

Abstract
The bifunctional ligand p-SCN-Bn-HOPO, which has four 1,2-hydroxypyridinone groups on a spermine backbone with an isothiocyanate linker, has been shown to be an efficient and stable chelator for Zr(IV) and, more importantly, the radioisotope 89Zr for use in radiolabeling antibodies for positron emission tomography (PET) imaging. Previous studies of 89Zr-HOPO-trastuzumab in mice showed low background, good tumor to organ contrast, and very low bone uptake which show p-SCN-Bn-HOPO to be an important next-generation bifunctional chelator for radioimmunoPET imaging with 89Zr. However, the reported synthesis of p-SCN-Bn-HOPO involves nine steps and multiple HPLC purifications with an overall yield of about 1.4%. Herein we report an improved and efficient synthesis of p-SCN-Bn-HOPO in four steps with 14.3% overall yield which will improve its availability for further biological studies and wider application in PET imaging. The new synthetic route also allows variation in linker length and chemistries which may be helpful in modifying in vivo clearance behaviors of future agents.
Introduction
Positron Emission Tomography (PET) is a molecular imaging modality that provides physiological data non-invasively and quantitatively. PET imaging relies on the decay of positron emitting radionuclides that are chemically bound to ligands or targeting vectors designed to probe biochemical phenomena in vivo. Traditionally, short-lived PET isotopes, such as 18F, 15O, 11C, and 13N, have been used for labelling peptides and small molecules that accumulate in target tissue and clear the body rapidly. For PET imaging with antibodies, called immunoPET, longer lived radionuclides are required to match the residence time of the antibodies in vivo. The radionuclide 89Zr has become a key to the success of immunoPET and the field is rapidly growing.1, 2
89Zr is ideally suited for pairing with monoclonal antibodies because its physical half-life of 78.4 h matches the residence time, generally several days, of intact mAbs in the body. The low positron energy, βmax:897 keV, βavg: 396.9 keV, provides high resolution PET images. The production of 89Zr can be accomplished easily and inexpensively using medical cyclotrons via proton bombardment on natural yttrium targets (100% abundance 89Y) by the 89Y(p,n)89Zr reaction.
Zr(IV) is an oxophilic, octacoordinate ion, so octadentate ligands comprised of oxygen donor atoms would be advantageous. Presently, the only bifunctional chelator used in the clinic for linking 89Zr to an antibody is desferrioxamine (DFO), most commonly in the form of commercially available p-SCN-Bn-DFO3 (Figure 1). DFO consists of three hydroxamate moieties and is a hexadentate chelator. The two open coordination sites result in demetallation of 89Zr from the chelate in vivo that is manifested in high bone uptake in preclinical studies in mice.
Figure 1.
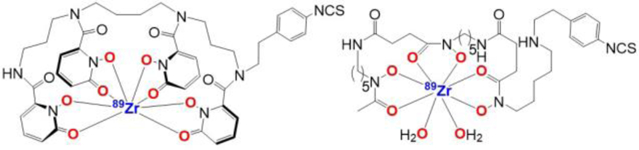
Chemical structures of the 89Zr complexes with bifunctional p-SCN-Bn-HOPO (left) and p-SCN-Bn-DFO (right) ligands.
Several chelators were reported with the goal of increasing 89Zr complex stability in vivo. Some chelators contain hydroxamate moieties; for example, the linear and macrocyclic tetrahydroxamate chelators reported by Guerrad et al.4, 5, Vugts and Mindt6, 7 and branched tetrahydroxymates by Rousseau8. Trihydroxamate chelators,9, 10 and a trihydroxamate possessing an additional cyclic hydroxamate (DFOcyclo*)11 giving it 8-oxygen donor atoms have been reported.
We reported the potential applications of the 3,4,3-LI(1,2-HOPO) or HOPO ligand,12 which has four 1,2-hydroxypyridinone groups for metal binding and comes from the actinide sequestration literature.13 The 89Zr-HOPO complex showed equal or greater stability compared to 89Zr-DFO in both in vitro and in vivo assays.12
The development of a bifunctional derivative of the HOPO ligand was the next logical step and led to the design and synthesis of p-SCN-Bn-HOPO.14 The p-SCN-Bn-HOPO conjugated to trastuzumab achieved similar specific activity, ~2 mCi/mg, and remained stable (~90%) in human serum (7 d). The greatest distinction between the two compounds was the amount of bone uptake seen in the imaging and biodistribution studies. The activity measured in the bone for 89Zr-HOPO-trastuzumab was more than 7 times lower than for 89Zr-DFO-trastuzumab. While the absolute uptake 89Zr-DFO-trastuzumab by BT474 breast cancer tumors was just over twice that of 89Zr-HOPO-trastuzumab, the tumor to bone ratio was more than 3-fold greater for the HOPO complex. This improved contrast between tumor and bone is advantageous for the detection of bone metastasis and for the general clarity of the images as well as reducing the radiation dose to sensitive non-target tissues. Furthermore, the lower bone uptake is the ultimate proof that the p-SCN-Bn-HOPO ligand forms a more stable complex with 89Zr4+ than p-SCN-Bn-DFO and drastically reduces the bone accumulation of free 89Zr4+ in vivo.
We reported the 9-step synthesis of p-SCN-Bn-HOPO that required multiple HPLC purifications with a 1.4% overall yield.14 The long reaction sequence and low yield limits the applications of this important ligand and is impractical for the synthesis of p-SCN-Bn-HOPO in large scale. Therefore, a more efficient synthesis is needed to develop cost effective applications. Herein we report an improved synthesis consisting of four steps with a 10-fold improvement in overall yield (14.3%). We also report modified conditions that improve the yields in our previously reported synthesis.
Results and Discussion
In order to achieve p-SCN-Bn-HOPO (compound 3 Scheme 1)14 in high yield, we adopted two strategies. In the first approach, we designed a new 4-step synthetic route requiring only HPLC purification of the final product (Scheme 1). In the second approach, we reinvestigated the reaction conditions and solvents to improve the yields in the original 9-step synthesis (Scheme 2).
Scheme 1.
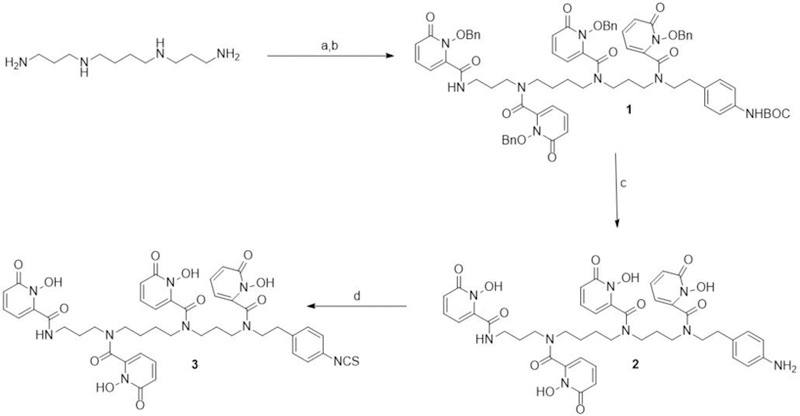
Four step synthesis of p-SCN-Bn-HOPO. (a) 2-(4-teritiarybutyl aminophenyl)ethyl 4-methylbenzenesulfonate, K2CO3, CH3CN, reflux, 8–10 h (b) 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-2-carboxylic acid chloride, NEt3, DMAP, dichloromethane, 0−25 °C, 24 h, (42% over 2 steps); (c) BCl3 in p-xylene, dichloromethane, −20 °C to RT, overnight, 95%; (d) di-2-pyridyl thiocarbonate, NEt3, CH3CN, H2O, r.t, 1 h, 36%.
Scheme 2.
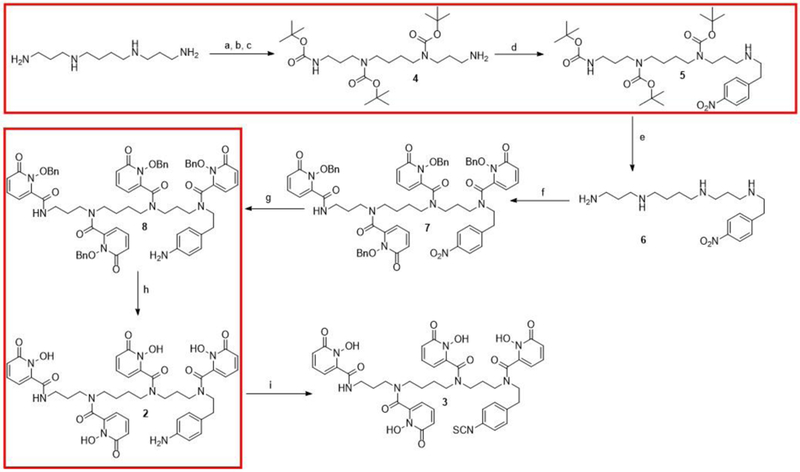
Improvements to the original 9-step synthesis of p-SCN-Bn-HOPO are outlined in red. (a) Ethyl trifluoroacetate, MeOH, −78 °C, 1 h; (b) (BOC)2O, MeOH, r.t, 18 h; (c) conc. NH4OH, r.t, 15 h (52% over 3 steps); (d) 4-nitrophenethyl 4-methylbenzenesulfonate, K2CO3, CH3CN, reflux, 12 h, 46%; (e) 4 M HCl in Dioxane, r.t, 2 h; (f) 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-2-carboxylic acid chloride, NEt3, DMAP, dichloromethane, 0−25 °C, 12 h, 56% (over 2 steps); (g) Raney Ni, H2, MeOH, 3 h; (h) BCl3 in p-xylene, dichloromethane, −20 °C to RT, 15 h, 99%; (i) di-2-pyridyl thiocarbonate, NEt3, CH3CN, H2O, r.t, 1 h, 36%.
The new strategy to make p-SCN-Bn-HOPO starts with commercially available spermine, which was reacted with 2-(4-tertiarybutyl-aminophenyl)ethyl tosylate (S2b, supporting information) under reflux for 8–10 h resulting in a mixture of free spermine and the mono substituted 2-(4-teritiarybutyl-aminophenyl)ethyl group on spermine (Scheme 1). The mono substituted spermine was easily isolated using preparative silica gel TLC (GF254) with methanol as the eluent. Though free spermine is not visible, the mono substituted spermine can be visualized using a UV lamp. The product was collected by scraping the preparative silica gel plates followed by washing the methanol. We observed that there was some quantity of free spermine obtained along with the substituted product. Unreacted spermine was recovered from the methanol wash and can be recycled. Compound S2b was synthesized by BOC protection of 2-(4-aminophenyl)ethan-1-ol followed by tosylation of the hydroxide group obtaining compound S2b in quantitative yield (Scheme S1, supporting information).
Mono-substituted spermine-Bn-NH-BOC, was treated with 4.2 equivalents of 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-2-carbonyl chloride under nitrogen to obtain compound 1 in good yield (42%). However, there was about 20% benzyl protected-3,4,3-(LI-1,2-HOPO)12 that can be separated from the reaction mixture as a side product, which suggests that some unreacted spermine remained from the previous step. The O-benzyl protecting groups and BOC protecting group on compound 1 were all deprotected in one step using the mild Lewis acid, BCl3 in toluene, to obtain compound 2 in quantitative yield (95%). Compound 2 precipitates as the reaction proceeds, and was filtered and washed with acetone several times obviating the need for HPLC purification. Finally, bifunctional (p-SCN-Bn-HOPO) was obtained from compound 2 using the published procedure including reaction with di-2-pyridyl thiocarbonate and purification by preparative HPLC.14 The overall yield obtained was 14.3%. It is important to note that about 20% 3,4,3-(LI-1,2-HOPO) and unreacted spermine are side products. HOPO is a useful ligand and can be isolated for testing with radiometals. The unreacted spermine can be recycled.
The second approach improves several key steps in the prior 9-step synthesis of p-SCN-Bn-HOPO (Scheme 2). Compound 4 was synthesized in a three-step reaction sequence starting from the commercially available spermine. We studied the effect of reaction time and reagent stoichiometry to improve the reaction yields (Table 1). Optimum yields (46–50%) were obtained by using conditions in entry 4, Table 1. In this case, the reaction of ethyl trifluoroacetate with amine groups on the spermine led to the mono-, di-, and tri- substituted spermine derivatives. To avoid the formation of these undesirable side products, ethyl trifluoroacetate was added by aid of syringe pump (2 h at 20 mL/h), followed by addition of five equivalents of di-tert-butyldicarbonate with prolonged reaction time (2 d) to ensure the protection all free amines. Ammonium hydroxide was then added until a pH of 14 was reached and stirred for two days to complete the de-protection of the ethyl trifluoroacetate group.
Table 1.
Optimizing the synthesis of tert-butyl(4-((3-aminopropyl)(tert-butoxycarbonyl)amino)butyl)(3-((tertbutoxycarbonyl)amino)propyl)-carbamate, compound 4, Scheme 2
| Entry | step 1a | Step 2 | Step 3 | Yield (%) | |||
|---|---|---|---|---|---|---|---|
| Insertion of trifluoroacetate | Insertion of Boc | Cleavage of COCF3 | |||||
| Time (h) | Equiv. | Time (h) | Equiv. | Time (h) | pH | ||
| 1 | 1 | 1 | 24 | 6 | 24 | 11 | 28 |
| 2 | 1 | 1 | 48 | 8 | 48 | 11 | 38.5 |
| 3 | 2 | 1 | 72 | 3 | 48 | 14 | 50 |
| 4 | 2 | 1 | 96 | 5 | 48 | 14 | 46 |
−78 0C
Although the reaction of an amine with a primary alkyl halide is well-established, the elimination product is often formed along with the desired substituted products. By following the previously published procedure for the synthesis of compound 5, the reaction of compound 4 with 4-nitrophenylethylbromide, we observed the formation 4-nitrostyrene as major byproduct drastically reducing the yield of compound 5. This prompted us to investigate the effect of solvents in the particular reaction. We screened several polar aprotic solvents at different temperatures (Table 2) in preparation of compound 5. Using 2 equiv. of 4-nitrophenylethyl bromide and compound 4 containing 2 equivalents of potassium carbonate in acetonitrile at r.t. for 7 days, afforded the expected compound 5 in yields of 40% (Entry 6 in Table 2) and no elimination product was observed. In attempts to reduce the reaction time the reaction was carried out in a microwave reactor at various temperatures (40–153 °C) and times (10–60 min), but the yields significantly decreased (supporting information, Table S1).
Table 2.
Trials of synthesis of tert-butyl(4-((tert-butoxycarbonyl)(3-((4-nitrophenethyl)amino)propyl)amino)butyl)(3-(tert-butoxycarbonyl)amino)-propyl)carbamate, compound 5, Scheme 2.
| Entry | Solvent | Temp ( °C ) | Linker | Reaction Time (h) | % Yield |
|---|---|---|---|---|---|
| Reporteda | DMF | 60 | Br | 12 | 38 |
| 1 | DMF | 20 | Br | 12 | 14 |
| 2 | MeCN | 20 | Br | 96 | 33 |
| 3 | DMF/MeCN | 20 | Br | 96 | 15 |
| 4 | THF | 80 | Br | 48 | 1 |
| 5 | MeCN | 55 | Br | 168 | 1 |
| 6 | MeCN | 40 | Br | 168 | 40 |
| 7 | DMF | 120–153 | Br | 1 microwave | 1 |
| 8 | MeCN | reflux | Tosyl | 24 | 46 |
| 9 | MeCN | 35 | Br | 96–168 | 33 −40 |
ref 13. The yield of this reaction is inconsistent and varies from a few % to ca. 38%
We found that using 4-nitrophenethyl tosylate S2a (Scheme S1) instead of 4-nitrophenylethylbromide under reflux conditions with excess K2CO3 over 24 h, resulted in the desired product 5 in 46% yields. This approach (Entry 8 in Table 2) provides the optimal way to obtain product 5.
Compounds 6, 7 and 8 in Scheme 2 were synthesized using conditions reported previously. We then optimized the synthesis of compound 2 where we reacted compound 8 with a weak Lewis acid (BCl3 in toluene) at room temperature overnight under nitrogen resulting in a precipitate which was centrifuged, filtered, and washed with acetone to obtain the desired benzyl deprotected product (compound 2) as a colourless solid in quantitative yields. Compound 2 was converted to the final product (p-SCN-Bn-HOPO) using the published procedure. The overall yield using the synthetic approach in Scheme 2 was improved to 8.1% compared to the 1.4% as previously published.14
Experimental section
General.
1H and 13C NMR spectra were obtained using Bruker Avance III 400 MHz, Bruker Avance DRX 500 MHz, or Bruker Avance III 600 MHz instruments; chemical shifts are expressed in ppm relative to CDCl3 (7.26 ppm, 1H; 77.0 ppm, 13C), (CD3)2CO (2.05 ppm, 1H; 29.84 and 206.26 ppm, 13C), CD3OD (3.31 and 4.78 ppm, 1H; 49.2 ppm, 13C). Mass analyses were conducted at the CUNY Mass Spectrometry Facility at Hunter College on an Agilent iFunnel 6550 Q-ToF LC/MS System (for HRMS-ESI). The electrospray ionization was run in 95% methanol, with 0.1% formic acid. Reactions were monitored by TLC with Analtech Uniplate silica gel G/UV 254 precoated plates (0.2 mm). TLC plates were visualized by UV (254 nm), and by iodine vapour. Preparative thin-layer chromatography was carried out on 20×20 cm glass plates coated with silica gel (1 mm thick). Preparative HPLC was performed on a Agilent 1200 Series HPLC equipped on a Phenomenex Luna C18(2) 100Å, 250 cm x 21.20 mm I.D. 10 µm reverse phase column (00G-4253-P0 AX). Purification by Flash chromatography system from Biotag-IsoleraTM was performed with Biotage ZIP Sphere 30-gram spherical silica 60 um. Analytical, reverse phase UPLC−MS was performed on a Waters Acquity H class HPLC/SQD mass spectrometer and a Acquity UPLC BEH 1.7 μm C18, 100 mm × 2.1 mm i.d. column (186002352), with a 10 min 5−95% H2O/ acetonitrile (MeCN) (0.1% FA) gradient and a flow rate of 0.3 mL/ min.
1-(Benzyloxy)-N-(3-(1-(benzyloxy)-6-oxo-1,6-dihydropyridine-2-arboxamido)propyl)-N-(4-(1-(benzyloxy)-N-(3-(1-(benzyloxy)-N-(4-teritiarybutyl-aminophenethyl)-2-oxo-1,2-dihydropyridine-3-carboxamido)propyl)-2-oxo-1,2-dihydropyridine-3-carboxamido)-butyl)-6-oxo-1,6-dihydropyridine-2-carboxamide (1)
To spermine (1 g, 5 mmol) and potassium carbonate (1.38 g, 10 mmol) in 150 mL acetonitrile (MeCN) was added compound S2b (977 mg, 2.5 mmol) in 150 mL acetonitrile drop wise at 0 °C. Then the reaction mixture was heated at reflux for 8–10 h (monitored via TLC dichloromethane:hexanes 3:1) until no compound S2b is observed. Then the reaction mixture was filtered to remove potassium carbonate and the solvent was evaporated resulting in viscous liquid, which was dissolved in minimum amount of methanol and loaded on preparative TLC and run in methanol as eluent. The compound that is UV active on preparative TLC was obtained. HRMS (ESI) m/z calcd for C23H43N5O2 ([M+H]+), 422.3490, found 422.3483; calcd for C23H43N5O2 ([M+Na]+), 444.3309, found 444.3282.
To the above purified mixture 0.7 g (~1.8 mmol), DMAP (0.0105 g, 0.087 mmol), triethylamine (1.46 mL, 10.5 mmol), in 100 mL dry dichloromethane under inert N2 atmosphere was added 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-2-carbonyl-chloride (1.78 g, 7.56 mmol) in dichloromethane (100 mL) at 0 °C. Then the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was washed with 10% NaHCO3 solution, followed by water. The organic phase was dried over anhydrous Na2SO4 and was then removed with a rotary evaporator. The crude product was purified through silica gel column chromatography using a 6% methanol in dichloromethane eluent to give the product as brownish yellow foam (overall yield, 42% (1.4 g, 1.05 mmol)). Note: Using excess of 1-(benzyloxy)-6-oxo-1,6-dihydropyridine-2-carbonyl chloride results in significant amounts of the penta substituted HOPO derivative. 1H NMR (500 MHz, CDCl3) δ 4.91–7.45 (m, 44H), 2.35–3.24 (m, 16H), 1.35–1.84 (m, 19 H); 13C NMR (125 MHz, CDCl3) δ; 157.19, 157.14, 157.11, 157.07, 156.99, 142.04, 141.75, 141.72, 141.60, 141.55, 141.48, 141.32, 137.39, 137.30, 137.27, 137.13, 132.38, 132.35, 132.32, 129.81, 129.52, 129.40, 129.37, 129.28, 129.25, 129.23, 129.20, 129.12, 128.44, 128.41, 128.24, 128.18, 127.61, 127.57, 127.50, 127.49, 121.87, 121.83, 121.72, 121.69, 121.67, 117.92, 117.84, 78.51, 78.47, 78.37, 78.35, 78.32, 78.28, 27.32, 27.31;. HRMS (ESI) m/z calcd for C75H79N9O14 ([M + H]+), 1330.5825, found 1330.5818. calcd for C75H79N9O14 ([M+Na]+), 1352.5644, found 1352.564.
4-(11,15-Bis(1-hydroxy-2-oxo-1,2-dihydropyridine-3-carbonyl)-1-(1-hydroxy-6-oxo-1,6-dihydropyridin-2-yl)-6-(1-hydroxy-6-oxo-1,6-dihydropyridine-2-carbonyl)-1-oxo-2,6,11,15-tetraazaheptadecan-17-yl)benzenaminium chloride (2, scheme 1)
To compound 1 (62.5 mg, 0.05 mmol) in 20 mL dry dichloromethane at −20 °C under N2 atmosphere was added 1 M BCl3 in p-xylene (5 mL, 5 mmol) dropwise. Then the reaction was stirred at room temperature overnight. The reaction mixture containing precipitate was centrifuged followed by washing and centrifuging with acetone (three times) resulting in a white solid compound 2 in 95% yield (41.5 mg, 0.048 mmol). 1H NMR (500 MHz, CD3OD) δ 6.07–7.34 (m, 16H), 2.62–3.54 (m, 16H), 0.99–1.96 (m, 8H); 13C NMR (125 MHz, CDCl3) δ 163.36, 160.22, 143.02, 142.99, 140.47, 140.43, 140.40, 132.01, 130.54, 124.52, 124.32, 120.38, 120.31, 120.26, 120.22, 120.20, 106.62, 34.68, 29.04, 27.84, 26.32, 26.24;. HRMS (ESI) m/z calcd for C42H47N7O12 ([M + H]+), 870.33, found 870.3413.
4-(11,15-bis(1-hydroxy-2-oxo-1,2-dihydropyridine-3-carbonyl)-1-(1-hydroxy-6-oxo-1,6-dihydropyridin-2-yl)-6-(1-hydroxy-6-oxo-1,6-dihydropyridine-2-carbonyl)-1-oxo-2,6,11,15-tetraazaheptadecan-17-yl)benzenaminium chloride (2, scheme 2)
Compound 8 (43 mg, 0.035 mmol) was dissolved in 20 mL of dry dichloromethane under N2 atmosphere. BCl3 in p-xylene were added dropwise to the reaction mixture in −20 °C bath. A white precipitate was observed during the addition. Then the reaction mixture was stirred at room temperature for 15 h. The reaction mixture containing precipitate was centrifuged followed by washing and centrifuging (2500 rpm, 3 min) with dry acetone three times to remove impurities resulting in a white solid. The yield was quantitative (99% yield). LCMS (ESI) Calcd. for ([M+H]+), C42H48N9O12 870.34 found 870.28.
1-Hydroxy-N-(3-(1-hydroxy-6-oxo-1,6-dihydropyridine-2-carboxamido)propyl)-N-(4-(1-hydroxy-N-(3-(1-hydroxy-N-(4-isothiocyanatophenethyl)-2-oxo-1,2-dihydropyridine-3-carboxamido)propyl)-2-oxo-1,2-dihydropyridine-3-carboxamido)butyl)-6-oxo-1,6-dihydropyridine-2-carboxamide (3 Scheme 1).
To a solution of 2 (38 mg, 0.043 mmol) in (8:2) acetonitrile and water (4 mL) was added NEt3 (5 mg, 0.048 mmol) followed by di-2-pyridyl thionocarbonate (0.044 g, 0.2 mmol) then the reaction mixture was stirred vigorously at room temperature for 1 h. The crude reaction solution was directly purified by preparative HPLC using a gradient of 10−90% MeCN in water (both containing 0.05% TFA) starting from 10% MeCN then a ramp to 90% MeCN over 35 min (12 mL/min). The product peak was collected from 18.5 min to 20.5 min and the eluted solution was lyophilized to recover the product as a white solid. The purified ligand was collected in multiple small batches with an approximate combined yield of 36% (12.9 mg, 0.014 mmol). HRMS (ESI) m/z calcd for C43H45N9O12S ([M + H]+), 912.2987, found 912.2978. calcd for C43H45N9O12S ([M+Na]+), 934.2806, found 934.2769.
N1, N4, N9 – Tri-tert-butoxycarbonyl)-1,12-di-amino-4,9-diazadodecane (4)
Spermine (5 g, 24.72 mmol) was dissolved in 100 mL of dried methanol under anhydrous nitrogen. To this reaction mixture a solution made of ethyltrifluoroacetate (3.51 g, 24.72 mmol) in 100 mL of methanol was added dropwise using syringe pump (120 mL/h) at −78 °C under N2. The mixture was stirred for an additional 30 min. Then di-tert-butyldicarbonate (26.98 g, 123.6 mmol) in 100 mL methanol was added over a period of 1 h at 0 °C in an ice bath, and the reaction mixture was stirred at room temperature for 48 h. Concentrated ammonium hydroxide (13M, 135 mL) was added until pH 14 and the reaction was stirred for an additional 48 h. The solvent was evaporated under reduce pressure and the resulting oily compound was dissolved in dichloromethane, washed with water, dried over sodium sulfate and evaporated under reduced pressure. The crude compound was then purified by column chromatography on silica using dichloromethane:methanol: NH4OH 92:8:1 of as eluent to obtain colourless oil. (Yield: 46%). 1HNMR (600 MHz, CDCl3): δ 8.21 (bs, 2H), 3.44 (s, 4H), 3.11–3.32 (m, 10H), 2.91(t, 2H), 1.87 (bs, 2H), 1.63 (bs, 2H), 1.41–1.46 (m, 27H). 13C NMR (150 MHz, CDCl3): δ 162.49, 162.26, 81.21, 80.03, 79.74, 79.11, 50.71, 47.09, 46.85, 44.31, 43.85, 42.51, 37.76, 37.41, 36.45, 28.97, 28.50, 25.96, 25.66, 25.43. HRMS calculated for C25H50N4O6 ([M + H]+), 503.38, found 503.3817; C25H50N4O6 ([M + Na]+), 525.28, found 525.3625.
tert-butyl (4-((tert-butoxycarbonyl)(3-((4-nitrophenethyl) amino)propyl)amino)butyl)(3-((tert-butoxycarbonyl) amino)propyl)carbamate (5, Scheme 2, Table 2, entry 6)
A suspension of compound 4 (1 g, 1.99 mmol) and K2CO3 (1.37 g, 9.94 mmol) in 100 mL acetonitrile under N2 was prepared. To this mixture a solution of 4-nitrophenylethyl bromide (0.92 g, 3.98 mmol) in 100 mL of acetonitrile was added dropwise at 0 °C. The resulting reaction mixture was stirred at 40 °C for 7 days. K2CO3 was filtrated and the solvent was removed under vacuum and the resulting residue was dissolved in methylene chloride, washed with water, dried over anhydrous sodium sulfate, and evaporated to dryness. The crude compound was purified by silica column chromatography using dichloromethane: methanol 92:8 as eluent to obtain light-brown gummy compound. Yield: 40% (0.52 g, 0.79 mmol).
tert-butyl (4-((tert-butoxycarbonyl)(3-((4-nitrophenethyl) amino)propyl)amino)butyl)(3-((tert-butoxycarbonyl)amino) propyl)carbamate (5, Scheme 2, Table 2, entry 8)
4-nitrophenethyl 4-methylbenzenesulfonate (S2a) (0.75 g, 2 mmol) in 100 mL of acetonitrile was added dropwise to a mixture of compound 3 (0.751 g, 1.5 mmol) and potassium carbonate (1.3 g, 9.4 mmol) in 100 mL of acetonitrile under N2 atmosphere at 0 °C. The resulting reaction mixture was refluxed for 24 h., then cooled to r.t. potassium carbonate was filtered and the filtrate was diluted with water and extracted with dichloromethane. The compound was purified over silica gel column chromatography using 5% methanol in dichloromethane to yield = 46% (450 mg, 0.6mmol). 1H NMR (600 MHz, CDCl3): (mixture of rotamers) δ 8.15–8.13 (d, 2H), 7.45–7.44 (d, 2H), 3.21– 2.63 (m, 20H), 1.93 (bs, 2H), 1.62 (bs, 2H), 1.42–1.34 (m, 27H, Boc). 13C NMR (150 MHz, CDCl3): (mixture of rotamers) δ 156.1, 155.5, 147.1, 129.9, 124.1, 80.6, 79.6, 53.5, 49.8, 47.2, 46.8, 46.3, 45.8, 44.4, 43.9, 43.4, 37.8, 37.5, 28.5, 26.1, 25.5. HRMS calculated for C33H57N5O8 ([M + H]+), 652.4207, found 652.4284, C33H57N5O8 ([M + Na]+), 674.3207, found 674.4102.
Conclusions
We developed new efficient synthesis of p-SCN-Bn-HOPO in 14.3% overall yield that is an improvement over our original 9-step synthesis. This improved synthesis uses four steps, eliminates the cumbersome protection and deprotection steps, and eliminates HPLC purification of intermediates from our previously reported preparation.13 Preparative HPLC is only performed on the final compound. We also optimized the reaction conditions to improve the yield of the previous report from 1.4% to 8.1%. The intermediate and side products obtained from both strategies, such as compounds 1 and 2, spermine and 3,4,3-(LI-1,2-HOPO)Bn, can be used to develop new ligands or can be recycled.
Supplementary Material
Acknowledgements
Supported by the U.S. National Science Foundation CHE-1610755 to C.M.D.; NSF DGE 0965983 (IGERT program) to L.C.F.; National Cancer Institute at the National Institutes of Health R21 CA201999 to J.S.L. & L.C.F.; MSKCC core facilities are supported by National Institutes of Health Cancer Center Support Grant (grant number P30 CA08748).
Footnotes
Conflicts of interest
The authors declare no competing financial interest.
Notes and references
- 1.Bhatt NB, Pandya DN and Wadas TJ, Molecules, 2018, 23, 638/631–638/624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deri MA, Zeglis BM, Francesconi LC and Lewis JS, Nuclear Medicine and Biology, 2013, 40, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perk LR, Vosjan MJWD, Visser GWM, Budde M, Jurek P, Kiefer GE and Dongen GAMS, European Journal of Nuclear Medicine and Molecular Imaging, 2010, 37, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guérard F, Lee Y-S and Brechbiel MW, Chemistry – A European Journal, 2014, 20, 5584–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerard F, Lee Y-S, Tripier R, Szajek LP, Deschamps JR and Brechbiel MW, Chemical Communications, 2013, 49, 1002–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vugts DJ, Klaver C, Sewing C, Poot AJ, Adamzek K, Huegli S, Mari C, Visser GWM, Valverde IE, Gasser G, Mindt TL and van Dongen GAMS, European Journal of Nuclear Medicine and Molecular Imaging, 2017, 44, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patra M, Bauman A, Mari C, Fischer CA, Blacque O, Haussinger D, Gasser G and Mindt TL, Chemical Communications, 2014, 50, 11523–11525. [DOI] [PubMed] [Google Scholar]
- 8.Rousseau J, Zhang Z, Dias GM, Zhang C, Colpo N, Benard F and Lin K-S, Bioorganic & Medicinal Chemistry Letters, 2017, 27, 708–712. [DOI] [PubMed] [Google Scholar]
- 9.Boros E, Holland JP, Kenton N, Rotile N and Caravan P, ChemPlusChem, 2016, 81, 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhai C, Summer D, Rangger C, Franssen GM, Laverman P, Haas H, Petrik M, Haubner R and Decristoforo C, Molecular Pharmaceutics, 2015, 12, 2142–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raave R, Sandker G, Rijpkema M, Boerman O, Heskamp S, Adumeau P, Mangin F, Meyer M, Moreau M, Bernhard C, Da Costa L, Dubois A, Goncalves V, Chambron J-C, Denat F, Jacobsen Christian B, Gustafsson M and Chambron J-C, Eur J Nucl Med Mol Imaging, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deri MA, Ponnala S, Zeglis BM, Pohl G, Dannenberg JJ, Lewis JS and Francesconi LC, Journal of Medicinal Chemistry, 2014, 57, 4849–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorden AEV, Xu J, Raymond KN and Durbin P, Chemical Reviews, 2003, 103, 4207–4282. [DOI] [PubMed] [Google Scholar]
- 14.Deri MA, Ponnala S, Kozlowski P, Burton-Pye BP, Cicek HT, Hu C, Lewis JS and Francesconi LC, Bioconjugate Chemistry, 2015, 26, 2579–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.