SERT Ala56 Mouse. Proc. Natl. Acad. Sci. 2018, 115 (43), E10245–E10254. 10.1073/pnas.1809137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Margolis KG; Li Z; Stevanovic K; Saurman V; Israelyan N; Anderson GM; Snyder I; Veenstra-VanderWeele J; Blakely RD; Gershon MD Serotonin Transporter Variant Drives Preventable Gastrointestinal Abnormalities in Development and Function. J. Clin. Invest. 2016, 126 (6), 2221–2235. 10.1172/JCI84877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ramamoorthy S; Samuvel DJ; Buck ER; Rudnick G; Jayanthi LD Phosphorylation of Threonine Residue 276 Is Required for Acute Regulation of Serotonin Transporter by Cyclic GMP. J. Biol. Chem. 2007, 282 (16), 11639–11647. 10.1074/jbc.M611353200. [DOI] [PubMed] [Google Scholar]
- (16).Zhu CB; Carneiro AM; Dostmann WR; Hewlett W. a.; Blakely RD P38 MAPK Activation Elevates Serotonin Transport Activity via a Trafficking-Independent, Protein Phosphatase 2A-Dependent Process. J. Biol. Chem. 2005, 280 (16), 15649–15658. 10.1074/jbc.M410858200. [DOI] [PubMed] [Google Scholar]
- (17).Zhu C-B; Hewlett WA; Feoktistov I; Biaggioni I; Blakely RD Adenosine Receptor, Protein Kinase G, and P38 Mitogen-Activated Protein Kinase-Dependent Up-Regulation of Serotonin Transporters Involves Both Transporter Trafficking and Activation. Mol. Pharmacol. 2004, 5 (10), 1462–1474. 10.1124/mol.65.6.1462. [DOI] [PubMed] [Google Scholar]
- (18).Kristensen AS; Andersen J; Jorgensen TN; Sorensen L; Eriksen J; Loland CJ; Stromgaard K; Gether U SLC6 Neurotransmitter Transporters: Structure, Function, and Regulation. Pharmacol. Rev. 2011, 63 (3), 585–640. 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- (19).Forrest LR; Zhang Y-W; Jacobs MT; Gesmonde J; Xie L; Honig BH; Rudnick G Mechanism for Alternating Access in Neurotransmitter Transporters. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (30), 10338–10343. 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Adhikary S; Deredge DJ; Nagarajan A; Forrest LR; Wintrode PL; Singh SK Conformational Dynamics of a Neurotransmitter:Sodium Symporter in a Lipid Bilayer. Proc. Natl. Acad. Sci. 2017, 114 (10), E1786–E1795. 10.1073/pnas.1613293114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen J-G; Rudnick G Permeation and Gating Residues in Serotonin Transporter. Proc. Natl. Acad. Sci. 2000, 97 (3), 1044–1049. 10.1073/pnas.97.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Coleman JA; Green EM; Gouaux E X-Ray Structures and Mechanism of the Human Serotonin Transporter. Nature 2016, 532 (7599), 334–339. 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fenollar-Ferrer C; Stockner T; Schwarz TC; Pal A; Gotovina J; Hofmaier T; Jayaraman K; Adhikary S; Kudlacek O; Mehdipour AR; et al. Structure and Regulatory Interactions of the Cytoplasmic Terminal Domains of Serotonin Transporter. Biochemistry 2014, 53 (33), 5444–5460. 10.1021/bi500637f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sweeney CG; Tremblay BP; Stockner T; Sitte HH; Melikian HE Dopamine Transporter Amino and Carboxyl Termini Synergistically Contribute to Substrate and Inhibitor Affinities. J. Biol. Chem. 2017, 292 (4), 1302–1309. 10.1074/jbc.M116.762872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Razavi AM; Khelashvili G; Weinstein H How Structural Elements Evolving from Bacterial to Human SLC6 Transporters Enabled New Functional Properties. BMC Biol. 2018, 16 (1). 10.1186/s12915-018-0495-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sucic S; Dallinger S; Zdrazil B; Weissensteiner R; Jørgensen TN; Holy M; Kudlacek O; Seidel S; Cha JH; Gether U; et al. The N-Terminus of Monoamine Transporters Is a Lever Required for the Action of Amphetamines. J. Biol. Chem. 2010, 285 (14), 10924–10938. 10.1074/jbc.M109.083154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Khoshbouei H; Sen N; Guptaroy B; Johnson L; Lund D; Gnegy ME; Galli A; Javitch JA N-Terminal Phosphorylation of the Dopamine Transporter Is Required for Amphetamine-Induced Efflux. PLoS Biol. 2004, 2 (3), E78 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kern C; Erdem FA; El-Kasaby A; Sandtner W; Freissmuth M; Sucic S The N-Terminus Specifies the Switch between Transport Modes of the Human Serotonin Transporter. J. Biol. Chem. 2017, 292 (9), 3603–3613. 10.1074/jbc.M116.771360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Khelashvili G; Stanley N; Sahai MA; Medina J; LeVine MV; Shi L; De Fabritiis G; Weinstein H Spontaneous Inward Opening of the Dopamine Transporter Is Triggered by PIP2-Regulated Dynamics of the N-Terminus. ACS Chem. Neurosci. 2015, 6 (11), 1825–1837. 10.1021/acschemneuro.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sulzer D; Sonders MS; Poulsen NW; Galli A Mechanisms of Neurotransmitter Release by Amphetamines: A Review. Prog. Neurobiol. 2005, 75 (6), 406–433. 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- (31).Fischer JF; Cho AK Chemical Release of Dopamine from Striatal Homogenates: Evidence for an Exchange Diffusion Model. J. Pharmacol. Exp. Ther. 1979, 208 (2), 203–209. [PubMed] [Google Scholar]
- (32).Sitte HH; Hiptmair B; Zwach J; Pifl C; Singer EA; Scholze P Quantitative Analysis of Inward and Outward Transport Rates in Cells Stably Expressing the Cloned Human Serotonin Transporter: Inconsistencies with the Hypothesis of Facilitated Exchange Diffusion. Mol. Pharmacol. 2001, 59 (5), 1129–1137. [DOI] [PubMed] [Google Scholar]
- (33).Rothman RB; Baumann MH; Dersch CM; Romero DV; Rice KC; Carroll FI; Partilla JS Amphetamine-Type Central Nervous System Stimulants Release Norepinephrine More Potently Than They Release Dopamine and Serotonin. Synapse 2001, 39, 32–41. [DOI] [PubMed] [Google Scholar]
- (34).Hilber B; Scholze P; Dorostkar MM; Sandtner W; Holy M; Boehm S; Singer EA; Sitte HH Serotonin-Transporter Mediated Efflux: A Pharmacological Analysis of Amphetamines and Non-Amphetamines. Neuropharmacology 2005, 49 (6), 811–819. 10.1016/j.neuropharm.2005.08.008. [DOI] [PubMed] [Google Scholar]
- (35).Rudnick G; Wall SC P-Chloroamphetamine Induces Serotonin Release through Serotonin Transporters. Biochemistry 1992, 31 (29), 6710–6718. 10.1021/bi00144a010. [DOI] [PubMed] [Google Scholar]
- (36).Gobbi M; Frittoli E; Uslenghi A; Mennini T Evidence of an Exocytotic-Like Release of [3H]5-Hydroxytryptamine Induced by d-Fenfluramine in Rat Hippocampal Synaptosomes. Eur. J. Pharmacol. 1993, 238 (1), 9–17. 10.1016/0014-2999(93)90499-8. [DOI] [PubMed] [Google Scholar]
- (37).Balcioglu A; Wurtman RJ Effects of Fenfluramine and Phentermine (Fen-Phen) on Dopamine and Serotonin Release in Rat Striatum: In Vivo Microdialysis Study in Conscious Animals. Brain Res. 1998, 813 (1), 67–72. 10.1016/S0006-8993(98)01003-8. [DOI] [PubMed] [Google Scholar]
- (38).Rocher C; Jacquot C; Gardier AM Simultaneous Effects of Local Dexfenfluramine Application on Extracellular Glutamate and Serotonin Levels in Rat Frontal Cortex: A Reverse Microdialysis Study. Neuropharmacology 1999, 38 (4), 513–523. 10.1016/S0028-3908(98)00212-3. [DOI] [PubMed] [Google Scholar]
- (39).Guptaroy B; Zhang M; Bowton E; Binda F; Shi L; Weinstein H; Galli A; Javitch JA; Neubig RR; Gnegy ME A Juxtamembrane Mutation in the N-Terminus of the Dopamine Transporter Induces Preference for an Inward-Facing Conformation. Mol. Pharmacol. 2009, 75 (3), 514–524. 10.1124/mol.108.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Binda F; Dipace C; Bowton E; Robertson SD; Lute BJ; Fog JU; Zhang M; Sen N; Colbran RJ; Gnegy ME; et al. Syntaxin 1A Interaction with the Dopamine Transporter Promotes Amphetamine-Induced Dopamine Efflux. Mol. Pharmacol. 2008, 74 (4), 1101–1108. 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dipace C; Sung U; Binda F; Blakely RD; Galli A Amphetamine Induces a Calcium/Calmodulin-Dependent Protein Kinase II-Dependent Reduction in Norepinephrine Transporter Surface Expression Linked to Changes in Syntaxin 1A/Transporter Complexes. Mol. Pharmacol. 2007, 71 (1), 230–239. 10.1124/mol.106.026690. [DOI] [PubMed] [Google Scholar]
- (42).Jacobs MT; Zhang YW; Campbell SD; Rudnick G Ibogaine, a Noncompetitive Inhibitor of Serotonin Transport, Acts by Stabilizing the Cytoplasm-Facing State of the Transporter. J. Biol. Chem. 2007, 282 (40), 29441–29447. 10.1074/jbc.M704456200. [DOI] [PubMed] [Google Scholar]
- (43).Staley JK; Ouyang Q; Pablo J; Hearn WL; Flynn DD; Rothman RB; Rice KC; Mash DC Pharmacological Screen for Activities of 12-Hydroxyibogamine: A Primary Metabolite of the Indole Alkaloid Ibogaine. Psychopharmacology (Berl). 1996, 127 (1), 10–18. 10.1007/BF02805969. [DOI] [PubMed] [Google Scholar]
- (44).Bulling S; Schicker K; Zhang Y-W; Steinkellner T; Stockner T; Gruber CW; Boehm S; Freissmuth M; Rudnick G; Sitte HH; et al. The Mechanistic Basis for Noncompetitive Ibogaine Inhibition of Serotonin and Dopamine Transporters. J. Biol. Chem. 2012, 287 (22), 18524–18534. 10.1074/jbc.M112.343681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Koldsø H; Autzen HE; Grouleff J; Schiøtt B Ligand Induced Conformational Changes of the Human Serotonin Transporter Revealed by Molecular Dynamics Simulations. PLoS One 2013, 8 (6), e63635 10.1371/journal.pone.0063635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cheng Y-C; Prusoff WH Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22 (23), 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- (47).Piston DW; Kremers GJ Fluorescent Protein FRET: The Good, the Bad and the Ugly. Trends in Biochemical Sciences. 2007, pp 407–414. 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- (48).Berney C; Danuser G FRET or No FRET: A Quantitative Comparison. Biophys. J. 2003, 84 (6), 3992–4010. 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Schicker K; Uzelac Z; Gesmonde J; Bulling S; Stockner T; Freissmuth M; Boehm S; Rudnick G; Sitte HH; Sandtner W Unifying Concept of Serotonin Transporter-Associated Currents. J. Biol. Chem. 2012, 287 (1), 438–445. 10.1074/jbc.M111.304261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Karlin A; Akabas MH; Karl A; Akabas MH Substituted-Cysteine Accessibility Method; Academic Press, 1998; Vol. 293 10.1016/S0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- (51).Henry LK; Iwamoto H; Field JR; Kaufmann K; Dawson ES; Jacobs MT; Adams C; Felts B; Zdravkovic I; Armstrong V; et al. A Conserved Asparagine Residue in Transmembrane Segment 1 (TM1) of Serotonin Transporter Dictates Chloride-Coupled Neurotransmitter Transport. J. Biol. Chem. 2011, 286 (35), 30823–30836. 10.1074/jbc.M111.250308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Henry LK; Field JR; Adkins EM; Parnas ML; Vaughan R. a; Zou M-F; Newman AH; Blakely RD Tyr-95 and Ile-172 in Transmembrane Segments 1 and 3 of Human Serotonin Transporters Interact to Establish High Affinity Recognition of Antidepressants. J. Biol. Chem. 2006, 281 (4), 2012–2023. 10.1074/jbc.M505055200. [DOI] [PubMed] [Google Scholar]
- (53).Rudnick G Cytoplasmic Permeation Pathway of Neurotransmitter Transporters. Biochemistry. 2011, pp 7462–7475. 10.1021/bi200926b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhang YW; Rudnick G Cysteine-Scanning Mutagenesis of Serotonin Transporter Intracellular Loop 2 Suggests an Alpha-Helical Conformation. J. Biol. Chem. 2005, 280 (35), 30807–30813. 10.1074/jbc.M504087200. [DOI] [PubMed] [Google Scholar]
- (55).Sato Y; Zhang YW; Androutsellis-Theotokis A; Rudnick G Analysis of Transmembrane Domain 2 of Rat Serotonin Transporter by Cysteine Scanning Mutagenesis. J. Biol. Chem. 2004, 279 (22), 22926–22933. 10.1074/jbc.M312194200. [DOI] [PubMed] [Google Scholar]
- (56).Zhang YW; Rudnick G The Cytoplasmic Substrate Permeation Pathway of Serotonin Transporter. J. Biol. Chem. 2006, 281 (47), 36213–36220. 10.1074/jbc.M605468200. [DOI] [PubMed] [Google Scholar]
- (57).Chen JG; Liu-Chen S; Rudnick G External Cysteine Residues in the Serotonin Transporter. Biochemistry 1997, 36 (6), 1479–1486. 10.1021/bi962256g. [DOI] [PubMed] [Google Scholar]
- (58).Ramamoorthy S; Baumant AL; Mooret KIMR; Han H; Yang-Feng T; Chang AS; Ganapathy V; Blakelyt RD; Bauman AL; Moore KR; et al. Antidepressant-and Cocaine-Sensitive Human Serotonin Transporter: Molecular Cloning, Expression, and Chromosomal Localization. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2542–2546. 10.1073/PNAS.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Fontana A; De Laureto PP; Spolaore B; Frare E; Picotti P; Zambonin M Probing Protein Structure by Limited Proteolysis. In Acta Biochimica Polonica; 2004; Vol. 51, pp 299–321. https://doi.org/035001299. [PubMed] [Google Scholar]
- (60).Bailey DM; Catron MA; Kovtun O; Macdonald RL; Zhang Q; Rosenthal SJ Single Quantum Dot Tracking Reveals Serotonin Transporter Diffusion Dynamics Are Correlated with Cholesterol-Sensitive Threonine 276 Phosphorylation Status in Primary Midbrain Neurons. ACS Chemical Neuroscience. American Chemical Society; May 24, 2018, p acschemneuro.8b00214. 10.1021/acschemneuro.8b00214. [DOI] [PubMed] [Google Scholar]
- (61).Zhang Y-W; Turk BE; Rudnick G Control of Serotonin Transporter Phosphorylation by Conformational State. Proc. Natl. Acad. Sci. 2016, 113 (20), E2776–E2783. 10.1073/pnas.1603282113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Blakely RD; Ramamoorthy S; Schroeter S; Qian Y; Apparsundaram S; Galli A; DeFelice LJ Regulated Phosphorylation and Trafficking of Antidepressant-Sensitive Serotonin Transporter Proteins. Biol. Psychiatry 1998, 44 (3), 169–178. 10.1016/S0006-3223(98)00124-3. [DOI] [PubMed] [Google Scholar]
- (63).Ramamoorthy S; Giovanetti E; Qian Y; Blakely RD Phosphorylation and Regulation of Antidepressant-Sensitive Serotonin Transporters. J. Biol. Chem. 1998, 273 (4), 2458–2466. [DOI] [PubMed] [Google Scholar]
- (64).Sørensen L; Strømgaard K; Kristensen AS Characterization of Intracellular Regions in the Human Serotonin Transporter for Phosphorylation Sites. ACS Chem. Biol. 2014, 9 (4), 935–944. 10.1021/cb4007198. [DOI] [PubMed] [Google Scholar]
- (65).Koban F; El-Kasaby A; Häusler C; Stockner T; Simbrunner BM; Sitte HH; Freissmuth M; Sucic S A Salt Bridge Linking the First Intracellular Loop with the C Terminus Facilitates the Folding of the Serotonin Transporter. J. Biol. Chem. 2015, 290 (21), 13263–13278. 10.1074/jbc.M115.641357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ansah T-A; Ramamoorthy S; Montañez S; Daws LC; Blakely RD Calcium-Dependent Inhibition of Synaptosomal Serotonin Transport by the Alpha 2-Adrenoceptor Agonist 5-Bromo-N-[4,5-Dihydro-1H-Imidazol-2-Yl]-6-Quinoxalinamine (UK14304). J. Pharmacol. Exp. Ther. 2003, 305 (3), 956–965. 10.1124/jpet.102.047134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.