

Abstract
Previous studies suggest that reducing the numbers of adult-born neurons in the dentate gyrus (DG) of the mouse increases susceptibility to severe continuous seizures (status epilepticus; SE) evoked by systemic injection of the convulsant kainic acid (KA). However, it was not clear if the results would be the same for other ways to induce seizures, or if SE-induced damage would be affected. Therefore, we used pilocarpine, which induces seizures by a different mechanism than KA. Also, we quantified hippocampal damage after SE. In addition, we used both loss-of-function and gain-of-function methods in adult mice. We hypothesized that after loss-of-function, mice would be more susceptible to pilocarpine-induced SE and SE-associated hippocampal damage, and after gain-of-function, mice would be more protected from SE and hippocampal damage after SE.
For loss-of-function, adult neurogenesis was suppressed by pharmacogenetic deletion of dividing radial glial precursors. For gain-of-function, adult neurogenesis was increased by conditional deletion of pro-apoptotic gene Bax in Nestin-expressing progenitors. Fluoro-Jade C (FJ-C) was used to quantify neuronal injury and video-electroencephalography (video-EEG) was used to quantify SE.
Pilocarpine-induced SE was longer in mice with reduced adult neurogenesis, SE had more power and neuronal damage was greater. Conversely, mice with increased adult-born neurons had shorter SE, SE had less power, and there was less neuronal damage. The results suggest that adult-born neurons exert protective effects against SE and SE-induced neuronal injury.
Keywords: Adult-born neurons, Epilepsy, Pilocarpine, Progenitor, Status epilepticus
INTRODUCTION
Neurogenesis in the adult brain (adult neurogenesis) occurs throughout life in mammals (Taupin, 2006; Gage et al., 2008; Altman, 2011; Kempermann, 2012; Kazanis, 2013). Adult-born neurons are generated primarily in two locations: the subgranular zone of the DG of the hippocampus and the subventricular zone of the lateral ventricles (Altman and Das, 1965; Kaplan and Hinds, 1977). In the DG of the hippocampus, adult-born neurons primarily mature into the principal cell type, the granule cell [GC; (Cameron et al., 1993; Kuhn et al., 1996)]. Although the numbers of the adult-born neurons are relatively small (Cameron and McKay, 2001; Snyder et al., 2009), they appear to have substantial effects on functions related to behaviors associated with the DG, such as spatial memory, pattern separation, and contextual fear conditioning (Nilsson et al., 1999; Saxe et al., 2006; Clelland et al., 2009; Drew et al., 2010; Sahay et al., 2011; Burghardt et al., 2012; Denny et al., 2012; Nakashiba et al., 2012; Niibori et al., 2012; Aimone et al., 2014; McAvoy et al., 2015; Anacker and Hen, 2017; Huckleberry et al., 2018).
How the adult-born neurons exert their effect in the DG is an active area of research. Most methods have used approaches that decrease or increase adult-born GCs to understand their role. These studies have typically shown that reduction of adult-born neurons can increase neural activity (firing of action potentials) of the GCs. For example, when adult-born GCs were ablated by focal X-irradiation to the DG, spontaneous bursts of GC firing were recorded (Lacefield et al., 2012). Using voltage-sensitive dyes in hippocampal slices Ikrar et al. (2013) showed that a reduction in adult-born neurons increased the response of GCs to stimulation of the perforant path, and increasing adult hippocampal neurogenesis had the opposite effect. In a behavioral study, expression of the activity-dependent immediate early gene Arc in the GC layer (GCL) was examined after a behavioral task that assessed cognitive flexibility (Burghardt et al., 2012); the upregulation of Arc was greater when there was a reduction in adult-born neurons. Notably, the mice with reduced neurogenesis had significantly more foot shocks compared to controls. In other behavioral studies, upregulation of immediate early genes was not necessarily found in the GCL after reduced adult neurogenesis (Glover et al., 2017; Seib et al., 2018).
Similar to normal physiological conditions, a reduction in adult-born neurons also appears to increase hippocampal neural activity under pathological conditions. For example, Iyengar et al. (2015) showed that mice with reduced adult neurogenesis had more severe hippocampal seizures in response to the systemic convulsant KA. Remarkably, the seizures both in and outside the hippocampus were increased (Iyengar et al., 2015). Further studies suggest that at least under normal physiological conditions, the mechanism is likely to be due to the ability of adult-born neurons to strongly activate local GABAergic interneurons (Toni et al., 2008; Drew et al., 2016). These local interneurons innervate GCs and therefore the DG output to CA3. Ordinarily, mature GCs activate local GABAergic neurons as well as CA3, but the young adult-born neurons are likely to activate the GABAergic neurons more because of their high excitability compared to mature GCs (Scharfman and McCloskey, 2009; Scharfman and Bernstein, 2015).
Neuronal injury in the hippocampus that is caused by severe seizures occurs primarily in the hilus, area CA3, and CA1 (Cavazos and Sutula, 1990; Mello et al., 1993; Cavazos et al., 1994; Poirier et al., 2000; Ben-Ari, 2012; Henshall and Meldrum, 2012; Huusko et al., 2015) a pattern called hippocampal or Ammon’s horn sclerosis (AHS) in humans [or the more recent term, mesial temporal lobe sclerosis, which includes other areas of damage (Wieser and Epilepsy, 2004; Scharfman and Pedley, 2006)]. This injury has been suggested to be important because it is typically followed by chronic seizures (epilepsy) in rodents and in humans, and therefore it has been suggested that this neuronal injury causes the epilepsy (acquired temporal lobe epilepsy; (Cavalheiro et al., 1996; Herman, 2002; Mathern et al., 2008; Dudek and Staley, 2012; Dingledine et al., 2014). In addition, neuronal damage in the hippocampus is considered to be a contributing factor in cognitive deficits that typically accompany acquired temporal lobe epilepsy (Kleen et al., 2012; Rattka et al., 2013) or are identified in other conditions (Shaw and Alvord, 1997; Nelson et al., 2013; Kalaria, 2016). Notably, induction of severe continuous seizures (SE) by administration of the pilocarpine is a reliable method to induce neuronal damage in the hilus, CA3, and CA1. Therefore, we asked if adult-born neurons would modulate hippocampal neuronal injury following pilocarpine-induced SE.
One might expect that a study of this kind would use KA instead of pilocarpine, because it was historically the first chemoconvulsant to produce a pattern of damage like AHS (Nadler, 1981; Scharfman and Pedley, 2006; Mathern et al., 2008; Ben-Ari, 2012). This early work about KA was in the rat, and when investigators began to use mice they found that KA-induced SE did not usually lead to AHS or epilepsy, and often there was mortality. We found this also in a previous study conducted in our laboratory (Iyengar et al., 2015), where C57BL6/J mice were injected with KA. Neuronal injury was not evident after SE until the dose was high (25 mg/kg), but this was accompanied by mortality, which had been noted (McKhann et al., 2003; Tse et al., 2014). Therefore, to understand whether adult-born neurons influence neuronal damage we turned to pilocarpine, which induces SE as well as neuronal injury in mice (Cavalheiro et al., 1996; Borges et al., 2003; Wang et al., 2008; do Nascimento et al., 2012). We still found some mortality but after adopting methods described before, the mortality was greatly reduced.
We chose to manipulate adult-born neurons up to 6 weeks of age because 4–6 weeks of age is when they have unique intrinsic properties which lead to increased excitability (Schmidt-Hieber et al., 2004; Ge et al., 2007; Ge et al., 2008; Drew et al., 2013; Anacker and Hen, 2017). The unique properties include high input resistance and lower action potential threshold than mature GCs (Ambrogini et al., 2004; Esposito et al., 2005; Mongiat et al., 2009; Dieni et al., 2013). In addition, immature adult-born neurons exhibit a depolarizing rather than hyperpolarizing response to GABA (Karten et al., 2006; Markwardt and Overstreet-Wadiche, 2008; Marin-Burgin et al., 2012) and there are many other distinguishing features (Snyder et al., 2001; Van Praag et al., 2002; Zhao et al., 2008; Scharfman and McCloskey, 2009; Gu et al., 2012; Beining et al., 2017). Another reason to select 6 weeks is that prominent inhibitory effects of adult-born neurons were evident when they were 6- to 7-weeks-old (Iyengar et al., 2015; Drew et al., 2016) although there are also excitatory effects (Drew et al., 2016) which appear to be particularly strong if the adult-born neurons are 4-weeks-old rather than up to 6- or 7-weeks-old (Temprana et al., 2015).
To evaluate neuronal injury and quantify seizures, we used two approaches that are considered to be the “gold standard”: FJ-C histochemistry (Poirier et al., 2000; Schmued and Hopkins, 2000; Schmued et al., 2005) and video-EEG (Rensing et al., 2012; Kadam et al., 2017; Moyer et al., 2017). We found more neuronal damage, longer SE duration, and more power during SE with a loss-of-function approach, i.e., suppressing adult neurogenesis. There was less neuronal damage, shorter SE duration, and less power during SE with a gain-of-function approach, i.e., increasing adult neurogenesis. These data suggest that adult-born neurons, when examined collectively at ages up to 6-weeks-old, exert protective effects against SE and neuronal injury in the hippocampus.
MATERIALS AND METHODS
I. General information
Animal care and use followed the guidelines set by the National Institute of Health and the New York State Department of Health. Mice were housed in standard mouse cages, with a 12 h light/dark cycle, with food (Laboratory rodent diet 5001; W.F. Fisher & Sons, Somerville, NJ) and water ad libitum. Mice were bred in-house. During gestation and until weaning, mice were fed chow formulated for breeding (Formulab diet 5008; W.F. Fisher & Sons). Mice were weaned at 23–25 days of age and then separated based on their sex. Littermates were housed together (maximum 4 mice per cage).
II. Methods to modify adult neurogenesis
A. Suppression of adult neurogenesis
Suppression of adult neurogenesis was produced by pharmacogenetic deletion of dividing radial glial precursors (Sofroniew et al., 1999; Schloesser et al., 2009) because this method has been validated (Garcia et al., 2004; Schloesser et al., 2009; Snyder et al., 2011; Iyengar et al., 2015). Mice with herpes simplex virus (HSV)-thymidine kinase (TK) in glial fibrillary acidic protein (GFAP)-expressing progenitor cells (GFAP-TK mice) were used (Jackson Laboratories, Stock number: 005698, Research Resource Identifiers (RRID):IMSR_JAX:005698). These mice were treated with chow containing anti-viral prodrug valganciclovir (VGCV, Custom Animal Diets, Bangor, PA). The HSV-TK in GFAP-expressing progenitor cells phosphorylates ganciclovir (GCV) to toxic nucleotide analogues which induce cell death (Sofroniew et al., 1999; Fischer et al., 2005; Schloesser et al., 2009).
Starting at 6 weeks of age, mice were fed chow containing VGCV (L-valyl ester of GCV; 165 mg/kg chow) Monday to Friday for 6 weeks. Mice had standard chow (Laboratory rodent diet 5001) on weekends to avoid adverse effects of GCV on the gastrointestinal tract (Iyengar et al., 2015). We could not detect abnormalities of the intestines in 3 mice that were euthanized immediately after the cessation of VGCV chow. In prior work we found a 7 day/week VGCV diet led to the abnormal appearance of the intestines (in ~25% of mice), similar to images of the intestines of mice injected with GCV (Bush et al., 1998). In a previous study with 5 day treatment, we found no significant difference in food intake of GFAP-TK+/− (called GFAP-TK+ below) and GFAP-TK−/− (called GFAP-TK- below) mice that were fed VGCV-treated chow (Iyengar et al., 2015). The general appearance of mice, such as fur (well groomed or not), eyes (open or squinted), posture (normal or hunched), and activity level (active or sluggish) did not appear to differ between genotypes.
The background strain was C57BL6/J (Jackson Laboratories; Stock# 005698). GFAP-TK+ female mice (2.5–5.0 months old) were bred with wild type C57BL6/J male mice and progeny were genotyped (see Supporting Information Table S1) in-house for the presence of TK. GFAP-TK+ male mice were not used because they are infertile (https://www.jax.org/strain/005698).
B. Enhancement of adult neurogenesis
To enhance neurogenesis, a method was used that depends on deletion of Bax, the major regulator of programmed cell death in adult-born neurons (Sun et al., 2004; Sahay et al., 2011; Ikrar et al., 2013; Adlaf et al., 2017). Enhancement of neurogenesis was induced by conditional deletion of Bax from Nestin-expressing progenitors because it has been well-characterized (Sahay et al., 2011). These mice were created by crossing mice that have loxP sites flanking the pro-apoptotic gene Bax (Baxf/f) with a NestinCreERT2 mouse line in which tamoxifen-inducible Cre recombinase (CreERT2) is expressed under the control of the rat Nestin promoter (Sahay et al., 2011). The NestinCreERT2Baxf/f mouse line was kindly provided by Drs. Amar Sahay and Rene Hen.
The background strain of these mice was a combination of C57BL6 and Sv129 (Sahay et al., 2011; Bermudez-Hernandez et al., 2017). For breeding, Baxf/f female mice were bred with NestinCreERT2+/−Baxf/f male mice and the progeny were genotyped (See Supporting Information Table S1) in-house for the presence of Cre recombinase.
Starting at 6 weeks of age, mice were injected subcutaneously (s.c.) either with tamoxifen (Cat# T5648, Sigma-Aldrich, St. Louis, MO, 100 mg/kg, 1/day for 4 days) or vehicle. Vehicle was corn oil (Cat# C8267, Sigma-Aldrich) with added absolute alcohol so that the final concentration of alcohol was 10% (i.e., 1 ml per 9 ml of corn oil). For vehicle, an identical volume of corn oil with 10% absolute alcohol was injected 1/day for 4 days. Tamoxifen was administered from a stock solution (20 mg/ml in corn oil containing 10% absolute alcohol). Solutions were stored at 4oC. Tamoxifen is light sensitive, so it was stored in an aluminum foil-wrapped container.
III. Stereotaxic surgery
A. General information
For all surgical procedures, mice were anesthetized by isoflurane inhalation (3% isoflurane for induction and 1.75 – 2% isoflurane for maintenance during the surgery) and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). Prior to surgery, the analgesic Buprenex (Buprenorphine hydrochloride, NDC# 12496–0757-5, Reckitt Benckiser, Richmond, VA) was diluted in saline (0.03 mg/ml in sterile 0.9% sodium chloride solution, Vedco Inc., St. Joseph, MO) and injected (0.2 mg/kg, s.c.). For surgery, hair over the skull was shaved and then a midline incision was made to expose the skull.
B. Viral injections
Injections of virus [pAAV5-hSyn-DIO-hM4D(Gi)-mCherry; 4×1012 vg/ml; Addgene, Cambridge, MA] were made bilaterally (230 nl per injection) in the dorsal DG using a Hamilton syringe (Cat# 86259, Hamilton, Reno, NV), a 33 gauge needle, and a flow rate of 40 nl/min. Coordinates were anterior-posterior (AP, relative to Bregma) −2.5 mm, mediolateral (ML, from the midline) ±1.5 mm, and dorsoventral (DV, relative to the skull surface) −1.7 mm. After each injection, the needle was left at the injection site for 5 min and then the needle was pulled up half-way and left for an additional 5 min. The incision was closed with Vetbond (Cat# 1469SB, 3M Animal Care Products, St. Paul, MN) and the mouse was injected with 50 ml/kg warm (31°C) lactated Ringer’s solution (NDC# 099355000476, Aspen Veterinary Resources Ltd, Liberty, MO, s.c.). Mice were housed individually in a clean cage on a heating blanket (37°C; Cat# 507220F, Harvard Apparatus, Holliston, MA) until mice were fully ambulatory.
C. Surgery to implant EEG electrodes
If electrodes were implanted, they were implanted 1 week after VGCV treatment ended. This delay of 1 week was intended to allow mice to clear any residual VGCV so that GFAP-expressing cells that divided around the tips of the implanted electrodes would be the same in GFAP-TK+ and TK- mice. To implant subdural screw electrodes (Cat# 8209, 0.10” stainless steel screw, Pinnacle Technology, Lawrence, KA) skull was exposed and 4 holes were drilled. The screws used as electrodes were mostly located in the skull and subdural space, but often the tips pressed on the brain surface or penetrated the very outer most layers of the cortex.
The coordinates were: right occipital cortex (Rt OC; AP −3.5 mm, ML 2.0 mm); left frontal cortex (Lt FC, AP −0.5 mm; ML −1.5 mm); left hippocampus (Lt HC, AP −2.5 mm; ML −2.0 mm) and right hippocampus (Rt HC, AP −2.5 mm; ML 2.0 mm). An additional screw was placed over the right olfactory bulb as ground (AP 2.3 mm; ML 1.8 mm) and another screw over the cerebellum at the midline as reference (relative to Lambda: AP −1.5 mm; ML −0.5 mm). Here, “ground” refers to the earth ground and “reference” refers to the reference for all 4 screw electrode recordings (Moyer et al., 2017). An 8-pin connector (Cat# ED85100-ND, Digi-Key Corporation, Thief River Falls, MN) was placed over the skull and secured with dental cement (Dental Cement Kit; Cat# 51459, Stoelting Co., Wood Dale, IL).
After surgery, animals were placed in a cage on a heating blanket and injected with Ringer’s solution as described above. Mice were housed in the room where video-EEG would be conducted so that they would acclimate to the recording environment. Another 2 weeks elapsed while animals recovered. This duration of time was necessary because we have found that mice can have different seizure thresholds if they injected with a convulsant within 1 week of surgery (unpublished data).
IV. Pilocarpine-induced SE
A. General procedures
The first cohort of mice was treated with pilocarpine and not recorded by video-EEG because the EEG electrodes interfered with optimal FJ staining. Therefore, another cohort of mice was implanted with electrodes to quantify seizures by video-EEG.
Two days before pilocarpine injection, mice were handled by the experimenter and pinched in the lower back where the experimenter was planning to inject pilocarpine. This acclimation was intended to reduce stress during the day of pilocarpine treatment because stress influences seizures (Cain and Corcoran, 1984; Sawyer and Escayg, 2010; Maguire, 2014) and adult hippocampal neurogenesis is involved in the normal stress response (Snyder et al., 2011). In addition, increasing adult neurogenesis can reduce the behavioral response to stress (Culig et al., 2017). These studies demonstrate that stress, seizures, and adult neurogenesis interact in a complex manner. The results presented here should be considered with these complex interactions in mind.
B. Procedure for unimplanted mice
On the day of pilocarpine injection, there was an initial injection of pretreatments and then one injection of pilocarpine. The pretreatments were a solution of scopolamine methyl nitrate (Cat# S2250, Sigma-Aldrich, 1 mg/kg of 0.2 mg/ml in sterile 0.9% sodium chloride solution, s.c.) and terbutaline hemisulfate (Cat# T2528, Sigma-Aldrich,1 mg/kg of 0.2 mg/ml in sterile 0.9% sodium chloride solution, s.c.). Scopolamine methyl nitrate is a muscarinic antagonist that does not cross the blood-brain barrier and was administered to minimize peripheral cholinergic side effects of pilocarpine. Terbutaline hemisulfate, a β2-adrenergic receptor agonist, is a bronchodilator and was injected to support respiration during seizures (Cho et al., 2015). Thirty min after the pretreatment, pilocarpine hydrochloride was injected (Cat# P6503, Sigma-Aldrich, 260 mg/kg of 50 mg/ml in sterile 0.9% sodium chloride solution, s.c.).
Mice were placed in a heated cage (Cat# S12781, Animal Intensive Care Unit, ThermoCare, Paso Robles, CA) 30 min before the injections to acclimate. For each injection, the mouse was removed from the cage and immediately returned to the cage following the injection. The cage temperature was maintained at 31oC. Two h after pilocarpine injection, the benzodiazepine diazepam (NDC# 0409–3213-12, 10 mg/kg of 5 mg/ml, Hospira Inc., Lake Forest, IL, s.c.) was administered to decrease the severity of SE. While sedated with diazepam, animals were injected with warm (31oC) lactated Ringer’s solution. Approximately 3 h later, mice were injected again with lactated Ringer’s solution and moistened-chow with water was placed at the base of the cage. Mice remained in the heated cage overnight. Starting on the next day, mice were housed in their home cage on a heating blanket, and the cage temperature was 31oC.
Note that all mice were injected with pilocarpine at a similar time of day (late morning) and perfused at a similar time of day (late morning, 3 days later). The day of pilocarpine injection was considered day 0 with 72 h (3 × 24 h) as day 3.
C. Procedures for mice with implanted electrodes
1). Pilocarpine-induced SE
Implanted mice were treated the same with the following exceptions. First, we added an injection of ethosuximide to the pretreatments (Cat# E7138, Sigma-Aldrich, 150 mg/kg of 84 mg/ml in phosphate buffered saline, s.c.) because we found some mortality in the first mice that were used, which were for FJ staining and unimplanted. Prior work in our laboratory showed that ethosuximide decreases mortality after KA-induced SE (Iyengar et al., 2015), so it was incorporated into the pilocarpine procedure. The cages were also different for implanted mice, so that the EEG cables did not interfere with the lid of the cage. Third, based on initial observations that implanted mice had a lower incidence of SE than unimplanted mice, we used 20 mg/kg more pilocarpine for implanted mice. The pilocarpine dose was 260 mg/kg in unimplanted mice and 280 mg/kg in mice with implanted electrodes.
To record video-EEG, the pin connector on the head of the mouse was attached to a preamplifier (Cat# 8406, Pinnacle Technology) which was attached to a commutator [Cat# 8408, Mouse Swivel/Commutator (4-channel), Pinnacle Technology] to allow freedom of movement. Signals were acquired at a 500 Hz sampling rate, and band-pass filtered at 1–100 Hz using Sirenia Acquisition software (https://www.pinnaclet.com, RRID:SCR_016183). Video was captured with a high-intensity infrared LED camera (Cat# PE-605EH, Pecham, El Monte, CA) and was synchronized to the EEG record. Baseline EEG was recorded for at least 20 min before injections began. Twenty minutes was chosen because mice passed through most behavioral states during this time (exploring, grooming, quiet wakefulness or sleep), and the artifacts associated with different behavioral states could be identified; these artifacts needed to be recorded before SE so that they could be discriminated from abnormal EEG after pilocarpine (Lehmkuhle et al., 2009; Fisher et al., 2014; Pearce et al., 2014; Moyer et al., 2017).
2). Video-EEG analysis
EEG was analyzed offline with Sirenia Seizure Pro, V1.7.4 (Pinnacle Technology, RRID:SCR_016184). A seizure was defined as a period of rhythmic (>3 Hz) deflections that were >2x the standard deviation of baseline mean noise and lasted at least 10 secs [Supporting Information Fig. S1; (Fisher et al., 2014; Cho et al., 2015; Iyengar et al., 2015; Trinka et al., 2015; Hosford et al., 2016)]. Seizures were rated as convulsive if an electrographic seizure was accompanied by a behavioral convulsion, defined by observation during SE (and confirmed later by video playback), as stages 3–5 using the Racine scale (Racine, 1972) where stages 1–2 involve subtle movements of the facial muscles or ears, too subtle in our experience to be sure the movement is abnormal [see also (Pearce et al., 2014)]. In contrast, stages 3–5 were accompanied by convulsive behavior that was so unusual that >1 experimenter in our group, blinded to the other opinions, would all judge the behavior to be convulsive. For this purpose, we defined stage 3 by unilateral forelimb clonus, stage 4 by bilateral forelimb clonus with rearing, and stage 5 by bilateral forelimb clonus followed by rearing and falling (Racine et al., 1972). A seizure was defined as non-convulsive when there was electrographic evidence of a seizure but there were no stage 3–5 behaviors.
Latency to the onset of a seizure was defined as the time from pilocarpine injection to the time when the peak-to-peak amplitude of the EEG exceeded 2x the standard deviation of the noise, measured peak-to-peak at 3 times during the baseline and averaged. The end of a seizure was defined as the time when peak-to-peak amplitude of the EEG returned to the baseline mean or a value lower than the baseline mean (in the case of post-ictal depression).
SE was defined as continuous severe seizures (Chen and Wasterlain, 2006). Here we defined the onset of SE as the time when seizures occurred continually for at least 5 min because if this occurred the animal did not resume normal behavior for several hours afterwards, consistent with definitions of SE elsewhere (Chen and Wasterlain, 2006; Trinka and Kalviainen, 2017). The time from pilocarpine injection to the onset of continuous seizures lasting for at least 5 min was considered as the latency to the onset of SE.
We observed that SE waned but did not end, as shown in the Results. Therefore, we defined the end of SE as the timepoint when the amplitude of the EEG (measured peak-to-trough or maximal positivity to maximal negativity) during SE was reduced to approximately 2 times the amplitude of the baseline noise. Our definition required this decline in amplitude to be in at least 3 channels and remain depressed for at least 10 min (Supporting Information Fig. S2).
Power spectral analyses were computed for the hippocampal channel (left) only for the recorded frequencies (1–100 Hz), using Spike2 software (Cambridge Electronic Design, Milton, Cambridge). To calculate power, a Fast Fourier Transform (FFT) was used with a Hanning window and 1 Hz resolution, similar to Phelan and colleagues Phelan et al. (2015).
V. Tissue processing
A. General timeline
Notably, hippocampal vulnerability to SE can vary depending on the time after SE that is chosen for study (Fujikawa, 1996; Covolan and Mello, 2000; Poirier et al., 2000; Weise et al., 2005; Wang et al., 2008; do Nascimento et al., 2012). The damage typically peaks 3–5 days after SE in mice (Wang et al., 2008). However, hilar damage can be rapid, occurring within 24 h of SE (Babb and Pretorius, 1993; Covolan and Mello, 2000; Borges et al., 2003; Choi et al., 2007; Wang et al., 2008; do Nascimento et al., 2012) or other insults (Kirino, 1982; Crain et al., 1988; Schmidt-Kastner and Hossmann, 1988; Scharfman and Schwartzkroin, 1990; Lowenstein et al., 1992; Mody et al., 1995; Kharatishvili et al., 2006). Based on these studies, we chose a time after SE that would not be too long to capture hilar cell death, but not too soon to capture death that occurs more slowly in other areas of the hippocampus: 3 days after pilocarpine injection.
B. Perfusion-fixation and sectioning
Mice were deeply anesthetized by isoflurane inhalation (NDC# 07–893-1389, Patterson Veterinary, Devens, MA) followed by urethane (Cat# U2500, Sigma-Aldrich, 250 mg/kg of 250 mg/ml in 0.9% sodium chloride, intraperitoneal, i.p.). The heart cavity was opened, and a 25-gauge needle inserted into the heart, followed by perfusion with 10 ml saline (0.9% sodium chloride in double-distilled water (ddH2O) using a peristaltic pump (Minipuls 1; Gilson, Middleton, WI) followed by 30 ml of cold (4oC) 4% paraformaldehyde (PFA, Cat# 19210, Electron Microscopy Sciences, Hatfield, PA) in 0.1 M phosphate buffer (PB; pH 7.4). The brains were removed immediately, hemisected, and post-fixed for at least 24 h in 4% PFA at 4oC. After post-fixation, one hemisphere was cut in the coronal plane and the other in the horizontal plane (50 μm-thick sections) using a vibratome (TPI-3000, Vibratome Co., St. Louis, MO). Sections were collected sequentially to process serial sections that were from similar septotemporal levels and always 300 μm apart.
C. Fluoro-Jade C (FJ-C)
1). Procedures for staining
FJ is a fluorescent dye that is the ‘gold-standard’ to stain degenerating neurons (Schmued and Hopkins, 2000; Schmued et al., 2005). The sections were stained with FJ based on methods of Schmued and colleagues (Schmued et al., 2005). First, sections were mounted on gelatin-coated slides (1% porcine gelatin in ddH2O, Cat# G1890, Sigma-Aldrich) and dried on a hot plate (Model # HP18325, Nuova II, Thermolyne, Dubuque, IA) at 50–55oC for 1 h. Then, slides were placed in a staining rack and immersed in a basic ethanol solution (1% sodium hydroxide (Cat# 3722–01, JT Baker, Phillipsburg, NJ in 80% ethanol) for 5 min, then in 70% ethanol for 2 min, followed by a <10 sec wash in ddH2O. Slides were then incubated in 0.06% potassium permanganate solution (Cat# P-279, Fisher Scientific, Boston, MA) for 10 min on a shaker (Model# BDRAA115S, “Belly Dancer”, Stovall Life Science Inc., Greensboro, NC) with gentle agitation, followed by washes in ddH2O (2 × 1 min). Slides were then incubated for 20 min in a 0.0002% solution of FJ-C (Histo-Chem Inc., Jefferson, AR) dissolved in 0.1% acetic acid (Cat# UN2789, Fisher Scientific) in ddH2O, in the dark. The stock solution of FJ-C was 0.01% in ddH2O and was stored at 4oC for up to 3 months. To prepare a working solution, 6 ml of stock solution was added to 294 ml of 0.1% acetic acid in ddH2O and used within 10 min of preparation. Slides were subsequently protected from direct light. First, they were washed in ddH2O (3 × 1 min) and dried overnight at room temperature. On the next day, slides were cleared in Xylene (2 × 3 min, Cat# 534056, Sigma-Aldrich) and coverslipped with DPX mounting medium (Cat# 44581, Sigma-Aldrich). Sections were photographed with an epifluorescence microscope (Model BX51; Olympus of America, Center Valley, PA) and analysis used images made through the thickness of the section with a confocal microscope (Model LSM 510 Meta; Carl Zeiss Microimaging, Thornwood, NY) as described further below.
2). FJ Analysis
a). Hilus
FJ staining of hilar cells were counted manually from stacks of images (1 μm-thick optical sections) throughout a tissue section. In each tissue section, the hilus was traced first using ImageJ. The definition of the hilus was based on Amaral [Amaral, (1978); Supporting Information Fig. S3]. Then cells were counted in each section, omitting cut cells at the section surfaces, and taking care not to count the same cell more than once when it overlapped onto adjacent optical sections.
b). Cell layers
Manual counting of FJ-positive (FJ+) cells was not possible in the cell layers (CA1, CA3, and GCL) because there could be so many FJ+ cells that were overlapping. Instead, FJ staining in cell layers was quantified by first outlining the cell layer as a region of interest (ROI) at 10x magnification in ImageJ (Image Analysis software; Bioquant; Nashville, TN). To make the borders of the cell layers as accurate as possible, a tissue section was photographed using ImageJ and the borders of the cell layers were outlined using the background as a guide. This was possible because the cell layer was a distinct shade of green. In addition, cresyl violet-stained sections adjacent to the sections processed for FJ were used as a guide for the outlines of the cell layer.
To outline the CA1 cell layer, the border with CA2 was defined as the point where the cell layer changed width, a sudden change that could be appreciated by the background in FJ-stained section and cresyl violet-stained section. The border of CA1 and the subiculum was defined as the location where the normally compact CA1 cell layer suddenly dispersed. To outline CA3, the border with CA2 and CA3 was defined by the point where stratum lucidum of CA3 terminated. This location was distinct in its background in FJ-stained sections. The border of CA3 and the hilus was defined according to Amaral (1978). This location was also possible to detect in FJ-stained sections because the background in the hilus was relatively dark compared to area CA3.
After defining ROIs, a threshold fluorescence level was selected so that all cells that had very bright immunofluorescence were above threshold but other cells that were similar in fluorescence to background staining were not (Iyengar et al., 2015). To make the setting of threshold more objective, the histogram used to analyze fluorescence intensities of an image, provided in ImageJ, was used, and the point where the histogram falls to approximately zero was selected as threshold. After selecting this point as threshold, the section was reevaluated by eye to confirm that cells which were bright were selected but not background areas such as stratum radiatum of CA1 and CA3. Stratum radiatum was useful because there was a relatively high background, probably reflecting degeneration of pyramidal cell dendrites. ImageJ was then used to calculate the area within the ROI that was above threshold relative to the entire area of the ROI.
This measurement is referred to as area fraction in the Results and expressed as a percent. For a given animal, the area fraction was determined for 3 coronal sections in the dorsal hippocampus between AP −1.94 to −2.06 mm and 3–4 horizontal sections in the ventral hippocampus between DV 0.84 to 1.08 mm, with sections spaced 300 μm apart. These area fractions were averaged so that a mean area fraction was defined for each animal. For these and other analyses described below, the investigator was blinded.
D. Cresyl violet
To confirm that FJ+ cells represented degenerating neurons, adjacent sections were stained with cresyl violet as described previously (Duffy et al., 2011). The FJ+ cells were confirmed as degenerating neurons if adjacent sections showed abnormal somata with cresyl violet stain (Supporting Information Fig. S4). When there was no FJ staining, the absence of degeneration was also confirmed as the absence of abnormalities with cresyl violet stain (Supporting Information Fig. S4). For these purposes, abnormalities often were reflected by a very dark cresyl violet stain and/or shrunken, angular nucleus/cell body shape (Supporting Information Fig. S4). In the past these cells have shown abnormal, degenerating ultrastructure using electron microscopy (Duffy et al., 2011).
E. Doublecortin
1). Procedures for staining
Doublecortin (DCX), a microtubule-associated protein (Gleeson et al., 1999), was used to identify immature adult-born neurons (Brown et al., 2003; Couillard-Despres et al., 2005), and was stained after antigen retrieval (Botterill et al., 2015). First, free floated sections were washed in 0.1 M Tris buffer (TB, 3 × 5 min). Sections were then incubated in sodium citrate buffer (Cat# S4641, Sigma-Aldrich, 2.94 mg/ml in ddH2O, pH 6.0 adjusted with HCl) in a preheated water bath at 85oC for 30 min. After allowing sections to cool to room temperature the sections were washed with 0.1 M TB (3 × 5 min), blocked in 5% goat serum (Cat# S-1000, RRID:AB_2336615, Vector Laboratories, Burlingame, CA) in 0.1 M TB with 0.5% (v/v) Triton X-100 and 1% (w/v) bovine serum albumin for 30 min and then incubated overnight with primary antibody (monoclonal anti-doublecortin made in rabbit, 1:1000 diluted in blocking serum, Cat# 4604S, RRID:AB_10693771, Cell Signaling Technology, Danvers, MA) on a rotator (“Belly Dancer”) at room temperature. On the next day, sections were washed in 0.1 M TB (3 × 5 min), treated with 2.5% hydrogen peroxide for 30 min to block endogenous peroxides, and then washed with 0.1 M TB (3 × 5 min). Sections were then incubated in secondary antibody (biotinylated anti-rabbit IgG made in goat; 1:500; Cat# BA-1000, RRID:AB_2313606, Vector Laboratories) for 1 h in 0.1 M TB, followed by washes with 0.1 M TB (3 × 5 min), blocked in avidin-biotin complex (1:500 in 0.1 M Tris buffer, Cat# PK-6100, Vector Laboratories) for 1 h, washed in 0.1 M TB (1 × 5 min) and then in 0.175 M sodium acetate (Cat# S8750, 14.36 mg/ml in ddH2O, pH 6.8, adjusted with glacial acetic acid, 2 × 5 min). Sections were reacted in 0.5 mg/ml 3, 3′-diaminobenzidine (DAB, Cat# D5905, Sigma-Aldrich) with 40 μg/ml ammonium chloride (Cat# A4514, Sigma-Aldrich), 3 μg/ml glucose oxidase (Cat# G2133, Sigma-Aldrich), 2 mg/ml (D+)-glucose (Cat# G5767, Sigma-Aldrich) and 25 mg/ml ammonium nickel sulfate (Cat# A1827, Sigma-Aldrich) in 0.175 M sodium acetate. Sections were washed in 0.175 M sodium acetate (2 × 5 min) and 0.1 M TB (5 min), mounted on gelatin-coated slides (1% bovine gelatin, Cat# G9391, Sigma-Aldrich) and dried overnight at room temperature. On the next day, sections were dehydrated with increasing concentrations of ethanol, cleared in Xylene, and coverslipped with Permount (Cat# 17986–01, Electron Microscopy Sciences).
Sections were viewed with a brightfield microscope (Model BX51; Olympus of America, Center Valley, PA). Photographs were taken with a digital camera (Model RET 2000R-F-CLR-12, Q-Imaging, Surrey, BC) and acquired using ImagePro Plus, v.7.0 (Media Cybernetics, Bethesda, MD).
2). DCX Analysis
Quantification of DCX immunoreactivity was not done by counting cell bodies because in control mice the adult-born GCs were so numerous that they overlapped extensively. Moreover, dendritic information that is relevant to quantifying the extent that there is suppression or increased adult neurogenesis, such as the degree that the dendrites are developed, is not acquired by counting cell bodies. To circumvent these limitations, DCX was quantified by first defining an ROI that included the adult-born cells and the majority of their DCX-labeled dendrites: the subgranular zone, GCL, and inner molecular layer. The subgranular zone was defined as a region that extended from the GCL into the hilus for a width of 100 μm because this wide an area included the vast majority of the DCX immunoreactivity [see the Figures and (Iyengar et al., 2015)]. Then a threshold was selected where DCX-immunoreactive cells were above, but the background was below threshold, with the methods already described above. Three dorsal and 3 horizontal sections were used to calculate area fraction, also using Methods described above. Then the mean for each animal was used to compare mice with intact vs. suppressed neurogenesis or control vs. increased neurogenesis.
To confirm the degree of suppression was high, we manually counted cell DCX-immunoreactive cell bodies as well. This was possible in mice with suppressed neurogenesis because cells were non-overlapping.
F. mCherry
Immunofluorescence was conducted as described previously (Bermudez-Hernandez et al., 2017). Free-floating sections were washed in 0.1 M TB, incubated in blocking serum for 1 h (10% goat serum, 0.025% (v/v) Triton X-100 and 0.005% (v/v) bovine serum albumin in 0.1 M Tris buffer), followed by washes in 0.1 M TB. Next, sections were incubated overnight at 4°C with a primary antibody (Rabbit polyclonal anti-mCherry 1:500, Cat# ab167453, RRID:AB_2571870; Abcam, Cambridge, MA) diluted in blocking serum. The next day, sections were washed in 0.1 M TB, followed by a 2 h-long incubation in secondary antibody (anti-rabbit IgG made in goat, Alexa Fluor 568; 1:1000; Cat# A11036, RRID:AB_10563566, Thermo Fisher Scientific) diluted in blocking serum. Sections were then washed in 0.1 M TB. Sections were mounted on gelatin-coated slides and coverslipped with Citifluor™ AF1 mounting solution (Cat# 17970–25, Electron Microscopy Sciences). Sections were viewed with a confocal microscope (LSM 510 Meta). Photography was conducted as described above for FJ.
VI. Statistical Analysis
All data are presented as the mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism Software (https://www.graphpad.com/scientific-software/prism/, RRID: SCR_002798). Statistical significance was set at p < 0.05.
Parametric tests were used when data fit a normal distribution, determined by the D’Agostino and Pearson or Shapiro-Wilk’s normality tests, and there was homoscedasticity of variance (confirmed by a F-test). A Student’s unpaired two-tailed t-test was used to assess differences between two groups. A Fisher’s exact test was used for comparing proportions of binary data (yes/no). One-way Analysis of Variance (ANOVA), two-way ANOVA, and two-way repeated measures ANOVA (RMANOVA) were performed when there were multiple groups and were followed by Bonferroni’s multiple comparison post-hoc test (Bonferroni’s test).
For data that did not follow a normal distribution, typically some data had a 0 value. In these cases, non-parametric tests were selected. The Mann-Whitney U test was used to compare two groups, and a Kruskal-Wallis test followed by post-hoc Dunn’s test was used for multiple groups. In one instance the data did not fit a normal distribution although all data were integers. This was the data from hilar FJ-stained sections for control or mice with increased adult neurogenesis. The differences between groups were assessed by a Mann-Whitney U test and were insignificant. A parametric test was also conducted after all data in both groups were log transformed, which led to a data set that followed a normal distribution, determined as described above. A parametric test (Student’s t-test) showed that the difference between groups were insignificant. Therefore, the choice of parametric or non-parametric statistics did not influence the Results. The Results only report the statistics for the Mann-Whitney U test.
RESULTS
The results are presented in two major parts: mice with suppressed adult neurogenesis and then increased adult neurogenesis. In each major part, mice were evaluated with FJ staining in the hippocampus 3 days after pilocarpine-induced SE and there was also an analysis of the EEG during SE. After both major parts, we show the confirmation that mice with reduced or enhanced adult neurogenesis did indeed have a suppression or augmentation of adult neurogenesis, respectively.
I. Effects of reduced adult neurogenesis
A. FJ staining was greater in mice with reduced adult neurogenesis
Fig. 1A shows the experimental timeline for the studies of neurodegeneration after SE, using FJ staining in mice with reduced adult neurogenesis (GFAP-TK+ mice) and intact adult neurogenesis (GFAP-TK- mice).
Figure 1. Greater Fluoro-Jade (FJ) staining in the dorsal hippocampus of mice with reduced adult neurogenesis.
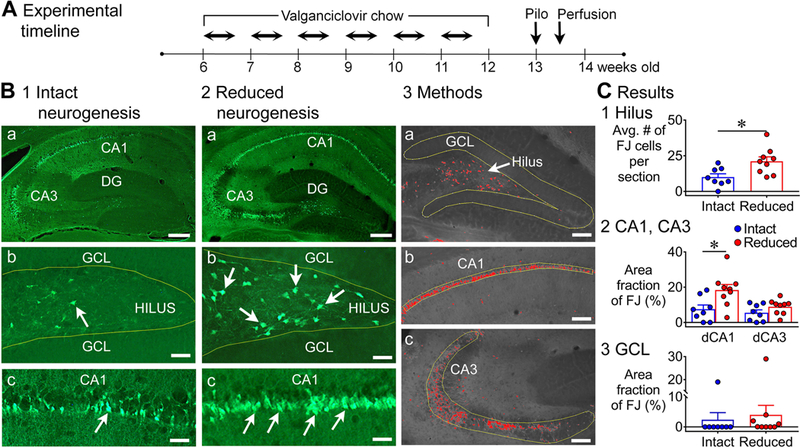
A. The experimental timeline is shown. Chow containing valganciclovir (VGCV) was given for 6 weeks (Monday to Friday) and standard chow on weekends. One week later, mice were injected with pilocarpine (Pilo) to induce status epilepticus (SE). Three days after pilocarpine injection, mice were transcardially-perfused. GFAP-TK-: intact neurogenesis (n=8); GFAP-TK+: reduced neurogenesis (n=9).
B. Representative examples of FJ staining in the dorsal hippocampus in mice with intact (1) and reduced (2) neurogenesis are shown. Calibration, 75 μm (a); 50 μm (b, c). 3. The tracing and thresholding of the region of interest (ROI) used to quantify FJ in the granule cell layer (GCL, a), CA1 (b), and CA3 (c) are shown. Calibration, 50 μm (a-c).
C. 1. There were significantly more FJ-positive (FJ+) hilar cells in mice with reduced neurogenesis than intact neurogenesis (p = 0.010). 2. There was a main effect of reduced neurogenesis on FJ staining in dorsal CA1 and CA3 (p = 0.009). Dunn’s test showed significantly more damage in mice with reduced neurogenesis in area CA1 (p = 0.014), but not in CA3 (p = 0.535). 3. The differences in area fraction of the GCL with FJ staining were not significant (p = 0.064).
1). FJ in the dorsal hippocampus
As shown in Fig. 1B, FJ staining was robust in the hippocampus after pilocarpine-induced SE, and it appeared to be greater in GFAP-TK+ mice relative to TK- mice. FJ staining was most evident in the hilus, CA1 and CA3 pyramidal cell layers (Fig. 1B1–2), consistent with the previous studies of SE in rats (Fujikawa, 1996; Covolan and Mello, 2000; Poirier et al., 2000; Dinocourt et al., 2003; Weise et al., 2005) and mice (Cavalheiro et al., 1996; Borges et al., 2003; Wang et al., 2008; do Nascimento et al., 2012). Within the CA1 cell layer, the subset of neurons near stratum radiatum showed the most FJ staining (Fig. 1B1–2), consistent with prior studies showing that they are more vulnerable than the pyramidal cells near stratum oriens (Mizuseki et al., 2011; Soltesz and Losonczy, 2018).
FJ staining was significantly greater in mice with suppressed adult neurogenesis. In Fig. 1C, the areas used for FJ measurements of the cell layers are shown (for the hilus, see Supporting Information Fig. S3; for additional information see Methods). In the hilus, mice with reduced adult neurogenesis had approximately twice the mean number of FJ+ neurons as mice with intact adult neurogenesis (intact: 10.0 ± 2.3; reduced: 21.1 ± 3.1; Mann-Whitney U test, U = 10, p = 0.010; Fig. 1C1). In CA1 and CA3, a Kruskal-Wallis test showed that there was a significant effect of genotype (H = 11.4, p = 0.009; Fig. 1C2). Dunn’s test showed significantly more damage in CA1 of mice with reduced adult neurogenesis (intact: 7.5 ± 2.4%; reduced: 18.4 ± 3.1%; p = 0.014), but not in area CA3 (intact: 5.5 ± 1.7%; reduced: 8.8 ± 1.4%; p = 0.535).
GCs do not show much degeneration after pilocarpine-induced SE (Cavalheiro et al., 1996; Fujikawa, 1996; Wang et al., 2008) because they are relatively resistant to insults and injury (Margerison and Corsellis, 1966; Sloviter, 1989; Meldrum, 1991; Scharfman, 1999; Choi et al., 2007). Nevertheless, there was some FJ staining of GCs, so this was compared (Fig. 1C3). The FJ staining had an unusual appearance: there were clusters of FJ+ cells that formed dense FJ+ areas next to evidently normal GCs (Supporting Information Fig. S4). There was only 1/8 GFAP-TK- mice that showed clusters, whereas there were 4/9 GFAP-TK+ mice. However, this difference in incidence was not significant (Fisher’s exact test, p = 0.294). When the mean area fraction of the GCL with FJ staining was compared by a Mann-Whitney U test, the differences between genotypes did not reach significance (U = 18.5, p = 0.064; Fig. 1C3). In the mice that did not show clustering (7 GFAP-TK-, 5 GFAP-TK+), there were a small number of FJ+ cells scattered throughout the GCL. Notably, these scattered cells were present only in mice with reduced neurogenesis, but the difference in the incidence of scattered FJ+ cells was not significant (intact: 0/7 mice; reduced: 2/5 mice; Fisher’s exact test, p = 0.151). Note that the clustering and scattered FJ+ cells in the GCL did not lead to significant differences here, but it did in other comparisons (see below), which is why we include it here.
Taken together, the data show that there was significantly more FJ staining in the dorsal hippocampus of GFAP-TK+ mice relative to TK- mice, and the significant differences were in the hilus and area CA1.
2). FJ in the ventral hippocampus
The ventral hippocampus was examined separately from the dorsal hippocampus because the neuronal loss and other aspects of neuropathology after SE differs between dorsal and ventral hippocampus (Scharfman et al., 2002; Ekstrand et al., 2011; Haussler et al., 2012; Uemori et al., 2017). Notably, FJ staining was always greater when adult neurogenesis was suppressed.
In the hilus (Fig. 2A1–3), mice with reduced adult neurogenesis had more than twice the mean number of FJ+ neurons as mice with intact adult neurogenesis (intact: 13.9 ± 4.2 cells/section; reduced: 32.6 ± 3.2 cells/section; Mann-Whitney U test, U = 6.5, p = 0.005; Fig. 2B1). There was no effect of genotype in FJ staining in areas CA1 and CA3 (CA1: intact: 4.2 ± 1.4%; reduced: 6.3 ± 1.2%; CA3: intact: 6.1 ± 1.3%; reduced: 8.3 ± 1.8%; Kruskal-Wallis test, H = 3.3, p = 0.343; Fig. 2B2).
Figure 2. Greater FJ staining in the ventral hippocampus of mice with reduced adult neurogenesis.
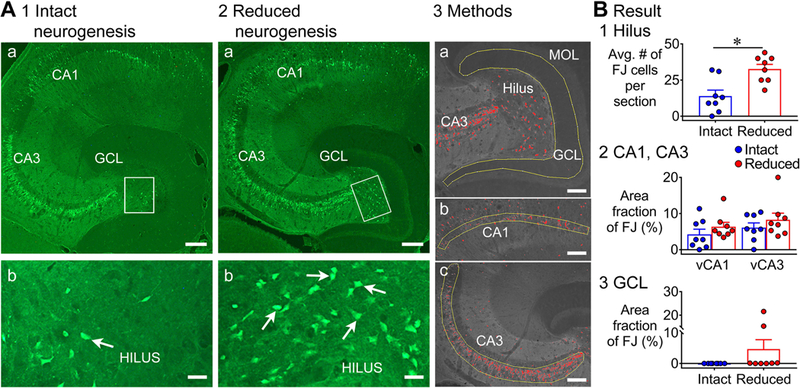
A. Representative examples of FJ staining in the ventral hippocampus in mice with intact (1) and reduced (2) neurogenesis are shown. Calibration, 75 μm (a); 50 μm (b). GFAP-TK-: intact neurogenesis (n=8); GFAP-TK+: reduced neurogenesis (n=8). 3. The tracing and thresholding of the ROI used to quantify FJ in the GCL (a), CA1 (b), and CA3 (c) are shown. Calibration, 50 μm (a-c).
B. 1. There were significantly more FJ+ hilar cells in mice with reduced neurogenesis than intact neurogenesis (p = 0.005). 2. There was no effect of reduced neurogenesis on FJ staining in ventral CA1 and CA3 (p = 0.343). 3. There was a significantly greater area fraction of FJ staining in the GCL of mice with reduced neurogenesis relative to intact neurogenesis (p = 0.025).
In the ventral GCL, there was no detectable FJ staining in 8 mice with intact neurogenesis, but 2/8 mice with reduced adult neurogenesis showed clusters of FJ staining, like the dorsal GCL. Although the incidence of clusters was not significantly different (Fisher’s exact test, p = 0.467; Fig. 2B3), the greatest area fraction in the GCL was in mice with reduced adult neurogenesis (Mann-Whitney U test, U = 12, p = 0.025; Fig. 2B3).
In addition to area fraction, we conducted a second measurement. The second measurement was counting individual FJ+ GCs that were scattered throughout the GCL. Only 6 of the 8 TK+ mice were used for this measurement. Notably, scattered FJ+ GCs were only detected in the mice with reduced adult neurogenesis. The mean number of scattered FJ+ GCs was significantly different (intact: 0 FJ+ cells/section; n=8, reduced: 2.0 ± 0.7 cells/section; n=6; Mann-Whitney U test, U = 8, p = 0.015).
Taken together, the data show that there was significantly more FJ staining in the ventral hippocampus of GFAP-TK+ mice relative to TK- mice, and the significant differences were in the hilus and GCL.
B. Differences in SE in mice with and without reduced adult neurogenesis
1). Incidence of SE, mortality, and sex differences
In unimplanted mice used to study FJ, there were no statistical differences in the incidence of SE (intact: 15/18 mice, 83.3%; reduced: 17/26 mice, 65.4%; Fisher’s exact test, p = 0.303). The mortality (death during or shortly after SE) was similar also (intact: 34.6%, reduced: 33.3%, Fisher’s exact test, p >0.999). In mice implanted with electrodes to record EEG, the incidence of SE was similar between the genotypes (intact: 18/19, 94.7%; reduced: 16/16, 100%; Fisher’s exact test, p > 0.999). Mortality was also similar (intact: 5/19, 26.3%; reduced, 3/16, 18.8%, p = 0.700).
We next confirmed that mice with and without electrodes were similar. By two-way ANOVA (with genotype and electrode implantation as main factors) there was no effect of electrode implantation on the incidence of SE [F(1, 1) = 13.1, p = 0.171], and the same was true for mortality [F(1, 1) = 13.3, p = 0.170]. Mice that had SE were further evaluated for convulsive seizures (occurring between pilocarpine and diazepam injection; Fig. 3, Supporting Information Fig. S5) and there was no effect of electrode implantation on the latency to the first convulsive seizure [two-way ANOVA, F(1, 39) = 3.9, p = 0.055]. We also counted the total number of convulsive seizures and this was similar: each animal had approximately 3 or 4 seizures after pilocarpine injection (intact, unimplanted: 3.8 ± 0.2; intact, implanted: 3.4 ± 0.3; suppressed, unimplanted: 4.2 ± 0.3; suppressed, implanted: 3.4 ± 0.2). However, by two-way ANOVA there were slightly more convulsive seizures in unimplanted mice (a difference of 1–2 seizures) and the effect of implantation was significant [F(1,39) = 5.1, p = 0.030], but not the effect of genotype [F(1,39) = 0.6, p = 0.449]. Note that for the NestinCreERT2Baxf/f mice the total number of convulsive seizures in implanted and unimplanted mice were not significantly different (see below).
Figure 3. EEG recording of SE in mice with intact and reduced adult neurogenesis.
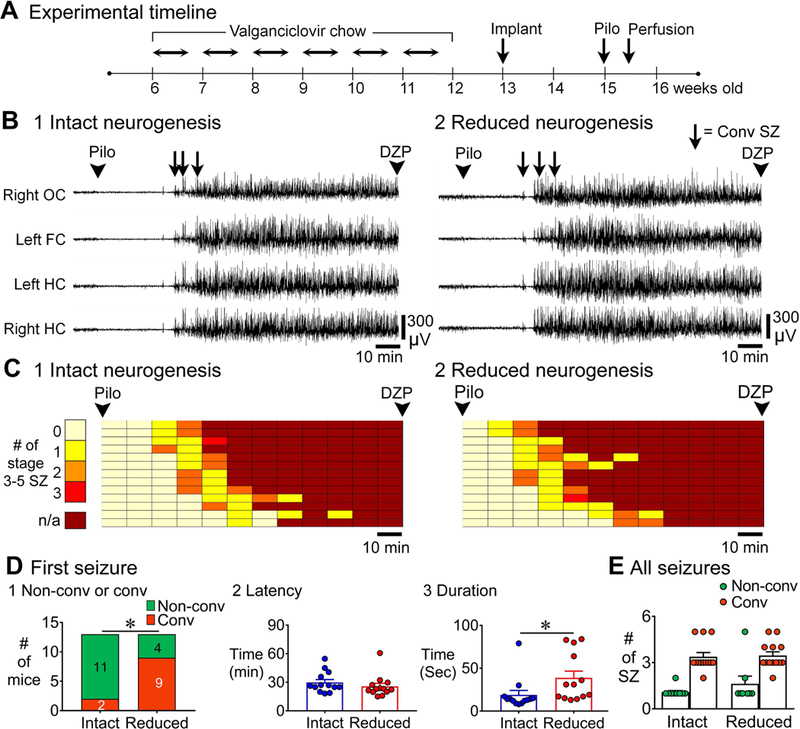
A. The experimental timeline for EEG recording of SE is shown. One week after cessation of chow, mice were implanted with electrodes. Two weeks later, mice were injected with pilocarpine to induce SE. Video-EEG was recorded for 24 h (see Methods). GFAP-TK-: intact neurogenesis (n=13); GFAP-TK+: reduced neurogenesis (n=13).
B. Representative examples of 2 h-long EEG for the time between pilocarpine and diazepam injections for mice with intact (1) and reduced (2) neurogenesis are shown.
C. The 2 h between pilocarpine and diazepam injection was divided into 10 min-long bins. The number of discrete convulsive seizures per bin is plotted with colors indicating lesser (light colors) and greater (darker red) numbers of seizures until SE (continuous seizures, darkest red). Each row is for a different mouse and arranged in order to the latency of first convulsive seizure. Convulsive seizures were seizures between stages 3–5 (see Methods).
D. 1. The first seizure before SE was more often convulsive (rather than non-convulsive) in mice with reduced neurogenesis (9/13) compared to intact neurogenesis (2/13, p = 0.015). 2. The latency to the onset of first seizure was similar in both genotypes (p = 0.217). 3. The duration of the first seizure was longer in mice with reduced neurogenesis relative to intact neurogenesis (p = 0.004).
E. There were differences in the total numbers of non-convulsive vs. convulsive seizures in all animals (p < 0.001) but there was no effect of genotype (Dunn’s test, p > 0.05).
In this figure and all others, OC = Occipital Cortex, FC = Frontal Cortex, HC = Hippocampus, DZP = Diazepam, SZ = Seizure.
The results discussed above only included male mice, because very few females developed SE when the initial cohorts were tested (which were unimplanted mice used for FJ staining). Also, those that had SE rarely survived, with mortality during SE or shortly afterwards. Specifically, only 28% of female mice developed SE and there was no significant effect of genotype (intact: 3/9, 25.0%; reduced: 4/9, 30.8%; Fisher’s exact test, p > 0.999). Of the 7 mice that had SE, 4 died in SE or afterwards (57.1%). The mortality was similar in both genotypes (2 mice in each genotype died; Fisher’s exact test, p > 0.999). Note that the results are consistent with prior studies showing that female rats rarely develop pilocarpine-induced SE (Scharfman et al., 2005; D’Amour et al., 2015; Scharfman and MacLusky, 2017) and there is considerable morbidity (Scharfman et al., 2005). They are also consistent with studies of female mice that were less likely to develop pilocarpine-induced SE, and mortality was high (Buckmaster and Haney, 2012).
2). Seizures during SE
In Fig. 3A, the specific timeline for GFAP-TK+ and TK- mice that were implanted with electrodes and then examined during SE is shown. Examples of the EEG from the time of pilocarpine injection to the time of diazepam injection are shown in Fig. 3B. The data in Fig. 3B show that the onset of SE was sudden and occurred in all electrodes almost simultaneously, and this occurred independent of genotype. It also occurred for the NestinCreERT2Baxf/f mice discussed below, and therefore seems to be a common finding in all mice, independent of the state of adult neurogenesis. To examine the EEG in more detail, the time between pilocarpine and diazepam injection was divided into 10 min-long bins and number of convulsive seizures (any seizures between stage 3–5, see Methods) in each 10 min bin was denoted (Fig. 3C). There were no discrete convulsive seizures in the 3–4 h after diazepam injection.
The overall pattern did not appear to be distinct, but there were differences after quantification (Fig. 3D). For the first seizure after pilocarpine injection, most were non-convulsive in controls, but they were convulsive in mice with reduced neurogenesis, a significant difference (Fisher’s exact test, p = 0.015, Fig. 3D1). The latency of the first seizure was not different between genotypes (intact: 29.8 ± 3.0 min; reduced: 25.7 ± 3.2 min; Mann-Whitney U test, U = 60, p = 0.217; Fig. 3D2). However, the duration of that first seizure was more than twice as long in mice with reduced adult neurogenesis [intact: 18.9 ± 5.2 sec; reduced: 38.7 ± 7.8 sec; Student’s t-test, t(24) = 3.2, p = 0.004; Fig. 3D3]. For the total number of seizures between pilocarpine injection and diazepam, there were differences between non-convulsive and convulsive seizures (Kruskal-Wallis test, p < 0.001; Fig. 3E), but there was no effect of genotype (Dunn’s test, non-convulsive seizures in control vs. suppressed conditions, p = 0.859; convulsive seizures in control vs. suppressed conditions, p > 0.999; Fig. 3E).
Thus, the numbers of seizures and their latency appeared to be similar in mice with intact and suppressed adult neurogenesis, but the duration of the first seizure and its severity were greater in mice with reduced adult neurogenesis.
3). Longer SE duration in mice with reduced adult neurogenesis
To examine the duration of SE we examined the time before and after diazepam injection because generalized electrographic seizures decreased in severity after diazepam but continued (Fig. 4A-B). In the example shown in Fig. 4A, the electrographic activity after diazepam injection appeared to decrease the EEG activity more in the GFAP-TK- mouse (intact adult neurogenesis) compared to the TK+ mouse (reduced adult neurogenesis). However, there was no consistent effect of diazepam to reduce electrographic activity preferentially in TK- mice relative to TK+ mice. In summary, diazepam reduced the amplitude of electrographic activity but did not stop it, and this occurred in both genotypes.
Figure 4. Longer duration of SE in mice with reduced adult neurogenesis compared to intact adult neurogenesis.
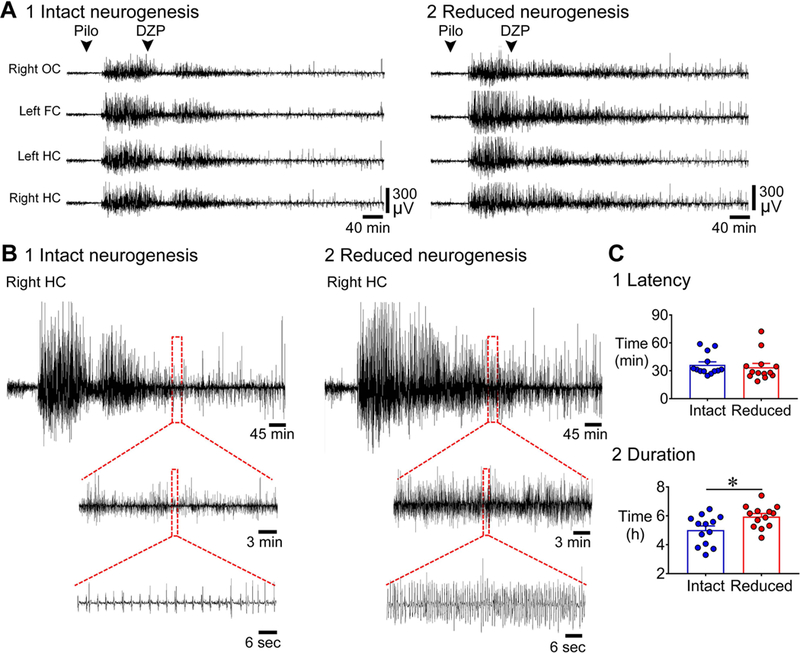
A. Representative examples of 10 h-long EEG are shown, beginning with pilocarpine injection, for mice with intact (1) and reduced (2) neurogenesis. GFAP-TK-: intact neurogenesis (n=13); GFAP-TK+: reduced neurogenesis (n=13).
B. The EEG recording of the right hippocampus at 5 h from the onset of SE is expanded in mice with intact (1) and reduced (2) neurogenesis. The EEG of the mouse with intact neurogenesis shows much less activity than the mouse with reduced neurogenesis.
C. 1. The latency to the onset of SE was similar in the two genotypes (p = 0.243). 2. The duration of SE (defined in Supporting Information Fig. S2 and Methods) was longer (by ~1 h) in mice with reduced neurogenesis compared to intact neurogenesis (p = 0.015).
We defined the end of SE as the time from the start to the end of the most intense period of SE in the EEG, using the EEG amplitude as a measure of intensity (described further in the Methods and Supporting Information Fig. S2). The latency to SE was not different (intact: 36.4 ± 3.2 min; reduced: 33.7 ± 4.1 min; Mann-Whitney U test, U = 61, p = 0.243, Fig. 4C1), but the duration of SE was longer by approximately 1 h in mice with reduced adult neurogenesis [intact: 5.0 ± 0.2 h; reduced: 5.9 ± 0.2 h; Student’s t-test, t(24) = 2.6, p = 0.015; Fig. 4C2]. Thus, like the first seizure, SE appeared to be similar in latency but longer in mice with reduced adult neurogenesis.
4). Greater power during SE in mice with reduced adult neurogenesis
Spectral analysis of the EEG showed periods of enhanced power in mice with reduced adult neurogenesis (Fig. 5). The examples of spectrograms in Fig. 5 are from the EEG traces shown in Fig. 4, with Fig. 5A-B showing 1–30 Hz and Fig. 5C-D showing 30–100 Hz. To evaluate group differences, power was compared for discrete epochs: 1) the baseline period, 2) time between pretreatment with ethosuximide and pilocarpine injection, 3) time between pilocarpine injection and SE onset, and then 4) every 30 min after the onset of SE. Frequency ranges were selected for comparison as delta, 1–4 Hz; theta, 4–8 Hz; beta, 8–30 Hz; low gamma, 30–80 Hz; high gamma, 80–100 Hz [(Kadam et al., 2017); Fig. 5A-D].
Figure 5. More power during SE in mice with reduced adult neurogenesis relative to intact adult neurogenesis.
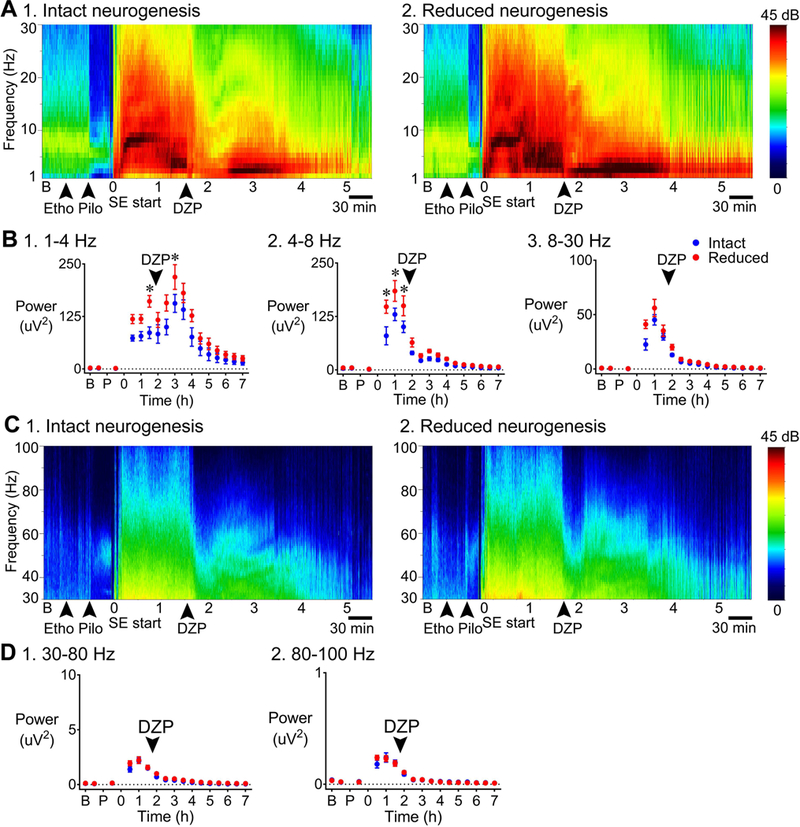
A. Representative examples of a 7 h-long spectrogram are shown for mice with intact (1) and reduced (2) neurogenesis, for the frequency ranges 1–30 Hz. Left: Colors are used to indicate the magnitude of power, ranging from 0 (dark blue) to 45 decibels (dB; dark red). GFAP-TK-: intact neurogenesis (n=7); GFAP-TK+: reduced neurogenesis (n=7).
B. Power was calculated for 30 min-long epochs: a) the baseline period, b) time between pretreatment with ethosuximide and pilocarpine injection, c) the time between pilocarpine injection and SE onset, and then d) every 30 min after the onset of SE. 1. 1–4 Hz: Power in the delta band was greater in mice with reduced neurogenesis (p = 0.021) 1.5 h after the onset of SE (Bonferroni’s test, p = 0.004) and 3 h after the onset of SE (p = 0.033) compared to mice with intact neurogenesis. 2. 4–8 Hz: There was significantly greater power at theta frequency in mice with reduced neurogenesis (p = 0.012; Bonferroni’s test, 30 min to 1 h, p < 0.001; 1.5 h, p = 0.001) compared to mice with intact neurogenesis. 3. 8–30 Hz: There was a trend towards greater power during SE in mice with reduced neurogenesis, but the differences were not significant (p = 0.056).
C. Representative examples of a 7 h-long spectrogram are shown for mice with intact (1) and reduced (2) neurogenesis, for the frequency ranges 30–100 Hz.
D. There was no significant effect of genotype for low gamma or high gamma (low gamma: p = 0.187, high gamma: p = 0.844).
For delta, two-way RMANOVA showed that there was a main effect of genotype with greater power for mice with suppressed adult neurogenesis [F(1, 12) = 7.1, p = 0.021; Bonferroni’s test, 1.5 h, p = 0.004; 3 h, p = 0.033; Fig. 5B1]. For theta, genotype was also a significant factor with greater power for mice with suppressed adult neurogenesis [two-way RMANOVA, F(1, 12) = 8.6, p = 0.012; Bonferroni’s test, 30 min, p < 0.001; 1 h, p < 0.001; 1.5 h, p = 0.001; Fig. 5B2). The mean power for beta was increased in the same direction (higher in mice with reduced adult neurogenesis) and was close to significance [two-way RMANOVA, F(1, 12) = 4.4, p = 0.056; Fig. 5B3]. There was no significant effect of genotype for low gamma or high gamma using two-way RMANOVA [low gamma: F(1, 12) = 1.9, p = 0.187, high gamma: F(1, 12) = 0.04, p = 0.844; Fig. 5D1–2].
These results suggest that reduction of adult neurogenesis led to greater power during SE for delta and theta frequencies, especially in the first hours after the onset of SE.
II. Effects of increased adult neurogenesis
A. Subjects
NestinCreERT2+/−Baxf/f (called “NestinCre+”, below) mice were treated with tamoxifen to increase adult neurogenesis (Fig. 6A). There were three types of controls: 1) NestinCreERT2−/−Baxf/f (called “NestinCre-” below) mice treated with tamoxifen, 2) NestinCre+ mice treated with vehicle (corn oil), or 3) NestinCre- mice treated with vehicle. NestinCre+ mice treated with tamoxifen are the only mice that are referred to below as having increased adult neurogenesis.
Figure 6. Less FJ staining in the dorsal hippocampus of mice with increased adult neurogenesis.
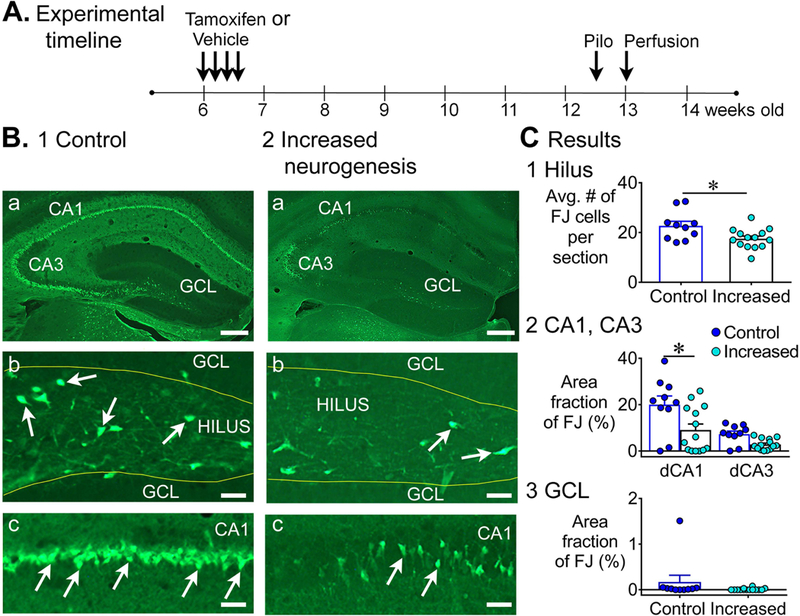
A. The experimental timeline is shown for NestinCreERT2Baxf/f mice. Tamoxifen or vehicle was injected for 4 consecutive days. Six weeks after last tamoxifen injection, mice were injected with pilocarpine to induce SE. Three days after pilocarpine injection, mice were transcardially-perfused. Controls: tamoxifen-treated NestinCre-, n=7 and vehicle-treated NestinCre+, n=3; Increased neurogenesis: tamoxifen-treated NestinCre+, n=14.
B. Representative examples of FJ staining in the dorsal hippocampus in 1) a control (tamoxifen-treated NestinCre-) mouse and 2) a mouse with increased neurogenesis (tamoxifen-treated NestinCre+) are shown. Calibration, 75 μm (a); 50 μm (b, c).
C. 1. There were significantly fewer FJ+ hilar cells in mice with increased neurogenesis than controls (p = 0.032). 2. There was a main effect of increased neurogenesis on FJ staining in dorsal CA1 and CA3 (p = 0.010). Dunn’s test showed significantly less damage in mice with increased neurogenesis in area CA1 (p = 0.046), but not in CA3 (p = 0.224). 3. The area fraction of FJ+ cells in the GCL was not significantly different (p = 0.072).
For the type 1 and 2 of control mice (as defined in the preceding paragraph) that were unimplanted, we found no differences in the numbers of FJ+ cells in the hilus or FJ area fraction in area CA1 and CA3 (Supporting Information Fig. S6A, B). For the type 1 and 3 of control mice that were used for EEG recordings, the latency to SE and the duration of SE were not different (Supporting Information Fig. S6C). For these reasons, the results from controls were pooled. Specific numbers of each type of control are listed in Supporting Information Table S2.
B. FJ staining was decreased in mice with increased adult neurogenesis
1). FJ in the dorsal hippocampus
The experimental timeline used to study SE-induced neurodegeneration with FJ staining in NestinCre+ and NestinCre- mice is shown in Fig. 6A. Representative examples of FJ staining in the dorsal hippocampus in controls and mice with increased adult neurogenesis shows less FJ staining in mice with increased adult neurogenesis (Fig. 6B). Quantification showed fewer FJ+ cells in the dorsal hilus in mice with increased adult neurogenesis relative to controls (control: 22.7 ± 1.8; increased: 17.5 ± 1.1; Mann-Whitney U test, U = 33.5, p = 0.032; Fig. 6C1). The area fraction of FJ staining in area CA1 and CA3 showed a main effect of increased adult neurogenesis (Kruskal-Wallis test; H = 11.3, p = 0.010). Dunn’s test showed significantly less damage in dorsal CA1 (control: 20.0 ± 3.7%; increased: 9.2 ± 2.6%; p = 0.046; Fig. 6C2), but not in CA3 (control: 7.3 ± 1.3%; increased: 2.8 ± 0.6%; p = 0.224; Fig. 6C2).
In the GCL, there was little FJ staining in controls, making it unlikely we would see protection in the mice with increased neurogenesis, and in fact there was little evidence for protection in mice with increased adult neurogenesis. Thus, 1/10 control mice had clusters of FJ+ cells and none of the 14 mice with increased adult neurogenesis exhibited this clustering of FJ+ cells (Fisher’s exact test, p = 0.440; Fig. 6C3). In addition, the area fraction of FJ+ staining in the GCL was not significantly different (Mann-Whitney U test, U = 44, p = 0.072), and the incidence of mice with scattered FJ+ cells in the GCL was not significantly different (control: 6/9; increased: 3/14; Fisher exact test, p = 0.077).
Thus, increased adult neurogenesis showed, in general, opposing effects to suppressed adult neurogenesis because mice with increased adult neurogenesis exhibited less FJ staining than their controls and mice with reduced adult neurogenesis showed more FJ staining than their controls.
2). FJ in the ventral hippocampus
Representative examples of FJ staining in the ventral hippocampus in controls and mice with increased adult neurogenesis showed a similar protective effect of increased adult neurogenesis as the dorsal hippocampus (Fig. 7A). Quantification of FJ staining showed that a similar number of FJ+ hilar neurons (control: 30.4 ± 1.9; increased: 34.1 ± 3.7; Mann-Whitney U test, U = 59.5, p = 0.841; Fig. 7B1). For area CA1 and CA3, a Kruskal-Wallis test showed a significant effect of genotype where mice with increased adult neurogenesis had less FJ staining (p = 0.04) in ventral CA3 (control: 7.9 ± 1.8%; increased: 3.1 ± 0.9%; Dunn’s test, p = 0.032; Fig. 7B2), but not in CA1 (control: 8.1 ± 2.0%; increased: 4.3 ± 1.0%; Dunn’s test, p = 0.353; Fig. 7B2).
Figure 7. Less FJ staining in the ventral hippocampus of mice with increased adult neurogenesis.
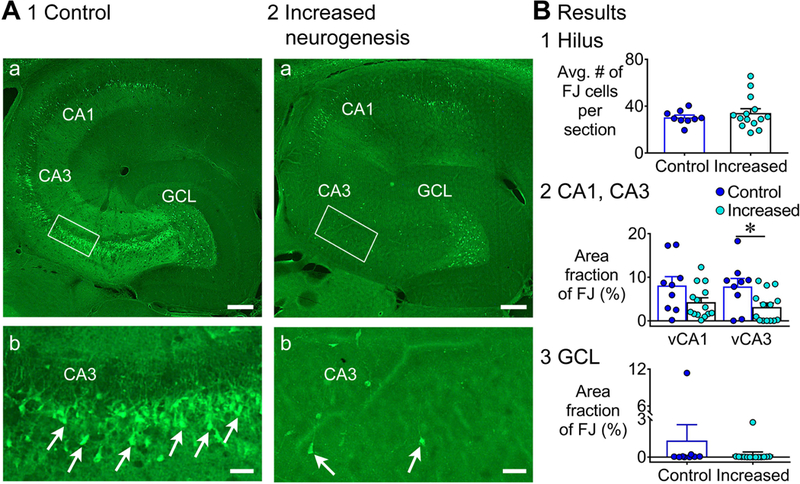
A. Representative examples of FJ staining in the ventral hippocampus are shown for 1) a control (tamoxifen-treated NestinCre-) mouse and 2) a mouse with increased neurogenesis (tamoxifen-treated NestinCre+). Calibration, 75 μm (a); 50 μm (b). Controls: tamoxifen-treated NestinCre-, (n=6) and vehicle-treated NestinCre+ (n=3); Increased neurogenesis: (tamoxifen-treated NestinCre+, n=14).
B. 1. There was no effect of increased neurogenesis on the numbers of FJ+ hilar cells (p = 0.841). 2. There was a main effect of increased neurogenesis on FJ staining in ventral CA1 and CA3 (p = 0.040). Dunn’s test showed significantly less damage in mice with increased neurogenesis in area CA3 (p = 0.032), but not in CA1 (p = 0.353). 3. There was no effect of increased neurogenesis on the area fraction of FJ staining in the GCL (p = 0.183).
In the GCL, FJ+ clusters were rare (control: 2/9 mice; increased: 1/14 mice; Fisher’s exact test, p = 0.537; Fig. 7B3). The area fraction of FJ+ cells was not significantly different (Mann-Whitney U test, U = 42, p = 0.183). The incidence of mice with scattered FJ+ cells was not different (control: 5/7; increased: 5/13; Fisher exact test, p = 0.350).
These data suggest that a protective effect of increased adult neurogenesis occurred in both dorsal and ventral hippocampus but the subfields showing the protection were distinct, with hilus and CA1 in dorsal but CA3 in ventral hippocampus.
B. Differences in SE in mice with and without increased adult neurogenesis
1). Incidence, mortality, and sex differences
To evaluate FJ staining, a cohort of NestinCreERT2Baxf/f mice did not have EEG electrodes, like GFAP-TK mice. In the unimplanted mice, there was a similar incidence of SE (controls: 11/18, 61.1%; increased adult neurogenesis: 14/19, 73.7%, Fisher’s exact test, p = 0.495). There also was no difference in mortality (control: 1/18, 5.2%; increased: 0/19, 0%; Fisher’s exact test, p > 0.999).
Like GFAP-TK mice, seizures during SE were analyzed in another cohort that was implanted with electrodes. These mice were implanted 4 weeks after tamoxifen injections and therefore had the same time until pilocarpine injection relative to tamoxifen injections as unimplanted mice (6 weeks after tamoxifen injections). Like unimplanted mice, there were no differences in the incidence of SE of implanted mice (control: 9/15, 60.0%; increased: 8/9, 88.9%; Fisher’s exact test, p = 0.191). There was no mortality.
Next, we compared the incidence of SE in all mice that were implanted and unimplanted by two-way ANOVA with electrode implantation and genotype as main factors. Both factors had no significant effect on the incidence of SE [F(1, 1) = 0.7, p = 0.546] or mortality [F(1, 1) = 1, p = 0.500]. There was no effect of electrode implantation on the latency to the first convulsive seizure [Fig. 8, Supporting Information Fig. S7; F(1, 37) = 0.8, p = 0.380] or the total number of convulsive seizures [F(1, 37) = 0.6, p = 0.440]. In summary, we did not detect an effect of electrode implantation.
Figure 8. EEG recording of SE in controls and mice with increased adult neurogenesis.
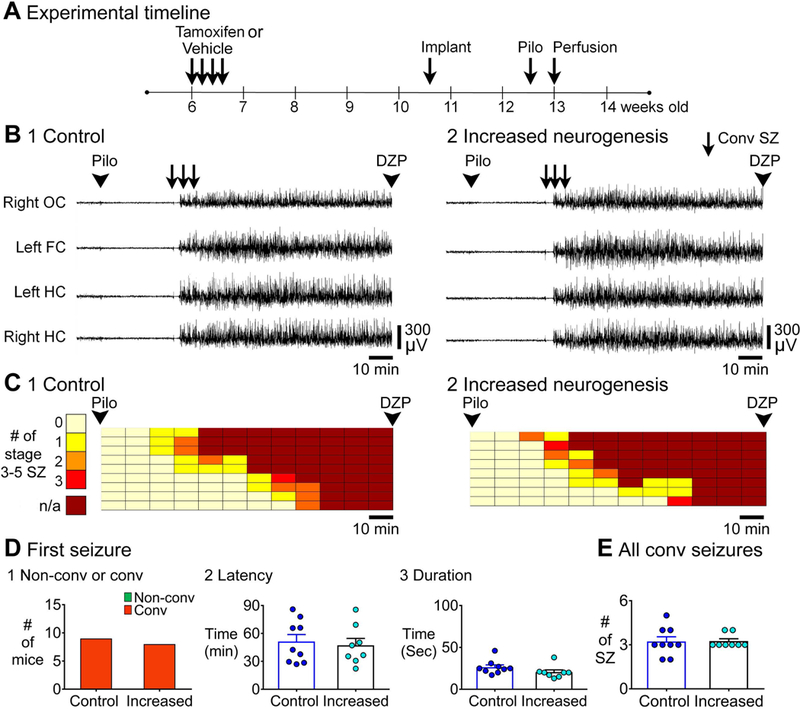
A. The experimental timeline to administer tamoxifen or vehicle is shown. Approximately 3–4 weeks after last tamoxifen or vehicle injection, mice were implanted. Two-three weeks after surgery, mice were injected with pilocarpine to induce SE. Controls: tamoxifen-treated NestinCre- (n=6) and vehicle-treated NestinCre- (n=3); Increased neurogenesis: tamoxifen-treated NestinCre+ (n=8).
B. Representative examples of 2 h-long EEG records after pilocarpine injection of 1) a control (tamoxifen-treated NestinCre-) mouse and 2) a mouse with increased neurogenesis (tamoxifen-treated NestinCre+) are shown.
C. The 2 h between pilocarpine and diazepam injection was divided into 10 min-long bins and each row is for a different mouse and arranged in order to the latency of first convulsive seizure (as shown in Fig. 3).
D. 1. The first SZ before SE was always convulsive so there were no genotypic differences. 2. The latencies to the onset of the first SZ were similar (p = 0.709). 3. Increased neurogenesis did not affect the duration of first seizure (p = 0.093).
E. Increased neurogenesis did not influence the total number of convulsive SZs (p = 0.887).
Unlike female GFAP-TK mice, female NestinCreERT2Baxf/f mice developed SE and survived. Thus, using type 1 and 2 control mice (described above), 8/13 (61.5%) female mice developed SE and 6 of the 8 (75.0%) with SE survived. In contrast, female mice with increased adult neurogenesis rarely had SE (2/13 mice, 15.3%), and the 2 mice with SE did not die, suggesting protection from SE. The incidence of SE in female controls and female mice with increased adult neurogenesis was significantly different (Fisher’s exact test, p = 0.041; Supporting Information Fig. S8A, B). Thus, there was a sex difference, with increased adult neurogenesis leading to a reduced incidence of SE in female NestinCreERT2Baxf/f mice but not in males. Moreover, mice with increased adult neurogenesis were protected from SE.
To clarify more about the female resistance to SE, the numbers of convulsive seizures were plotted in 10 min-long bins (Supporting Information Fig. S8A). These analyses showed that there were 4/13 mice with increased neurogenesis that had convulsive seizures, but they never had SE. In contrast, when control mice had convulsive seizures they always had SE afterwards (Fisher’s exact test, p = 0.015; Supporting Information Fig. S8). Thus, the females with increased neurogenesis seemed to resist the transition from convulsive seizures to SE. In contrast, the onset of seizures seemed similar because there were no differences in the latency to onset of the first convulsive seizure [control: 46.8 ± 2.7 min; increased: 56 ± 6.1 min; Student’s t-test, t(12) = 1.5, p = 0.160; Supporting Information Fig. S8C].
2). Seizures during SE
The experimental timeline for mice that were examined using EEG is shown in Fig. 8A. Examples of the EEG are presented in Fig. 8B. In Fig. 8C-D the EEG is divided into 10 min-long bins and numbers of convulsive seizures are plotted and quantified.. The first seizure was convulsive in both genotypes (Fig. 8D1). There was no difference in the latency to the onset of the first seizure between controls and mice with increased adult neurogenesis [control: 51.2 ± 7.6 min; increased: 47.1 ± 7.5 min; Student’s t-test, t(15) = 0.4, p = 0.709; Fig. 8D2]. There also was no difference in the duration of the first seizure [control: 26.3 ± 2.8 sec; increased: 20.5 ± 2.7 sec; Student’s t-test, t(15) = 1.8, p = 0.093; Fig. 8D3]. In both genotypes, all seizures that were detected by EEG during the time between pilocarpine and diazepam injection were convulsive. The total number of these seizures were not affected by increased adult neurogenesis (Mann-Whitney U test, U = 34, p = 0.887; Fig. 8E).
Thus, there appeared to be similar initial seizures and total numbers of seizures in mice with and without increased adult neurogenesis.
3). Shorter SE duration in mice with increased adult neurogenesis
Examples of the compressed EEG, to show the 10 h after pilocarpine injection, are presented in Fig. 9A. The latency to the onset of SE was not different between groups [control: 54.7 ± 7.7 min; increased: 50.3 ± 7.3 min; Student’s t-test, t(15) = 0.4, p = 0.690; Fig. 9B1]. However, the duration of SE was reduced by more than 1 h in mice with increased adult neurogenesis [control: 5.7 ± 0.4 h; increased 4.4 ± 0.4 h; Student’s t-test, t(15) = 2.3, p = 0.032; Fig. 9B2].
Figure 9. Shorter duration of SE in mice with increased adult neurogenesis compared to control.
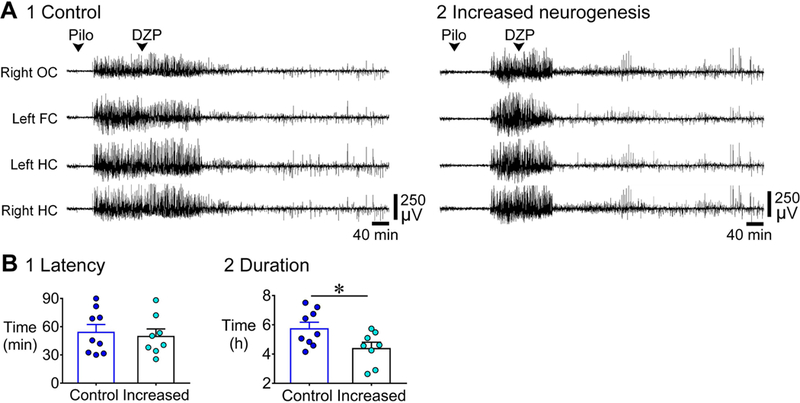
A. Representative examples of a 10 h-long EEG are shown, beginning with pilocarpine injection for 1) a control (tamoxifen-treated NestinCre-) mouse and 2) a mouse with increased neurogenesis (tamoxifen-treated NestinCre+). Controls: tamoxifen-treated NestinCre- (n=6) and vehicle-treated NestinCre- (n=3); Increased neurogenesis: tamoxifen-treated NestinCre+ (n=8).
B. 1. The latency to the onset of SE was similar (p = 0.690). 2. The duration of SE was shorter (by ~1 h) in mice with increased neurogenesis compared to controls (p = 0.032).
Thus, mice with increased adult neurogenesis showed an opposing effect on SE duration relative to mice with suppressed adult neurogenesis.
4). Reduced power during SE in mice with increased adult neurogenesis
Power was calculated from the onset of SE to 7 h later, analogous to GFAP-TK mice (Fig. 10A,C). Power in the delta band was reduced in mice with increased adult neurogenesis at 2.5 and 3 h [two-way RMANOVA, F(1, 12) = 8.4, p = 0.013; Bonferroni’s test, 2.5 h, p < 0.001; 3 h, p < 0.001; Fig. 10B1]. Theta also showed significantly less power in mice with increased adult neurogenesis at these times, as well as for 30 min and 1 h [two-way RMANOVA, F(1, 12) = 9.3, p = 0.010; Bonferroni’s test, 30 min, p = 0.012; 1 h, p = 0.002; 2.5 h, p = 0.003; 3 h, p < 0.001; Fig. 10B2]. Beta power was significantly reduced in mice with increased adult neurogenesis [two-way RMANOVA, F(1, 12) = 6.5, p = 0.025; Bonferroni’s test, 30 min, p = 0.017; 1 h, p < 0.001; 1.5 h, p = 0.007; Fig. 10B3]. Low gamma power was reduced in mice with increased adult neurogenesis [two-way RMANOVA, F(1, 12) = 16.7, p = 0.001; Bonferroni’s test, 30 min to 1.5 h, p < 0.001; 2 h, p = 0.029] and high gamma showed less power also [two-way RMANOVA, F(1, 12) = 19.4, p < 0.001; Bonferroni’s test, 30 min to 1.5 h, p < 0.001; Fig. 10D].
Figure 10. Less power during SE in mice with increased adult neurogenesis compared to control.
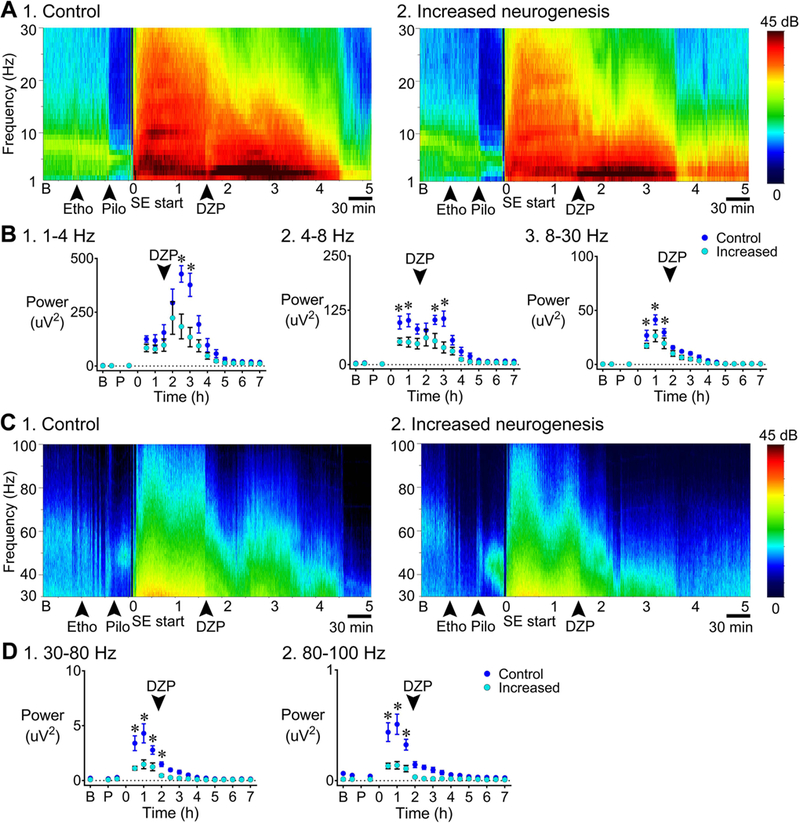
A. Representative examples of a 7 h-long spectrogram of 1) a control (tamoxifen-treated NestinCre-) mouse and 2) a mouse with increased neurogenesis (tamoxifen-treated NestinCre+), for the frequency range 1–30 Hz are shown. Left: Colors are used to indicate the magnitude of power, ranging from 0 (dark blue) to 45 decibels (dB; dark red). Controls: tamoxifen-treated NestinCre- (n=5) and vehicle-treated NestinCre- (n=2); Increased neurogenesis: tamoxifen-treated NestinCre+ (n=8).
B. Power was calculated for different time points as described in Fig. 5. 1. 1–4 Hz: Power in the delta band was decreased in mice with increased neurogenesis (p = 0.013) 2.5 h after the onset of SE (Bonferroni’s test, p < 0.001) and 3 h after the onset of SE (p < 0.001) relative to control. 2. 4–8 Hz: There was significantly less power at theta frequency in mice with increased neurogenesis (p = 0.010; Bonferroni’s test, 30 min, p = 0.012; 1 h, p = 0.002; 2.5 h, p = 0.003; 3 h, p < 0.001) than control. 3. 8–30 Hz: There was significantly less beta power in mice with increased neurogenesis (p = 0.025; Bonferroni’s test, 30 min, p = 0.017; 1 h, p < 0.001; 1.5 h, p = 0.007) than controls.
C. Representative examples of a 7 h-long spectrogram of 1) a control and 2) a mouse with increased neurogenesis, for the frequency range 30–100 Hz are shown.
D. There was a main effect of increased neurogenesis on 1) low gamma power (p = 0.001; Bonferroni’s test, 30 min to 1.5 h, p < 0.001; 2 h, p = 0.029) and 2) high gamma power (p < 0.001; Bonferroni’s test, 30 min to 1.5 h, p < 0.001).
These results suggested opposing effects of increased adult neurogenesis and suppressed adult neurogenesis on power, and the effects on the power were broader in scope in mice with increased adult neurogenesis because there was reduced power across all frequency bands.
III. Confirmation of suppressed and increased adult neurogenesis
A. Suppression of adult neurogenesis
GFAP-TK+ and TK- mice were perfusion-fixed at ages when pilocarpine would have been injected (Fig. 11A). For each group, 2 mice were perfused at 1 week and 2 mice at 3 weeks following cessation of chow containing VGCV (n=4/group). The 1-week timepoint was used because the mice that had FJ staining received pilocarpine 1 week after cessation of VGCV-treated chow; the 3-week timepoint was for mice that had EEG and had an additional 2 week-long recovery period following electrode implantation (Fig. 1A and 3A).
Figure 11. Confirmation of suppression and enhancement of adult neurogenesis.
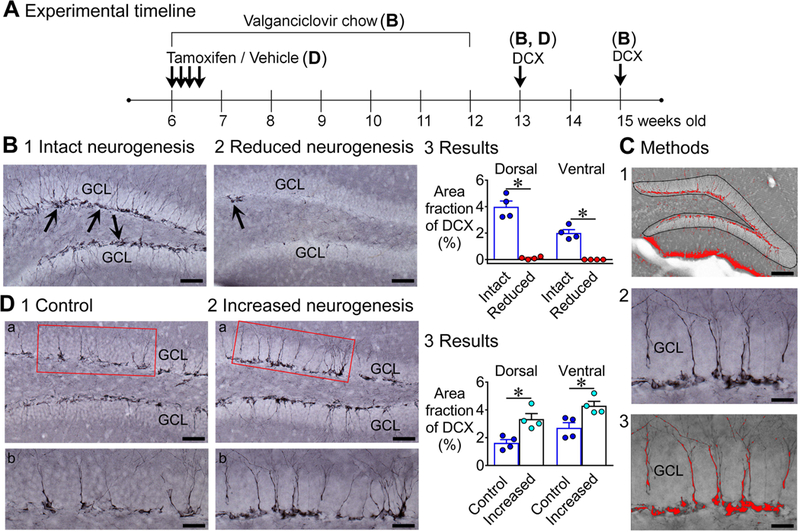
A. The experimental timeline for suppression and enhancement of neurogenesis. Details are in the text.
B. Doublecortin (DCX) immunoreactivity is shown in mice with intact (1) and reduced (2) neurogenesis. Calibration, 30 μm (1, 2). 3. DCX immunoreactivity in mice with reduced neurogenesis was significantly decreased relative to mice with intact neurogenesis in both dorsal and ventral hippocampus (p < 0.001; Dunn’s test, p = 0.021 for dorsal; p = 0. 047 for ventral; n = 4/group).
C. 1. The ROI used to quantify DCX immunoreactivity is shown. 2, 3. DCX immunoreactivity before (2) and after thresholding (3). The area above threshold is red. Calibration, 50 μm (1); 15 μm (2, 3).
D. 1, 2. DCX immunoreactivity in control (1a-b) and mice with increased (2a-b) neurogenesis, shows a significantly greater area fraction for DCX immunoreactivity in mice with increased neurogenesis. Calibration, 30 μm (a); 15 μm (b). 3. Mice with increased neurogenesis had a significantly greater area fraction of DCX immunoreactivity compared to control mice and this occurred in both the dorsal and ventral hippocampus (p < 0.001; Bonferroni’s test, p = 0.007 for dorsal; p = 0.012 for ventral; n = 4/group).
DCX immunoreactivity confirmed robust adult neurogenesis throughout the hippocampus in GFAP-TK- mice (Fig. 11B1) but almost no immunoreactivity in GFAP-TK+ mice (Fig. 11B2–3,C; Kruskal-Wallis test, H = 13.9, p < 0.001; Dunn’s test, p = 0.021 for dorsal, p = 0.047 for ventral). When there were residual cells in GFAP-TK+ mice, they were in dorsal hippocampus (3.2 ± 1.2 residual cells per section) whereas there was no detectable DCX-immunoreactive cell in ventral hippocampus.
B. Enhancement of adult neurogenesis
Mice were perfusion-fixed at ages when pilocarpine would have been injected, that is, 6 weeks after the last injection of tamoxifen or vehicle (n = 4/group; Fig. 11A). DCX immunoreactivity confirmed a significant increase of immunoreactivity in both dorsal and ventral hippocampus by one-way ANOVA [F(3, 12) = 11.0, p < 0.001; Bonferroni’s test, p = 0.007 for dorsal; p = 0.012 for ventral; Fig. 11D1–3]. The mean area fraction of DCX immunoreactivity was increased by approximately 2-fold in dorsal hippocampus (control, 1.6 ± 0.2%; increased, 3.3 ± 0.4%) and 1.6 times in ventral hippocampus (control, 2.7 ± 0.4%; increased, 4.3 ± 0.3%). These increases in DCX immunoreactivity are similar in magnitude to previous studies that used these mice (Sahay et al., 2011; Anacker et al., 2018).
Note that the DCX immunoreactivity appeared to be greater in control GFAP-TK mice (C57BL6; Fig. 11B1) than control NestinCreERT2Baxf/f mice (C57BL6/Sv129; Fig. 11D1). This strain difference is likely to be due to the differences between C57BL6 mice and Sv129 mice where C57BL6 mice have more adult neurogenesis (Kempermann et al., 1997; Kim et al., 2017).
C. Specificity
We reported previously that the effects of pharmacogenetic deletion of adult-born neurons in the GFAP-TK+ mouse appeared to be specific for adult-born neurons because GFAP-TK+ and TK- mice had similar GFAP-immunoreactivity throughout the hippocampus; in addition, immunoreactivity for Iba1, a marker of microglia, showed no microglial invasion of the DG in GFAP-TK+ mice or shrinkage of the DG (Iyengar et al., 2015). For the NestinCreERT2Baxf/f mice, prior studies in other labs demonstrated specificity (Sahay et al., 2011). We also examined specificity by injecting a virus expressing mCherry into the DG so that infected cells would express mCherry if they expressed Cre recombinase (see Methods). Mice (n=2) were perfused 4 weeks later to provide time for expression. Cells expressing mCherry were only found in the DG and had the morphology of GCs (Supporting Information Fig. S9).
DISCUSSION
I. General summary
This study demonstrates that reduction of adult-born neurons increases the hippocampal neurodegeneration after pilocarpine-induced SE in the adult mouse and increased adult neurogenesis has an opposite effect, reducing hippocampal neurodegeneration. In addition, reduced adult neurogenesis increased the duration of pilocarpine-induced SE and its power, whereas increased adult neurogenesis has the opposite effect.
II. Effects of adult neurogenesis on hippocampal degeneration after SE
Mice with suppressed adult-born neurons had significantly more FJ staining than controls. The areas where this was significant were the hilus and CA1 in dorsal hippocampus and the hilus in ventral hippocampus. Remarkably, there was significantly more ventral GCL degeneration in mice with reduced adult neurogenesis, which is notable because FJ staining is not often observed in the GCL after SE.
Conversely, mice with increased adult neurogenesis showed significantly less FJ staining than controls. In the mice with increased adult neurogenesis, the areas that were significant were dorsal hilus and CA1, like mice with reduced adult neurogenesis. However, ventral CA3 was significantly protected when adult neurogenesis was increased and ventral hilus and the GCL were not protected.
The general opposing effects of mice with reduced neurogenesis vs. mice with increased neurogenesis, but differences in affected subfields, could be due to differences in the degree of adult neurogenesis in background strains of mice that were used (Kempermann et al., 1997; Kim et al., 2017). However, it is not clear how the degree of adult neurogenesis in the background strain would influence subfields differentially. For suppressed adult neurogenesis, the background was C57BL6 and for increased adult neurogenesis it was C57BL6/Sv129. Notably, it has been reported previously that C57BL6 mice have less damage after KA than C57BL6/Sv129 mice (McKhann et al., 2003). This is consistent with the high levels of adult neurogenesis in the C57BL6 mice relative to Sv129 mice (Kempermann et al., 1997; Kim et al., 2017).
The strain effect on neurodegeneration is important because, for example, when an area is extensively damaged in controls, worsening of damage in mice with reduced adult neurogenesis is harder to prove than the situation where there is relatively little damage in controls. Conversely, protection in mice with increased neurogenesis is hard to show if the controls sustain little damage. For example, CA1 of ventral hippocampus of the background strain for the NestinCreERT2Baxf/f mice (NestinCre-) showed little damage after SE, and protection was not shown. Protection in mice with increased adult neurogenesis (NestinCre+) might have been shown if the controls showed more damage.
Although the effects were not exactly the opposite for the loss-of-function and gain-of-function models, the fact that there were opposing effects, in general, is striking. Thus, all of the genotypic differences we detected were consistent with worse SE and SE-induced damage in mice with suppressed neurogenesis and protected SE and SE-induced damage in mice with increased neurogenesis. This argues that the specific aspects of the experimental methods to reduce or enhance adult neurogenesis did not cause the results - although factors such as strain probably influenced the results. Instead, the generally opposing results suggest that the vulnerability and protection were mediated by a common phenomenon, and the data support the hypothesis that the common mechanism is more or less adult neurogenesis. This interpretation is consistent with other studies showing that adult neurogenesis can improve DG-dependent functions when it is selectively increased (Sahay et al., 2011) but have an opposing effect when it is suppressed (Clelland et al., 2009; Drew et al., 2010; Burghardt et al., 2012; Denny et al., 2012).
III. Effects of adult neurogenesis on SE
Mice with reduced adult neurogenesis had a greater response to pilocarpine because there was a longer duration of the first seizure and of SE, and the first seizure was more likely to be convulsive. There was more delta and theta power at specific times during SE. In mice with enhanced adult neurogenesis, the effects were generally the opposite, because the duration of SE was shorter. There were no significant differences in the severity or duration of the first seizure but there was a decrease in power, not only in delta and theta, but also other frequencies. Here the lesser effects of enhanced adult neurogenesis regarding the first seizure and the greater effects on power, relative to suppression of adult neurogenesis, may be related to background strain (as discussed above), as well as other factors that are complex. For example, we found complex differences in the strains in females, because the GFAP-TK- females had pilocarpine-induced SE rarely and there was high mortality, but the NestinCreERT2Baxf/f control females had a higher incidence of SE and there was little mortality.
The greater severity of SE when adult neurogenesis is reduced is consistent with a previous study of GFAP-TK mice in our laboratory showing that there are more convulsive seizures and durations of seizures are longer after KA-induced SE (Iyengar et al., 2015). Interestingly, both studies showed that there is a ‘switch’ in the first seizure from being mainly non-convulsive in mice with intact neurogenesis to convulsive after neurogenesis is suppressed. The similarity suggests that effects are not specific to pilocarpine or KA but result from common effects of reduced adult-born neurons to make seizures worse.
The consistency of a duration effect for KA, pilocarpine, as well as the gain-vs. loss-of-function models suggests that adult-born neurons have their influence mainly by lengthening or curtailing seizures rather than facilitating or blocking the initiation of SE. This influence is consistent with the idea that after KA or pilocarpine, seizures can be initiated in areas outside the DG (Botterill, 2017; Connell et al., 2017). The idea is also consistent with the view that the DG is an inhibitory brake on seizures that already have been generated in cortex and are propagating into the DG (Heinemann et al., 1992; Lothman et al., 1992; Bonislawski et al., 2007; Hsu, 2007; Pathak et al., 2007; Patrylo et al., 2007; Krook-Magnuson et al., 2015; Dengler and Coulter, 2016; Scharfman and Myers, 2016). Indeed, the idea that normally the adult-born GCs support the DG inhibitory “gate” has been suggested before in the context of seizures (Murphy et al., 2011; Iyengar et al., 2015; Scharfman and Bernstein, 2015).
This interpretation is also consistent with what was observed in power in prior studies where power was examined after reducing adult neurogenesis. For example, Lacefield and colleagues (Lacefield et al., 2012) reported that there was an increase in gamma power (25–80 Hz) in the hippocampus in urethane-anesthetized mice after chronic ablation of adult-born neurons by GCV treatment for 4 weeks. Zhuo and colleagues (Zhuo et al., 2016) reported that optogenetic silencing of young adult-born neurons increased power in multiple frequency bands and, interestingly, did so in the hippocampus contralateral to the silencing.
Another study is important to consider in light of our findings. Korn and colleagues (Korn et al., 2016) used mice with partial deletion of adult-born neurons (reduced by approximately 1/3) caused by conditional deletion of the adaptor protein for reelin disabled-1 (DAB-1) in Nestin-expressing precursors. The Nestin-expressing cells that survived were likely to have been abnormal due to the DAB-1 deletion, and indeed mismigration was identified because there were hilar (ectopic) GCs. The authors found reduced threshold and latency to pilocarpine-induced SE which is consistent with prior studies showing that hilar ectopic GCs are associated with increased excitability (Scharfman et al., 2000; Parent and Lowenstein, 2002; Scharfman, 2004; Zhan et al., 2010; Pun et al., 2012; Myers et al., 2013; Cho et al., 2015). The study by Korn and colleagues (Korn et al., 2016) is important because it suggests that complete ablation of adult-born GCs may not be required to influence seizures. That is significant because there are many environmental conditions that reduce adult neurogenesis, but don’t completely do so. In addition, the study further supports the idea that mismigration of adult-born GCs can lead to increased excitability (Scharfman et al., 2000; Parent and Lowenstein, 2002; Scharfman, 2004; Zhan et al., 2010; Pun et al., 2012; Myers et al., 2013; Cho et al., 2015).
IV. Mechanisms
Hippocampal neuron loss after SE is thought to be due to excitotoxic effects of SE (Ingvar et al., 1988; Meldrum, 2002; Pitkanen et al., 2002), and hippocampal degeneration after SE is greater when SE is longer (Siesjo and Wieloch, 1986; Mazzuferi et al., 2012; Brandt et al., 2015). Therefore, it seems likely that the effects of adult-born neurons to make SE shorter or longer led to the effects on neurodegeneration.
Why adult-born neurons inhibit SE is currently unclear. Drew and colleagues (2016) suggested that young adult-born neurons normally support GABAergic inhibition of the mature GC network, and therefore diminish the output of the DG. The work presented here is consistent with the hypothesis that young adult-born neurons increase inhibition of mature GCs because reduced activity of mature GCs would reduce the excitotoxic effects of seizures on the hilar neurons and pyramidal neurons “downstream” of the GCs (Sloviter, 1994; Scharfman, 1999; Scharfman and Bernstein, 2015). In turn, reduced activity in CA3 pyramidal cells would reduce CA1 activity and protect that region.
The mechanism by which adult-born neurons inhibit GCs is consistent with the innervation of DG GABAergic neurons by young adult-born GCs (Toni et al., 2008), and highly excitable young adult-born GCs driving the GABAergic neurons stronger than GCs born in early life.. Other studies are consistent with this hypothesis. For example, (Ikrar et al., 2013) showed that when young adult-born neurons are reduced there is a larger spread of excitatory activity in the DG. On the other hand, Temprana et al. (2015) used optogenetics to activate 4-week-old GCs, showed that they have weak inhibitory effects on mature GCs. That result would seem at odds with the data presented here and in other studies (Drew et al., 2016). One explanation is that the current study and the work of Drew and colleagues (Drew et al., 2016) suppressed adult neurogenesis for 6–7 weeks so neurons that were over 4-weeks-old were suppressed as well as younger cells. Temprana and colleagues (Temprana et al., 2015) used retrovirus so they could influence only 4-week-old cells. Also, it is important to bear in mind that Drew and colleagues (Drew et al., 2016) found the major effects of young adult-born GCs were inhibitory, but excitatory effects were also found. Therefore, young adult-born GCs probably have a range of excitatory and inhibitory effects and removing adult-born neurons that are up to 6- or 7-weeks-old could have more inhibitory effects than selectively manipulating only those at 4 weeks of age. How might this occur? A clue comes from studies of the long-term synaptic plasticity in 4- and 8-week-old male rats, where GC axons (mossy fibers) of 8-week-old male had much less long-term synaptic plasticity than 4-week-old males (Skucas et al., 2013). Therefore at 4-weeks-old, the mossy fiber system may be primed to excite CA3 but then decline somewhat.
One caveat is that there could have been compensatory effects such as alterations in circuitry in response to deletion or augmentation of adult-born GCs. This was suggested by a study showing that the supramammillary input to the DG is greater in another TK model where Nestin was the promoter instead of GFAP (Singer et al., 2011). This effect, as well as others, appeared to favor excitation (Singer et al., 2011). However, there were several differences between the studies of Singer et al. (2011) and ours, in addition to the use of a Nestin-TK mouse rather than a GFAP-TK mouse.
Another consideration is that Bax deletion in adult-born neurons did more than block apoptosis, because functions unrelated to apoptosis have been reported (Jiao and Li, 2011; Adlaf et al., 2017). However, because of the opposing nature of the loss-of-function and gain-of-function models, in general, it seems unlikely to explain the findings.
The presented data appear to conflict with an earlier report (Cho et al., 2015) that treatment of Nestin-TK+ or TK- mice with VGCV for 4 weeks did not change pilocarpine-induced SE. Furthermore, the authors did not find differences in FJ staining in dorsal hippocampus 3 days after SE (Cho et al., 2015). However, there were several technical differences between that study and the one presented here. Some of the technical differences provide a possible explanation, based on studies of Singer et al. (2011). Singer et al. (2011) used Nestin-TK mice and showed compensation after ablation of adult-born neurons. This compensation might not have been the same in Nestin-TK mice and our GFAP-TK mice because GFAP-TK mice are a different transgenic mouse, and because the timing of our methods (to ablate adult neurogenesis, to examine effects, etc.) was different from those of Singer et al. (2011) and Cho et al. (2015).
V. Implications
One implication of the present study is that adult neurogenesis in the DG normally will benefit adult animals by protecting them from excitotoxic effects of SE. Notably, SE does occur in the normal population, so this is potentially significant (Hesdorffer et al., 1998; Sanchez and Rincon, 2016; Lv et al., 2017). In addition, the pattern of damage in the hippocampus following SE also occurs after traumatic brain injury (TBI), early life infection (ELI) and febrile seizures (FS) in both laboratory animals and humans (Baram and Shinnar, 2001; Swartz et al., 2006; England et al., 2012; Frankowski et al., 2018). Since the causes of the neuronal loss in the hilus, CA1 and CA3 are generally associated with seizures in response to the insults or injuries (Siesjo and Wieloch, 1986; Wasterlain et al., 1993; Meldrum, 2002), it is possible that adult neurogenesis would modulate the damage observed in TBI, ELI, and FS. That would be significant because they are considered risk factors for temporal lobe epilepsy, like SE (Sloviter, 1994; Fountain, 2000; Baram and Shinnar, 2001; England et al., 2012; Feng and Chen, 2016; Tubi et al., 2018). Given all these insults occur in humans, the ability of adult-born neurons to protect against hippocampal damage appears to be a valuable asset. However, these insults occur only in a fraction of the population (Hesdorffer et al., 1998; Stafstrom, 2002; England et al., 2012). In addition, adult-born neurons also support normal cognition and behavior as mentioned above. Therefore, an alternative interpretation to the idea that adult-born neurons are valuable because they protect against damage after insults or injury is that adult-born neurons support normal cognition and behavior.
Notably, the proposed protective effects of adult neurogenesis may not be present unless there are normal conditions to support normal migration and integration of new neurons into the preexisting circuitry. The reason, as mentioned above, is that in the days and weeks after SE in rodents, adult-born neurons often mismigrate (becoming ectopic) and integrate abnormally, increasing excitability (Scharfman et al., 2000; Parent and Lowenstein, 2002; Scharfman, 2004; Scharfman and Hen, 2007; Parent and Murphy, 2008; Scharfman and McCloskey, 2009; Zhan et al., 2010; Pun et al., 2012; Myers et al., 2013; Cho et al., 2015). On the other hand, some adult-born neurons migrate to the GCL correctly and do not have increased neuronal activity (Jakubs et al., 2006). These adult-born neurons in the GCL of the epileptic brain appear to have reduced excitatory synaptic inputs and decreased excitability (Jakubs et al., 2006; Jessberger and Parent, 2015). These newborn neurons that migrate into the GCL in the epileptic brain may change their intrinsic properties to compensate for the hyperexcitability (Kempermann, 2006). Taken together, these studies suggest that adult hippocampal neurogenesis is a complex process and the neural activity of adult-born neurons may depend on whether they are born in the normal brain or pathological conditions.
Importantly, the results have implications for the effects of commonly used drugs that influence adult neurogenesis or are influenced by it. For example, adult neurogenesis in mice is essential to improve behavioral outcomes of antidepressants (Santarelli et al., 2003; Pollak et al., 2008). Those findings, and the results of the current study, provide a potential explanation for the observation that some antidepressants that increase adult neurogenesis, like fluoxetine (Santarelli et al., 2003), can reduce seizures in patients with epilepsy (Thome-Souza et al., 2007; Hamid and Kanner, 2013; Ribot et al., 2017). This might occur because as adult neurogenesis increases, inhibition in the DG increases. Other drugs (Taupin, 2008; Apple et al., 2017) that increase neurogenesis may be protective also. Conversely, drugs that reduce adult neurogenesis could increase the risk that brain damage will be worse after insult or injury.
The results also support studies which suggested that electroconvulsive therapy (ECT) which can increase hippocampal neurogenesis (Madsen et al., 2000; Scott et al., 2000; Bouckaert et al., 2014; Olesen et al., 2017) also reduces seizure susceptibility in patients with epilepsy (Lunde et al., 2006; Yang and Wang, 2015). These findings are consistent with studies showing that P7C3, a drug that enhances adult hippocampal neurogenesis (Pieper et al., 2014; Latchney et al., 2015) is neuroprotective (Tesla et al., 2012; Blaya et al., 2014; Walker et al., 2015) and ameliorates cognitive impairments in a mouse model of Alzheimer’s disease (Choi et al., 2018).
VI. Conclusions
We conclude that adult neurogenesis modulates pilocarpine-induced SE and SE-induced neuronal damage. It is not likely in our view that our data only pertain to our specific methods, since effects were the opposite in the loss-of-function and gain-of-function models. It also seems unlikely in our view that the effects were due specifically to pilocarpine because prior studies with KA are consistent with those shown here. It seems likely that the mechanisms are related to up- or down-regulation of DG inhibition, and the DG inhibitory gate. The implications are important: regulation of adult neurogenesis modifies severe seizures and the ability of severe seizures to induce neuronal damage in the adult hippocampus. As such, they suggest new potential ways to protect the brain from severe seizures and seizure-induced neuronal damage.
Supplementary Material
SIGNIFICANCE STATEMENT.
This study demonstrates that reduction of adult-born neurons increases the severity of severe seizures and hippocampal damage after those seizures. Conversely, augmentation of adult-born neurons protects the hippocampus from severe seizures and associated neuronal damage. The implication is that normal adult-born neurons in the DG protect the hippocampus. This is significant because the hippocampus is vulnerable to many insults and injuries and until now the mechanisms underlying this vulnerability have not been related to adult neurogenesis.
Acknowledgements:
This study was supported by NIH R01 NS-081201, R01 AG-055328, the Savoy Foundation, Natural Sciences and Engineering Research Council of Canada (NSERC) and the New York State Department of Health. We thank Drs. Amar Sahay and Rene Hen for the NestinCreERT2Baxf/f mice; and Drs. Sloka Iyengar and Korey Kam for assistance with techniques.
REFERENCES
- Adlaf EW, Vaden RJ, Niver AJ, Manuel AF, Onyilo VC, Araujo MT, . . . Overstreet-Wadiche L (2017). Adult-born neurons modify excitatory synaptic transmission to existing neurons. Elife, 6, e19886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH (2014). Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev, 94(4), 991–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J, Das GD (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol, 124(3), 319–335. [DOI] [PubMed] [Google Scholar]
- Altman J (2011). The discovery of adult mammalian neurogenesis In Seki T, Sawamoto K, Parent JM, Alvarez-Buylla A (Eds.), Neurogenesis in the Adult Brain I: Neurobiology (Vol. I, pp. 3–46): Springer. [Google Scholar]
- Amaral DG (1978). A Golgi study of cell types in the hilar region of the hippocampus in the rat. J Comp Neurol, 182, 851–914. [DOI] [PubMed] [Google Scholar]
- Ambrogini P, Lattanzi D, Ciuffoli S, Agostini D, Bertini L, Stocchi V, . . . Cuppini R (2004). Morpho-functional characterization of neuronal cells at different stages of maturation in granule cell layer of adult rat dentate gyrus. Brain Res, 1017(1–2), 21–31. [DOI] [PubMed] [Google Scholar]
- Anacker C, Hen R (2017). Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat Rev Neurosci, 18(6), 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anacker C, Luna VM, Stevens GS, Millette A, Shores R, Jimenez JC, . . . Hen R (2018). Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature, 559(7712), 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apple DM, Fonseca RS, Kokovay E (2017). The role of adult neurogenesis in psychiatric and cognitive disorders. Brain Res, 1655, 270–276. [DOI] [PubMed] [Google Scholar]
- Babb TL, Pretorius JK (1993). Pathologic substrates of epilepsy In Wyllie E (Ed.), The Treatment of Epilepsy: Principles and Practice (2nd ed., pp. 55–70). Baltimore, MD: Williams & Wilkins. [Google Scholar]
- Baram TZ, Shinnar S (2001). Febrile Seizures. San Diego, CA: Academic Press. [Google Scholar]
- Beining M, Jungenitz T, Radic T, Deller T, Cuntz H, Jedlicka P, Schwarzacher SW (2017). Adult-born dentate granule cells show a critical period of dendritic reorganization and are distinct from developmentally born cells. Brain Struct Funct, 222(3), 1427–1446. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y (2012). Kainate and temporal lobe epilepsies: 3 decades of progress In Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies (4th ed., pp. 639–671). Bethesda, MD: Oxford University Press. [PubMed] [Google Scholar]
- Bermudez-Hernandez K, Lu YL, Moretto J, Jain S, LaFrancois JJ, Duffy AM, Scharfman HE (2017). Hilar granule cells of the mouse dentate gyrus: effects of age, septotemporal location, strain, and selective deletion of the proapoptotic gene Bax. Brain Struct Funct, 222(7), 3147–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD (2014). Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J Neurotrauma, 31(5), 476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonislawski DP, Schwarzbach EP, Cohen AS (2007). Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis, 25(1), 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R (2003). Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol, 182(1), 21–34. [DOI] [PubMed] [Google Scholar]
- Botterill JJ, Brymer KJ, Caruncho HJ, Kalynchuk LE (2015). Aberrant hippocampal neurogenesis after limbic kindling: Relationship to BDNF and hippocampal-dependent memory. Epilepsy Behav, 47, 83–92. [DOI] [PubMed] [Google Scholar]
- Botterill JJ, LaFrancois JJ, Scharfman HE (2017). Relative timing of the initial neocortical and hippocampal seizure activity preceding kainic acid-induced status epilepticus in adult rats. American Epilepsy Society annual meeting https://www.aesnet.org/meetings_events/annual_meeting_abstracts/view/345796 [Google Scholar]
- Bouckaert F, Sienaert P, Obbels J, Dols A, Vandenbulcke M, Stek M, Bolwig T (2014). ECT: its brain enabling effects: a review of electroconvulsive therapy-induced structural brain plasticity. J ECT, 30(2), 143–51. [DOI] [PubMed] [Google Scholar]
- Brandt C, Tollner K, Klee R, Broer S, Loscher W (2015). Effective termination of status epilepticus by rational polypharmacy in the lithium-pilocarpine model in rats: Window of opportunity to prevent epilepsy and prediction of epilepsy by biomarkers. Neurobiol Dis, 75, 78–90. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG (2003). Transient expression of doublecortin during adult neurogenesis. J Comp Neurol, 467(1), 1–10. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Haney MM (2012). Factors affecting outcomes of pilocarpine treatment in a mouse model of temporal lobe epilepsy. Epilepsy Res, 102(3), 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghardt NS, Park EH, Hen R, Fenton AA (2012). Adult-born hippocampal neurons promote cognitive flexibility in mice. Hippocampus, 22(9), 1795–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, . . . Sofroniew MV (1998). Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell, 93(2), 189–201. [DOI] [PubMed] [Google Scholar]
- Cain DP, Corcoran ME (1984). Intracerebral beta-endorphin, met-enkephalin and morphine: kindling of seizures and handling-induced potentiation of epileptiform effects. Life Sci, 34(25), 2535–2542. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Woolley CS, McEwen BS, Gould E (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience, 56(2), 337–344. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD (2001). Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol, 435(4), 406–417. [DOI] [PubMed] [Google Scholar]
- Cavalheiro EA, Santos NF, Priel MR (1996). The pilocarpine model of epilepsy in mice. Epilepsia, 37(10), 1015–1019. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Sutula TP (1990). Progressive neuronal loss induced by kindling: a possible mechanism for mossy fiber synaptic reorganization and hippocampal sclerosis. Brain Res, 527(1), 1–6. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Das I, Sutula TP (1994). Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci, 14(5 Pt 2), 3106–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Wasterlain CG (2006). Status epilepticus: pathophysiology and management in adults. Lancet Neurol, 5(3), 246–256. [DOI] [PubMed] [Google Scholar]
- Cho KO, Lybrand ZR, Ito N, Brulet R, Tafacory F, Zhang L, . . . Hsieh J (2015). Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun, 6(6606). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Lin SL, Lee B, Kurup P, Cho HY, Naegele JR, . . . Obrietan K (2007). Status epilepticus-induced somatostatinergic hilar interneuron degeneration is regulated by striatal enriched protein tyrosine phosphatase. J Neurosci, 27(11), 2999–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD Jr., Fragniere A, Tyers P, . . . Bussey TJ (2009). A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science, 325(5937), 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, Bayat A, Joshi S, Koubeissi MZ (2017). Acute and spontaneous seizure onset zones in the intraperitoneal kainic acid model. Epilepsy Behav, 68, 66–70. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, . . . Aigner L (2005). Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci, 21(1), 1–14. [DOI] [PubMed] [Google Scholar]
- Covolan L, Mello LE (2000). Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res, 39(2), 133–152. [DOI] [PubMed] [Google Scholar]
- Crain BJ, Westerkam WD, Harrison AH, Nadler JV (1988). Selective neuronal death after transient forebrain ischemia in the Mongolian gerbil: a silver impregnation study. Neuroscience, 27(2), 387–402. [DOI] [PubMed] [Google Scholar]
- Culig L, Surget A, Bourdey M, Khemissi W, Le Guisquet AM, Vogel E, . . . Belzung C (2017). Increasing adult hippocampal neurogenesis in mice after exposure to unpredictable chronic mild stress may counteract some of the effects of stress. Neuropharmacology, 126, 179–189. [DOI] [PubMed] [Google Scholar]
- D’Amour J, Magagna-Poveda A, Moretto J, Friedman D, LaFrancois JJ, Pearce P, . . . Scharfman HE (2015). Interictal spike frequency varies with ovarian cycle stage in a rat model of epilepsy. Exp Neurol, 269, 102–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler CG, Coulter DA (2016). Normal and epilepsy-associated pathologic function of the dentate gyrus. Prog Brain Res, 226, 155–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny CA, Burghardt NS, Schachter DM, Hen R, Drew MR (2012). 4- to 6-week-old adult-born hippocampal neurons influence novelty-evoked exploration and contextual fear conditioning. Hippocampus, 22(5), 1188–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieni CV, Nietz AK, Panichi R, Wadiche JI, Overstreet-Wadiche L (2013). Distinct determinants of sparse activation during granule cell maturation. J Neurosci, 33(49), 19131–19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Varvel NH, Dudek FE (2014). When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol, 813, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinocourt C, Petanjek Z, Freund TF, Ben-Ari Y, Esclapez M (2003). Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J Comp Neurol, 459(4), 407–425. [DOI] [PubMed] [Google Scholar]
- do Nascimento AL, Dos Santos NF, Campos Pelagio F, Aparecida Teixeira S, de Moraes Ferrari EA, Langone F (2012). Neuronal degeneration and gliosis time-course in the mouse hippocampal formation after pilocarpine-induced status epilepticus. Brain Res, 1470, 98–110. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Fusi S, Hen R (2013). Adult neurogenesis in the mammalian hippocampus: why the dentate gyrus? Learn Mem, 20(12), 710–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew LJ, Kheirbek MA, Luna VM, Denny CA, Cloidt MA, Wu MV, . . . Hen R (2016). Activation of local inhibitory circuits in the dentate gyrus by adult-born neurons. Hippocampus, 26(6), 763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew MR, Denny CA, Hen R (2010). Arrest of adult hippocampal neurogenesis in mice impairs single- but not multiple-trial contextual fear conditioning. Behav Neurosci, 124(4), 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Staley KJ (2012). The time course and circuit mechanisms of acquired epileptogenesis In Noebels J, Avoli M, Rogawski M, Olsen R, Delgado-Escueta A (Eds.), Jasper’s Basic Mechanisms of the Epilepsies (4th ed., pp. 595–610). Bethesda, MD: Oxford University Press. [PubMed] [Google Scholar]
- Duffy AM, Schaner MJ, Wu SH, Staniszewski A, Kumar A, Arevalo JC, . . . Scharfman HE (2011). A selective role for ARMS/Kidins220 scaffold protein in spatial memory and trophic support of entorhinal and frontal cortical neurons. Exp Neurol, 229(2), 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand JJ, Pouliot W, Scheerlinck P, Dudek FE (2011). Lithium pilocarpine-induced status epilepticus in postnatal day 20 rats results in greater neuronal injury in ventral versus dorsal hippocampus. Neuroscience, 192, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England MJ, Liverman CT, Schultz AM, Strawbridge LM (2012). Epilepsy Across the Spectrum: Promoting Health and Understanding. Washington, DC: National Academies Press. [PubMed] [Google Scholar]
- Esposito MS, Piatti VC, Laplagne DA, Morgenstern NA, Ferrari CC, Pitossi FJ, Schinder AF (2005). Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J Neurosci, 25(44), 10074–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Chen Z (2016). Generation of Febrile Seizures and Subsequent Epileptogenesis. Neurosci Bull, 32(5), 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Steffens S, Frank S, Rainov NG, Schulze-Osthoff K, Kramm CM (2005). Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/ 5-fluorocytosine suicide gene therapy-induced cell death in glioma cells. Oncogene, 24(7), 1231–1243. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Scharfman HE, deCurtis M (2014). How can we identify ictal and interictal abnormal activity? Adv Exp Med Biol, 813, 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountain NB (2000). Status epilepticus: risk factors and complications. Epilepsia, 41 Suppl 2, S23–30. [DOI] [PubMed] [Google Scholar]
- Frankowski JC, Kim YJ, Hunt RF (2018). Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiol Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG (1996). The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Res, 725(1), 11–22. [DOI] [PubMed] [Google Scholar]
- Gage F, Kempermann G, Song H (2008). Adult Neurogenesis. New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV (2004). GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci, 7(11), 1233–1241. [DOI] [PubMed] [Google Scholar]
- Ge S, Yang CH, Hsu KS, Ming GL, Song H (2007). A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron, 54(4), 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Sailor KA, Ming GL, Song H (2008). Synaptic integration and plasticity of new neurons in the adult hippocampus. J Physiol, 586(16), 3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999). Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron, 23(2), 257–271. [DOI] [PubMed] [Google Scholar]
- Glover LR, Schoenfeld TJ, Karlsson RM, Bannerman DM, Cameron HA (2017). Ongoing neurogenesis in the adult dentate gyrus mediates behavioral responses to ambiguous threat cues. PLoS Biol, 15(4), e2001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Arruda-Carvalho M, Wang J, Janoschka SR, Josselyn SA, Frankland PW, Ge S (2012). Optical controlling reveals time-dependent roles for adult-born dentate granule cells. Nat Neurosci, 15(12), 1700–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid H, Kanner AM (2013). Should antidepressant drugs of the selective serotonin reuptake inhibitor family be tested as antiepileptic drugs? Epilepsy Behav, 26(3), 261–265. [DOI] [PubMed] [Google Scholar]
- Haussler U, Bielefeld L, Froriep UP, Wolfart J, Haas CA (2012). Septotemporal position in the hippocampal formation determines epileptic and neurogenic activity in temporal lobe epilepsy. Cereb Cortex, 22(1), 26–36. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL (1992). The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl, 7, 273–280. [PubMed] [Google Scholar]
- Henshall DC, Meldrum BS (2012). Cell death and survival mechanisms after single and repeated brief seizures In Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies (4th ed., pp. 527–550). Bethesda, MD: Oxford University Press. [PubMed] [Google Scholar]
- Herman ST (2002). Epilepsy after brain insult: targeting epileptogenesis. Neurology, 59(9 Suppl 5), S21–26. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA (1998). Incidence of status epilepticus in Rochester, Minnesota, 1965–1984. Neurology, 50(3), 735–741. [DOI] [PubMed] [Google Scholar]
- Hosford BE, Liska JP, Danzer SC (2016). Ablation of Newly Generated Hippocampal Granule Cells Has Disease-Modifying Effects in Epilepsy. J Neurosci, 36(43), 11013–11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu D (2007). The dentate gyrus as a filter or gate: a look back and a look ahead. Prog Brain Res, 163, 601–613. [DOI] [PubMed] [Google Scholar]
- Huckleberry KA, Shue F, Copeland T, Chitwood RA, Yin W, Drew MR (2018). Dorsal and ventral hippocampal adult-born neurons contribute to context fear memory. Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko N, Romer C, Ndode-Ekane XE, Lukasiuk K, Pitkanen A (2015). Loss of hippocampal interneurons and epileptogenesis: a comparison of two animal models of acquired epilepsy. Brain Struct Funct, 220(1), 153–191. [DOI] [PubMed] [Google Scholar]
- Ikrar T, Guo N, He K, Besnard A, Levinson S, Hill A, . . . Sahay A (2013). Adult neurogenesis modifies excitability of the dentate gyrus. Front Neural Circuits, 7, 204–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvar M, Morgan PF, Auer RN (1988). The nature and timing of excitotoxic neuronal necrosis in the cerebral cortex, hippocampus and thalamus due to flurothyl-induced status epilepticus. Acta Neuropathol, 75(4), 362–369. [DOI] [PubMed] [Google Scholar]
- Iyengar SS, LaFrancois JJ, Friedman D, Drew LJ, Denny CA, Burghardt NS, . . . Scharfman HE (2015). Suppression of adult neurogenesis increases the acute effects of kainic acid. Exp Neurol, 264, 135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O (2006). Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron, 52(6), 1047–1059. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Parent JM (2015). Epilepsy and Adult Neurogenesis. Cold Spring Harb Perspect Biol, 7(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Li Z (2011). Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron, 70(4), 758–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam SD, D’Ambrosio R, Duveau V, Roucard C, Garcia-Cairasco N, Ikeda A, . . . Kelly KM (2017). Methodological standards and interpretation of video-electroencephalography in adult control rodents. A TASK1-WG1 report of the AES/ILAE Translational Task Force of the ILAE. Epilepsia, 58 Suppl 4, 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN (2016). Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol, 131(5), 659–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MS, Hinds JW (1977). Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science, 197(4308), 1092–1094. [DOI] [PubMed] [Google Scholar]
- Karten YJ, Jones MA, Jeurling SI, Cameron HA (2006). GABAergic signaling in young granule cells in the adult rat and mouse dentate gyrus. Hippocampus, 16(3), 312–320. [DOI] [PubMed] [Google Scholar]
- Kazanis I (2013). Neurogenesis in the Adult Mammalian Brain: How Much Do We Need, How Much Do We Have? In Belzung C, Wigmore P (Eds.), Neurogenesis and neural plasticity (Vol. 15, pp. 3–30). New York: Springer. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH (1997). Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Natl Acad Sci U S A, 94(19), 10409–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G (2006). They are not too excited: the possible role of adult-born neurons in epilepsy. Neuron, 52(6), 935–937. [DOI] [PubMed] [Google Scholar]
- Kempermann G (2012). Adult Hippocampal Neurogenesis In Kempermann G (Ed.), Adult Neurogenesis 2 (2 ed.). New York: Oxford University Press. [Google Scholar]
- Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A (2006). A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience, 140(2), 685–697. [DOI] [PubMed] [Google Scholar]
- Kim JW, Nam SM, Yoo DY, Jung HY, Kim IY, Hwang IK, . . . Yoon YS (2017). Comparison of Adult Hippocampal Neurogenesis and Susceptibility to Treadmill Exercise in Nine Mouse Strains. Neural Plast, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T (1982). Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res, 239(1), 57–69. [DOI] [PubMed] [Google Scholar]
- Kleen JC, Scott RC, Lenck-Santini PP, Holmes GL (2012). Cognitive and behavioral co-morbidities of epilepsy In Noebels J, Avoli M, Rogawski M, Olsen R, Delgado-Escueta A (Eds.), Jasper’s Basic Mechanisms of the Epilepsies (4th ed., pp. 1355–1376). Bethesda, MD: Oxford University Press. [Google Scholar]
- Korn MJ, Mandle QJ, Parent JM (2016). Conditional Disabled-1 Deletion in Mice Alters Hippocampal Neurogenesis and Reduces Seizure Threshold. Front Neurosci, 10, Article 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, Soltesz I (2015). In vivo evaluation of the dentate gate theory in epilepsy. J Physiol, 593(10), 2379–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci, 16(6), 2027–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacefield CO, Itskov V, Reardon T, Hen R, Gordon JA (2012). Effects of adult-generated granule cells on coordinated network activity in the dentate gyrus. Hippocampus, 22(1), 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchney SE, Jaramillo TC, Rivera PD, Eisch AJ, Powell CM (2015). Chronic P7C3 treatment restores hippocampal neurogenesis in the Ts65Dn mouse model of Down Syndrome [Corrected]. Neurosci Lett, 591, 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmkuhle MJ, Thomson KE, Scheerlinck P, Pouliot W, Greger B, Dudek FE (2009). A simple quantitative method for analyzing electrographic status epilepticus in rats. J Neurophysiol, 101(3), 1660–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH (1992). The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl, 7, 301–313. [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci, 12(12), 4846–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde ME, Lee EK, Rasmussen KG (2006). Electroconvulsive therapy in patients with epilepsy. Epilepsy Behav, 9(2), 355–9. [DOI] [PubMed] [Google Scholar]
- Lv RJ, Wang Q, Cui T, Zhu F, Shao XQ (2017). Status epilepticus-related etiology, incidence and mortality: A meta-analysis. Epilepsy Res, 136, 12–17. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A (2000). Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry, 47(12), 1043–9. [DOI] [PubMed] [Google Scholar]
- Maguire J (2014). Stress-induced plasticity of GABAergic inhibition. Front Cell Neurosci, 8(157). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margerison JH, Corsellis JA (1966). Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain, 89(3), 499–530. [DOI] [PubMed] [Google Scholar]
- Marin-Burgin A, Mongiat LA, Pardi MB, Schinder AF (2012). Unique processing during a period of high excitation/inhibition balance in adult-born neurons. Science, 335(6073), 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt S, Overstreet-Wadiche L (2008). GABAergic signalling to adult-generated neurons. J Physiol, 586(16), 3745–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Wilson CL, Beck H (2008). Hippocampal sclerosis In Engel J, Pedley TA, Aicardi J (Eds.), Epilepsy: A Comprehensive Textbook (2nd ed., pp. 121–136). Philadelphia, PA: Lippincott Williams & Wilkins. [Google Scholar]
- Mazzuferi M, Kumar G, Rospo C, Kaminski RM (2012). Rapid epileptogenesis in the mouse pilocarpine model: video-EEG, pharmacokinetic and histopathological characterization. Exp Neurol, 238(2), 156–167. [DOI] [PubMed] [Google Scholar]
- McAvoy K, Besnard A, Sahay A (2015). Adult hippocampal neurogenesis and pattern separation in DG: a role for feedback inhibition in modulating sparseness to govern population-based coding. Front Syst Neurosci, 9, Article 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA (2003). Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience, 122(2), 551–561. [DOI] [PubMed] [Google Scholar]
- Meldrum B (1991). Excitotoxicity and epileptic brain damage. Epilepsy Res, 10(1), 55–61. [DOI] [PubMed] [Google Scholar]
- Meldrum BS (2002). Concept of activity-induced cell death in epilepsy: historical and contemporary perspectives. Prog Brain Res, 135, 3–11. [DOI] [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM (1993). Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia, 34(6), 985–995. [DOI] [PubMed] [Google Scholar]
- Mizuseki K, Diba K, Pastalkova E, Buzsaki G (2011). Hippocampal CA1 pyramidal cells form functionally distinct sublayers. Nat Neurosci, 14(9), 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Otis TS, Bragin A, Hsu M, Buzsaki G (1995). GABAergic inhibition of granule cells and hilar neuronal synchrony following ischemia-induced hilar neuronal loss. Neuroscience, 69(1), 139–150. [DOI] [PubMed] [Google Scholar]
- Mongiat LA, Esposito MS, Lombardi G, Schinder AF (2009). Reliable activation of immature neurons in the adult hippocampus. PLoS One, 4(4), e5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JT, Gnatkovsky V, Ono T, Otahal J, Wagenaar J, Stacey WC, . . . Galanopoulou AS (2017). Standards for data acquisition and software-based analysis of in vivo electroencephalography recordings from animals. A TASK1-WG5 report of the AES/ILAE Translational Task Force of the ILAE. Epilepsia, 58 Suppl 4, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BL, Pun RY, Yin H, Faulkner CR, Loepke AW, Danzer SC (2011). Heterogeneous integration of adult-generated granule cells into the epileptic brain. J Neurosci, 31(1), 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CE, Bermudez-Hernandez K, Scharfman HE (2013). The influence of ectopic migration of granule cells into the hilus on dentate gyrus-CA3 function. PLoS One, 8(6), e68208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV (1981). Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci, 29(20), 2031–42. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, . . . Tonegawa S (2012). Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell, 149(1), 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Smith CD, Abner EL, Wilfred BJ, Wang WX, Neltner JH, . . . Schmitt FA (2013). Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol, 126(2), 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niibori Y, Yu TS, Epp JR, Akers KG, Josselyn SA, Frankland PW (2012). Suppression of adult neurogenesis impairs population coding of similar contexts in hippocampal CA3 region. Nat Commun, 3(1253). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS (1999). Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J Neurobiol, 39(4), 569–578. [DOI] [PubMed] [Google Scholar]
- Olesen MV, Wortwein G, Folke J, Pakkenberg B (2017). Electroconvulsive stimulation results in long-term survival of newly generated hippocampal neurons in rats. Hippocampus, 27(1), 52–60. [DOI] [PubMed] [Google Scholar]
- Parent JM, Lowenstein DH (2002). Seizure-induced neurogenesis: are more new neurons good for an adult brain? Prog Brain Res, 135, 121–131. [DOI] [PubMed] [Google Scholar]
- Parent JM, Murphy GG (2008). Mechanisms and functional significance of aberrant seizure-induced hippocampal neurogenesis. Epilepsia, 49 Suppl 5, 19–25. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA (2007). Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci, 27(51), 14012–14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrylo PR, Tyagi I, Willingham AL, Lee S, Williamson A (2007). Dentate filter function is altered in a proepileptic fashion during aging. Epilepsia, 48(10), 1964–1978. [DOI] [PubMed] [Google Scholar]
- Pearce PS, Friedman D, Lafrancois JJ, Iyengar SS, Fenton AA, Maclusky NJ, Scharfman HE (2014). Spike-wave discharges in adult Sprague-Dawley rats and their implications for animal models of temporal lobe epilepsy. Epilepsy Behav, 32, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan KD, Shwe UT, Williams DK, Greenfield LJ, Zheng F (2015). Pilocarpine-induced status epilepticus in mice: A comparison of spectral analysis of electroencephalogram and behavioral grading using the Racine scale. Epilepsy Res, 117, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, McKnight SL, Ready JM (2014). P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chem Soc Rev, 43(19), 6716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Nissinen J, Nairismagi J, Lukasiuk K, Grohn OH, Miettinen R, Kauppinen R (2002). Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res, 135, 67–83. [DOI] [PubMed] [Google Scholar]
- Poirier JL, Capek R, De Koninck Y (2000). Differential progression of Dark Neuron and Fluoro-Jade labelling in the rat hippocampus following pilocarpine-induced status epilepticus. Neuroscience, 97(1), 59–68. [DOI] [PubMed] [Google Scholar]
- Pollak DD, Monje FJ, Zuckerman L, Denny CA, Drew MR, Kandel ER (2008). An animal model of a behavioral intervention for depression. Neuron, 60(1), 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, . . . Danzer SC (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron, 75(6), 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol, 32(3), 281–294. [DOI] [PubMed] [Google Scholar]
- Rattka M, Brandt C, Loscher W (2013). The intrahippocampal kainate model of temporal lobe epilepsy revisited: epileptogenesis, behavioral and cognitive alterations, pharmacological response, and hippoccampal damage in epileptic rats. Epilepsy Res, 103(2–3), 135–152. [DOI] [PubMed] [Google Scholar]
- Rensing NR, Guo D, Wong M (2012). Video-EEG monitoring methods for characterizing rodent models of tuberous sclerosis and epilepsy In Weichhart T (Ed.), Mtor: Methods in Molecular Biology (pp. 373–391). New York: Humana Press. [DOI] [PubMed] [Google Scholar]
- Ribot R, Ouyang B, Kanner AM (2017). The impact of antidepressants on seizure frequency and depressive and anxiety disorders of patients with epilepsy: Is it worth investigating? Epilepsy Behav, 70(Pt A), 5–9. [DOI] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, . . . Hen R (2011). Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature, 472(7344), 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez S, Rincon F (2016). Status Epilepticus: Epidemiology and Public Health Needs. J Clin Med, 5(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, . . . Hen R (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science, 301(5634), 805–809. [DOI] [PubMed] [Google Scholar]
- Sawyer NT, Escayg A (2010). Stress and epilepsy: multiple models, multiple outcomes. J Clin Neurophysiol, 27(6), 445–452. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, . . . Drew MR (2006). Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A, 103(46), 17501–17506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Schwartzkroin PA (1990). Responses of cells of the rat fascia dentata to prolonged stimulation of the perforant path: sensitivity of hilar cells and changes in granule cell excitability. Neuroscience, 35(3), 491–504. [DOI] [PubMed] [Google Scholar]
- Scharfman HE (1999). The role of nonprincipal cells in dentate gyrus excitability and its relevance to animal models of epilepsy and temporal lobe epilepsy. Adv Neurol, 79, 805–820. [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL (2000). Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci, 20(16), 6144–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Smith KL, Jackson MB, Goodman JH (2002). Structural and functional asymmetry in the normal and epileptic rat dentate gyrus. J Comp Neurol, 454(4), 424–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (2004). Functional implications of seizure-induced neurogenesis. Adv Exp Med Biol, 548, 192–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Rigoulot MA, Berger RE, Walling SG, Mercurio TC, . . . Maclusky NJ (2005). Seizure susceptibility in intact and ovariectomized female rats treated with the convulsant pilocarpine. Exp Neurol, 196(1), 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Pedley TA (2006). Temporal Lobe Epilepsy In Gilman A (Ed.), The Neurobiology of Disease (pp. 349–369). New York: Academic Press. [Google Scholar]
- Scharfman HE, Hen R (2007). Neuroscience. Is more neurogenesis always better? Science, 315(5810), 336–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, McCloskey DP (2009). Postnatal neurogenesis as a therapeutic target in temporal lobe epilepsy. Epilepsy Res, 85(2–3), 150–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Bernstein HL (2015). Potential implications of a monosynaptic pathway from mossy cells to adult-born granule cells of the dentate gyrus. Front Syst Neurosci, 9, Article 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Myers CE (2016). Corruption of the dentate gyrus by “dominant” granule cells: Implications for dentate gyrus function in health and disease. Neurobiol Learn Mem, 129, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, MacLusky NJ (2017). Sex differences in hippocampal area CA3 pyramidal cells. J Neurosci Res, 95(1–2), 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloesser RJ, Manji HK, Martinowich K (2009). Suppression of adult neurogenesis leads to an increased hypothalamo-pituitary-adrenal axis response. Neuroreport, 20(6), 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J (2004). Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature, 429(6988), 184–7. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Hossmann KA (1988). Distribution of ischemic neuronal damage in the dorsal hippocampus of rat. Acta Neuropathol, 76(4), 411–421. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ (2000). Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res, 874(2), 123–130. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L (2005). Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res, 1035(1), 24–31. [DOI] [PubMed] [Google Scholar]
- Scott BW, Wojtowicz JM, Burnham WM (2000). Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp Neurol, 165(2), 231–6. [DOI] [PubMed] [Google Scholar]
- Seib DR, Espinueva D, Princz-Lebel O, Chahley E, Floresco SB, Snyder JS (2018). Hippocampal neurogenesis promotes preference for future rewards. bioRxiv. [DOI] [PubMed] [Google Scholar]
- Shaw CM, Alvord EC Jr. (1997). Neuropathology of the limbic system. Neuroimaging Clin N Am, 7(1), 101–142. [PubMed] [Google Scholar]
- Siesjo BK, Wieloch T (1986). Epileptic brain damage: pathophysiology and neurochemical pathology. Adv Neurol, 44, 813–847. [PubMed] [Google Scholar]
- Singer BH, Gamelli AE, Fuller CL, Temme SJ, Parent JM, Murphy GG (2011). Compensatory network changes in the dentate gyrus restore long-term potentiation following ablation of neurogenesis in young-adult mice. Proc Natl Acad Sci U S A, 108(13), 5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skucas VA, Duffy AM, Harte-Hargrove LC, Magagna-Poveda A, Radman T, Chakraborty G, . . . Scharfman HE (2013). Testosterone depletion in adult male rats increases mossy fiber transmission, LTP, and sprouting in area CA3 of hippocampus. J Neurosci, 33(6), 2338–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS (1989). Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol, 280(2), 183–196. [DOI] [PubMed] [Google Scholar]
- Sloviter RS (1994). The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol, 35(6), 640–654. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Kee N, Wojtowicz JM (2001). Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol, 85(6), 2423–2431. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Choe JS, Clifford MA, Jeurling SI, Hurley P, Brown A, . . . Cameron HA (2009). Adult-born hippocampal neurons are more numerous, faster maturing, and more involved in behavior in rats than in mice. J Neurosci, 29(46), 14484–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA (2011). Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature, 476(7361), 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Bush TG, Blumauer N, Lawrence K, Mucke L, Johnson MH (1999). Genetically-targeted and conditionally-regulated ablation of astroglial cells in the central, enteric and peripheral nervous systems in adult transgenic mice. Brain Res, 835(1), 91–95. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Losonczy A (2018). CA1 pyramidal cell diversity enabling parallel information processing in the hippocampus. Nat Neurosci, 21(4), 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE (2002). The incidence and prevalence of febrile seizures In Baram TZ, Shinnar S (Eds.), Febrile Seizures (pp. 1–21). San Diego, CA: Elsevier Science. [Google Scholar]
- Sun W, Winseck A, Vinsant S, Park OH, Kim H, Oppenheim RW (2004). Programmed cell death of adult-generated hippocampal neurons is mediated by the proapoptotic gene Bax. J Neurosci, 24(49), 11205–11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz BE, Houser CR, Tomiyasu U, Walsh GO, DeSalles A, Rich JR, Delgado-Escueta A (2006). Hippocampal cell loss in posttraumatic human epilepsy. Epilepsia, 47(8), 1373–82. [DOI] [PubMed] [Google Scholar]
- Taupin P (2006). Adult Neurogenesis and Neural Stem Cells in Mammals. New York: Nova Science Publishers, Inc. [Google Scholar]
- Taupin P (2008). Adult neurogenesis pharmacology in neurological diseases and disorders. Expert Rev Neurother, 8(2), 311–320. [DOI] [PubMed] [Google Scholar]
- Temprana SG, Mongiat LA, Yang SM, Trinchero MF, Alvarez DD, Kropff E, . . . Schinder AF (2015). Delayed coupling to feedback inhibition during a critical period for the integration of adult-born granule cells. Neuron, 85(1), 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, . . . Pieper AA (2012). Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A, 109(42), 17016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome-Souza MS, Kuczynski E, Valente KD (2007). Sertraline and fluoxetine: safe treatments for children and adolescents with epilepsy and depression. Epilepsy Behav, 10(3), 417–425. [DOI] [PubMed] [Google Scholar]
- Toni N, Laplagne DA, Zhao C, Lombardi G, Ribak CE, Gage FH, Schinder AF (2008). Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat Neurosci, 11(8), 901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, . . . Lowenstein DH (2015). A definition and classification of status epilepticus--Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia, 56(10), 1515–23. [DOI] [PubMed] [Google Scholar]
- Trinka E, Kalviainen R (2017). 25 years of advances in the definition, classification and treatment of status epilepticus. Seizure, 44, 65–73. [DOI] [PubMed] [Google Scholar]
- Tse K, Puttachary S, Beamer E, Sills GJ, Thippeswamy T (2014). Advantages of repeated low dose against single high dose of kainate in C57BL/6J mouse model of status epilepticus: behavioral and electroencephalographic studies. PLoS One, 9(5), e96622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubi MA, Lutkenhoff E, Blanco MB, McArthur D, Villablanca P, Ellingson B, . . . Zimmermann L (2018). Early seizures and temporal lobe trauma predict post-traumatic epilepsy: A longitudinal study. Neurobiol Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemori T, Toda K, Seki T (2017). Seizure severity-dependent selective vulnerability of the granule cell layer and aberrant neurogenesis in the rat hippocampus. Hippocampus, 27(10), 1054–1068. [DOI] [PubMed] [Google Scholar]
- Van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002). Functional neurogenesis in the adult hippocampus. Nature, 415(6875), 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Rivera PD, Wang Q, Chuang JC, Tran S, Osborne-Lawrence S, . . . Zigman JM (2015). The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatry, 20(4), 500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu YH, Huang YG, Chen LW (2008). Time-course of neuronal death in the mouse pilocarpine model of chronic epilepsy using Fluoro-Jade C staining. Brain Res, 1241, 157–167. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Fujikawa DG, Penix L, Sankar R (1993). Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia, 34 Suppl 1, S37–53. [DOI] [PubMed] [Google Scholar]
- Weise J, Engelhorn T, Dorfler A, Aker S, Bahr M, Hufnagel A (2005). Expression time course and spatial distribution of activated caspase-3 after experimental status epilepticus: contribution of delayed neuronal cell death to seizure-induced neuronal injury. Neurobiol Dis, 18(3), 582–590. [DOI] [PubMed] [Google Scholar]
- Wieser HG, Epilepsy I. C. o. N. o. (2004). ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia, 45(6), 695–714. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang X (2015). Potential mechanisms and clinical applications of mild hypothermia and electroconvulsive therapy on refractory status epilepticus. Expert Rev Neurother, 15(2), 135–44. [DOI] [PubMed] [Google Scholar]
- Zhan RZ, Timofeeva O, Nadler JV (2010). High ratio of synaptic excitation to synaptic inhibition in hilar ectopic granule cells of pilocarpine-treated rats. J Neurophysiol, 104(6), 3293–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH (2008). Mechanisms and functional implications of adult neurogenesis. Cell, 132(4), 645–660. [DOI] [PubMed] [Google Scholar]
- Zhuo JM, Tseng HA, Desai M, Bucklin ME, Mohammed AI, Robinson NT, . . . Han X (2016). Young adult born neurons enhance hippocampal dependent performance via influences on bilateral networks. Elife, 5, e22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.