

Abstract
Modulating protein interaction pathways may lead to the cure of many diseases. Known protein-protein inhibitors bind to large pockets on the protein-protein interface. Such large pockets are detected also in the protein-protein complexes without known inhibitors, making such complexes potentially druggable. The inhibitor-binding site is primary defined by the side chains that form the largest pocket in the protein-bound conformation. Low-resolution ligand docking shows that the success rate for the protein-bound conformation is close to the one for the ligand-bound conformation, and significantly higher than for the apo conformation. The conformational change on the protein interface upon binding to the other protein results in a pocket employed by the ligand when it binds to that interface. This proof-of-concept study suggests that rather than using computational pocket-opening procedures, one can opt for an experimentally determined structure of the target co-crystallized protein-protein complex as a starting point for drug design.
Keywords: molecular recognition, drug design, conformational properties, molecular modeling, ligand-receptor interaction
Introduction
Most biological functions in a cell are mediated through protein-protein interactions (PPI). Modulating PPI pathways may lead to the cure of many diseases such as cancer. For example, a novel drug Venetoclax, the inhibitor of anti-apoptotic protein Bcl-2, recently was approved by the FDA to treat refractory chronic lymphocytic leukemia [1]. In addition, a number of drugs undergo stage III of clinical trials and show promise to increase the survival rate of various aggressive cancers [2]. The estimated size of human interactome is 300,000 – 650,000 PPIs [3,4]. Consequently, the number of potential PPI targets far surpasses the number of “classical” druggable targets (enzymes, receptors, transporters, etc.) in the human cell [5]. However, despite the large numbers of potential targets and their importance, there are only ~ 30 examples of PPIs that were successfully disturbed by competitive small inhibitor and less than a dozen of small ligands that reached the late stages of clinical trials [2,5–7].
The two approaches to disturb the PPI are allosteric and competitive inhibition. The allosteric inhibition is binding of a small ligand away from the interface. The binding leads to conformational changes at the interface, thus disturbing complementarity of the protein partners. Predicting propagation of conformational change to the protein interface from a distant ligand-binding site is problematic [8,9]. Currently, the competitive inhibition of PPI appears to be a more straightforward approach to the drug design, since it (a) does not require modeling of the global conformational change, and (b) reduces the search for the ligand binding site from the whole surface to the interface or even to small patches of the interface [9–14].
Rational drug design tools and small molecule libraries were developed for enzymes, transporters, ion channels and receptors - proteins that evolved to have a pocket matching a particular substrate or signaling molecule [15–17]. Generally, protein interfaces are large (1200 – 2000 Å2) [18,19], without well-defined pockets, which make them difficult for the design of inhibitors.
Studies showed that a small molecule inhibitor mimics most important interaction fragments on the protein-protein interface [10]. To block the PPI, the inhibitor must bind to the hot spot residues that have the largest energetic contribution to the binding affinity (≥ 2 kcal/mol) [10–12]. Another important criterion for inhibition is the presence of a well-defined pocket in the proximity of the hot spot that could accommodate the small non-peptidic ligand (volume > 300 Å3) [20–22]. These pockets are supposed to be closed in the protein’s unbound conformation [20–22].
It was observed that these pockets are opened by conformational change in the side chains and minor movement of the backbone (Cα Root Mean Square Deviation [RMSD] between unbound and ligand-bound conformation ~2.7 Å). This led to a conclusion that these pockets could be present in the ensemble of the unbound low energy conformations. It was shown that the pockets open spontaneously during molecular dynamics simulation, although with low probability [20,23]. This method of pocket opening requires significant computational time for the 6 ns simulation to yield an ensemble of 1000 snapshots [21]. To accelerate the computation, a biased simulation could be performed. However, it requires the knowledge of the hot spot residues [22].
Ligand-receptor docking is an essential tool in the design of PPI inhibitors [24]. The low-resolution approaches showed that, similarly to the protein-protein docking [25], ligand docking can be successfully applied to the low-resolution/modeled structures of the protein receptors [26–28].
A serious difficulty in studying PPI inhibitors is a very limited set of solved structures of interfaces in protein-bound and ligand-bound conformation. Currently, there are only 27 protein interfaces solved crystallographically in both protein-bound and ligand-bound conformations [7]. Inspired by work of Koes and Camacho [12], we assumed that the current PDB should contain many more PPI inhibitors that were not included in the list because of the lack of medical use. Still, the physical basis of the medically irrelevant protein-ligand complexes should be the same as that of the complexes with known PPI inhibitors. Thus, accounting for such complexes would increase the statistical significance of the structure set.
A recent study showed that a protein-bound conformation is generally closer to the ligand-bound conformation than to the unbound one [29]. In our study, we generated an expanded set of protein-ligand complexes, where the proteins are co-crystallized both with another protein and with the ligand at the protein-protein interface. We showed that the inhibitor site significantly overlaps with the largest pocket in the protein-bound conformation. We determined that such large pockets also exist in the protein-protein complexes in a generic set built with no regard to PPI inhibition, making such complexes potentially druggable. The results showed that the transition from protein-bound to ligand-bound conformation is small enough for the low-resolution docking to have similar success rate for both. This proof-of-concept study shows that the protein-bound conformation is almost as good as the ligand-bound one as a starting point for drug design.
Methods
Structural and sequence alignment
Structural alignment of whole proteins was performed by TM-align [30]. Minimizing RMSD for the superposition of interface and pocket atoms was done by Newton-Raphson quaternion-based method suggested by Liu et al. [31]. Sequence alignment was performed by BLAST [32] with BLOSUM-62 substitution matrix and the gap penalty 10.
Docking
Protein-ligand modeling was by rigid-body docking, based on the systematic search for geometric complementarity by correlation using Fast Fourier Transform (FFT), implemented in the GRAMM software [33,34]. GRAMM was used at low resolution, with translational grid step 3.5 Å and angular interval 10°, which are the optimal parameters for the GRAMM docking of unbound proteins. Docking matches were restricted to those having ligand in contact with the protein-protein interface residues (defined as those with any heavy atom within 6.0 Å from any heavy atom of the other protein). Matches with big clashes with the receptor (> 50% ligand volume overlap) were removed. According to the paradigm of this study to investigate the difference of protein steric complementarity to the ligand, between protein-bound and ligand-bound conformations of the protein, scoring of the matches was by surface complementarity only.
Pocket volume calculation
Protein-bound pockets (“lead pockets,” according to the paradigm of this study) and ligand-bound pockets are similar enough for the lower-resolution rigid-body ligand docking (see Results). However, the protein-bound pockets are generally more shallow than the ligand-bound ones. Thus, our pocket detection procedure was designed for the “shallow lead pocket” search.
To characterize cavities on the protein surface, we implemented grid-based algorithm [35]. The protein is projected on a rectangular grid with 0.5 Å step. Each point of the grid is marked as protein or solvent, depending on the distance to the center of the closest atom. If the distance is > 3.0 Å (solvent accessible radius of a carbon atom) then the point is marked as solvent. All solvent points that are completely surrounded by protein are marked as protein. At the next step, we marked protein points as surface if they are adjacent to the solvent point. To check if the surface points encompass the solvent points, rays were casted from each surface point of the grid in 26 grid-based directions (vertices of a single-step cubic grid around the point). A solvent point was marked as a cavity if the surface points in at least three directions enclosed it.
To remove the shallow cavities and the procedure artifacts, we removed all cavity points within 1 Å from the solvent points. The grid was converted to a graph representation where vertices of the graph were the grid points, and two vertices were connected by an edge if they were adjacent grid points of the same type. The graph that consisted of cavity points was divided into communities by an algorithm suggested by Clauset et al. [36]. Edges that connected the resulting communities were removed. The graph splitting procedure was repeated until each connected subgraph consisted of only one community. Small subgraphs with < 80 nodes (10 Å3) were removed. Each resulting subgraph was treated as a separate cluster. Points of the grid in a cluster were marked as pocket if at least one point in that cluster was within 3 Å of an interface atom. To form lead pockets, small shallow pockets were combined if the distance between them was < 2 Å. The volumes of the resulting pockets were calculated and the pocket with the largest volume was marked as a potential binding pocket. The steps of the procedure are illustrated in Supplemental Figure S1.
Protein-ligand complexes
We designed a procedure to extract from Protein Data Bank (PDB) [37] the structures of proteins that are both co-crystallized with another protein and with a ligand at the protein-protein interface in the absence of the other protein. The method is similar to the procedure of Koes and Camacho [12]. We obtained all protein complexes, including NMR and complexes with short peptides ≤ 10 residues, from PDB. All multimeric proteins from biological assemblies and NMR structures were split into binary complexes. The interface residues were designated as those with any heavy atom within 6.0 Å from any heavy atom of the binding partner. Retaining structures with higher than 3.5 Å resolution, 30 or more residues long, and with at least six residues at the interface, resulted in 1,016,532 interfaces. To eliminate redundancy, we performed all-to-all structural alignment by TM-align. Two proteins were considered having the same interface if the TM-score between the two structures was ≥ 0.95, and the interface residues fully overlapped (Cα RMSD ≤ 1.0 Å). To speed up calculation, we aligned only structures with similar sequence length (≤ 100 amino acids difference), because proteins with a larger difference in length do not achieve such high structural similarity score by the TM-align algorithm. The resulting set contained 24,131 unique interfaces. To find all interfaces occupied by non-peptidic ligands, we structurally aligned each protein from our set to all structures in PDB. The interfaces from the query structures where mapped onto similar structures in PDB (TM-score ≥ 0.9). Then we selected structures with HETATM-tagged atoms, and with no ATOM-tagged atoms, in the vicinity of the mapped residues (within 6.0 Å from any heavy atom of the mapped residues). This resulted in 2,459 unique interfaces and 17,980 corresponding holo structures. Because small ligands (< 6 atoms) are common solvents and ions that most likely are artifacts of the crystallographic procedure, they were discarded from the set. It was observed that PPI inhibitors, while bound to a protein, tend to be more exposed compared to classical drugs [16]. We removed structures with ligands in deep, well-defined pockets, in which ligands are well encompassed by the protein (≤ 25% of the ligand solvent accessible area exposed), assuming that such deep pockets are enzymatic sites, which as “traditional” drug targets are outside the scope of this study. The resulting set had 788 protein-protein complexes (1,194 unique interfaces). Each of these unique interfaces was co-crystallized with non-peptidic ligands (in total, 2,526 such protein-ligand complexes). We further narrowed the protein-protein set to 87 complexes by eliminating ones that have residue differences in the sequence at their interfaces and the respective parts of the corresponding holo structures. This set corresponded to 194 protein holo structures co-crystallized with 107 ligands. Since we used a more recent version of PDB, and the structure, rather than sequence, similarity to extract similar interfaces, our set is larger than a similar set of Koes and Camacho (51 protein complexes [12]).
We analyzed protein structures in a complex with known PPI inhibitors, solved in both protein-bound (bound) and ligand-bound (holo) conformation. There were only 27 such examples in 2P2I database [7]. To avoid over-representation of protein interfaces co-crystallized with more than one inhibitor, we chose structures with the most potent inhibitor. In addition, we excluded complexes with identical interfaces (≥ 95% sequence identity) and structures that have more than one interface residue mutated. That left 19 proteins, which were solved in both bound and holo states, and 14 of those solved in the unbound (apo) state.
To identify complexes in our set (87 protein-protein complexes and corresponding complexes with ligands, see above) that are similar to those with known PPI inhibitors, we analyzed the changes in geometric properties of the interfaces (RMSD, Solvent Accessible Surface Area [SASA], and volume of the pocket) upon ligand binding. The interface side chains conformations were not significantly different in the protein-bound and inhibitor-bound states. On average, the side chains that participated in forming of the binding pocket in the known inhibitors shifted by 1.4 Å RMSD. These minor shifts slightly increased solvent accessibility of the pocket residues, on average, by 4.5 Å2. Seven complexes from our set had bound/holo conformational differences at the interface similar to the ones in complexes with known PPI inhibitors, and were added to the set (a 30% increase).
One of the proteins from our additional set is present in the latest update of the 2P2Idb set of proteins with known PPI inhibitors (in complex with a different ligand) [7]. It involves the bromodomain module of histone acetyltransferase complex (HAC) (Figure 1). Acetylation of the histone lysine is a common post-translational modification performed by HAC. The bromodomain module is responsible for specific recognition of acetyl-lysine residues located on the histone tails, and thus plays an important role in regulation of gene transcription.
Figure 1. The protein from the generated set of complexes, which is also present in the set of protein complexes with known PPI inhibitors (in complex with a different ligand).
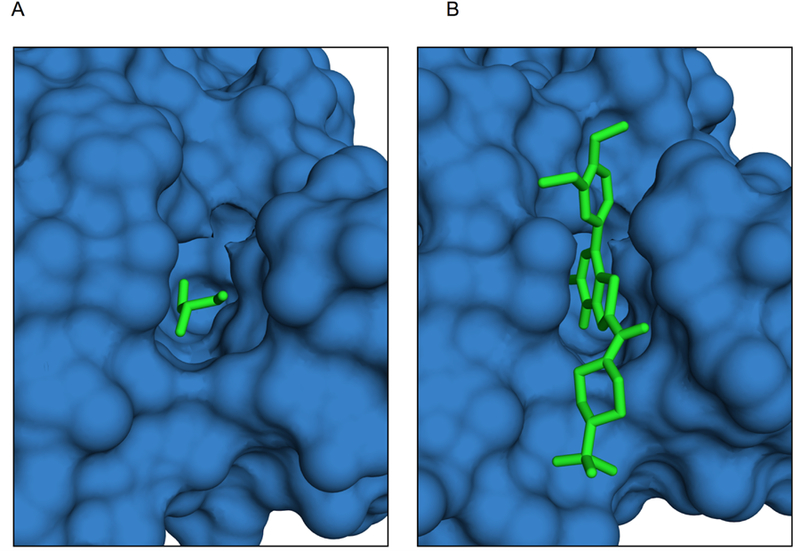
(A) Bromodomain in the general set cocrystallized with glycerol (4ldf). (B) Bromodomain cocrystallized with its known inhibitor N1D (4uit).
The resulting full set for further analysis consisted of 25 complexes - 19 complexes from the 2P2I database and 6 complexes from our generated set (7 minus one that was already present in 2P2I). The results in this study were obtained for the full set, as well as just for the set of 19 complexes with known PPI inhibitors.
Pockets at protein-protein interface
Large pockets exist in bound conformations of protein-protein complexes without known inhibitors
To obtain a set of general protein-protein complexes without known PPI inhibitors (reference set), we started with the set of 4,450 complexes from our previous study [38]. The set consists of binary complexes (crystallographic resolution ≤ 3.5 Å) with well-defined interface (surface area ≥ 250 Å2). We eliminated complexes in which both proteins were annotated as enzyme or one of the proteins was marked as kinase/phosphatase or hydrolase (E.C. numbers 2.7. and 3). We excluded complexes where one protein encapsulated the other, because for those, the whole interface became a pocket. We also excluded complexes in which ≥ 82% of protein surface is in contact with the other protein. The 82% value was chosen because it is the maximal one in the set of complexes with known PPI inhibitors. The resulting set consisted of 1,451 protein-protein complexes and 2,875 interfaces (one protein can participate in more than one complex).
We compared the volume of the largest pocket at a protein-protein interface in both monomers in the protein-bound conformation in our reference set, with the one in proteins from the protein-ligand set. A well-defined pocket was present in all protein-ligand interfaces. The average volume was 543 ± 71 Å3 in the full set of 25 complexes (19 complexes with known PPI inhibitors and 6 complexes generated in this study), and 588 ± 72 Å3 in the set of 19 complexes with known PPI inhibitors. Although the average size of the largest pocket in the reference set was smaller (152 ± 13 Å3), a significant number of pockets at protein-protein interfaces in the reference set were comparable in size with the ones at the protein-protein interfaces in the set where proteins are also co-crystallized with ligands. Figure 2 shows comparison of the pocket size distribution in the protein-protein reference set and that in the set of 19 protein-protein complexes with known PPI inhibitors (the distribution in the full set of 25 protein-ligand complexes is similar). The results below show that the interface pockets on protein bound structures in the protein-ligand set are comparable with those in the holo structures. Thus, the proteins in the general protein-protein set with pockets comparable to ones in the protein-ligand set can potentially be druggable as well.
Figure 2. The volume of the largest pocket at the protein-protein interface in the set with known PPI inhibitors vs. reference protein-protein set.
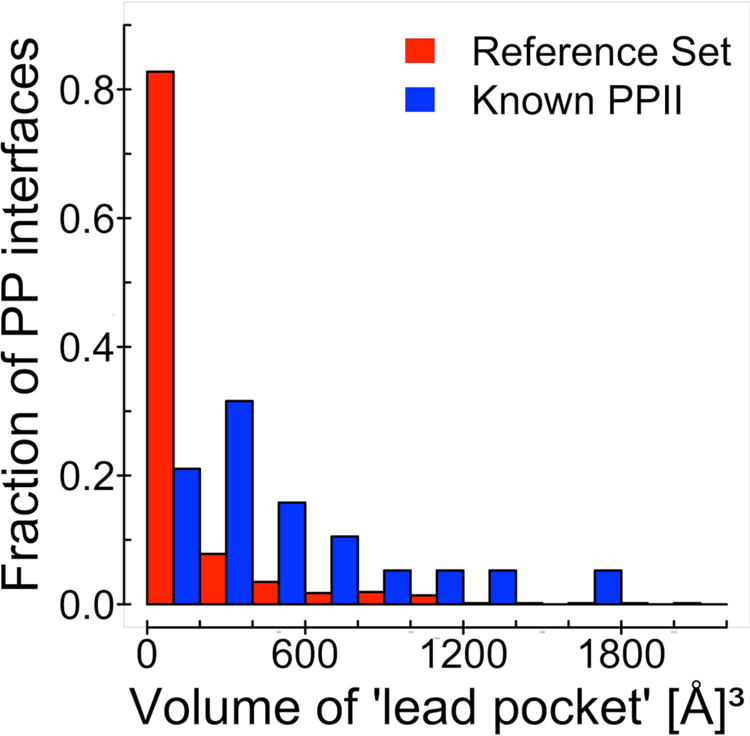
The pocket at the protein-protein interface in 19 protein complexes with known PPI inhibitors (blue) is on average larger than the pocket at the protein-protein interface of the general/reference set (red). However, a significant number of protein interfaces in the reference set is comparable in size with the ones in the set with known PPI inhibitors.
Such proteins fall into four groups, illustrated in Figure 3: (1) Modulator proteins, common in signaling and apoptosis pathways, where the binding/unbinding of the protein controls if it can be reached by an enzyme; (2) stabilizers - chaperones ensuring correct folding of a protein or helping it pass through the membrane; (3) parts of a larger complex, e.g. exon junction complex; and (4) other. Enzyme - protein inhibitor, enzyme - substrate, and antigen - antibody complexes were excluded from this classification.
Figure 3. Examples of four groups of proteins with large pockets on the interface.
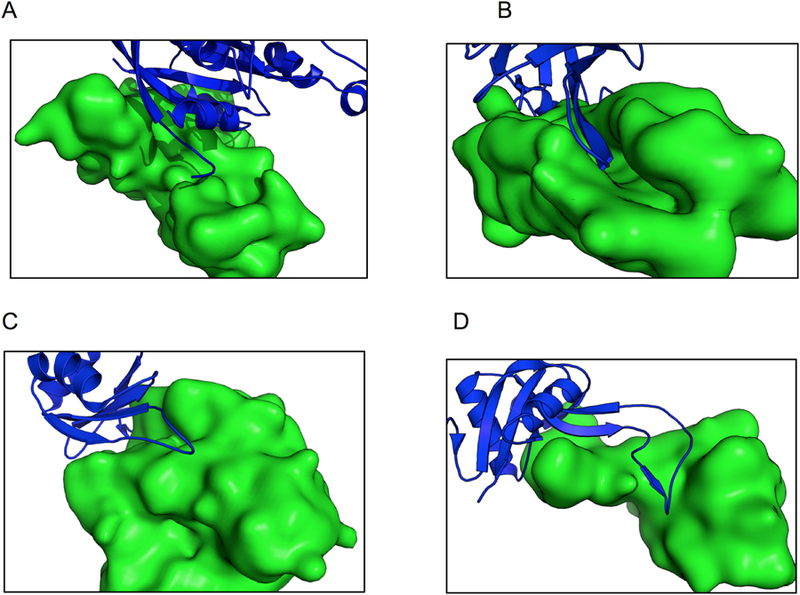
(A) Modulator proteins - 3gcg is a complex of Mitochondria associated protein (MAP, green) and Cell division control protein 42 (CDC42, blue). CDC42 belongs to signaling networks that control cell division. The protein can exist in GTP-bound (active) and GDP-bound (inactive) states. Guanine nucleotide exchange factor (GEF) switches CDC42 from inactive to active state. MAP mimics bindings of GEF and thus prevents CDC42 from activating [59]. (B) Stabilizers - 2wmp is chaperone PapD (green) in complex with pilin domain of PapG adhesin (blue). PapD temporary stabilizes the structure of pilin domain of PapG before it fully assembles. PapG is present on bacterial surface and its function is to allow bacteria to bind to other cells and surfaces (in this case kidney cells). In its functional form, it must form a dimer and the pilin domain play an important role in the dimerization. To ensure that the beta strand of PapG will not bind to something else or form a non-functional complex, it is stabilized by a chaperon before its full assembly [60]. (C) Part of a larger complex - 1oo0 is Mago Nashi (green) in complex with Y14 (blue), an essential component of Exon Junction Complex (EJC), which controls the lifespan of the spliced mRNA performing its quality control, export and degradation. Mago and Y14 are two out of eight identified EJC proteins located in the core of EJC. They are essential for the correct localization of mRNA [61]. (D) Other - 1sb2 is a complex of Rhodocetin beta (blue) and alpha (green) subunits, a snake toxin that inhibits collagen-induced aggregation, active only in the dimer form [62].
Inhibitor site overlaps with the largest pocket in the protein-bound conformation
Our analysis of the set of 19 complexes with known PPI inhibitors showed that 74% of residues in the interface largest pocket in protein-bound conformation are in contact with the ligand in the holo conformation. At the same time, 62% of the ligand-binding pocket residues in the holo conformation are from the largest pocket in the protein-bound conformation (Figure 4). In the full set of 25 complexes the observation was similar: 72% and 60% correspondingly. Even though protein interface typically consists of more than a dozen residues, the inhibitor-binding site is primary defined by the side chains that form the largest pocket in the protein-bound conformation (examples in Figure 5).
Figure 4. Percent of residues common to the largest pockets in ligand-bound and protein-bound conformations.
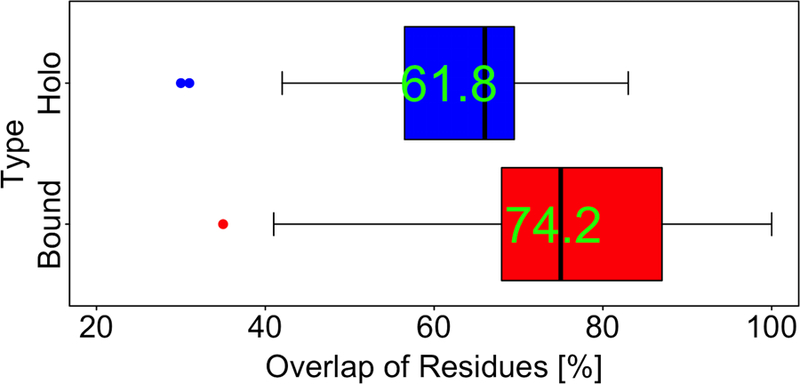
The interface largest pocket in protein-bound conformation typically is the same pocket that participates in ligand binding. On average, 74% of side-chains of the largest pocket in the protein-bound conformation bind to the ligand in the holo conformation, and 62% of side chains that are in contact with the inhibitor in the holo conformation are present in the pocket in the protein-bound conformation.
Figure 5. Examples of largest pockets in protein-bound vs. ligand-bound conformations.
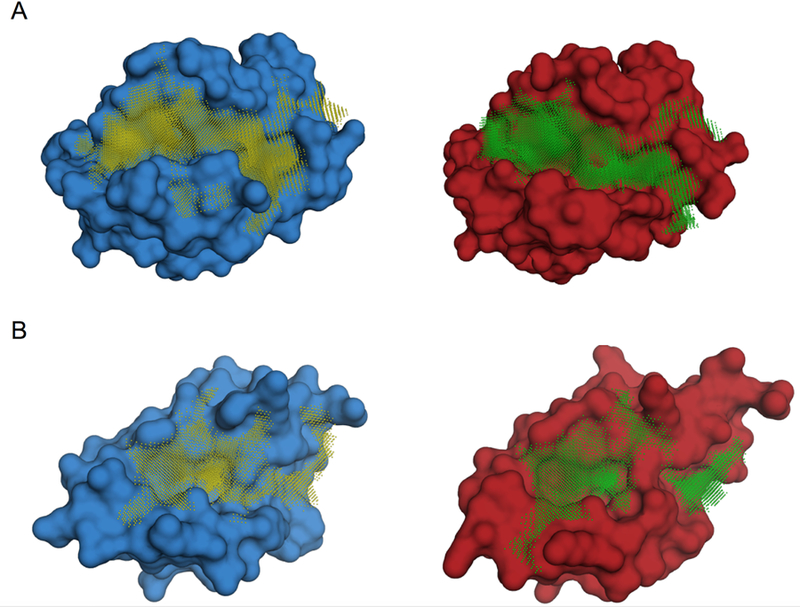
The ligand-bound conformations are in red and the protein-bound ones are in blue. (A) The apoptosis regulator protein BCL2 - its inhibition triggers apoptosis of tumor cells - in ligand-bound (4ltv, blue) and protein-bound conformations (2xa0, red). (B) The negative regulator of the p53 tumor suppresser protein MDMX in ligand-bound (3lbj, blue) and protein-bound conformations (3dab, red). The largest interface pockets are shown by the grid.
Docking
Conformational change upon binding is the physical basis of protein function. A number of proteins have structures experimentally determined in both unbound and bound states [39,40]. When proteins bind other proteins, the conformational changes sometimes are significant (e.g. loop movements, domain shifts), but most often are rather small, primarily involving the side chains [39]. Binding of a small ligand at the protein-protein interface typically leads to a small conformational change as well. Protein interfaces in unbound and holo states in our study were different, on average, by 2.7 Å Cα RMSD, and in bound and holo states, by 1.9 Å Cα RMSD.
A ligand-binding pocket at the protein-protein interface, which is usually the largest at the interface, is key to ligand binding. As shown above, these pockets typically are already present in the protein-bound conformation and are structurally similar to those in the ligand-bound conformation. Although structural metrics like RMSD quantitatively capture this similarity, the ultimate test of it should come from the ligand docking. To that end, we docked the ligand in the biologically active (receptor-bound) conformation to protein-bound and ligand-bound conformations of the protein, as well as to its apo form.
It is important to emphasize, that the goal of this study was not to develop a protocol for drug design, but to determine the structural difference of the protein-bound and the ligand-bound pockets. Thus, we intentionally did not use flexible docking with sophisticated sampling/scoring but employed a rigid-body procedure with a basic steric complementarity scoring. The rigid-body docking of the biologically active ligand conformation excludes the effect of conformational sampling of the ligand, which introduces another degree of uncertainty. Thus, it allows one to pinpoint the exact role of the protein-bound vs. ligand-bound protein conformational difference.
The fitting was performed by the low-resolution FFT docking GRAMM (see Methods). The FFT method is dominant in protein-protein docking [41,42], but also has been used in other biomolecular applications, like protein-RNA docking [43], and in a number of small ligand-receptor modeling approaches, including prediction and mapping of the ligand site [44,11], and protein-peptide [45] and ligand-receptor docking [33,34,46–49]. The low resolution provides tolerance to the structural mismatches that are similar in the rigid-body docking of unbound proteins and docking of ligand co-crystallized with the protein holo structure to the protein bound structure (conformational variation of the side chains and small backbone movements). According to the low-resolution docking paradigm [34,25], the measure of the success was based on the position of the ligand’s center of mass. Metrics like ligand RMSD (between ligand in predicted and experimentally determined positions, with receptors superimposed) are inherently sensitive to the ligand angular orientation, which require high resolution details [50]. The ligand’s center of mass provides the location of the binding site on the protein interface, which can be used as the starting point for further modeling.
The docking was performed on the following protein conformations: bound to the other protein (bound), bound to the ligand (holo) and unbound (apo). The prediction was considered correct, if the distance of the ligand’s center of mass in the predicted position from the one co-crystalized with the protein (with the proteins superimposed) was < 3.5 Å (the average diameter of a carbon atom).
The docking showed that for ligands docked to holo proteins, where conformational differences should not play a role, not all complexes were predicted correctly. Such behavior is common in low-resolution docking of proteins [25], and is not surprising in the docking of small ligands to large and relatively flat protein interfaces, where the ligands can find other locations providing low-resolution shape complementarity. Complexes shown in Figure 6 are examples of successful docking. Figure 7 shows examples of predictions away from the experimentally determined site, due to large structural characteristics of the interface providing alternative low-resolution ligand/protein complementarity (a typical case in protein-protein docking [25]).
Figure 6. Examples of correctly predicted complexes.
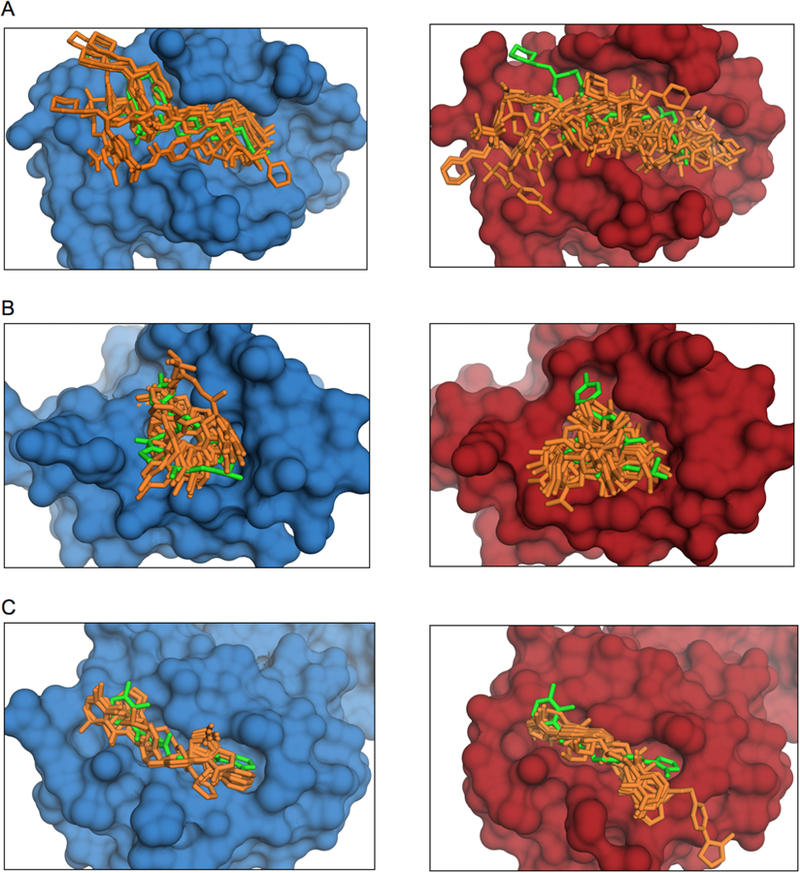
Holo is in blue and bound is in red. The co-crystallized ligand position is in green and the predicted ones are in orange. (A) Bcl2 in ligand-bound (4lvt) and protein-bound conformation (2xa9). (B) Mdmx in ligand-bound (3lbj) and protein-bound conformation (3dab). (C) The von Hippel–Lindau protein in ligand-bound (4b9k) and protein-bound conformation (4ajy).
Figure 7. Examples of predictions away from the experimentally determined site.
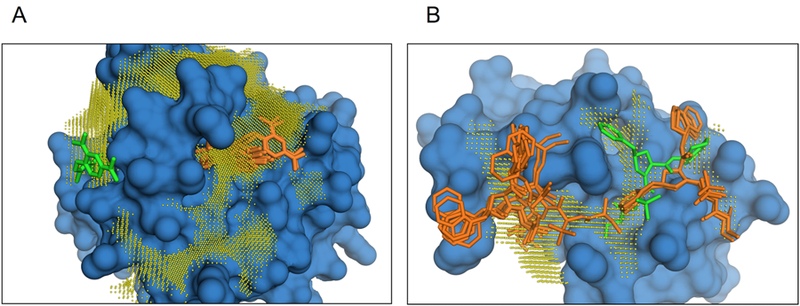
The co-crystallized ligand position is in green and the predicted ones are in orange. (A) Ras oncoprotein in ligand-bound conformation (2lwi). Big well-defined enzymatic pocket located on the far side of interface, away from the inhibitor binding side. (B) BIR3 domain of XIAP protein (X-inked inhibitor of Apoptosis) bound to its inhibitor (1tft). Top predictions are spread over the elongated groove on the XIAP-caspase 9 interface, which connects to a deep pocket away from the inhibitor site.
The docking results on the set of 19 complexes with known PPI inhibitors (Figure 8; results on the full set of 25 complexes were similar) showed that the success rate of ligand docking to the protein-bound conformation is close to the one for the docking to the ligand-bound conformation. Both are significantly higher than the success rate for the protein apo conformation. This supports the observation of the interface pocket similarity in protein-bound and holo states, from the ligand docking perspective. The conformational change on the protein interface upon binding to the other protein results in a pocket that is the one used by the ligand when it binds to that interface. The results suggest that the protein-bound conformation of the receptor is a significantly better starting point for drug design than the apo structure.
Figure 8. Success rate of rigid-body low-resolution ligand-receptor docking.
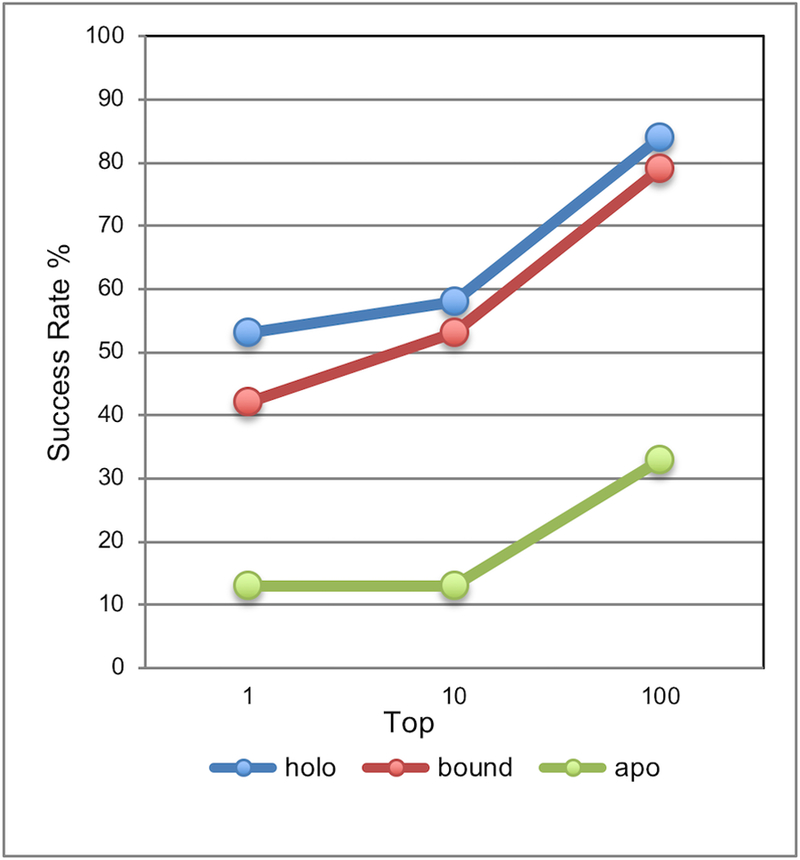
The ligand is in the conformation co-crystallized with the holo protein. A match was considered correct if the distance of the ligand’s center of mass in the predicted position from the one co-crystalized with the protein (with the proteins superimposed) was < 3.5 Å. The complex was predicted successfully if a correct match was in top N predictions (N = 1, 10, and 100).
One consequence of this study is that a common notion of “opening pockets” on the protein-protein interface for binding of a ligand, and the corresponding procedures developed for that purpose [20,21,51], actually may not be needed when the protein-protein co-crystallized structure of the target is available. Computational opening of a pocket has significant accuracy limitations, inherent to such a challenging modeling task. Thus, an experimentally determined structure that has that pocket in a conformation close to the actual opened one in the holo structure may be a preferred option.
Overall, the number of protein-protein complexes in PDB is comparable to the number of monomers. For about half of the monomers, a homologous structure exists in the protein-protein complex as well (Figure S2). For the entire genomes of model organisms such as Escherichia coli or yeast, PDB offers homology modeling templates for a significant part of soluble proteins [52]. Comparative docking templates can be found for protein-protein complexes representing almost all known PPI, provided the components themselves have an experimentally-determined structure or can be homology-built [53]. Thus, the structural characterization of PPI, which can be used as a starting point for PPI inhibition, is quite significant.
A number of proteins interact with different protein partners at the same interface [54–57]. This poses an intriguing question of selectively inhibiting some of such binding proteins, while permitting binding of the others. Given tight structural packing of the protein-protein interfaces [58], the selective inhibition of PPI, in principle, may be feasible if the different protein binders had significantly different pockets at the same interface. We will investigate this issue in the future study.
Conclusion
The ability to inhibit protein-protein interactions is important for curing diseases. An expanded set of protein-ligand complexes was generated, with proteins co-crystallized with another protein and with the ligand at the protein-protein interface. Known PPI inhibitors bind to large pockets on the protein-protein interface. We detected such large pockets also in the protein-protein complexes in a generic protein-protein set without known inhibitors, making such complexes potentially druggable. In proteins from the protein-protein complexes also co-crystallized with PPI inhibitors, even though the protein-protein interface consists of more than a dozen residues, the inhibitor-binding site is primary defined by the side chains that form the largest pocket in the protein-bound conformation. Low-resolution docking was performed on the ligand-receptor set showing that the success rate for the protein-bound conformation is close to the one for the ligand-bound conformation (and far better than for the apo conformation). The conformational change on the protein interface upon binding to the other protein results in a pocket used by the ligand when it binds to that interface. Our proof-of-concept study suggests that rather than performing a challenging modeling task of pocket-opening, one can opt for an experimentally determined structure of the target co-crystallized protein-protein complex as a starting point for druggability assessment and design of inhibitors.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health grant R01GM074255 and National Science Foundation grants DBI1262621, DBI1565107 and CNS1337899.
References
- 1. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm495351.htm.
- 2.Kang MH, Reynolds CP (2009) Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15:1126–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stumpf MP, Thorne T, de Silva E, Stewart R, An HJ, Lappe M, Wiuf C (2008) Estimating the size of the human interactome. Proc Natl Acad Sci USA 105:6959–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, Maniatis T, Califano A, Honig B (2012) Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 490:556–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cierpicki T, Grembecka J (2015) Targeting protein–protein interactions in hematologic malignancies: Still a challenge or a great opportunity for future therapies? Immunol Rev 263:279–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higueruelo AP, Schreyer A, Bickerton GRJ, Pitt WR, Groom CR, Blundell TL (2009) Atomic interactions and profile of small molecules disrupting protein–protein interfaces: The TIMBAL database. Chem Biol Drug Des 74:457–467 [DOI] [PubMed] [Google Scholar]
- 7.Basse MJ, Betzi S, Bourgeas R, Bouzidi S, Chetrit B, Hamon V, Morelli X, Roche P (2013) 2P2Idb: A structural database dedicated to orthosteric modulation of protein–protein interactions. Nucl Acid Res 41:D824–D827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorczynski MJ, Grembecka J, Zhou Y, Kong Y, Roudaia L, Douvas MG, Newman M, Bielnicka I, Baber G, Corpora T, Shi J, Sridharan M, Lilien R, Donald BR, Speck NA, Brown ML, Bushweller JH (2007) Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFβ. Chem Biol 14:1186–1197 [DOI] [PubMed] [Google Scholar]
- 9.Smith MC, Gestwicki JE (2012) Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert Rev Mol Med 14:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acuner Ozbabacan SE, Gursoy A, Keskin O, Nussinov R (2010) Conformational ensembles, signal transduction and residue hot spots: Application to drug discovery. Curr Opin Drug Discov Devel 13:527–537 [PubMed] [Google Scholar]
- 11.Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, Beglov D, Pelletier J, Whitty A, Vajda S (2011) Structural conservation of druggable hot spots in protein–protein interfaces. Proc Natl Acad Sci USA 108:13528–13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koes DR, Camacho CJ (2012) Small-molecule inhibitor starting points learned from protein–protein interaction inhibitor structure. Bioinformatics 28:784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.London N, Raveh B, Movshovitz-Attias D, Schueler-Furman O (2010) Can self-inhibitory peptides be derived from the interfaces of globular protein–protein interactions? Proteins 78:3140–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaidman D, Wolfson HJ (2016) PinaColada: Peptide–inhibitor ant colony ad-hoc design algorithm. Bioinformatics 32:2289–2296 [DOI] [PubMed] [Google Scholar]
- 15.Fuller JC, Burgoyne NJ, Jackson RM (2009) Predicting druggable binding sites at the protein-protein interface. Drug Discov Today 14:155–161 [DOI] [PubMed] [Google Scholar]
- 16.Gowthaman R, Deeds EJ, Karanicolas J (2013) Structural properties of non-traditional drug targets present new challenges for virtual screening. J Chem Inf Model 53:2073–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoichet BK, Walters WP, Jiang H, Bajorath J (2016) Advances in computational medicinal chemistry: A reflection on the evolution of the field and perspective going forward. J Med Chem 59:4033–4034 [DOI] [PubMed] [Google Scholar]
- 18.Janin J, Bahadur RP, Chakrabarti P (2008) Protein-protein interaction and quaternary structure. Quart Rev Biophys 41:133–180 [DOI] [PubMed] [Google Scholar]
- 19.Gao M, Skolnick J (2012) The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. Proc Natl Acad Sci USA 109:3784–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eyrisch S, Helms V (2007) Transient pockets on protein surfaces involved in protein-protein interaction. J Med Chem 50:3457–3464 [DOI] [PubMed] [Google Scholar]
- 21.Metz A, Pfleger C, Kopitz H, Pfeiffer-Marek S, Baringhaus KH, Gohlke H (2011) Hot spots and transient pockets: Predicting the determinants of small-molecule binding to a protein-protein interface. J Chem Inf Model 52:120–133 [DOI] [PubMed] [Google Scholar]
- 22.Johnson DK, Karanicolas J (2013) Druggable protein interaction sites are more predisposed to surface pocket formation than the rest of the protein surface. PLoS Comp Biol 9:e1002951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ulucan O, Eyrisch S, Helms V (2012) Druggability of dynamic protein-protein interfaces. Curr Pharm Design 18:4599–4606 [DOI] [PubMed] [Google Scholar]
- 24.Kuenemann MA, Sperandio O, Labbe CM, Lagorce D, Miteva MA, Villoutreix BO (2015) In silico design of low molecular weight protein-protein interaction inhibitors: Overall concept and recent advances. Prog Biophys Mol Biol 119:20–32 [DOI] [PubMed] [Google Scholar]
- 25.Vakser IA, Matar OG, Lam CF (1999) A systematic study of low-resolution recognition in protein-protein complexes. Proc Natl Acad Sci USA 96:8477–8482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brylinski M, Skolnick J (2008) Q-Dock: Low-resolution flexible ligand docking with pocket-specific threading restraints. J Comput Chem 29:1574–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Dundas J, Kachalo S, Ouyang Z, Liang J (2011) Accuracy of functional surfaces on comparatively modeled protein structures. J Struct Funct Genomics 12:97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skolnick J, Zhou H, Gao M (2013) Are predicted protein structures of any value for binding site prediction and virtual ligand screening? Curr Opin Struct Biol 23:191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohnuud T, Kozakov D, Vajda S (2014) Evidence of conformational selection driving the formation of ligand binding sites in protein-protein interfaces. PLoS Comp Biol 10:e1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Skolnick J (2005) TM-align: A protein structure alignment algorithm based on the TM-score. Nucl Acid Res 33:2302–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu P, Agrafiotis DK, Theobald DL (2010) Fast determination of the optimal rotational matrix for macromolecular superpositions. J Comput Chem 31:1561–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: Architecture and applications. BMC Bioinformatics 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA (1992) Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA 89:2195–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vakser IA (1995) Protein docking for low-resolution structures. Protein Eng 8:371–377 [DOI] [PubMed] [Google Scholar]
- 35.Hendlich M, Rippmann F, Barnickel G (1997) LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Mod 15:359–363 [DOI] [PubMed] [Google Scholar]
- 36.Clauset A, Newman MEJ, Moore C (2004) Finding community structure in very large networks. Phys Rev E 70:66111. [DOI] [PubMed] [Google Scholar]
- 37.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA (2015) Structural templates for comparative protein docking. Proteins 83:1563–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruvinsky AM, Kirys T, Tuzikov AV, Vakser IA (2011) Side-chain conformational changes upon protein-protein association. J Mol Biol 408:356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruvinsky AM, Kirys T, Tuzikov AV, Vakser IA (2013) Ensemble-based characterization of unbound and bound states on protein energy landscape. Protein Sci 22:734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Camacho CJ, Vajda S (2002) Protein-protein association kinetics and protein docking. Curr Opin Struct Biol 12:36–40 [DOI] [PubMed] [Google Scholar]
- 42.Lensink MF, Wodak SJ (2013) Docking, scoring, and affinity prediction in CAPRI. Proteins 81:2082–2095 [DOI] [PubMed] [Google Scholar]
- 43.Tuszynska I, Bujnicki JM (2011) DARS-RNP and QUASI-RNP: New statistical potentials for protein-RNA docking. BMC Bioinformatics 12:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bliznyuk AA, Gready JE (1999) Simple method for locating possible ligand binding sites on protein surfaces. J Comput Chem 20 (9):983–988 [Google Scholar]
- 45.Porter KA, Xia B, Beglov D, Bohnuud T, Alam N, Schueler-Furman O, Kozakov D (2017) ClusPro PeptiDock: Efficient global docking of peptide recognition motifs using FFT. Bioinformatics 33:3299–3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bamborough P, Cohen F (1996) Modeling protein-ligand complexes. Curr Opin Struct Biol 6:236–241 [DOI] [PubMed] [Google Scholar]
- 47.Blom NS, Sygusch J (1997) High resolution fast quantitative docking using fourier domain correlation techniques. Proteins 27:493–506 [PubMed] [Google Scholar]
- 48.Kufareva I, Rueda M, Katritch V, participants GD, Stevens RC, Abagyan R (2011) Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 19:1108–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Padhorny D, Hall DR, Mirzaei H, Mamonov AB, Moghadasi M, Alekseenko A, Beglov D, Kozakov D (2018) Protein-ligand docking using FFT based sampling: D3R case study. J Comput Aded Mol Design 32:225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vakser IA (1996) Low-resolution docking: Prediction of complexes for underdetermined structures. Biopolymers 39:455–464 [DOI] [PubMed] [Google Scholar]
- 51.Johnson DK, Karanicolas J (2015) Selectivity by small-molecule inhibitors of protein interactions can be driven by protein surface fluctuations. PLoS Comp Biol 11:e1004081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levitt M (2009) Nature of the protein universe. Proc Natl Acad Sci USA 106:11079–11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kundrotas PJ, Zhu Z, Janin J, Vakser IA (2012) Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci USA 109:9438–9441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinha R, Kundrotas PJ, Vakser IA (2010) Docking by structural similarity at protein-protein interfaces. Proteins 78:3235–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sinha R, Kundrotas PJ, Vakser IA (2012) Protein docking by the interface structure similarity: How much structure is needed? PloS One 7:e31349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kundrotas PJ, Vakser IA (2013) Global and local structural similarity in protein-protein complexes: Implications for template-based docking. Proteins 81:2137–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kundrotas PJ, Anishchenko I, Dauzhenka T, Vakser IA (2018) Modeling CAPRI targets 110–120 by template-based and free docking using contact potential and combined scoring function. Proteins 86:302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vakser IA (2014) Protein-protein docking: From interaction to interactome. Biophys J 107:1785–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Z, Sutton SE, Wallenfang AJ, Orchard RC, Wu X, Feng Y, Chai J, Alto NM (2009) Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nature Struct Mol Biol 16:853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ford B, Verger D, Dodson K, Volkan E, Kostakioti M, Elam J, Pinkner J, Waksman G, Hultgren S (2012) The structure of the PapD-PapGII pilin complex reveals an open and flexible P5 pocket. J Bacteriol 194:6390–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi H, Xu R (2003) Crystal structure of the Drosophila Mago nashi – Y14 complex. Genes Dev 17:971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paaventhan P, Kong C, Joseph JS, Chung MCM, Kolatkar PR (2005) Structure of rhodocetin reveals noncovalently bound heterodimer interface. Protein Sci 14:169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.