

SUMMARY
Barley yellow mosaic virus (BaYMV), the type species of the genus Bymovirus in the family Potyviridae in the picornavirus‐like superfamily, causes a yellow mosaic disease of winter barley with significant yield losses in Europe and East Asia. Until now, infectious in vitro transcripts for the bipartite plus‐sense RNA genome of any bymovirus species have not been available, rendering molecular analyses of bymovirus pathogenicity and the host resistance mechanisms difficult. In this study, we constructed the first cDNA clones of BaYMV RNA1 and RNA2, from which infectious RNA can be transcribed in vitro. Using in vitro transcripts, we showed that RNA1, which encodes eight proteins, including a viral proteinase NIa‐Pro, the RNA‐dependent RNA polymerase NIb, genome‐linked viral protein VPg and the capsid protein CP, replicated autonomously in barley mesophyll protoplasts in the absence of RNA2 optimally at 15 °C, a temperature similar to the optimum for causing disease in barley fields. For systemic infection of barley plants, RNA1 alone was not sufficient and RNA2 was also required. Of the two proteins encoded on RNA2 (P1 with cysteine proteinase activity and P2 with unknown functions), P1 was essential and P2 was dispensable for systemic infectivity. The expression of both P1 and P2, but not the precursor polyprotein, together with RNA1 increased systemic infection and caused mosaic leaf symptoms. The infectious cDNA clones of BaYMV will be vital for future studies of bymovirus–host–vector interactions at the molecular level.
INTRODUCTION
The genus Bymovirus in the family Potyviridae includes at least six species of agriculturally important viruses infecting barley, wheat, rice and oat plants, which typically cause mosaic diseases with different degrees of severity depending on the virus strains and plant cultivars (Berger et al., 2005; Kanyuka et al., 2003; Kühne, 2009). All known bymoviruses are transmitted in soil by zoospores of Polymyxa graminis, an obligate parasite belonging to Plasmodiophoraceae, and can be retained inside the thick‐walled resting spores for years (Adams et al., 1988; Kanyuka et al., 2003). As a result of the long viability of resting spores in soil and root debris, it is impossible to eliminate bymovirus from fields once infested by viruliferous P. graminis. The growth of resistant cultivars is the only practical way to avoid crop losses caused by bymovirus infection in such fields (Kühne, 2009).
Barley yellow mosaic virus (BaYMV) is the type species of the genus Bymovirus (Berger et al., 2005). It has only one natural host, barley (Hordeum vulgare), and causes a yellow mosaic disease at 13–16 °C in fields (Kusaba et al., 1971). BaYMV was first reported in Japan in 1940 (Ikata and Kawai, 1940) and, since then, has been reported in China and Korea, as well as in European countries including Germany and the UK (Chen et al., 1999; Davidson et al., 1991; Kashiwazaki et al., 1989; Kühne et al., 2003; Lee et al., 2006; Nishigawa et al., 2008; Peerenboom et al., 1992; Shi et al., 1996). The virus has a bipartite positive‐sense RNA genome, composed of 7.6‐kb RNA1 and 3.6‐kb RNA2, which are separately encapsidated in slightly flexuous virions of two different lengths. RNA1 has a long open reading frame (ORF), translated into a polyprotein, which is believed to be cleaved by nuclear inclusion protein a‐proteinase (NIa‐Pro) into eight final products [P3/6K1/cytoplasmic inclusion protein (CI)/6K2/genome‐linked viral protein (VPg)/NIa‐Pro/nuclear inclusion protein b (NIb)/capsid protein (CP)] (Adams et al., 2005). In addition, there is an overlapping gene in the P3‐coding region, termed pipo, translated by a +2 frame‐shift to express a short polypeptide fused with the N‐terminal region of P3 (Chung et al., 2008). RNA2 is translated into a single polyprotein, which is believed to be cleaved by P1 proteinase into two final proteins (P1/P2) (Adams et al., 2005). Except for CP encoded on RNA1, all the other products are putative, identified by sequence similarities and motif comparisons with other well‐characterized potyviruses (Adams et al., 2005; Chung et al., 2008; Urcuqui‐Inchima et al., 2001).
Previously, no system of reverse genetics for any bymovirus has been well characterized. Infectious cDNA clones for RNA1 and RNA2 of Barley mild mosaic virus (BaMMV), another species in the genus Bymovirus, using the Cauliflower mosaic virus 35S promoter for the transcription of the infectious RNA in host nuclei have been described, but the systemic infectivity to barley plants was lower than 10% (Meyer and Dessens, 1997). Infectious BaMMV RNA2 transcripts from a full‐length cDNA clone have also been described (Timpe and Kühne, 1995), but not for RNA1. Unfortunately, these cDNA clones have not been used successfully to study the molecular biological aspects of interactions between the virus, host plants and transmission vector (Habekuss et al., 2008; Jacobi et al., 1995; Kanyuka et al., 2004; Kashiwazaki and Hibino, 1996; Peerenboom et al., 1997). For BaYMV, there are no infectious cDNA clones available to study aspects of the biology of BaYMV, such as P. graminis‐mediated soil transmission (Toyama and Kusaba, 1970), the requirement for low temperature for disease manifestations (Kusaba et al., 1971) and virus pathogenicity. At present, four pathological types of BaYMV (I, II, III and IV) have been identified in Japan based on their responses to different barley cultivars (Kashiwazaki et al., 1989; Konishi et al., 1997; Nishigawa et al., 2008; Okada et al., 2004). In Europe, two strains have been distinguished by their responses to cultivars carrying the rym4 resistance gene (Kühne et al., 2003); an interaction between the host eIF4E encoded by rym4 and virus VPg has been suggested for susceptibility (Kanyuka et al., 2005; Stein et al., 2005).
In this article, we report the construction of full‐length cDNA clones for both RNA1 and RNA2 of the Japanese K05 isolate of BaYMV (BaYMV‐JK05), designated as pBY1 and pBY2, respectively. Using in vitro transcripts from these clones, we show that RNA1 replicates autonomously in barley protoplasts most efficiently at 15 °C, that both RNA1 and RNA2 are required for the systemic infection of barley plants (cv. Ryofu), and that RNA2‐encoded P1 protein is essential for systemic infection, whereas P2 may facilitate virus movement and symptom development.
RESULTS
Genome characterization of the BaYMV‐JK05 isolate
Complete nucleotide sequences of BaYMV‐JK05 RNA1 and RNA2 were determined directly from reverse transcriptase‐polymerase chain reaction (RT‐PCR) products derived from viral RNA using a panel of primers (Table S1A, see Supporting Information). RNA1 was 7642 nucleotides long, excluding its 3′ poly(A) tail. A long (7239 nucleotides) ORF started at base 172 and terminated at base 7410. It encoded a 2412‐amino‐acid, 271‐kDa polyprotein which is supposedly cleaved into 38‐kDa P3, 7‐kDa 6K1, 73‐kDa CI, 14‐kDa 6K2, 22‐kDa VPg, 25‐kDa NIa‐Pro, 60‐kDa NIb and 32‐kDa CP, as shown in Fig. 1A. The 3′ untranslated region (UTR) was 232 nucleotides long. In addition, a +2 reading frame (bases 681–944) led by a GGAAAAAA motif (bases 676–683) was present within the P3 cistron on RNA1, which may be translated into an 87‐amino‐acid PIPO protein, as predicted by Chung et al. (2008). RNA2 was 3586 nucleotides long, excluding the 3′ poly(A) tail. It contains a single ORF (2673 nucleotides long, bases 155–2827), which encodes the 890‐amino‐acid (98‐kDa) precursor polyprotein P12. This polyprotein is proposed to be cleaved into the 255‐amino‐acid (28‐kDa) P1 and 635‐amino‐acid (70‐kDa) P2 proteins, as shown in Fig. 1B. The 3′ UTR was 759 nucleotides long. Phylogenetic analysis of the polyproteins encoded on RNA1 (Fig. S1A, see Supporting Information) and RNA2 (Fig. S1B, see Supporting Information) showed that BaYMV‐JK05 was included in the East Asian cluster, but distinct from all other isolates characterized so far.
Figure 1.
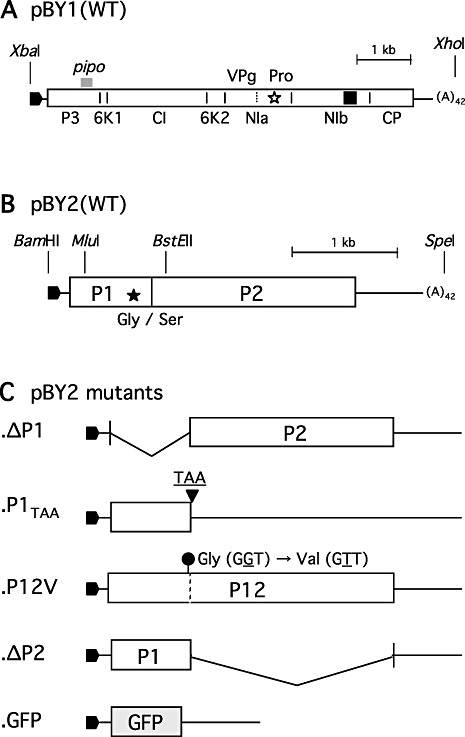
The genome organizations of Barley yellow mosaic virus (BaYMV) RNA1, RNA2 and RNA2 mutants shown as their full‐length cDNA constructs. (A) pBY1, the full‐length cDNA clone for wild‐type (WT) RNA1. A rectangular box indicates a 271‐kDa polyprotein, which is cleaved into eight proteins by a serine proteinase activity residing in the C‐terminal region of nuclear inclusion protein a (NIa) (indicated by an open star). A thick grey bar above P3 indicates a putative pipo gene expressed by a +2 frame‐shift at GGAAAAAA (nucleotides 676–683) (Chung et al., 2008). Putative cleavage sites are shown by short vertical bars, and the names of the cleaved products are written below the box. A dotted vertical bar in NIa indicates an internal cleavage into the N‐terminal genome‐linked viral protein (VPg) and the C‐terminal nuclear inclusion protein a‐proteinase (NIa‐Pro). A filled square in nuclear inclusion protein b (NIb) indicates the position of the GDD RNA‐dependent RNA‐polymerase motif. (B) pBY2, the full‐length cDNA clone for WT RNA2. A rectangular box indicates a 98‐kDa polyprotein, which is cleaved into P1 and P2 at a glycine–serine bond by a cysteine proteinase activity residing in the C‐terminal region of P1 (indicated by a filled star). (C) Five pBY2 mutants, which were prepared so that P1, P2, P12 or green fluorescent protein (GFP) could be expressed as mentioned in Experimental procedures. Rightward filled pentagons at the 5′ end indicate the T7 promoter.
Infectivity of in vitro transcripts in barley mesophyll protoplasts
The 32‐kDa CP could be detected from protoplasts transfected with RNA1 transcripts (Fig. 2A, α‐CP) using anti‐CP serum. The accumulation level of CP increased several fold when protoplasts were transfected with both RNA1 and RNA2 transcripts, indicating that RNA2 or products from RNA2 enhance the expression of CP. Except for the 32‐kDa CP, other larger proteins corresponding to precursor polyproteins were not detectable, indicating that cleavage between the C‐terminus of NIb and the N‐terminus of CP was complete. Using anti‐VPg serum, 22‐kDa, 36‐kDa and 61‐kDa proteins were detected from protoplasts transfected with RNA1 transcripts alone, which were identified by size as VPg, 6K2/VPg and 6K2/NIa, respectively (Fig. 2A, α‐VPg). The detection of these precursor polyproteins suggested that processing at 6K2/VPg and VPg/NIa‐Pro cleavage sites was incomplete. Similar incomplete digestion by NIa‐Pro has also been reported for Tobacco etch virus (Carrington et al., 1993). The 47‐kDa NIa (consisting of N‐terminal VPg and C‐terminal Pro) was probably masked by co‐migration with the large subunit of ribulose bisphosphate carboxylase/oxygenase (Rubisco) at 53 kDa (NCBI Reference Sequence: YP_874661) in sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). The presence or absence of RNA2 transcripts in the inoculum did not alter the accumulation of these three proteins. We speculate that the level of NIa expression may be increased in the presence of RNA2, as was the case for CP, but could not be seen as a result of co‐migration with the Rubisco large subunit, as indicated by an asterisk in Fig. 2A (α‐VPg). From protoplasts transfected with both RNA1 and RNA2 transcripts, 28‐kDa P1 was detected using anti‐P1 serum (Fig. 2A, α‐P1), but the precursor P12 was not. P1 was not detected in protoplasts infected with RNA2 alone (data not shown). These results indicate that RNA1 replicates autonomously in cells and that the replication of RNA2 is dependent on RNA1. The transfection efficiency for barley mesophyll protoplasts (cv. Minorimugi) was about 30%, as indicated by green fluorescent protein (GFP) fluorescence after co‐transfection with RNA1 transcripts and GFP‐encoding RNA2 transcripts (Fig. S2A, see Supporting Information).
Figure 2.
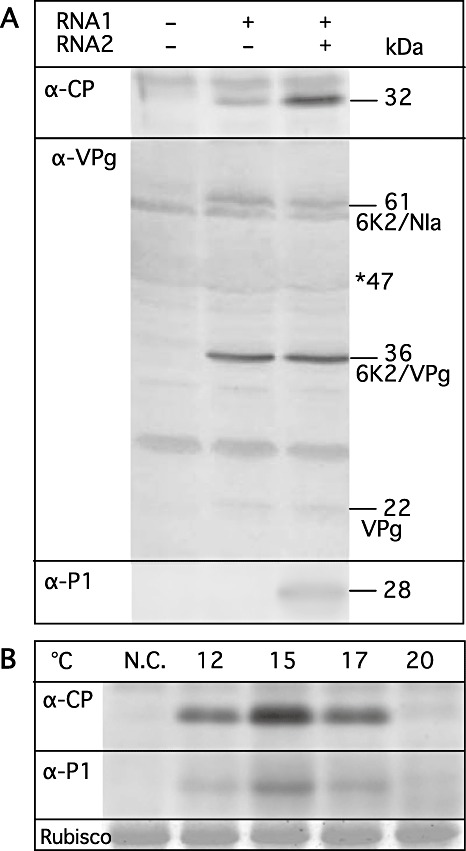
Viral protein expression in transcript‐transfected barley protoplasts. (A) Protoplasts were transfected with mock, RNA1 transcripts alone, or both RNA1 and RNA2 transcripts and incubated at 15 °C for 48 h. Total proteins were subjected to Western blot analysis. α‐CP, anti‐capsid protein serum; α‐VPg, anti‐genome‐linked viral protein serum; α‐P1, anti‐P1 serum. The sizes of the proteins are indicated in the right margin in kilodaltons. An asterisk indicates the position of the 47‐kDa nuclear inclusion protein a (NIa), which may co‐migrate with the large subunit of ribulose bisphosphate carboxylase/oxygenase (Rubisco). (B) Protoplasts were transfected with both RNA1 and RNA2 transcripts and incubated at 12, 15, 17 or 20 °C for 60 h. N.C. indicates mock‐infected protoplasts which were incubated at 15 °C for 60 h. Total proteins were subjected to Western blot analysis using anti‐CP serum (α‐CP) and anti‐P1 serum (α‐P1). ‘Rubisco’ is the Rubisco large subunit stained by Coomassie Brilliant Blue G‐250.
Optimal temperature for BaYMV infection in host cells is 15 °C
In barley fields, BaYMV disease always occurs favourably between 13 and 16 °C (Kusaba et al., 1971); therefore, we used 15 °C for the initial protoplast experiments described above. To determine the optimal temperature for BaYMV RNA replication in host cells between 12 °C and 20 °C, protoplasts were transfected with RNA1 and RNA2 transcripts and incubated at 12, 15, 17 or 20 °C for 60 h. The conditions of the protoplasts after incubation at different temperatures looked the same under a light microscope. By Western blot analysis, the largest amounts of CP and P1 were detected at 15 °C, followed by 17 °C, with lesser amounts at 12 °C and the lowest amounts at 20 °C (Fig. 2B), indicating that the optimal temperature for BaYMV RNA replication is 15 °C, corresponding closely to the optimum for disease manifestation in fields, and similar to the case of Soil‐borne wheat mosaic furovirus for which the optimal temperature for RNA replication is 17 °C (Ohsato et al., 2003).
Systemic infectivity of in vitro transcripts from pBY1 and pBY2
Barley plants (cv. Ryofu) were mechanically inoculated with RNA1 and RNA2 transcripts at the three‐ to four‐leaf stage. After growth for 3 weeks at 15 °C, yellow patches, streaks and mosaic patterns appeared on uninoculated upper leaves [Fig. S2B, see Supporting Information; Fig. 5, wild‐type (WT)]. Leaves with mosaic symptoms were sampled at 4 weeks and 3 months after inoculation and analysed by Western blot to detect CP, VPg‐related proteins and P1 using their respective antisera. The 32‐kDa CP, 22‐kDa VPg, 36‐kDa 6K2/VPg, 61‐kDa 6K2/NIa and 28‐kDa P1 migrated at the same positions as those from transcript‐transfected protoplasts (data not shown). Progeny virions were purified from leaves of systemically infected plants and viral RNA1 and RNA2 were extracted. The entire genome was RT‐PCR amplified as overlapping fragments and the nucleotide sequence was determined using a panel of primers (Table S1A, see Supporting Information). The results showed that the progeny viral RNA1 and RNA2 have identical nucleotide sequences as cDNA inserts of pBY1 and pBY2, respectively, with no deletions or substitutions.
Figure 5.
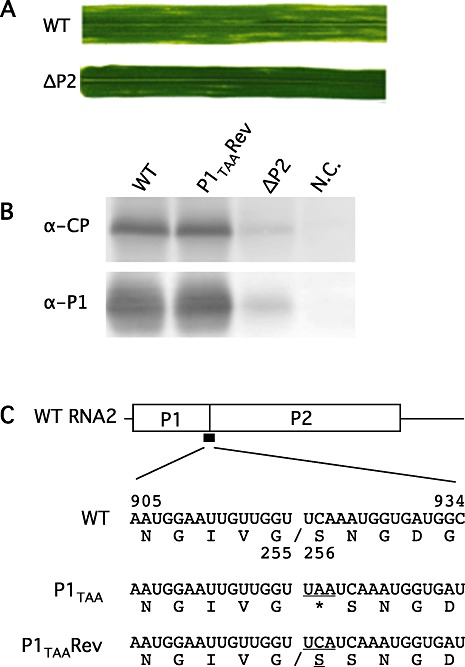
(A) Upper leaf symptoms after inoculation with wild‐type RNA1 and RNA2 transcripts (WT) and with WT RNA1 and ΔP2 RNA2 transcripts (ΔP2) at 4 weeks and 6 weeks post‐inoculation, respectively. (B) Immunoblot detection of capsid protein (CP) and P1 from leaves infected, from the left, with WT RNA1 and RNA2, WT RNA1 and P1TAA revertant RNA2, WT RNA1 and ΔP2 RNA2 and uninfected. (C) Genome organization of RNA2 and nucleotide and translated amino acid sequences at the boundary of the P1/P2 cleavage site. WT, bases 905–934 encoding amino acids 221–260 of polyprotein P12. Cleavage occurs between Gly255 and Ser256. P1TAA, a UAA termination codon was inserted between the glycine and serine codons. P1TAA Rev, the inserted UAA codon was substituted with a UCA serine codon in this revertant.
More than one‐half of Ryofu barley plants were systemically infected after mechanical inoculation using RNA1 and RNA2 transcripts (Table 1). When RNA1 transcripts were inoculated without RNA2 transcripts, none of the plants developed symptoms and CP was not detected from the upper leaves by Western blotting. Therefore, both RNA1 and RNA2 are required for systemic infection of barley plants, and RNA2 must encode one or more proteins essential for systemic infection.
Table 1.
Infectivity to barley plants using in vitro transcripts.*
| Experiment | RNA2 | WT RNA1 + | |||||
|---|---|---|---|---|---|---|---|
| None | WT | ΔP1 | P1TAA † | P12V | ΔP2‡ | ||
| 1 | 0/8§ | 3/6 | 0/7 | 0/7 | 0/6 | 0/8 | |
| 2 | 0/8 | 5/8 | 0/7 | 1/7 | 0/6 | 1/5 | |
| 3 | 0/7 | 3/8 | 0/8 | 0/6 | 0/8 | 1/8 | |
| 4 | 0/8 | 4/6 | 0/5 | 0/8 | 0/7 | 3/8 | |
| Total | 0/31 | 15/28 (53.5%) | 0/27 | 1/28 (3.6%) | 0/27 | 5/29 (17.2%) | |
Plants were subjected to Western blot analysis to detect capsid protein (CP) from upper leaves.
Progeny virions were purified from CP‐positive leaf tissues, and mutated regions in the genome were sequenced from reverse transcriptase‐polymerase chain reaction (RT‐PCR) products.
Number of CP‐positive plants/number of plants inoculated.
Translation of RNA2 mutants in cell‐free system
RNA2 encodes a polyprotein, which is cleaved into P1 and P2 by cysteine proteinase activity residing in the C‐terminal region of P1. In this study, we constructed four RNA2 mutants, as shown in Fig. 1C: pBY2.ΔP1 with deletion of the P1 gene, pBY2.ΔP2 with deletion of the P2 gene, pBY2.P12V with the Gly255Val substitution at the P1/P2 cleavage site, and pBY2.P1TAA with insertion of a TAA termination codon after the P1 gene. In vitro transcripts from pBY2.ΔP1, pBY2.ΔP2, pBY2.P12V and pBY2.P1TAA were translated into wheat germ extract and rabbit reticulocyte lysate. As shown in Fig. 3A, P1, P2 and P12 were detected from WT RNA2 transcripts in wheat germ extract. As predicted, only P2 was detected from ΔP1 RNA2 transcripts, only P1 from P1TAA RNA2 or ΔP2 RNA2 transcripts, and only P12 from P12V RNA2 transcripts (Fig. 3A,B). The same sets of protein species were also detected in rabbit reticulocyte lysate (data not shown).
Figure 3.
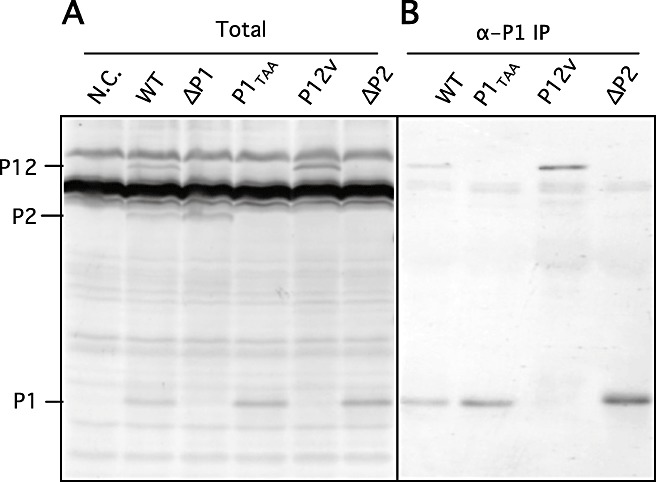
Cell‐free translation products from RNA2 mutants in wheat germ extract. (A) Total products after the translation reaction using precharged biotinylated lysine‐tRNA as a label. From the left, water control and RNA2 transcripts from pBY2, pBY2.ΔP1, pBY2.P1TAA, pBY2.P12V and pBY2.ΔP2. (B) Immunoprecipitated proteins using anti‐P1 serum. From the left, water control and RNA2 transcripts from pBY2, pBY2.P1TAA, pBY2.P12 and pBY2.ΔP2. The positions of P1, P2 and P12 are shown in the left margin.
Infectivity assay of RNA2 mutants in protoplasts
Barley mesophyll protoplasts were transfected with RNA1 transcripts, together with either WT or mutant RNA2 transcripts, and incubated at 15 °C for 60 h before harvesting. Protoplasts transfected with RNA1 and WT RNA2 transcripts gave strong bands of CP and P1 on Western blots (Fig. 4A,B). Similarly, protoplasts transfected with RNA1 and ΔP2 RNA2 transcripts gave strong bands of CP and P1. However, from protoplasts transfected with RNA1, together with ΔP1 RNA2, P1TAA RNA2 or P12V RNA2 transcripts, CP was only weakly detectable, similar to the situation in which protoplasts were transfected with RNA1 transcripts alone or in combination with GFP RNA2 transcripts (Fig. 4B,D), although, in the case of P12V RNA2 or GFP RNA2, the P12 protein or GFP was readily detected, respectively (Fig. 4A,C). In particular, the level of P12 accumulation in RNA1 plus P12V RNA2 transcript‐transfected cells was nearly identical to the level of P1 accumulation in RNA1 plus WT RNA2 or ΔP2 RNA2 transcript‐transfected cells (Fig. 4A). These results indicate that expression of P1 from RNA2 leads to increased expression of CP, whereas the level of expression of RNA2‐encoded protein is unaffected by the gene present between the 5′ and 3′ UTRs of RNA2. Interestingly, transfection of protoplasts with RNA1 and P1TAA RNA2 transcripts did not result in the accumulation of detectable P1 or increased levels of CP (Fig. 4A,B). As P1TAA RNA2 transcripts are translated as efficiently into P1 in cell‐free translation systems as are ΔP2 RNA2 transcripts, the low level of P1 expression in P1TAA RNA2‐transfected cells may be a result of decreased replication efficiency of this RNA in protoplasts.
Figure 4.
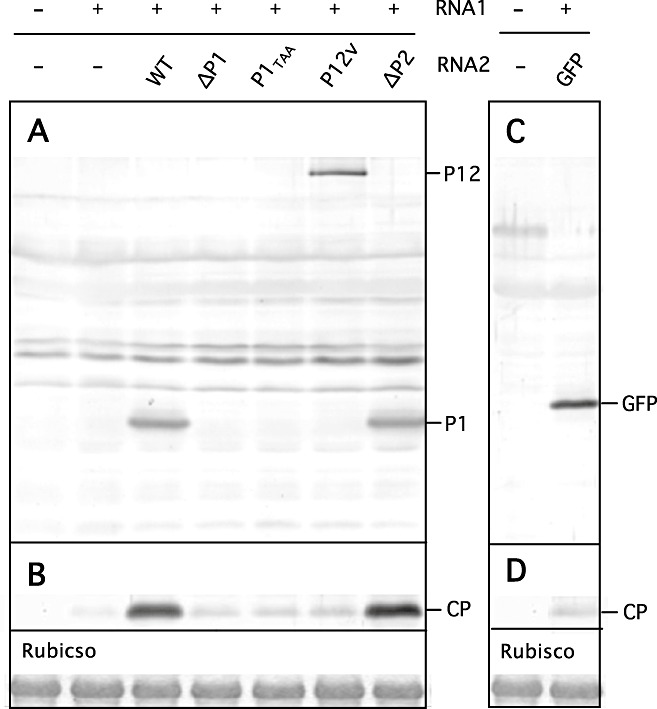
Protein expression profiles in protoplasts transfected with wild‐type (WT) RNA1 and RNA2 mutants analysed by Western blotting using anti‐P1 serum (A), anti‐capsid protein (anti‐CP) serum (B, D) and anti‐green fluorescent protein (anti‐GFP) serum (C). (A, B) From the left, water control, RNA1 transcripts only, RNA1 transcripts with WT RNA2 transcripts, ΔP1 RNA2 transcripts, P1TAA RNA2 transcripts, P12V RNA2 transcripts and ΔP2 RNA2 transcripts. The positions of P1, P12 and CP are indicated in the right margin. (C, D) Left, water control; right, RNA1 transcripts with GFP RNA2 transcripts. The positions of GFP and CP are indicated in the right margin. ‘Rubisco’ is the Rubisco large subunit stained by Coomassie Brilliant Blue G‐250.
Systemic infectivity of RNA2 mutants in barley plants
Mechanical inoculation of barley seedlings with RNA1 transcripts, plus either ΔP1 RNA2 or P12V RNA2 transcripts, did not result in any visible symptoms, nor was CP detectable in the upper leaves, indicating that neither P2 nor P12 alone is functional in systemic infection (Table 1). However, inoculation with RNA1 and ΔP2 RNA2 transcripts resulted in the formation of inconspicuous scattered small chlorotic spots on the upper uninoculated leaves at 5–6 weeks after inoculation (Fig. 5A, ΔP2), which is clearly different from the yellow mosaic symptoms caused by WT RNA1/RNA2 infection (Fig. 5A, WT). CP and P1 were detectable at much reduced levels from these plants whether or not chlorotic spots were present (Fig. 5B), indicating that the plants were systemically infected with RNA1 and ΔP2 RNA2 despite the fact that it replicated too inefficiently to cause typical symptoms. The infection rate was 17.2% (five of 29) (Table 1). When virions were purified from the upper leaves and RNA was RT‐PCR amplified for sequence confirmation, we found that the original deletion in ΔP2 RNA2 was retained in the recovered virus genome.
One plant inoculated with the RNA1 and P1TAA RNA2 transcripts started to show symptoms similar to those caused by WT virus at 3 weeks after inoculation. When progeny virions were purified and the RNA genome was amplified by RT‐PCR and sequenced at the mutated site, we found that the inserted UAA termination codon had been substituted with a UCA serine codon, which regenerated a glycine–serine dipeptide at the cleavage site. P2 produced from this revertant after processing should have two consecutive serines at the N‐terminus (Fig. 5C, P1TAARev).
DISCUSSION
The genetic content of bymovirus RNA1 is identical to that of monopartite potyvirus RNA, except that the 5′‐terminal P1/HC‐Pro genes are missing (Berger et al., 2005). Our results showed that BaYMV RNA1 can replicate autonomously in protoplasts, but that both RNA1 and RNA2 are required for systemic infection in plants. RNA2 encodes a polyprotein P12, which is processed into P1 and P2 by the P1‐cysteine proteinase activity. To determine which RNA2 product is required for systemic infection, we constructed four RNA2 mutants: two for expressing P1 but not P2, one for expressing P2 but not P1, and one for expressing polyprotein P12, but not P1 or P2 (Fig. 1C; Table 2). The results clearly indicated that the expression of P1 is the only essential requirement for systemic infectivity. In protoplasts, P1 expression resulted in increased accumulation of CP from RNA1, implying that P1 enhances RNA1 replication. However, P1 alone was not sufficient to increase the accumulation of CP to WT levels in systemically infected plants. The symptoms caused by the ΔP2 mutant expressing P1 alone were quite mild; the mutant produced only a limited number of small chlorotic spots on the leaves, whereas the WT virus formed large yellow streaks (Fig. 5A). The levels of accumulation of both CP and P1 were much reduced in the leaf tissue infected with the ΔP2 mutant compared with that infected with the WT virus (Fig. 5B). As neither P2 nor P12 cause systemic infectivity by themselves, the role of P2 in systemic infectivity may be to facilitate virus movement and symptom development. However, it has been suggested that P2 is required for transmission by P. graminis, as bymovirus P2 shares similarities with the capsid protein readthrough regions of viruses in the genera Furovirus, Pomovirus and Benyvirus, all of which are also transmitted in soil by zoospores of plasmodiophorids, in having two hydrophobic transmembrane domains (Adams et al., 2001). Furthermore, internal portions of P2 appear to be spontaneously deleted during serial mechanical passages (Jacobi et al., 1995; Kühne et al., 2003; Peerenboom et al., 1997; Zheng et al., 2002), and deletions in P2 of different bymoviruses differ in length and location. Deletions of about 300 amino acids in length were found in the central region of BaYMV P2 (Kühne et al., 2003), whereas all the C‐terminal 364 amino acids, except for the last six, were deleted in BaMMV P2 and the virus isolate lost P. graminis‐mediated transmissibility (Jacobi et al., 1995). In the case of Oat mosaic bymovirus, only the N‐terminal 28 amino acids and the C‐terminal 27 amino acids were retained in the deleted P2, but the isolate with a shorter P2 was still systemically infectious by mechanical inoculation (Zheng et al., 2002). Obviously, the roles of BaYMV P2 in P. graminis‐mediated transmission and in enhancing systemic infectivity should be tested using infectious transcripts in the future.
Table 2.
Expression of RNA2‐encoded proteins in vitro and in vivo.
| RNA2 | In vitro translation* | In protoplasts† | In planta ‡ | |||||
|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P12 | P1 | P12 | CP | P1 | CP | |
| WT | +§ | + | + | + | − | ++ | ++ | ++ |
| ΔP1 | − | + | − | − | − | + | − | − |
| ΔP2 | + | − | − | + | − | ++ | + | + |
| P12V | − | − | + | − | + | + | − | − |
| P1TAA | + | − | − | − | − | + | − | − |
Wheat germ extract incubated at 23°C for 1 h or rabbit reticulocyte lysate incubated at 30 °C for 1 h.
Barley mesophyll protoplasts (cv. Minorimugi) incubated at 15 °C for 60 h.
Barley plants (cv. Ryofu) grown at 15 °C for 6 weeks.
+, detected; ++, detected in abundance; −, not detected.
Mutant P1TAA RNA2 gave a result inconsistent with those from the other three mutants. This mutant was constructed by inserting a TAA termination codon between the P1 C‐terminal glycine codon and the P2 N‐terminal serine codon, so that P1 could be translated but not P2. Protein P1 was translated in vitro from P1TAA RNA2 in both wheat germ extract and rabbit reticulocyte lysate. However, P1 was not detectable in protoplasts transfected with P1TAA RNA2 and plants were not infected systemically by P1TAA RNA2 as they were when inoculated with ΔP2 RNA2 (Table 2). The only difference between P1TAA RNA2 and ΔP2 RNA2 is that the former still retains the full‐length P2‐coding sequence as part of the long 3′ UTR, whereas the latter has the authentic shorter 3′ UTR. The additional sequence in the 3′ UTR in P1TAA RNA2 may have deleterious effects in RNA2 replication by interfering with the formation of functional RNA replication complexes. A similar result has been found with Tobacco etch virus, where translation of the CP gene located at the 3′‐terminal region is required for efficient RNA replication, implying that genome translation and replication are coupled (Mahajan et al., 1996). The appearance of a revertant virus with the UAA to UCA mutation, which resulted in WT‐like virus infection in barley plants, indicates that the P1TAA RNA2 is not absolutely nonviable, but replicates in the cells extremely slowly. Reversion occurred in only one of 28 inoculated plants. No internal deletions in the P2‐coding region were found in the revertant genome, implying that the reversion occurred very early after infection.
When P1 was expressed from a construct containing the authentic 3′ UTR, the replication of RNA1 was enhanced, as shown by the increased level of CP accumulation (Table 2). However, the efficiency of RNA2 replication did not appear to be affected by P1 expression. P12 was expressed in protoplasts in P12V RNA2‐transfected cells as well as P1 was expressed in WT or ΔP2 RNA2‐transfected cells (Fig. 4A). GFP expression also appeared to be efficient in GFP RNA2‐transfected cells (Fig. 4C). These results indicate that RNA2 replicates in cells at WT efficiency if the 5′ and 3′ UTRs are authentic, regardless of the gene inserted between them. At present, progeny viral RNA analyses have not been completed because of difficulties caused by a very low level and slow rate of replication in protoplasts at 15 °C. We need to confirm the above hypothesis by Northern blotting or by quantitative RT‐PCR later.
The role of P1 in addition to its proteinase activity to cleave the P1/P2 site could be as a suppressor of RNA silencing, as is the case for P1/HC‐Pro of potyviruses (Roth et al., 2004; Urcuqui‐Inchima et al., 2001). However, bymovirus P1 does not share amino acid sequence similarity with potyviral P1, and only the C‐terminal cysteine proteinase domain of bymovirus P1 has any similarity with the potyvirus HC‐Pro (Adams et al., 2005; Urcuqui‐Inchima et al., 2001). Among viruses in the family Potyviridae, Cucumber vein yellowing virus in the genus Ipomovirus does not have an HC‐Pro gene, but does have two P1 serine proteinases, the C‐terminal one of which has been shown to have RNA silencing suppressor activity (2006, 2008). However, Wheat streak mosaic virus in the genus Tritimovirus is still systemically infectious even after removal of the entire HC‐Pro gene (Stenger et al., 2005). In this regard, it will be interesting to investigate the putative RNA silencing suppressor activity of BaYMV P1 in the future.
Our results using protoplasts showed that the preference for lower temperatures of BaYMV in field infection (Kusaba et al., 1971) is primarily determined at the RNA replication level in infected cells, implying that the recovery of infected plants from disease symptoms in late spring is a result of the inability of the virus to replicate at higher temperatures.
This is the first report of the construction of cDNA clones of a bymovirus from which infectious transcripts can be generated in vitro. Using this system, the biology of bymoviruses, including viral pathogenicity and host resistance mechanisms, can now be studied in depth at the molecular level. The study of BaYMV is particularly important for agriculture because BaYMV is spreading widely in East Asia and Europe, causing significant yield losses in winter barley every year. More than 10 resistance genes have been mapped from barley germplasm, and some have been used to breed resistant cultivars (Ordon et al., 2004; Tyrka et al., 2008; Werner et al., 2003). However, resistance‐breaking BaYMV variants are continuing to emerge (Kashiwazaki et al., 1989; Kühne et al., 2003; Nishigawa et al., 2008). The set of infectious cDNA clones developed here will be important tools for studying the resistance mechanisms of BaYMV and for controlling this disease in the future.
EXPERIMENTAL PROCEDURES
Virus isolate and RNA
BaYMV‐infected barley leaves were collected in an experimental field of the Research Institute of Bioresources, Okayama University, Kurashiki, Japan in March 2005, and the virus isolate was designated as BaYMV‐JK05. Virions were purified by one cycle of differential ultracentrifugation from infected leaves and suspended in water (Shirako and Brakke, 1984). For viral RNA extraction, virions were treated with 100 µg/mL proteinase K in 10 mm Tris‐HCl, pH 8.0, 5 mm ethylenediaminetetraacetic acid (EDTA) and 0.5% SDS at 37 °C for 1 h, and viral RNA was extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany), precipitated in ethanol and resuspended in water.
Nucleotide sequence determination
The nucleotide sequences of RNA1 and RNA2 were determined directly from overlapping RT‐PCR products covering the entire genome. An initial set of primers was designed from the published nucleotide sequence of the Japanese II‐1 isolate of BaYMV [D01091 for RNA1 (Kashiwazaki et al., 1990) and D01092 for RNA2 (Kashiwazaki et al., 1991)], from which BaYMV‐JK05 RNA1 and RNA2 differed by 2.4% and 4.2%, respectively. Later primers were prepared from the sequence determined for JK05 to avoid base mismatching in RT‐PCR amplification and in sequencing reactions. AMV reverse transcriptase and Ex Taq DNA polymerase (TaKaRa Bio, Ohtsu, Japan) were used for cDNA synthesis and PCRs, respectively. The 5′‐terminal nucleotide sequences of RNA1 and RNA2 were determined from RT‐PCR inserts cloned in pGEM‐T (Promega, Madison, WI, USA) using a 5′ RACE method included in the SMART RACE cDNA Amplification Kit (Clontech, Mountain View, CA, USA). The 3′‐terminal nucleotide sequences of RNA1 and RNA2 were also determined from pGEM‐T clones containing RT‐PCR inserts derived with an oligo d(T)‐containing primer and an internal forward primer. The primers used to obtain the final sequence information are listed in Table S1A (see Supporting Information).
Construction of full‐length cDNA clones of BaYMV RNA1 and RNA2
On the basis of the determined nucleotide sequence, XbaI and XhoI sites and BamHI and SpeI sites were selected for the cloning of full‐length cDNA inserts for RNA1 and RNA2 into the 1.9‐kb vectors, pProXbXh and pProBmSp, respectively, both of which were derived from pBR322 with the tetracycline resistance gene removed (Rice et al., 1987). For the construction of RNA1 cDNA clones, cDNA was synthesized with a primer ctcCTCGAG(T)42 ATTACCTTC (XhoI site; 3′‐terminal nine bases) and PCR‐amplified together with the primer ctcTCTAGA TAATACGACTCACTATAG AAAATAAAACAACCCTAAAC (XbaI site; T7 promoter + G; 5′‐terminal 20 bases) using PrimeSTAR HS DNA polymerase (TaKaRa Bio). The 7.7‐kb product was digested with XbaI and XhoI and cloned into XbaI/XhoI‐digested pProXbXh. Twelve independent clones were initially chosen for infectivity assays using protoplasts. As for RNA2 cDNA clones, cDNA was synthesized with a primer cacACTAGT(T)42 VN (SpeI site; mixed bases of A, C and G followed by four mixed bases) and amplified together with a primer agaGGATCC TAATACGACTCACTATAG AAAATAAAACAACCCTAAACCA (BamHI site; T7 promoter + G; 5′‐terminal 22 bases) as performed for RNA1. The 3.6‐kb product was digested with BamHI and SpeI and cloned into BamHI/SpeI‐digested pProBmSp. Eight independent clones were chosen for protoplast infectivity assay.
The highest expression level of CP from protoplasts transfected with RNA1 transcripts alone was used as a criterion to chose the best cDNA clone for RNA1, clone #1‐351, from 12 independent cDNA clones. As for the best cDNA clone for RNA2, CP expression levels from protoplasts transfected with the #1‐351 RNA1 transcripts together with RNA2 transcripts from eight independent cDNA clones were compared, and a clone, #2‐624‐1.1, giving the highest expression level, was chosen. Clones #1‐351 and #2‐624‐1.1 were designated as pBY1 (Fig. 1A) and pBY2 (Fig. 1B), respectively. The complete nucleotide sequences of the cDNA inserts from pBY1 and pBY2 were determined, and deposited at the DNA Data Bank of Japan (DDBJ) with accession numbers AB500948 and AB500949, respectively.
RNA2 mutant constructs
RNA2 encodes P1 and P2, which are initially translated as the polyprotein and processed into two final products by a proteinase activity located in the C‐terminal region of P1. Four mutant constructs (Fig. 1C) were prepared from pBY2 using sets of mutagenic primers (Table S1B, see Supporting Information). Convenient restriction sites used for cloning, MluI at nucleotide 416 and BstEII at nucleotide 1093, are indicated in Fig. 1B. The four mutant constructs are as follows: (i) pBY2.ΔP1, in which nucleotides 158–919 were deleted so that the initiation codon for P1 (nucleotides 155–157) was connected to the P2 gene starting at nucleotide 920; (ii) pBY2.ΔP2, in which nucleotides 920–2824 were deleted so that the P1 gene was followed by the P2 termination codon at nucleotides 2825–2827; (iii) pBY2.P12V, in which a GGT glycine codon at nucleotides 917–919 was substituted with a GTT valine codon to prevent cleavage at the putative glycine–serine bond encoded at nucleotides 917–922; and (iv) pBY2.P1TAA, in which a TAA termination codon was inserted after nucleotide 919 so that translation of the P1 gene was terminated and no translation of P2 could occur. All mutations were confirmed by sequencing of these clones. In addition, a GFP gene derived from pQBI25 (Takara Shuzo) was cloned between the 5′ and 3′ UTRs to construct pBY2.GFP. Transcripts from pBY2.GFP were used with the pBY1 transcripts to determine the transfection efficiency to barley protoplasts by observing GFP fluorescence under a fluorescence light microscope (Olympus IX‐70, Tokyo, Japan).
In vitro transcription and transfection to barley mesophyll protoplasts
pBY1 and pBY2 DNA were propagated in Escherichia coli strain MC1061. Plasmid DNA was extracted by a modified boiling method and purified using the PureLink column kit from Invitrogen (Carlsbad, CA, USA). pBY1 and pBY2 were digested with XhoI or SpeI, respectively, and purified using NucleoSpin Extract II from Macherey‐Nagel (Düren, Germany), concentrated by ethanol precipitation and suspended in TE (10 mm Tris‐HCl, pH 7.5, 1 mm EDTA) to be used as templates for in vitro transcription. In vitro transcription reactions were performed as described previously (Yamamiya and Shirako, 2000), using T7 RNA Polymerase (TaKaRa Bio). A cap analogue was included in the transcription reaction. Barley mesophyll protoplasts were prepared from the primary leaves of barley plants (cv. Minorimugi and cv. Ryofu) and transfected with in vitro transcripts using a polyethyleneglycol (PEG) method, as described previously (Ohsato et al., 2003). Protoplasts were incubated at 15 °C for 48 or 60 h and harvested for the detection of expressed viral proteins by Western blotting.
Infectivity assay to barley plants using in vitro transcripts
Approximately 10 µg of transcripts from pBY1 and pBY2, or pBY2 derivatives, were mixed with 500 µL of RNA inoculation buffer (50 mm glycine, 30 mm K2HPO4, pH 9.2) containing 1% (w/v) bentonite and rubbed onto the leaves of barley plants (cv. Ryofu) at the three‐ to four‐leaf stage using carborundum as the abrasive. Inoculated plants were placed in the dark at 15 °C overnight and grown in a growth cabinet with 16 h of light and 8 h of dark daily at 15 °C.
Antisera production
The entire coding regions for CP, VPg and P1 were expressed as glutathione S‐transferase (GST) fusion proteins in E. coli cells (strain MC1061) using the pGEX‐6P‐1 vector (Pharmacia/GE Healthcare, Buckinghamshire, UK). The primers used for the construction of recombinant plasmids are listed in Table S1B (see Supporting Information). Insoluble fusion proteins were isolated from E. coli cells using B‐PER reagent (Pierce, Rockford, IL, USA), denatured in SDS and fractionated on 7.5% SDS‐PAGE gels. After staining the target band in ice‐cold 1 m KCl, fusion proteins were eluted from excised gel slices with 0.1% SDS, 50 mm Tris‐HCl, pH 9.0, 0.1 mm EDTA, 5 mm dithiothreitol (DTT) and 0.15 m NaCl, and concentrated to 1 mg/mL using AmiconUltra concentrators (Millipore, Billerica, MA, USA). One rabbit was immunized for each antigen with 2 mg of fusion protein.
Western blot analysis
Proteins from barley leaf tissue or barley mesophyll protoplasts were denatured in 5% SDS and 20 mm DTT, run on 12.5% SDS‐PAGE gels and blotted onto nitrocellulose membranes (Protran BA 85, Whatman, Piscataway, NJ, USA). Antisera against GST:CP, GST:VPg, GST:P1 or GFP (Yamaguchi and Shirako, 2002) were used as primary antibodies, and anti‐rabbit IgG goat IgG conjugated with alkaline phosphatase (Jackson Immunotechnology, West Grove, PA, USA) was used as the secondary antibody. Specific proteins were detected by the nitroblue tetrazolium/5‐bromo‐4‐chloroindol‐3‐yl phosphate (NBT/BCIP) method (Yamaguchi and Shirako, 2002).
In vitro translation and immunoprecipitation using anti‐GST:P1 serum
In vitro transcripts from pBY2 or pBY2 derivatives were precipitated in ethanol and suspended in water. Approximately 100 ng of transcripts were incubated in 10 µL of rabbit reticulocyte lysate or wheat germ extract (Promega) containing precharged biotinylated lysine‐tRNA for 1 h at 30 °C or 23 °C, respectively. For immunoprecipitation using anti‐GST:P1 serum, the translation mix was diluted by the addition of 200 µL of 0.5% SDS, 0.15 m NaCl, 5 mm EDTA, 50 mm Tris‐HCl, pH 7.5, 0.05% Triton X‐100 containing 2 µL of the antiserum and 20 µL of Protein G agarose suspension (Oncogene/EMD Chemicals, San Diego, CA, USA), and incubated at room temperature for 30 min. Protein G–antibody–antigen complexes were collected by centrifugation and washed twice by centrifugation with 0.15 m NaCl, 5 mm EDTA, 50 mm Tris‐HCl, pH 7.5, and 0.05% Triton X‐100. The resulting pellet was suspended in 1 × SDS‐PAGE sample buffer containing 20 mm DTT and heated at 95 °C for 3 min, and clarified by centrifugation. The supernatant was run on SDS‐PAGE gels, and the bands were transferred to nitrocellulose membranes, probed with streptavidin–alkaline phosphatase and detected by the NBT/BCIP method.
Supporting information
Fig. S1 Phylogenetic trees of Polyprotein1 encoded on RNA1 (A) and Polyprotein2 encoded on RNA2 (B). Neighbour‐joining method. Bootstrap = 1000. Polyprotein1: PP1_DE, German isolate, X69757; PP1_GB‐EST, UK strain1 (common) isolate EST, AJ515479; PP1_CN‐Y, Chinese isolate Yancheng, AJ132268; PP1_KR, Korean isolate, AF536958; PP1_I, Japanese Type I, AB430765; PP1_II, Japanese Type II, D01091; PP1_III, Japanese Type III, AB430767; PP1_IV, Japanese Type IV, AB430769. Polyprotein2: PP2_DE, German isolate, D01099; PP1_GB‐EST, UK strain1 (common) isolate EST, AJ515486; PP1_CN‐Y, Chinese isolate Yancheng, AJ132269; PP1_KR, Korean isolate, AF536957; PP1_I, Japanese Type I, AB430766; PP1_II, Japanese Type II, D01092; PP1_III, Japanese Type III, AB430768; PP1_IV, Japanese Type IV, AB4307770.
Fig. S2 Infectivity assay of pBY1 and pBY2 in vitro transcripts. (A) Green fluorescent protein (GFP) fluorescence from barley mesophyll protoplasts (cv. Minorimugi) transfected with in vitro transcripts from pBY1 and pBY2 GFP. Incubation for 48 h at 15 °C. (B) Symptoms of systemically infected barley plants (cv. Ryofu) after inoculation with in vitro transcripts from pBY1 and pBY2. Plants were grown at 15 °C for 8 weeks.
Table S1 (A) Primers used for genome characterization and for the construction of pBY1 and pBY2. (B) Primers used for the construction of plasmid for antigen preparation and of pBY2 mutants.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
ACKNOWLEDGEMENTS
We thank Drs Kazuyoshi Takeda and Kazuhiro Sato of the Research Institute for Bioresources, Okayama University, Japan, for providing us with the BaYMV‐infected barley materials used in this study. We are grateful to Dr Andrew Jackson for suggesting the buffer used for mechanical inoculation with in vitro transcripts. We are appreciative of Dr Ellen Strauss for critical reading of the manuscript. YY was supported by the University of Tokyo Fellowship for International Students. This work was supported by a research fund from the University of Tokyo, Japan.
REFERENCES
- Adams, M.J. , Swaby, A.G. and Jones, P. (1988) Confirmation of the transmission of barley yellow mosaic virus (BaYMV) by the fungus Polymyxa graminis . Ann. Appl. Biol. 112, 133–141. [Google Scholar]
- Adams, M.J. , Antoniw, F. and Mullins, J.G.L. (2001) Plant virus transmission by plasmodiophorid fungi is associated with distinctive transmembrane regions of virus‐encoded proteins. Arch. Virol. 146, 1139–1153. [DOI] [PubMed] [Google Scholar]
- Adams, M.J. , Antoniw, J.F. and Beaudoin, F. (2005) Overview and analysis of the polyprotein cleavage sites in the family Potyviridae . Mol. Plant Pathol. 6, 471–487. [DOI] [PubMed] [Google Scholar]
- Berger, P.H. , Adams, M.J. , Barnett, O.W. , Brunt, A.A. , Hammond, J. , Hill, J.H. , Jordan, R.L. , Kashiwazaki, S. , Rybicki, E. , Spence, N. , Stenger, D.C. , Ohki, S.T. , Uyeda, I. , Van Zaayen, A. , Valkonen, J. and Vetten, H. (2005) Family Potyviridae In: Virus Taxonomy. Eighth Report of the International Committee on Taxonomy of Viruses (Fauquet C.M., Mayo M.A., Maniloff J., Desselberger U. and Ball L.A., eds), pp. 819–841. San Diego, CA: Elsevier Academic Press. [Google Scholar]
- Carrington, J.C. , Haldeman, R. , Dolja, V.V. and Restrepo‐Hartwig, M.A. (1993) Internal cleavage and trans‐proteolytic activities of the VPg‐proteinase (NIa) of tobacco etch potyvirus in vivo. J. Virol. 67, 6995–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Shi, N. , Cheng, Y. , Diao, A. , Wilson, T.M. , Antoniw, J.F. and Adams, M.J. (1999) Molecular analysis of barley yellow mosaic virus isolates from China. Virus Res. 64, 13–21. [DOI] [PubMed] [Google Scholar]
- Chung, B.Y.‐W. , Miller, W.A. , Atkins, J.F. and Firth, A.E. (2008) An overlapping essential gene in the Potyviridae. Proc. Natl Acad. Sci. USA, 105, 5897–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, A.D. , Prols, M. , Schell, J. and Steinbiss, H.H. (1991) The nucleotide sequence of RNA 2 of barley yellow mosaic virus. J. Gen. Virol. 72, 989–993. [DOI] [PubMed] [Google Scholar]
- Habekuss, A. , Kühne, T. , Kramer, I. , Rabenstein, F. , Ehrig, F. , Ruge‐Wehling, B. , Huth, W. and Ordon, F. (2008) Identification of Barley mild mosaic virus isolates in Germany breaking rym5 resistance. J. Phytopathol. 156, 36–41. [Google Scholar]
- Ikata, S. and Kawai, I. (1940) Studies on wheat yellow mosaic disease (in Japanese). Noji Kaiyro Shiryo, 154, 1–123. [Google Scholar]
- Jacobi, V. , Peerenboom, E. , Schenk, P.M. , Antoniw, J.F. , Steinbiss, H.‐H. and Adams, M.J. (1995) Cloning and sequence analysis of RNA‐2 of a mechanically transmitted UK isolate of barley mild mosaic bymovirus (BaMMV). Virus Res. 37, 99–111. [DOI] [PubMed] [Google Scholar]
- Kanyuka, K. , Ward, E. and Adams, M.J. (2003) Polymyxa graminis and the cereal viruses it transmits: a research challenge. Mol. Plant Pathol. 4, 393–406. [DOI] [PubMed] [Google Scholar]
- Kanyuka, K. , McGrann, G. , Alhudaib, K. , Hariri, D. and Adams, M.J. (2004) Biological and sequence analysis of a novel European isolate of Barley mild mosaic virus that overcomes the barley rym5 resistance gene. Arch. Virol. 149, 1469–1480. [DOI] [PubMed] [Google Scholar]
- Kanyuka, K. , Druka, A. , Caldwell, D.G. , Tymon, A. , McCallum, N. , Waugh, R. and Adams, M.J. (2005) Evidence that the recessive bymovirus resistance locus rym4 in barley corresponds to the eukaryotic translation initiation factor 4E gene. Mol. Plant Pathol. 6, 449–458. [DOI] [PubMed] [Google Scholar]
- Kashiwazaki, S. and Hibino, H. (1996) Genomic reassortment of barley mild mosaic virus: evidence for the involvement of RNA1 in pathogenicity. J. Gen. Virol. 77, 581–585. [DOI] [PubMed] [Google Scholar]
- Kashiwazaki, S. , Ogawa, K. , Usugi, T. , Omura, T. and Tsuchizaki, T. (1989) Characterization of several strains of Barley yellow mosaic virus. Ann. Phytopathol. Soc. Jpn. 55, 16–25. [Google Scholar]
- Kashiwazaki, S. , Minobe, Y. , Omura, T. and Hibino, H. (1990) Nucleotide sequence of barley yellow mosaic virus RNA 1: a close evolutionary relationship with potyviruses. J. Gen. Virol. 71, 2781–2790. [DOI] [PubMed] [Google Scholar]
- Kashiwazaki, S. , Minobe, Y. and Hibino, H. (1991) Nucleotide sequence of barley yellow mosaic virus RNA 2. J. Gen. Virol. 72, 995–999. [DOI] [PubMed] [Google Scholar]
- Konishi, T. , Ban, T. , Iida, Y. and Yoshimi, R. (1997) Genetic analysis of disease resistance to all strains of BaYMV in a Chinese barley landrace, Mokusekko 3. Theor. Appl. Genet. 94, 871–877. [Google Scholar]
- Kühne, T. (2009) Soil‐borne viruses affecting cereals – known for long but still a threat. Virus Res. 141, 174–183. [DOI] [PubMed] [Google Scholar]
- Kühne, T. , Shi, N. , Proeseler, G. , Adams, M.J. and Kanyuka, K. (2003) The ability of a bymovirus to overcome the rym4‐mediated resistance in barley correlates with a codon change in the VPg coding region on RNA1. J. Gen. Virol. 84, 2853–2859. [DOI] [PubMed] [Google Scholar]
- Kusaba, T. , Toyama, A. , Yumoto, T. and Tatebe, Y. (1971) Studies on the ecology of Soil‐borne barley mosaic virus and its control in two‐row barleys. Spec. Bull. Tottori Agric. Exp. Stn. 2, 1–208. [Google Scholar]
- Lee, K.J. , Choi, M.K. , Lee, W.H. and Rajkumar, M. (2006) Molecular analysis of Korean isolate of barley yellow mosaic virus. Virus Genes, 32, 171–176. [DOI] [PubMed] [Google Scholar]
- Mahajan, S. , Dolja, V.V. and Carrington, J.C. (1996) Role of the sequence encoding Tobacco etch virus capsid protein in genome amplification: requirements for the translation process and a cis‐active element. J. Virol. 70, 4370–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, M. and Dessens, J.T. (1997) 35S promoter‐driven cDNAs of barley mild mosaic virus RNA1 and RNA2 are infectious on barley plants. J. Gen. Virol. 78, 3147–3151. [DOI] [PubMed] [Google Scholar]
- Nishigawa, H. , Hagiwara, T. , Yumoto, M. , Sotome, T. , Kato, T. and Natsuaki, T. (2008) Molecular phylogenetic analysis of Barley yellow mosaic virus. Arch. Virol. 153, 1783–1786. [DOI] [PubMed] [Google Scholar]
- Ohsato, S. , Miyanishi, M. and Shirako, Y. (2003) The optimal temperature for RNA replication in cells infected by Soil‐borne wheat mosaic virus is 17 °C. J. Gen. Virol. 84, 995–1000. [DOI] [PubMed] [Google Scholar]
- Okada, Y. , Kanatani, R. , Arai, S. and Ito, K. (2004) Interaction between barley yellow mosaic disease‐resistance genes rym1 and rym5, in the response to BaYMV strains. Breed. Sci. 54, 319–325. [Google Scholar]
- Ordon, F. , Friedt, W. , Scheurer, K. , Pellio, B. , Werner, K. , Neuhaus, G. , Huth, W. , Habekuss, A. and Graner, A. (2004) Molecular markers in breeding for virus resistance in barley. J. Appl. Genet. 45, 145–159. [PubMed] [Google Scholar]
- Peerenboom, E. , Prols, M. , Schell, J. , Steinbiss, H.‐H. and Davidson, A.D. (1992) The complete nucleotide sequence of RNA 1 of a German isolate of barley yellow mosaic virus and its comparison with a Japanese isolate. J. Gen. Virol. 73, 1303–1308. [DOI] [PubMed] [Google Scholar]
- Peerenboom, E. , Cartwright, E.J. , Foulds, I. , Adams, M.J. , Stratford, R. , Rosner, A. , Steinbiss, H.‐H. and Antoniw, J.F. (1997) Complete RNA1 sequences of two UK isolates of barley mild mosaic virus: a wild‐type fungus‐transmissible isolate and a non‐fungus‐transmissible derivative. Virus Res. 50, 175–183. [DOI] [PubMed] [Google Scholar]
- Rice, C.M. , Levis, R. , Strauss, J.H. and Huang, H.V. (1987) Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature‐sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61, 3809–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, B.M. , Pruss, G.J. and Vance, V.B. (2004) Plant viral suppressors of RNA silencing. Virus Res. 102, 97–108. [DOI] [PubMed] [Google Scholar]
- Shi, N.‐N. , Chen, J. , Wilson, M.T.A. , Macfarlane, S.A. , Antoniw, J.F. and Adams, M.J. (1996) Single‐strand conformation polymorphism analysis of RT‐PCR products of UK isolates of barley yellow mosaic virus. Virus Res. 44, 1–9. [DOI] [PubMed] [Google Scholar]
- Shirako, Y. and Brakke, M.K. (1984) Two purified RNAs of Soil‐borne wheat mosaic virus are needed for infection. J. Gen. Virol. 65, 119–127. [Google Scholar]
- Stein, N. , Perovic, D. , Kumlehn, J. , Pellio, B. , Stracke, S. , Streng, S. , Ordon, F. and Graner, A. (2005) The eukaryotic translation initiation factor 4E confers multiallelic recessive Bymovirus resistance in Hordeum vulgare (L.). Plant J. 42, 912–922. [DOI] [PubMed] [Google Scholar]
- Stenger, D.C. , French, R. and Gildow, F.E. (2005) Complete deletion of Wheat streak mosaic virus HC‐Pro: a null mutant is viable for systemic infection. J. Virol. 79, 12077–12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpe, U. and Kühne, T. (1995) In vitro transcripts of a full‐length cDNA of a naturally deleted RNA2 of barley mild mosaic virus (BaMMV) replicate in BaMMV‐infected plants. J. Gen. Virol. 76, 2619–2623. [DOI] [PubMed] [Google Scholar]
- Toyama, A. and Kusaba, T. (1970) Transmission of Soil‐borne barley yellow mosaic virus 2. Polymyxa graminis Led. as vector. Ann. Phytopathol. Soc. Jpn. 36, 223–229. [Google Scholar]
- Tyrka, M. , Perovic, D. , Wardynska, A. and Ordon, F. (2008) A new diagnostic SSR marker for selection of the Rym4/Rym5 locus in barley breeding. J. Appl. Genet. 49, 127–134. [DOI] [PubMed] [Google Scholar]
- Urcuqui‐Inchima, S. , Haenni, A.L. and Bernardi, F. (2001) Potyvirus proteins: a wealth of functions. Virus Res. 74, 157–175. [DOI] [PubMed] [Google Scholar]
- Valli, A. , Martín‐Hernández, A.M. , López‐Moya, J.J. and García, J.A. (2006) RNA silencing suppression by a second copy of the P1 serine protease of Cucumber vein yellowing ipomovirus, a member of the family Potyviridae that lacks the cysteine protease HCPro. J. Virol. 80, 10055–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli, A. , Dujovny, G. and García, J.A. (2008) Protease activity, self interaction, and small interfering RNA binding of the silencing suppressor p1b from cucumber vein yellowing ipomovirus. J. Virol. 82, 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, K. , Friedt, W. , Laubach, E. , Waugh, R. and Ordon, F. (2003) Dissection of resistance to soil‐borne yellow‐mosaic‐inducing viruses of barley (BaMMV, BaYMV, BaYMV‐2) in a complex breeders' cross by means of SSRs and simultaneous mapping of BaYMV/BaYMV‐2 resistance of var. ‘Chikurin Ibaraki 1’. Theor. Appl. Genet. 106, 1425–1432. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y. and Shirako, Y. (2002) Engineering of a Sagiyama Alphavirus RNA‐based transient expression vector. Microbiol. Immunol. 46, 119–129. [DOI] [PubMed] [Google Scholar]
- Yamamiya, A. and Shirako, Y. (2000) Construction of full‐length cDNA clones to Soil‐borne wheat mosaic virus RNA1 and RNA2, from which infectious RNAs are transcribed in vitro: virion formation and systemic infection without expression of the N‐terminal and C‐terminal extensions to the capsid protein. Virology, 277, 66–75. [DOI] [PubMed] [Google Scholar]
- Zheng, T. , Chen, J. , Chen, J.P. and Adams, M.J. (2002) The complete sequence of Oat mosaic virus and evidence for deletion and duplication in RNA2. Arch. Virol. 147, 635–642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Phylogenetic trees of Polyprotein1 encoded on RNA1 (A) and Polyprotein2 encoded on RNA2 (B). Neighbour‐joining method. Bootstrap = 1000. Polyprotein1: PP1_DE, German isolate, X69757; PP1_GB‐EST, UK strain1 (common) isolate EST, AJ515479; PP1_CN‐Y, Chinese isolate Yancheng, AJ132268; PP1_KR, Korean isolate, AF536958; PP1_I, Japanese Type I, AB430765; PP1_II, Japanese Type II, D01091; PP1_III, Japanese Type III, AB430767; PP1_IV, Japanese Type IV, AB430769. Polyprotein2: PP2_DE, German isolate, D01099; PP1_GB‐EST, UK strain1 (common) isolate EST, AJ515486; PP1_CN‐Y, Chinese isolate Yancheng, AJ132269; PP1_KR, Korean isolate, AF536957; PP1_I, Japanese Type I, AB430766; PP1_II, Japanese Type II, D01092; PP1_III, Japanese Type III, AB430768; PP1_IV, Japanese Type IV, AB4307770.
Fig. S2 Infectivity assay of pBY1 and pBY2 in vitro transcripts. (A) Green fluorescent protein (GFP) fluorescence from barley mesophyll protoplasts (cv. Minorimugi) transfected with in vitro transcripts from pBY1 and pBY2 GFP. Incubation for 48 h at 15 °C. (B) Symptoms of systemically infected barley plants (cv. Ryofu) after inoculation with in vitro transcripts from pBY1 and pBY2. Plants were grown at 15 °C for 8 weeks.
Table S1 (A) Primers used for genome characterization and for the construction of pBY1 and pBY2. (B) Primers used for the construction of plasmid for antigen preparation and of pBY2 mutants.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item