

SUMMARY
The co‐evolution of bacterial plant pathogens and their hosts is a complex and dynamic process. Plant resistance can impose stress on invading pathogens that can lead to, and select for, beneficial changes in the bacterial genome. The Pseudomonas syringae pv. phaseolicola (Pph) genomic island PPHGI‐1 carries an effector gene, avrPphB (hopAR1), which triggers the hypersensitive reaction in bean plants carrying the R3 resistance gene. Interaction between avrPphB and R3 generates an antimicrobial environment within the plant, resulting in the excision of PPHGI‐1 and its loss from the genome. The loss of PPHGI‐1 leads to the generation of a Pph strain able to cause disease in the plant. In this study, we observed that lower bacterial densities inoculated into resistant bean (Phaseolus vulgaris) plants resulted in quicker PPHGI‐1 loss from the population, and that loss of the island was strongly influenced by the type of plant resistance encountered by the bacteria. In addition, we found that a number of changes occurred in the bacterial genome during growth in the plant, whether or not PPHGI‐1 was lost. We also present evidence that the circular PPHGI‐1 episome is able to replicate autonomously when excised from the genome. These results shed more light onto the plasticity of the bacterial genome as it is influenced by in planta conditions.
INTRODUCTION
Plant–pathogen interactions are complex and dynamic systems, reflecting the co‐evolution of host and parasite. Pathogens evolve to overcome the plant's defence mechanisms and to cause disease, and the plant adapts to nullify bacterial virulence and to resist invasion. The bacterial pathogens manipulate their hosts by delivering effector proteins into the plant cell cytoplasm through the conserved type III secretion system (TTSS) (Casper‐Lindley et al., 2002). Plants have evolved resistance proteins that recognize a subset of effectors, called avirulence (Avr) proteins, which trigger a defensive hypersensitive reaction (HR), generating an antimicrobial environment which leads to restricted bacterial colonization (Jones and Dangl, 2006). The antimicrobial conditions provided by plants undergoing the HR presents a strongly selective environment for bacterial strains that can avoid triggering resistance. Mutations in effector function, either by mutation of the gene itself or by the loss of larger pieces of DNA from the genome carrying the effector gene(s), are common. For example, the avr gene, avrPphE (hopX1), is present in all nine races of the bean pathogen Pseudomonas syringae pv. phaseolicola (Pph), but only races 2, 4, 5 and 7 are avirulent on cultivars of bean with the corresponding R2 resistance gene (Stevens et al., 1998). The other AvrPphE homologues have been inactivated via either single base pair changes in races 1, 3, 6 and 9 or by insertion of 104 bp in the allele of race 8. Inactivation of gene function can also be caused by gene disruption by mobile genetic elements, such as insertion sequences and transposons. Analysis of the Pseudomonas syringae pv. tomato (Pto) DC3000 genome sequence revealed 31 effectors and seven additional proteins secreted by TTSS (Buell et al., 2003) which, when compared with orthologues in the draft genome of Pseudomonas syringae pv. syringae (Psy) B728A, showed at least four of these effectors to be disrupted by mobile genetic elements. Stavrinides et al. (2006) used computational and evolutionary approaches to identify numerous mosaic and truncated TTSS effectors among animal and plant pathogens. They proposed the term ‘terminal reassortment’ to describe effectors whose termini are mobilized within the genome, creating random genetic fusions that result in chimeric genes that are subsequently selected for as a result of improved host colonization.
The entire coding region of effector genes may also be lost from the bacterial cell if the gene is carried on mobile DNA elements that can be lost during cell replication or through rare spontaneous deletions (Arnold et al., 2007). Previously, we have shown such a change to occur in the genome of Pph through the loss of a section of bacterial DNA, described as a genomic island (GI) (Jackson et al., 2000; Pitman et al., 2005). GIs are areas of the genome that are present only in certain strains of a bacterial species. They are often flanked by specific DNA sequences that contain direct repeats or tRNA loci and also carry genes coding for genetic mobility, such as phage genes, insertion sequence elements, integrases and transposases (Hacker and Carniel, 2001; Hacker and Kaper, 2000; van der Meer and Sentchilo, 2003). The 106‐kb GI, PPHGI‐1, including the effector avrPphB, is lost from the chromosome of Pph strain 1302A at high frequency when inoculated in planta in bean cultivar (cv.) Tendergreen (TG), which expresses the R3 resistance protein that detects AvrPphB (Pitman et al., 2005). Loss of the island bypasses R3/AvrPphB‐based detection and subsequently leads to the proliferation of the adapted strain in planta, resulting in the onset of disease. We found that the antimicrobial environment of the R3/AvrPphB‐induced HR imposes strong selective pressure for bacteria to lose the avrPphB gene. We have also shown, through a process of in planta bacterial transformation, that a nonchromosomal circular form of PPHGI‐1 can be transferred from one Pph strain to another (Lovell et al., 2009).
The aim of this study was to expand the work of Pitman et al. (2005) and to further investigate: (i) whether bacterial cell density and the host plant can affect the rate of PPHGI‐1 loss; (ii) whether bacterial genome changes (in addition to GI movement) occur in the bacteria during repeated re‐inoculation through plants (referred to as passaging); and (iii) whether PPHGI‐1 is capable of independent replication when in a nonchromosomal circular form.
RESULTS AND DISCUSSION
Rate of loss of PPHGI‐1 from Pph 1302A is cell density dependent
In the original studies, Pitman et al. (2005) passaged 8 × 107 colony‐forming units (cfu)/mL of Pph 1302A through resistant bean cv. TG leaves five times, and showed that, after the fifth passage, 98% of the recovered bacterial population had lost the GI, PPHGI‐1. Here, we expand this study by reproducing the experiment, but using an additional range of bacterial cell densities. Our hypothesis is that, by altering the bacterial : plant cell ratio to modify the plant symptoms and bacterial spatial distribution within the plant tissue, altered rates of loss of PPHGI‐1 will be observed. Cell suspensions of Pph 1302A between 8 × 101 and 5 × 108 cfu/mL were inoculated into TG leaves, and the plants were incubated in a growth cabinet for 48 h before symptoms were observed (Fig. 1). Inocula of 5 × 108, 8 × 107, 8 × 106 and 8 × 105 cfu/mL were chosen to follow island loss, in order to ensure the recovery of sufficient numbers of bacteria for pod testing. At each of these concentrations, a visible HR developed after 48 h, but tissue collapse was patchy rather than confluent with the lowest level of inoculum. Pph 1302A suspensions at these four cell densities were passaged six times through resistant bean cv. TG, as described in Pitman et al. (2005). At each passage (7 days), the harvested bacterial cells were re‐diluted to the original starting cell density before being re‐inoculated into new leaves and incubated for a further 7 days. A sample from each passage was plated onto King's B (KB) agar and 200 colonies were tested on TG pods for the loss of PPHGI‐1, which was observed as a change from the HR to a disease response. Polymerase chain reaction (PCR) was used to confirm that this change in virulence was caused by a loss of PPHGI‐1 containing avrPphB (data not shown). The rate of PPHGI‐1 loss from the population was observed to be cell density dependent (Fig. 2). The lower the inoculum density, the more quickly PPHGI‐1 was lost. For example, after four passages at 8 × 107 cfu/mL, there was a 42% PPHGI‐1 loss from the population, compared with losses of 60% and 75% from 8 × 106 and 8 × 105 cfu/mL, respectively. By passage six, 5 × 108 cfu/mL showed only 35% loss of PPHGI‐1, whereas the other three concentrations recorded between 96% and 98% loss.
Figure 1.

Phenotypes exhibited by resistant bean cv. Tendergreen (TG) inoculated with various Pseudomonas syringae pv. phaseolicola (Pph) 1302A cell densities. Ten‐day‐old TG leaves were inoculated with 500 µL Pph 1302A, at concentrations of 5 × 108, 8 × 107, 8 × 106, 8 × 105 and 8 × 104 colony‐forming units (cfu)/mL. Hypersensitive reaction (HR) symptoms were visible at 8 × 105 cfu/mL and above after 72 h. Cell densities between 8 × 104 and 8 × 101 cfu/mL were also tested, but are not illustrated as they produced no visible HR symptoms.
Figure 2.
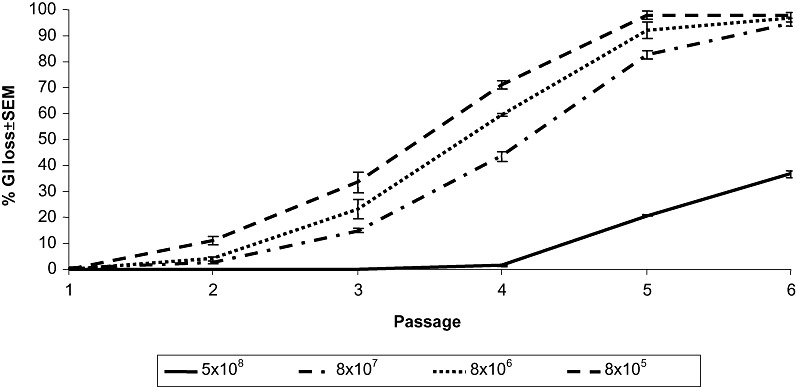
Loss of the Pseudomonas syringae pv. phaseolicola (Pph) genomic island (PPHGI‐1) from Pph 1302A is cell density dependent. Four cell densities of 5 × 108, 8 × 107, 8 × 106 and 8 × 105 colony‐forming units (cfu)/mL were passaged six times through the resistant bean cv. Tendergreen (TG). At each passage, 200 colonies were tested on TG pods and the number of disease‐causing colonies was recorded (i.e. colonies that had lost PPHGI‐1). The values are the means of three replicates ± standard error of the mean (SEM). At passage four, one way analysis of variance (anova) shows the rate of PPHGI‐1 loss to be significantly different (P < 0.05) for each inoculum concentration.
The model proposed by Arnold et al. (2007), describing the evolution of virulent bacteria within the leaf, may provide a theory to explain the findings in Fig. 2. On inoculation into TG leaves, Pph 1302A expresses AvrPphB, which triggers the HR; in turn, the antimicrobial conditions generated by the HR trigger the excision of PPHGI‐1. In a resistant host, there is selection for cells lacking avrPphB and therefore, under these conditions, PPHGI‐1 can be lost from the cell completely. Bacteria lacking avrPphB can come into contact with plant cells that have not been induced for HR activation and can divert the plant metabolism to favour bacterial growth, allowing virulent bacterial colonies to expand rapidly. However, if the HR is triggered in these plant cells by bacteria that retain avrPphB, the bacteria having lost avrPphB will still be subject to the effects of the HR and will die. This model supports the data in Fig. 2 by showing that the lower the density of bacterial cells, the higher the likelihood that evolved virulent Pph 1302A cells will be able to escape the effects of the HR. The ratio of infiltrated bacteria to plant cells is lower, thus enabling emerging virulent forms to proliferate rapidly.
The total loss of PPHGI‐1 from the population was not observed with any of the four inoculum cell densities tested. This raises the question: can PPHGI‐1 be lost from the genome of this remaining subpopulation of cells or has it become fixed in the chromosome in some way to ensure its survival? To address this question, eight colonies still causing the HR after six passages through TG were selected and grown overnight in Luria–Bertani (LB) broth. PCR was used to detect whether PPHGI‐1 could be excised from these cells by testing for the presence of a circular intermediate. Pitman et al. (2005) designed primers directed outwards from the internal boundaries of PPHGI‐1, which amplified the junction at which the two ends join to form the circular intermediate, and hence produced a product only when PPHGI‐1 was in its excised circular form. All eight bacterial cell suspensions tested had an amplifiable junction of the circular intermediate form of PPHGI‐1 (data not shown), indicating that PPHGI‐1 had not become fixed in the genome of this subpopulation of cells.
Race–cultivar HR drives selection for loss of PPHGI‐1
The rapid emergence of evolved virulent genotypes of Pph has been described previously following the expression of varietal resistance. However, we only observed a very low loss (less than 2%) of PPHGI‐1 when Pph 1302A was passaged through a susceptible cultivar of bean, presumably because there is no selection for a population without PPHGI‐1. Here, we expanded this work to test whether the stress of plant resistance encountered when Pph infects other plants could lead to GI loss. We used the stress induced by the basal resistance (no HR) of Arabidopsis thaliana ecotype Niedersenz (Nd‐0), nonhost HR of tobacco (White Burley) and, as a control, the race–cultivar HR of bean cv. TG. Both wild‐type Pph 1302A and 1302A::avrPphB (a knockout of avrPphB in Pph 1302A; Pitman et al., 2005) were passaged six times through all three plants. Only wild‐type Pph 1302A passaged through resistant bean cv. TG lost PPHGI‐1 from the majority of the population (Fig. 3). This suggests that only the stress generated by the race–cultivar HR is capable of selecting for cells that have lost PPHGI‐1 from the population. Pph 1302A::avrPphB generates no HR in bean, and hence there is very little loss of PPHGI‐1. Likewise, the two Pph 1302A strains do not generate an HR in Arabidopsis Nd‐0, and the conditions therefore do not ensure selection for cells which have lost PPHGI‐1. Nevertheless, after six passages, a very small proportion, 7%, had lost PPHGI‐1. An HR is generated in tobacco by an unknown nonhost resistance–avr gene interaction. PPHGI‐1 loss does not prevent the HR from being triggered in tobacco, and there is therefore no selection for disease‐causing strains. Under the conditions in tobacco, a very small number of bacteria, ∼4%, lost PPHGI‐1 after six passages. Although these levels of PPHGI‐1 loss are low, they are, however, higher than those observed in vitro, where PPHGI‐1 loss was almost never observed (only one in 12 000 colonies tested; Pitman et al., 2005). From these results, it can be concluded that only a race–cultivar HR, such as that generated by the AvrPphB–R3 interaction. provides the conditions needed to activate excision and, importantly, select for strains lacking PPHGI‐1.
Figure 3.
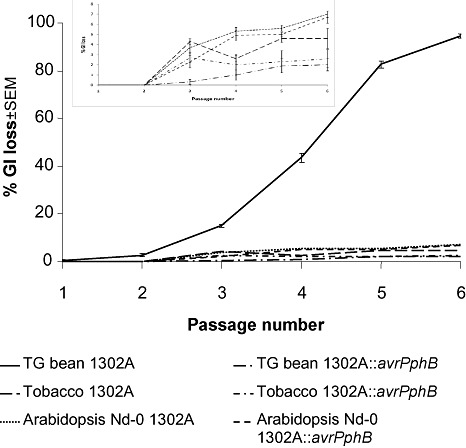
Loss of the Pseudomonas syringae pv. phaseolicola (Pph) genomic island (PPHGI‐1) is driven by selection caused by race–cultivar stress. Wild‐type Pph 1302A and Pph 1302A::avrPphB were passaged six times through resistant bean cv. Tendergreen (TG), Arabidopsis Nd‐0 and tobacco. At each passage, 200 colonies were tested on TG pods for the loss of PPHGI‐1. Only wild‐type Pph 1302A lost PPHGI‐1 over time because of the presence of the race–cultivar‐generated hypersensitive reaction (HR), which drives the selection of cells that have lost PPHGI‐1, and hence avrPphB. The inset is an enlarged version of the bottom five strains. The values are the means of three replicates ± standard error of the mean (SEM).
Multiple genome changes occur within Pph 1302A when passaged through the resistant host
Although the loss of PPHGI‐1 was identified during race–cultivar HR, it remained possible that other changes occurred to the Pph genome during plant colonization. We used genome analysis to investigate this possibility. Pph 1302A was passaged six times through bean cv. TG leaves and, after each passage, one strain still causing the HR (retaining PPHGI‐1) and one strain causing disease (that had lost PPHGI‐1) were obtained. DNA from each of these 12 strains was digested with restriction enzyme SwaI and subjected to pulsed‐field gel electrophoresis (PFGE; Rainey et al., 1994). Multiple differences in banding patterns were observed between wild‐type Pph 1302A and both the HR‐ and disease‐producing passaged strains (Fig. 4). Visual analysis of the fragments produced by PFGE revealed five distinct banding profiles. Wild‐type Pph 1302A was designated as profile 1. All HR‐causing (retaining PPHGI‐1) passaged strains exhibited profiles 1, 2 or 3, whereas the disease‐causing (lost PPHGI‐1) passaged strains exhibited profiles 2, 3, 4 and 5. No disease‐causing strain had the same fragment pattern as the wild‐type strain.
Figure 4.
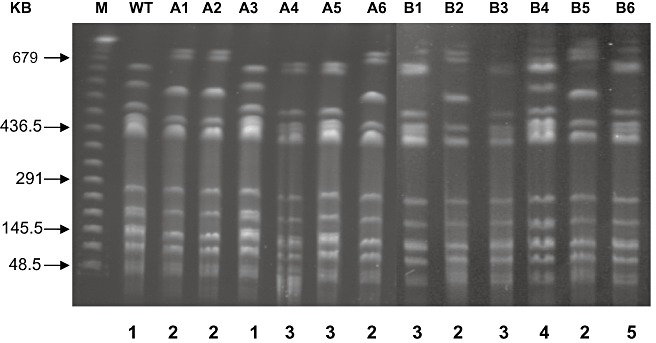
Pulsed‐field gel electrophoresis (PFGE) reveals genomic changes in Pseudomonas syringae pv. phaseolicola (Pph) 1302A that has been passaged through resistant bean cv. Tendergreen (TG). DNA of wild‐type Pph 1302A, together with one strain from each of six passages through TG that either caused a hypersensitive reaction (HR), or had lost PPHGI‐1 and hence caused disease, was subjected to PFGE, which revealed several genomic differences compared with the wild‐type. Lanes: M, DNA molecular marker; WT, wild‐type Pph 1302A; A, Pph 1302A passages 1–6, HR‐causing strains, B, Pph 1302A passages 1–6, disease‐causing strains. Numbers below the gels represent the different banding profiles assigned.
These results indicate that significantly more changes occur in the bacterial genome within the plant microenvironment than just the excision and loss of PPHGI‐1. Indeed, a number of changes occur when PPHGI‐1 remains in the strain. We also showed that just one passage through the plant, over 7 days, provides sufficient time for changes to occur within the Pph genome. In the future, next‐generation resequencing techniques should be employed on the evolved strains to document and quantify all genome changes that occur.
PPHGI‐1 appears as a plasmid in passaged strains of Pph 1302A
One possible cause of some of the genome changes observed may be alterations in the native plasmid content of the strains (Moulton et al., 1993). Plasmid profiles of the same 12 passaged Pph 1302A strains were examined to search for alterations. Wild‐type Pph 1302A has four plasmids, designated pAV505 (150 kb), pAV506 (50 kb), pAV507 (47 kb) and pAV508 (42 kb) (Jackson et al., 1999). The Pph 1302A strain used for these passaging experiments contained all four plasmids (Fig. 5A, B). No changes were observed in the native plasmid profiles of the disease‐causing strains, i.e. the strains that had lost PPHGI‐1; these strains retained the four native plasmids (Fig. 5A). This suggests that the genetic changes seen in PFGE of the disease‐causing strains are chromosomal changes or point mutations in SwaI sites in the plasmids. However, this is not the case for strains retaining PPHGI‐1 and therefore causing an HR (Fig. 5B). Five of the HR‐causing strains (1, 2, 4, 5 and 6) appeared to have lost the three largest native plasmids (pAV505, pAV506 and pAV507), with only the smallest native plasmid remaining. Intriguingly, they all also gained an extra plasmid of approximately 100 kb, a size expected for a circular form of PPHGI‐1 (106 kb). The DNA from the ∼100‐kb band was extracted from a plasmid profile gel and PCR was used to determine whether the four PPHGI‐1 genes were present in this band: these included soj (pph01), avrPphB (pph16), bacteriophytochrome (pph29) and xerC[pph100; open reading frame (ORF) numbers as in Pitman et al., 2005]. All four genes were amplifiable from the ∼100‐kb band DNA (data not shown). PCR was also used to amplify the PPHGI‐1 circular intermediate product from this band, i.e. the junction which is only present when PPHGI‐1 is in its circular form. To confirm that this band was not contaminated chromosomal DNA, primers for the amplification of the chromosomal avr gene avrPphE were also used and no amplification product was achieved (data not shown). These tests provide evidence that the observed ∼100‐kb band is PPHGI‐1 existing extrachromosomally as a circular molecule.
Figure 5.
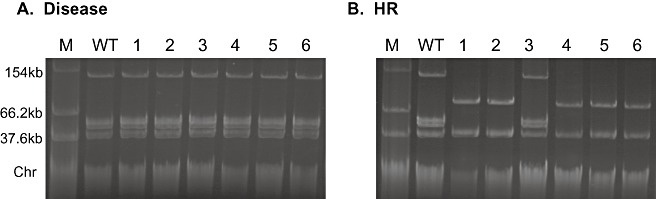
Changes in plasmid profiles of Pseudomonas syringae pv. phaseolicola (Pph) 1302A following passaging though resistant bean cv. Tendergreen (TG). Native plasmid profiles of wild‐type (WT) Pph 1302A, together with one strain from each of six passages through TG that either caused a hypersensitive reaction (HR), or had lost PPHGI‐1 and hence caused disease. (A) Disease‐causing strains retain the same native plasmid profile as WT Pph 1302A. (B) Native plasmid profiles of HR‐causing strains show that three native plasmids have been lost and an ∼100‐kb plasmid gained in five strains. Lanes: M, Escherichia coli 39R861 marker strain containing plasmids of known size; WT, wild‐type Pph 1302A; 1–6, Pph 1302A isolates from passages 1–6.
PCR was also used to determine whether the 52‐bp chromosomal att site from which PPHGI‐1 excises was occupied in each of the HR‐causing strains (1, 2, 4, 5 and 6). In all the disease‐causing strains, the att site was empty, as PPHGI‐1 had been lost from the bacteria (data not shown). In the HR‐causing strains, it was expected that the att site would be occupied as PPHGI‐1 is retained, except in those strains in which PPHGI‐1 appears as an extrachromosomal band. However, in all cases, the att site was occupied which, in the case of the HR passages 1, 2, 4, 5 and 6, indicated that something else had inserted into the att site. PCR was also used to amplify virPphA (Jackson et al., 1999), which is present on the largest native plasmid pAV505, and was amplifiable in all passaged strains. This was unexpected for the HR passages 1, 2, 4, 5 and 6 as they appeared to have lost the plasmid band for pAV505 in Fig. 5B.
These results suggest that pAV505, or either of the other pAV plasmids, may have integrated into the chromosome, possibly into the att site from which PPHGI‐1 was excised, which could explain why PCR shows the att site to be occupied in these strains. Plasmid integration has been observed in Pph previously; Szabo and Mills (1984) reported that a strain of Pph contained a plasmid, pMC7105, which could replicate autonomously or integrate into the bacterial chromosome. Interestingly, they also showed that imprecise excision of the integrated form of pMC7105 resulted in the formation of plasmids that ranged in size from approximately 35 to 270 kb, some of which contained large segments of chromosomal DNA.
PPHGI‐1 contains a previously unidentified plasmid origin of replication (oriV)
A question that remains is whether PPHGI‐1 can replicate independently of the host chromosome. Although bioinformatic analysis of the PPHGI‐1 sequence has failed to reveal a gene coding for a replication protein (Rep), further sequence comparisons have now revealed a putative origin of plasmid replication site (oriV) (Fig. 6A). The potential oriV can be found between ORFs pph08 and pph10, as designated by Pitman et al. (2005). This oriV site, and the ORFs surrounding it, have a similar structure to the putative oriVs described for P. aeruginosa C GI pKLC102 (Klockgether et al., 2004) and P. fluorescens Pf‐5 GI PFGI‐1 (Mavrodi et al., 2009). Between pph08 and pph10, there is an area of DNA that contains seven copies of a 36‐bp imperfect direct repeat, 5′CCcCtGgaATCACttccaGCacCgatCtgGGTGATg3′, that may act as iterons, which provide binding sites for Rep proteins and are involved in replication and partitioning. Other features of oriV sites include A + T‐rich regions in which host initiation factors bind and open DNA. There are also a number of inverted repeats in the PPHGI‐1 putative oriV site and an A + T‐rich region at the 3′ end of pph09. As with the other two putative oriVs, the oriV in PPHGI‐1 is also flanked by genes encoding proteins putatively involved in plasmid replication, including DnaB (pph03), ParB (pph07) and a single‐stranded binding protein (pph12). Taken together, this evidence suggests that PPHGI‐1 may be capable of replication outside of the host chromosome if a suitable Rep protein is available from elsewhere in the genome (chromosome or plasmids).
Figure 6.
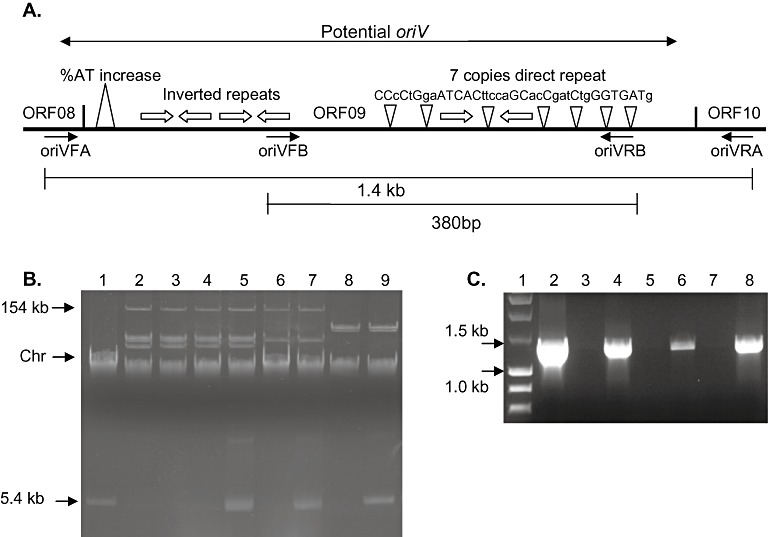
The Pseudomonas syringae pv. phaseolicola (Pph) genomic island (PPHGI‐1) contains a previously unidentified origin of plasmid replication. (A) Diagram of potential oriV on PPHGI‐1 and position of primers used to amplify and clone oriV. (B) Plasmid profiles of Pseudomonas strains transformed with cloned oriV. Lanes: 1, Escherichia coli containing pCR2.1 + oriVA; 2, Pph 1302A; 3, Pph 1302A transformed with pCR2.1 + oriVA; 4, Pph RJ3; 5, Pph RJ3 transformed with pCR2.1 + oriVA; 6, Pph 1448A; 7, Pph 1448A transformed with pCR2.1 + oriVA; 8, Pseudomonas syringae pv. tomato (Pto) DC3000; 9, Pto DC3000 transformed with pCR2.1 + oriVA. (C) The 5.4‐kb free plasmids from (B) are confirmed as containing cloned oriVA by PCR amplification using primers oriVFA and oriVRA from (A). Lanes: 1, DNA molecular marker; 2, Pph 1302A; 3, Pph RJ3; 4, 5.4‐kb plasmid from Pph RJ3; 5, Pph 1448A; 6, 5.4‐kb plasmid from Pph 1448A; 7, Pph DC3000; 8, 5.4‐kb plasmid from Pph DC3000. Chr, chromosome band.
Cloning of oriV suggests that PPHGI‐1 is able to replicate independently
To investigate whether the putative oriV identified was sufficient to allow independent replication of PPHGI‐1, two sets of PCR primers were designed for its amplification. One pair was used to amplify the entire oriV (oriVA) and a second pair to amplify only a section of the oriV (oriVB) truncating both the direct and inverted repeats (Fig. 6A). The PCR‐amplified fragments were then cloned into commercial vector pCR2.1 and electroporated into various Pseudomonas strains (Table 1). Vector pCR2.1 is unable to replicate in Pseudomonas; however, transformants of Pph RJ3 (1302A minus PPHGI‐1), Pph 1448A and Pto DC3000, harbouring pCR2.1 + oriVA, were obtained, and the construct was clearly visible on a plasmid profile gel (Fig. 6B). The detection of the plasmid construct indicates that pCR2.1 + oriVA is able to replicate in Pseudomonas strains. PCR was further used to confirm that the 5.4‐kb plasmid present in the transformed strains (Fig. 6B) did in fact contain oriVA (Fig. 6C).
Table 1.
Transfer frequency of a potential origin of replication from the Pseudomonas syringae pv. phaseolicola genomic island (PPHGI‐1) into various Pseudomonas strains.
| Strain | Transfer frequency* | |
|---|---|---|
| oriVA | oriVB | |
| Pph 1302A | 6.2 ± 0.8 | 0 |
| Pph RJ3 | 3.4 ± 0.7 | 0 |
| Pph 1448A | 3.8 ± 0.7 | 0 |
| Pto DC3000 | 1.6 ± 0.3 | 0 |
The whole potential oriV (oriVA), or a truncated fragment (oriVB), was cloned into pCR2.1. These constructs were electroporated into various Pseudomonas strains, and the transfer frequencies were calculated as the number of transformants per microgram of DNA used. A control of pCR2.1 containing a 150‐bp fragment of virB4 (pph84) and no oriV DNA was also tested, and produced no transformants in any Pseudomonas strain.
Colony‐forming units (cfu)/µg DNA ± SEM × 102.
Pto, Pseudomonas syringae pv. tomato; SEM, standard error of the mean.
Transformants of Pph 1302A (containing PPHGI‐1) were also obtained. However, the 5.4‐kb pCR2.1 + oriVA plasmid was not visible on the plasmid profile gel (Fig. 6B), indicating that the construct had probably recombined homologously with oriV present on PPHGI‐1 in the chromosome. The pCR2.1 + oriVB construct produced no transformants in any Pph strain, indicating that the truncated fragment of oriV, oriVB, is not sufficient to allow replication of pCR2.1. A control of vector pCR2.1 containing no oriV DNA produced no transformants in any of the four Pseudomonas strains tested, and nor did a small fragment of virB4 (pph84 of PPHGI‐1) cloned into pCR2.1. These results indicate that the putative oriV identified in PPHGI‐1 may be sufficient to allow the independent replication of PPHGI‐1 in Pph 1302A, although the mechanism of replication remains to be determined.
As mentioned previously, PPHGI‐1 loss is almost never observed in vitro (only one in 12 000 colonies tested; Pitman et al., 2005). However, in Pph 1302A containing vector pCR2.1 + oriVA integrated into the chromosomal oriV, oriV is disrupted. To investigate whether this disruption had any effect on PPHGI‐1 loss in vitro, Pph 1302A::oriVA, together with wild‐type Pph 1302A, were passaged four times through M9 minimal medium. Colonies were then replica plated onto KB agar and KB agar + kanamycin. Any colonies not growing on KB + kanamycin would have lost the pCR2.1 vector, indicating that they had also lost PPHGI‐1, which was confirmed by PCR (data not shown). No loss of PPHGI‐1 was seen from the wild‐type strain; however, in Pph 1302A::oriVA, the PPHGI‐1 loss rate was 4.7% ± 1.1%. The rates of loss detected indicate that disruption of oriV stops PPHGI‐1 being able to replicate outside of the chromosome, which, in turn, enables it to be lost from the bacterial cell during replication in vitro.
In conclusion, it can be seen that genome rearrangement in Pph is a highly dynamic process that can occur in a relatively short time frame. The rate of PPHGI‐1 loss depends on a number of factors, including the initial bacterial cell density and the temporal and spatial plant environments encountered by the bacteria. The shortest time point tested was 1 week (i.e. one passage) and genome changes could already be seen. Within 4 weeks, a GI can be lost from a majority of the population, thus leading to high levels of virulent individuals within it. It was also interesting to observe that PPHGI‐1 can form a circular episome containing genes which suggest that it can replicate independently of the bacterial chromosome. This ‘plasmid’ is rarely visualized, and we only observed it from strains that were recovered from resistant host plants. This suggests that the in planta conditions encountered during the HR drive the excision of PPHGI‐1 from the chromosome and that the plasmid oriV acts as a rescue system for the excised island during bacterial replication. However, when it is not under this stress, for example in vitro, PPHGI‐1 is generally stably integrated into the chromosome. In the future, it will be important to understand the mechanism and dynamics of a pathogen's genome plasticity if breeding for durable resistance is to be successful.
EXPERIMENTAL PROCEDURES
Bacterial strains and culture conditions
All bacterial strains and plasmids used in this study are listed in Table 2. Pseudomonas strains and Escherichia coli strains were cultured at 25 °C for 48 h on KB agar (Difco, Surrey, UK) or at 37 °C for 24 h on LB medium (Difco) containing 15 g/L Bacteriological No. 1 agar (Oxoid, Basingstoke, UK), respectively. Overnight cultures of both Pseudomonas and E. coli strains were cultured in LB broth for 18 h at the appropriate temperature. Medium was supplemented with 25 µg/mL kanamycin where appropriate.
Table 2.
Bacterial strains and plasmids.
| Strain | Description | Source/reference |
|---|---|---|
| Pseudomonas syringae pv. phaseolicola (Pph) 1302A | Cause of halo blight disease in bean. Race 4 | Taylor et al. (1996) |
| Pph 1448A | Race 6 | Mansfield et al. (1994) |
| Pph RJ3 | 1302A variant PPHGI‐1‐ | Jackson et al. (2000) |
| Pseudomonas syringae pv. tomato (Pto) DC3000 | Pathogenic to tomato and Arabidopsis | Cuppels (1986) |
| Pph 1302A::NCR* | NCR insertion mutant | Pitman et al. (2005) |
| Pph 1302A::avrPphB * | avrPphB insertion mutant | Pitman et al. (2005) |
| Pph 1302A HR—P1, P2, P3, P4, P5, P6† | 1302A HR‐causing passaged strains | This study |
| Pph 1302A D—P1, P2, P3, P4, P5, P6† | 1302A Disease‐causing passaged strains | This study |
| Pph 1302A::oriVA‡ | 1302A variant | This study |
| Escherichia coli 39R861 | Contains four plasmids used as size standards—154 kb, 66.2 kb, 37.6 kb and 7.2 kb | Threllfall et al. (1986) |
| Top10 | Chemically competent cells | Invitrogen |
| TOPO cloning vector pCR2.1 | KmR and ApR | Invitrogen |
Pph 1302A variant with cloning vector pCR2.1 inserted into selected region in PPHGI‐1, KmR.
Pph 1302A variants from each of six passages through Tendergreen (TG).
Pph variants with cloning vector pCR2.1 inserted into oriV in PPHGI‐1.
NCR, non‐coding region; KmR and ApR, kanamycin and ampicillin resistant.
Plant growth conditions and pathogenicity tests
Bean cultivars Tendergreen (TG), Canadian Wonder (CW) and tobacco (White Burley) were grown at 23 °C, 70% humidity, with a 16‐h photoperiod. Arabidopsis accession Niedersenz (Nd‐0) was grown at 20 °C, 70% humidity, with a 10‐h day. To test in planta excision, bacterial cultures were washed, resuspended in 10 mm MgCl2 and diluted to an optical density at 600 nm (OD600) of 0.1 (8 × 107 cfu/mL, unless otherwise stated) before being infiltrated into either bean leaves by syringe or Arabidopsis and tobacco by pressure infiltration. After 7 days, inoculated tissue was excised and homogenized in 10 mm MgCl2. The bacteria were recovered by brief centrifugation and diluted to the starting inoculum concentration with 10 mm MgCl2 before being re‐infiltrated into new leaves. This process was repeated six times (six passages). Serial dilutions of recovered bacteria were plated onto KB at each passage, and 200 single colonies from each passage were screened for disease symptoms or HR on TG pods. Screening of single colonies was performed by toothpick transfer from agar plate to surface piercing of pod. Inoculated pods were incubated on moist tissue for 4 days at room temperature and the symptoms were recorded daily. Disease was observed as water‐soaking around the inoculation site, whereas resistance (HR) was observed as tissue browning and often collapse. Pathogenicity tests were carried out on leaves of bean cultivars TG and CW using a bacterial concentration of 5 × 108 (unless otherwise stated) and incubated for 72 h before observation.
PFGE and plasmid profiling
Pph 1302A strains were subjected to PFGE following the method of Rainey et al. (1994) with the DNA digested with restriction enzyme SwaI (NEB, UK) at 25 °C for 6 h. PFGE was carried out at 6 V/cm with a ramped pulse time of 30–75 s for 21 h. Plasmid profiles were determined by extracting total uncut plasmid DNA from overnight cultures following the method of Moulton et al. (1993). DNA was extracted from excised bands from agarose gels, using a Qiaex II gel extraction kit (Qiagen, Crawley, UK), following the manufacturer's instructions.
PCR conditions
A standard 25‐µL PCR mix was used which consisted of the following: 40 ng total DNA or 1 µL overnight culture, 0.5 µm oligonucleotide primers (Table 3), 12.5 µL mastermix (Taq PCR mastermix; Qiagen) and 9.5 µL sterile deionized water. Standard PCR cycling conditions consisted of 94 °C for 10 min, followed by 30 cycles of 94 °C for 30 s (30 s at an annealing temperature appropriate to primers) and 72 °C for 1 min, followed by a final extension step of 72 °C for 10 min. For amplification of the circular intermediate, 40 cycles were used. After amplification, a 20‐µL sample was visualized on a 0.7% agarose gel with 2 µg/mL ethidium bromide in comparison with a DNA molecular marker (Hyperladder, Bioline, London, UK).
Table 3.
Oligonucleotide primers.
| Primer name | Primer sequence 5′–3′ | Target gene or location | Reference |
|---|---|---|---|
| HL1‐Fa | GGTGTCGGTAAAACCACTTTGGC | soj (PPHGI‐1, pph01) | Lovell et al. (2009) |
| HL1‐Ra | CCTCTACGGCGGATTCAGTCATC | ||
| HL4‐Fa | GGTCTGACAGTCCTTATCACAGC | Bacteriophytochrome (PPHGI‐1, pph29) | Lovell et al. (2009) |
| HL4‐Ra | GTTCTTACCCTCTCGACGCTCC | ||
| HL10‐Fc | ATTGCTGGGTGCCGGTATGG | virB4 (PPHGI‐1, pph84) | Lovell et al. (2009) |
| HL10‐Rc | GGCACAGGTTTGCAACATGC | ||
| IntF | GAACTGACCCAGGAGTACATCC | xerC (PPHGI‐1, pph100) | Pitman et al. (2005) |
| IntR | CGTTCCAGTCGATAGAACC | ||
| avrPphBA1 | GCGATTGCGTGTCCTTGA | avrPphB (PPHGI‐1, pph16) | Pitman et al. (2005) |
| avrPphBS1 | CTGTAAGACCTGAGCCTG | ||
| avrPphEF | ATCGTCATGGACCCGTGGTC | avrPphE | Lovell et al. (2009) |
| avrPphER | GACCGTCTTGACGCCTTCGG | ||
| virPphAF | CTCGGTGCTCGCTAACGAGC | virPphA | This study |
| virPphAR | GAGCTATATCCAAGGCTGCGC | ||
| LJatt | ATGTGCGATGAGGTCGAATATGC | Circular intermediate | Pitman et al. (2005) |
| Lratt | TATGCGTGGCCTCCAGTAGCTCTG | ||
| delF | CATTACAGCCGGTCACAGATG | att site | This study |
| delR | GGAGTCGCCCTCTGAGGGCA | ||
| oriVFA | ATGATGCACCGAATGCCGTGG | oriVA | This study |
| oriVRA | GATGCGGTTGAGGATGTTGGC | ||
| oriVFB | CCGCAAGATCGTGACGAGTC | oriVB | This study |
| oriVRB | GAATCACCCTGATCGGCGCT |
Cloning and transformation of oriV
PCR‐amplified oriV fragments, oriVA and oriVB, together with a small fragment of virB4 (pph84), were cloned into pCR2.1 and transformed into E. coli TOP10 chemically competent cells (TA cloning kit, Invitrogen, Paisley, UK). Plasmid DNA was extracted from successful transformants using the QIAprep spin miniprep kit (Qiagen), following the manufacturer's instructions, and quantified on a NanoDrop ND‐1000 spectrophotometer (Thermo Scientific, Wilmington, USA). Four Pseudomonas strains, including Pph 1302A, RJ3, 1448A and Pto DC3000, were electroporated (as Keen et al., 1992) with 50 ng plasmid DNA, and incubated at 25 °C with shaking for 3 h. Cells were plated onto KB + kanamycin and incubated at 25 °C for up to 72 h. Transformants were analysed by plasmid profiling and PCR.
ACKNOWLEDGEMENTS
We thank the Biotechnology and Biological Sciences Research Council for financial support (02972057). This work was carried out under the Department for Environment, Food and Rural Affairs' Plant Health and Seeds Inspectorate license number PHL 217A/6185.
REFERENCES
- Arnold, D.L. , Jackson, R.W. , Waterfield, N.R. and Mansfield, J.W. (2007) Evolution of microbial virulence: the benefits of stress. Trends Genet. 23, 293–300. [DOI] [PubMed] [Google Scholar]
- Buell, C.R. , Joardar, V. , Lindeberg, M. , Selengut, J. , Paulsen, I.T. , Gwinn, M.L. , Dodson, R.J. , Deboy, R.T. , Durkin, A.S. , Kolonay, J.F. , Madupu, R. , Daugherty, S. , Brinkac, L. , Beanan, M.J. , Haft, D.H. , Nelson, W.C. , Davidsen, T. , Zafar, N. , Zhou, L. , Liu, J. , Yuan, Q. , Khouri, H. , Fedorova, N. , Tran, B. , Russell, D. , Berry, K. , Utterback, T. , Van Aken, S.E. , Feldblyum, T.V. , D'Ascenzo, M. , Deng, W.L. , Ramos, A.R. , Alfano, J.R. , Cartinhour, S. , Chatterjee, A.K. , Delaney, T.P. , Lazarowitz, S.G. , Martin, G.B. , Schneider, D.J. , Tang, X. , Bender, C.L. , White, O. , Fraser, C.M. and Collmer, A. (2003) The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. USA, 100, 10 181–10 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper‐Lindley, C. , Dahlbeck, D. , Clark, E.T. and Staskawicz, B.J. (2002) Direct biochemical evidence for type III secretion‐dependent translocation of the AvrBs2 effector protein into plant cells. Proc. Natl. Acad. Sci. USA, 99, 8336–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuppels, D.A. (1986) Generation and characterisation of Tn5 insertion mutations in Pseudomonas syringae pv. tomato . Appl. Environ. Microbiol. 152, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker, J. and Carniel, E. (2001) Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker, J. and Kaper, J.B. (2000) Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679. [DOI] [PubMed] [Google Scholar]
- Jackson, R.W. , Athanassopoulos, E. , Tsiamis, G. , Mansfield, J.W. , Sesma, A. , Arnold, D.L. , Gibbon, M.J. , Murillo, J. , Taylor, J.D. and Vivian, A. (1999) Identification of a pathogenicity island, which contains genes for virulence and avirulence, on a large native plasmid in the bean pathogen Pseudomonas syringae pathovar phaseolicola . Proc. Natl. Acad. Sci. USA, 96, 10 875–10 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, R.W. , Mansfield, J.W. , Arnold, D.L. , Sesma, A. , Paynter, C.D. , Murillo, J. , Taylor, J.D. and Vivian, A. (2000) Excision from tRNA genes of a large chromosomal region, carrying avrPphB, associated with race change in the bean pathogen, Pseudomonas syringae pv. phaseolicola . Mol. Microbiol. 38, 186–197. [DOI] [PubMed] [Google Scholar]
- Jones, J.D.G. and Dangl, J.L. (2006) The plant immune system. Nat. Rev. 444, 323–329. [DOI] [PubMed] [Google Scholar]
- Keen, N.T. , Shen, H. and Cooksey, D.A. (1992) Introduction of cloned DNA into plant pathogenic bacteria In: Molecular Plant Pathology. A Practical Approach (Gurr S.J., McPherson M.J. and Bowles D.J., eds), pp. 45–50. Oxford: IRL Press. [Google Scholar]
- Klockgether, J. , Reva, O. , Larbig, K. and Tummler, B. (2004) Sequence analysis of the mobile genome island pKLC102 of Pseudomonas aeruginosa C. J. Bacteriol. 186, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell, H.C. , Mansfield, J.W. , Godfrey, S.A.C. , Jackson, R.W. , Hancock, J.T. and Arnold, D.L. (2009) Bacterial evolution by genomic island transfer occurs via DNA transformation in planta. Curr. Biol. 19, 1586–1590. [DOI] [PubMed] [Google Scholar]
- Mansfield, J. , Jenner, C. , Hockenhull, R. , Bennet, M.A. and Stewart, R. (1994) Characterisation of avrPphE, a gene for cultivar‐specific avirulence from Pseudomonas syringae pv. phaseolicola which is physically linked to hrpY, a new hrp gene identified in the halo blight bacterium. Mol. Plant–Microbe Interact. 7, 726–739. [DOI] [PubMed] [Google Scholar]
- Mavrodi, D.V. , Loper, J. , Paulsen, I.T. and Thomashow, L.S. (2009) Mobile genetic elements in the genome of the beneficial rhizobacterium Pseudomonas fluorescens Pf‐5. BMC Microbiol. 9, 8–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer, J.R. and Sentchilo, V. (2003) Genomic islands and the evolution of catabolic pathways in bacteria. Curr. Opin. Biotechnol. 14, 248–254. [DOI] [PubMed] [Google Scholar]
- Moulton, P.J. , Vivian, A. , Hunter, P.J. and Taylor, J.D. (1993) Changes in cultivar‐specificity toward pea can result from transfer of plasmid RP4 and other incompatibility group P1 replicons to Pseudomonas syringae pv. pisi . J. Gen. Microbiol. 139, 3149–3155. [DOI] [PubMed] [Google Scholar]
- Pitman, A. , Jackson, R.W. , Mansfield, J.W. , Kaitell, V. , Thwaites, R. and Arnold, D.L. (2005) Exposure to host resistance mechanisms drives evolution of bacterial virulence in plants. Curr. Biol. 15, 2230–2235. [DOI] [PubMed] [Google Scholar]
- Rainey, P.B. , Bailey, M.J. and Thompson, I.P. (1994) Phenotypic and genotypic diversity of fluorescent pseudomonads isolated from field‐grown sugar beet. Microbiology, 140, 2315–2331. [DOI] [PubMed] [Google Scholar]
- Stavrinides, J. , Ma, W. and Guttman, D.S. (2006) Terminal reassortment drives the quantum evolution of type III effectors in bacterial pathogens. PLoS Pathog. 2, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, C. , Bennett, M.A. , Athanassopoulos, E. , Tsiamis, G. , Taylor, J.D. and Mansfield, J.W. (1998) Sequence variations in alleles of the avirulence gene avrPphE.R2 from Pseudomonas syringae pv. phaseolicola lead to loss of recognition of the AvrPphE protein within bean cells and a gain in cultivar‐specific virulence. Mol. Microbiol. 29, 165–177. [DOI] [PubMed] [Google Scholar]
- Szabo, L.J. and Mills, D. (1984) Integration and excision of pMC7105 in Pseudomonas syringae pv. phaseolicola: involvement of repetitive sequences. J. Bacteriol. 157, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J.D. , Teverson, D.M. , Allen, D.J. and Pastor‐Corrales, M.A. (1996) Identification and origin of races of Pseudomonas syringae pv. phaseolicola from Africa and other bean growing areas. Plant Pathol. 45, 469–478. [Google Scholar]
- Threllfall, E.J. , Rowe, B. , Ferguson, J.L. and Ward, L.R. (1986) Characterisation of plasmids conferring resistance to gentamycin and apramycin in strains of Salmonella typhimurium phage type 204c isolated in Britain. J. Hyg. 97, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]