

SUMMARY
Tomato mosaic virus (ToMV) encodes a movement protein (MP) that is necessary for virus cell‐to‐cell movement. We have demonstrated previously that KELP, a putative transcriptional coactivator of Arabidopsis thaliana, and its orthologue from Brassica campestris can bind to ToMV MP in vitro. In this study, we examined the effects of the transient over‐expression of KELP on ToMV infection and the intracellular localization of MP in Nicotiana benthamiana, an experimental host of the virus. In co‐bombardment experiments, the over‐expression of KELP inhibited virus cell‐to‐cell movement. The N‐terminal half of KELP (KELPdC), which had been shown to bind to MP, was sufficient for inhibition. Furthermore, the over‐expression of KELP and KELPdC, both of which were co‐localized with ToMV MP, led to a reduction in the plasmodesmal association of MP. In the absence of MP expression, KELP was localized in the nucleus and the cytoplasm by the localization signal in its N‐terminal half. It was also shown that ToMV amplified normally in protoplasts prepared from leaf tissue that expressed KELP transiently. These results indicate that over‐expressed KELP interacts with MP in vivo and exerts an inhibitory effect on MP function for virus cell‐to‐cell movement, but not on virus amplification in individual cells.
INTRODUCTION
Plant viruses move from an initially infected cell to adjacent healthy cells through cytoplasmic channels called plasmodesmata (Lucas, 2006). In tobamoviruses, a 30‐kDa protein, known as movement protein (MP), is one of the viral protein mediators for this cell‐to‐cell movement (Okada, 1999; Waigmann et al., 2007). In the best‐studied tobamoviruses, Tobacco mosaic virus (TMV) and Tomato mosaic virus (ToMV), MP associates with plasmodesmata (Tagami and Watanabe, 2007; Tomenius et al., 1987) and increases plasmodesmal permeability (Wolf et al., 1989); MP is regulated by phosphorylation (Kawakami et al., 1999; Trutnyeva et al., 2005; Waigmann et al., 2000). MP functions through interactions with host factors, such as cytoskeletal elements, i.e. actin and tubulin (McLean et al., 1995), cell wall‐associated pectin methylesterase and calreticulin (Chen and Citovsky, 2003; Chen et al., 2005), microtubule‐associated protein MPB2C (Kragler et al., 2003) and a plasmodesmal‐associated protein kinase (Lee et al., 2005). The host factors may play a variety of roles. For instance, pectin methylesterase may contribute to efficient systemic movement of the virus (Chen and Citovsky, 2003), whereas MPB2C and calreticulin presumably act as negative regulators for MP function for virus cell‐to‐cell movement (Chen et al., 2005; Curin et al., 2007; Kragler et al., 2003). The identification and functional analysis of such MP‐interacting proteins have helped to elucidate the mechanisms of virus movement at a molecular level.
In recent years, we have reported several host factors that bind to ToMV MP in vitro, such as protein kinases and putative transcriptional coactivators (Matsushita et al., 2000, 2001, 2002, 2003; Yoshioka et al., 2004). One of the transcriptional coactivators of Arabidopsis thaliana, called KELP, binds in vitro to ToMV MP, but also the MPs of Cucumber mosaic virus (CMV) and a wasabi strain of a crucifer tobamovirus (Matsushita et al., 2001). KELP was originally shown to interact with another transcriptional coactivator, KIWI, by the yeast two‐hybrid system (Cormack et al., 1998). As the KIWI protein can interact with pathogenesis‐related homeodomain proteins PRHA and PRHP, which are possible transcriptional factors derived from A. thaliana and parsley, respectively, it has been argued that KELP is involved in gene regulation in defence responses against pathogens, including viruses (Cormack et al., 1998; Matsushita et al., 2001). However, the significance of KELP binding to MPs in virus movement remains to be elucidated.
In this study, we have investigated the effects of the transient over‐expression of KELP on virus movement and intracellular MP localization in Nicotiana benthamiana. The over‐expression of KELP inhibited the cell‐to‐cell movement of ToMV and interfered with the localization of ToMV MP to plasmodesmata. Furthermore, the N‐terminal half of KELP, which contained an MP‐binding domain, was sufficient for inhibitory effects on both virus movement and intracellular MP localization. KELP was found in the nucleus as well as the cytoplasm when expressed alone, indicating its role as a transcriptional coactivator. The N‐terminal half of KELP was shown to be necessary and sufficient for its nuclear and cytoplasmic localization. The over‐expression of KELP did not interfere with viral amplification in individual cells. Our current results suggest that KELP interacts with ToMV MP in vivo and, when over‐expressed, can function as an inhibitory factor for virus movement.
RESULTS
Over‐expression of KELP interfered with the cell‐to‐cell movement of ToMV in N. benthamiana
Previously, we have demonstrated that KELP is able to bind to ToMV MP and other virus MPs in vitro (Matsushita et al., 2001), suggesting the possibility that KELP may interact with MPs in vivo and play a role in virus movement. To address the issue, we employed a co‐bombardment method using expression plasmids for KELP and ToMV. For the expression of KELP, we constructed pTH2‐KELP‐DsRed, an expression plasmid containing a cDNA of KELP fused with DsRed, a monomeric red fluorescent protein (KELP‐DsRed). As a negative control, we also constructed pART7‐DsRed, an expression plasmid containing a cDNA of free DsRed. The use of the plasmids allowed us to visualize the expression of the proteins. To monitor virus movement, we utilized piL.erG3L, an expression plasmid containing a modified ToMV genomic cDNA in which the coat protein (CP) gene was replaced by the green fluorescent protein (GFP) gene. This plasmid allowed bombarded cells to produce viral replicases, MP and an endoplasmic reticulum (ER)‐targeted GFP. As the ER‐targeted GFP molecules did not move between cells (Tamai et al., 2003), GFP distribution from bombarded single cells to surrounding cells was an indicator of virus movement leading to multiple cell infection. Nicotiana benthamiana leaves were bombarded with piL.erG3L and pTH2‐KELP‐DsRed or pART7‐DsRed. GFP distribution at 1 and 2 days post‐bombardment (dpb) was examined at all infection sites with or without cells showing DsRed fluorescence.
When free DsRed was co‐expressed (Fig. 1A), multiple cell infection sites were observed in 75% (1 dpb) and 94% (2 dpb) of all infection sites, and in 76% (1 dpb) and 97% (2 dpb) of DsRed‐expressing infection sites (Table 1), showing no influence of DsRed expression on the percentage of multiple infection sites. In contrast, when KELP‐DsRed was co‐expressed, GFP was restricted to only initially bombarded single cells in most sites (Fig. 1B). Multiple cell infection sites were observed in 10% (1 dpb) and 20% (2 dpb) of all infection sites, and in 0% (1 dpb) and 6% (2 dpb) of infection sites showing DsRed fluorescence, indicating the co‐expression of KELP‐DsRed (Table 1). The remarkable decrease in the percentage of multiple cell infection sites suggests that the over‐expression of KELP‐DsRed interferes with the cell‐to‐cell movement of the modified ToMV.
Figure 1.
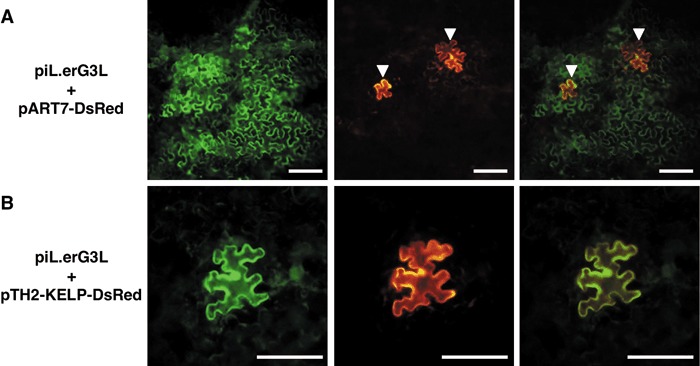
Green fluorescent protein (GFP)‐expressing sites with multiple cells (A) or a single cell (B). Typical fluorescence patterns are shown for epidermal cells after bombardment with piL.erG3L together with pART7‐DsRed (A) or pTH2‐KELP‐DsRed (B). Images were taken from the same field with filter units for GFP (left) and DsRed (middle). Superimposed images are shown on the right. In (A), initially bombarded cells surrounded by GFP‐expressing cells are indicated with arrowheads. In (B), a single cell expressing both GFP and KELP‐DsRed is shown. These images were taken at 2 days post‐bombardment. Scale bar, 200 µm.
Table 1.
Inhibition of the movement of Tomato mosaic virus (ToMV) by co‐expression of KELP or KELPdC.
| Co‐expressed protein* | dpb† | Number of infection sites‡ | Multiple cell infection sites (%)** | |
|---|---|---|---|---|
| Multiple cell§ | Single cell¶ | |||
| DsRed | 1 | 39 (49) | 12 (16) | 76 (75) |
| 2 | 56 (58) | 2 (4) | 97 (94) | |
| KELP‐DsRed | 1 | 0 (8) | 50 (70) | 0 (10) |
| 2 | 6 (31) | 98 (127) | 6 (20) | |
| KELPdC‐DsRed | 1 | 4 | 41 | 9 |
| 2 | 20 | 58 | 26 | |
| KELP | 1 | 15 | 62 | 19 |
| 2 | 45 | 49 | 48 | |
piL.erG3L was co‐bombarded with pART7‐DsRed, pTH2‐KELP‐DsRed, pART7‐KELPdC‐DsRed or pART7‐KELP.
Days post‐bombardment.
The numbers of infection sites consisting of a single cell or multiple (more than two) cells are shown. During the co‐expression with DsRed, KELP‐DsRed or KELPdC‐KELP, the numbers were determined by investigating the infection sites that contained a cell with DsRed fluorescence, whereas all infection sites were examined when the native KELP was co‐expressed. In the case of DsRed and KELP‐DsRed, all infection sites were also examined (numbers in parentheses). These data were obtained from at least three independent experiments.
At a multiple cell infection site, green fluorescent protein (GFP) fluorescence was distributed in cells surrounding an initially bombarded cell, indicating successful virus movement.
At a single cell infection site, GFP fluorescence was confined only to an initially bombarded cell, indicating failure of virus movement.
The percentage of multiple cell infection sites was determined on the basis of the number of multiple cell infection sites vs. the total number of infection sites. The percentages in parentheses were determined using the numbers of infection sites in parentheses.
As the N‐terminal half (amino acids 1–86) of a KELP homologue of Brassica campestris is sufficient to bind to ToMV MP in vitro (Matsushita et al., 2001), it was possible that the corresponding N‐terminal amino acids 1–86 of KELP (KELPdC) might interfere with virus movement. To test this possibility, we constructed a plasmid, pART7‐KELPdC‐DsRed, for expression of KELPdC fused with DsRed (KELPdC‐DsRed), bombarded N. benthamiana leaves with this plasmid, together with piL.erG3L, and examined only infection sites showing DsRed fluorescence. The co‐expression of KELPdC‐DsRed led to a decrease in the percentage of multiple cell infection sites (9% at 1 dpb and 26% at 2 dpb), although less effectively than KELP‐DsRed (Table 1). Thus, the N‐terminal half of KELP is sufficient to interfere with virus movement, and the C‐terminal half may contribute to some extent to the full inhibitory effect of KELP‐DsRed.
To confirm that native KELP could interfere with virus movement, we also constructed pART7‐KELP which contained an untagged KELP cDNA. In this experiment, all infection sites showing GFP fluorescence were examined because there was no indicator for the co‐expression of native KELP. During the co‐expression of native KELP, the percentages of multiple cell infection sites were 19% (1 dpb) and 48% (2 dpb), indicating a significant decrease in virus movement. However, the inhibitory effect of native KELP was less than that of the fused variant KELP‐DsRed (i.e. 10% and 20%).
Effects of the over‐expression of KELP on the intracellular localization of ToMV MP
The localization of MPs at plasmodesmata is believed to be one of the most important properties of MPs for the transport of a virus genome to adjacent cells. Assuming that an interaction between MP and KELP in vivo may affect MP function, we investigated whether the intracellular localization of ToMV MP could be altered by the over‐expression of KELP. To monitor the localization of MP, we used pTH2‐MP‐YFP, an expression plasmid that contained a cDNA for MP fused with yellow fluorescent protein (MP‐YFP). Nicotiana benthamiana leaves were bombarded with the plasmid, together with pART7‐DsRed, pTH2‐KELP‐DsRed or pART7‐KELPdC‐DsRed. Cells showing both YFP and DsRed fluorescence were examined at 1 dpb under a confocal microscope.
In most cells expressing DsRed, MP‐YFP was exclusively localized in many punctate structures along the cell periphery (Fig. 2A, B; Table 2), in agreement with recent observations for ToMV MP fused with GFP (Tagami and Watanabe, 2007). These punctate structures on the cell periphery represent the localization of virus MP at plasmodesmata (Blackman et al., 1998; Oparka et al., 1997). Thus, ToMV MP could target to and accumulate at plasmodesmata in the presence of DsRed. In fewer cases, MP‐YFP was largely seen in the peripheral cytoplasm or associated with filamentous structures (Table 2). These cells exhibited a few or several punctate structures on the cell periphery. In addition, MP‐YFP was often found in irregular structures of different sizes. Such an association with irregular and filamentous structures was also observed during the infection of protoplasts or leaf tissue with ToMV expressing GFP‐tagged MP (Tagami and Watanabe, 2007).
Figure 2.
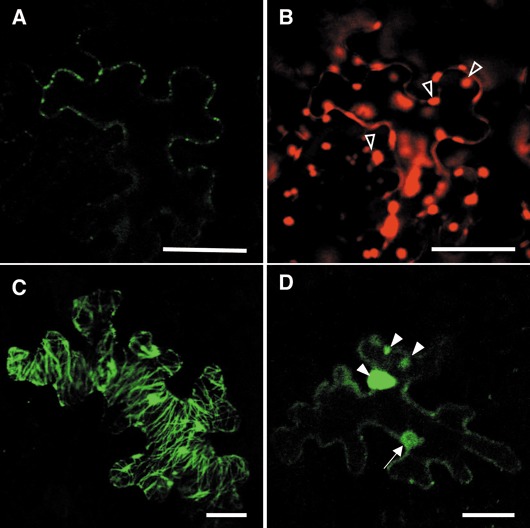
Effects of KELP‐DsRed on the intracellular localization of movement protein‐yellow fluorescent protein (MP‐YFP) in Nicotiana benthamiana epidermal cells. Epidermal cells expressing MP‐YFP together with DsRed (A and B) or KELP‐DsRed (C and D) were observed under a confocal microscope 24 h after bombardment. Green and red colours indicate fluorescence of YFP and DsRed, respectively. The localization of MP‐YFP in tiny punctate structures on the cell wall (A), filamentous structures (C) or the cytoplasm (D) is shown. During the observation of DsRed fluorescence (B), chlorophyll autofluorescence was detected (open arrowheads). The filled arrow and arrowheads in (D) indicate the localization of MP‐YFP in the nucleus and irregular structures, respectively. Scale bars, 25 µm.
Table 2.
Effects of co‐expression of KELP on subcellular localization of Tomato mosaic virus (ToMV) movement protein (MP).
| Co‐expressed protein* | Localization patterns of MP‐YFP† | ||
|---|---|---|---|
| Punctate structures | Cytoplasmic irregular aggregates | Filamentous structures | |
| DsRed | 55 (89%) | 5 (8%) | 2 (3%) |
| KELP‐DsRed | 13 (23%) | 27 (48%) | 16 (29%) |
| KELPdC‐DsRed | 12 (21%) | 25 (45%) | 19 (34%) |
pTH2‐MP‐YFP was bombarded together with pART7‐DsRed, pTH2‐KELP‐DsRed or pART7‐KELPdC‐DsRed.
The numbers of cells showing the indicated localization patterns of MP‐YFP were determined 24 h after bombardment under a confocal microscope. The percentages of the localization patterns are shown in parentheses. These data were collected from at least two independent experiments.
In contrast, when KELP‐DsRed or KELPdC‐DsRed was co‐expressed, the percentage of cells showing many punctate structures decreased significantly (Table 2). Instead, the percentage of cells showing cytoplasmic irregular aggregation and filamentous structures increased (Fig. 2C,D; Table 2). These results suggest that KELP interferes with the localization of MP to plasmodesmata.
Intracellular localization of KELP in the presence and absence of ToMV MP
Although our confocal microscope system captured both DsRed fluorescence and chlorophyll autofluorescence (Fig. 2B), a close examination of the images suggested the co‐localization of MP‐YFP with KELP‐DsRed in the nucleus or irregular/filamentous structures, but not in punctate structures (Supporting Fig. S1). To confirm the results, cells expressing MP‐YFP alone or MP‐YFP and KELP‐DsRed were examined at 1 dpb using an epifluorescence microscope with detection units for GFP and DsRed. In cells expressing MP‐YFP, only YFP fluorescence was captured with the GFP unit (Fig. 3A), and no background from chlorophyll autofluorescence was observed with the DsRed unit (Fig. 3B). However, MP‐YFP and KELP‐DsRed, when co‐expressed, were observed in the same irregular or filamentous structures (Fig. 3C,D). In addition, both of the proteins appeared to be within the nucleus (Fig. 3E,F). It should be noted that such a nuclear localization of MP‐YFP was not seen in the absence of KELP‐DsRed (data not shown). The relocation of MP‐YFP suggests that KELP may interact with ToMV MP in vivo.
Figure 3.
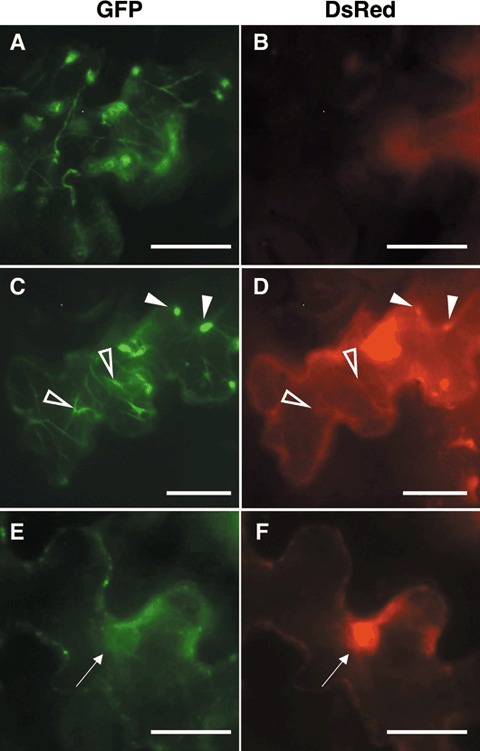
Localization of movement protein‐yellow fluorescent protein (MP‐YFP) and KELP‐DsRed in Nicotiana benthamiana epidermal cells. Epidermal cells expressing MP‐YFP only (A and B) or MP‐YFP and KELP‐DsRed (C–F) were observed under an epifluorescence microscope 24 h after bombardment. Left and right panels show fluorescence captured by detection units for green fluorescent protein (GFP) and DsRed, respectively. A cell expressing MP‐YFP shows filamentous structures with green fluorescence (A). No chlorophyll autofluorescence is detected with the unit for DsRed (B). Both MP‐YFP and KELP‐DsRed are detected in filamentous or irregular structures (C and D) and the nucleus (E and F). Open and filled arrowheads in (C) and (D) indicate filamentous and irregular structures, respectively. Arrows in (E) and (F) indicate the nucleus. Scale bars, 50 µm.
To obtain detailed images of the intracellular localization of KELP under the confocal microscope, we constructed an expression plasmid, pTH2‐KELP‐GFP, encoding GFP‐tagged KELP (KELP‐GFP). Simultaneously, the intracellular localization of KELPdC was also examined using an expression plasmid, pART7‐KELPdC‐YFP, that codes for YFP‐tagged KELPdC (KELPdC‐YFP). Nicotiana benthamiana leaves were bombarded with each of the plasmids and observed at 1 dpb under a confocal microscope. Intracellular localization patterns were indistinguishable between KELP‐GFP and KELPdC‐YFP (data not shown). In the majority of bombarded cells, the fluorescence of KELP‐GFP (70 of 72 cells) or KELPdC‐YFP (66 of 67 cells) was distributed in the cytoplasm and the nucleus (Fig. 4A), but not in the nucleolus (Fig. 4B). In the other cells, we observed many aggregates with bright fluorescence (data not shown). We did not observe any fluorescence in the filamentous structures that were seen during the co‐expression of KELP‐DsRed and MP‐YFP as mentioned above. To confirm the nuclear localization of KELP‐GFP, the cells of the bombarded leaves were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI). Coupled detections of GFP and DAPI under the epifluorescence microscope revealed that GFP was distributed in the nucleus stained with DAPI and also the cytoplasm (Fig. 4D–I). These results suggest that KELP may be a nuclear and cytoplasmic protein.
Figure 4.
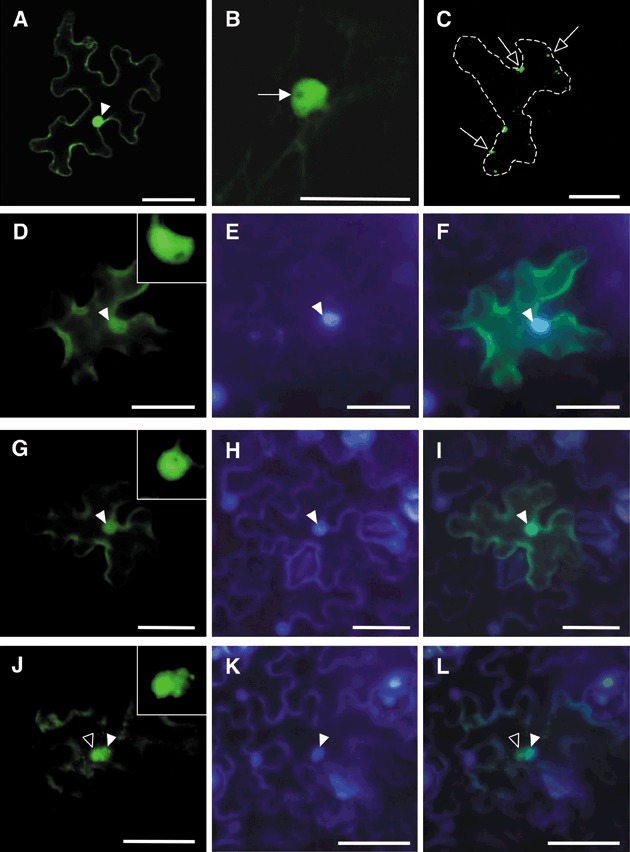
Subcellular localization of KELP‐GFP, KELPdC‐YFP and KELPdN‐YFP in Nicotiana benthamiana epidermal cells 24 h after bombardment. Confocal and epifluorescence microscopic images of epidermal cells are shown in (A)–(C) and (D)–(L), respectively. In most bombarded cells with KELP‐GFP, GFP fluorescence was observed in the nucleus (filled arrowhead) and the cytoplasm (A). In the nucleus, fluorescence was absent in the nucleolus (filled arrow) (B). Similar fluorescence patterns were observed for KELPdC‐YFP (data not shown). In contrast, in the case of KELPdN‐YFP, small protein aggregates (open arrows) were seen in the cytoplasm of a cell outlined by dots (C). Epidermal cells expressing KELP‐GFP (D–F), KELPdC‐YFP (G–I) and KELPdN‐YFP (J–L) were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) and observed under an epifluorescence microscope with a GFP filter unit (D, G and J) and a DAPI filter unit (E, H and K). Superimposed images are shown in (F), (I) and (L). KELP‐GFP and KELPdC‐YFP localized clearly in the nucleus (filled arrowheads), whereas KELPdN‐YFP sometimes formed aggregates (open arrowheads) around the nucleus. Small boxes in (D), (G) and (J) show enlarged images of the nucleus. Scale bars in (B) and the other panels represent 50 and 100 µm, respectively. GFP, green fluorescent protein; YFP, yellow fluorescent protein.
Free GFP and YFP localize in the cytoplasm and the nucleus, but not in the nucleolus (data not shown), as reported by Haseloff et al. (1997). It was possible that the observed intracellular localizations of KELP‐GFP and KELPdC‐YFP were governed by the localization signals of GFP and YFP. We examined the intracellular localization of the C‐terminal half (amino acids 87–165) of KELP (KELPdN) using pART7‐KELPdN‐YFP for expression of YFP‐tagged KELPdN. Observation of the cells expressing KELPdN‐YFP under a confocal microscope showed a different localization pattern from those of KELP‐GFP and KELPdC‐YFP. In all cells examined (32 cells), fluorescence of KELPdN‐YFP was observed as irregular cytoplasmic aggregates, but not in the nucleus (Fig. 4C). Furthermore, observation of DAPI‐stained, KELPdN‐YFP‐expressing cells under an epifluorescence microscope proved that large aggregates of KELPdN‐YFP gathered around the nucleus (Fig. 4J,L). The collective data indicate that the N‐terminal half, but not the C‐terminal half, of KELP contains a nuclear localization signal.
ToMV multiplied normally in N. benthamiana protoplasts over‐expressing KELP
According to Tamai et al. (2003), GFP encoded by piL.erG3L should be translated from subgenomic mRNAs produced during the course of viral multiplication. Thus, GFP signals observed in initially bombarded sites (Fig. 1B) indicate that virus multiplication actually occurs and may not be affected by the over‐expression of KELP‐DsRed. We confirmed this in the virus‐infected protoplasts. Nicotiana benthamiana leaves were infiltrated with agrobacteria carrying binary plasmid pART27‐KELP or pART27‐KELP‐DsRed for the transient expression of KELP or KELP‐DsRed, respectively. Western blot analysis of infiltrated leaves using anti‐KELP antibody showed that the expression of KELP and KELP‐DsRed reached peak levels between 2 and 3 days post‐infiltration (dpi) (data not shown). Infiltrated leaves at 2 dpi were used for the preparation of protoplasts. Red fluorescence was observed in approximately 85% of the protoplasts prepared from KELP‐DsRed‐expressing leaves (data not shown). Considering that the same 35S promoter was used for the over‐expression of KELP and KELP‐DsRed, we assumed that the untagged KELP could accumulate at similar levels to KELP‐DsRed in protoplasts. These protoplasts were then inoculated with virus RNA prepared from wild‐type ToMV virions. Protoplasts were also prepared from leaves infiltrated with untransformed agrobacteria and inoculated with the virus RNA or treated with buffer. Protoplasts were collected at 24 h post‐inoculation and used for Western blot analysis to detect ToMV CP, KELP and KELP‐DsRed.
As shown in Fig. 5 (top, lanes 2–4), ToMV CP accumulated at comparable levels in all the ToMV RNA‐inoculated protoplasts. KELP (Fig. 5, middle, lane 3) and KELP‐DsRed (Fig. 5, middle, lane 4) were detected in protoplasts infiltrated with the corresponding agrobacteria. No proteins corresponding to ToMV CP, KELP or KELP‐DsRed were detected in protoplasts infiltrated with untransformed agrobacteria and treated with buffer (Fig. 5, top and middle, lane 1). These data suggest that the over‐expression of KELP and KELP‐DsRed has no deleterious effects on virus multiplication in individual cells.
Figure 5.
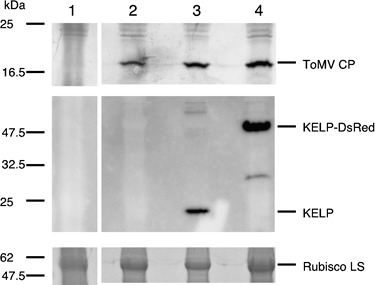
Accumulation of Tomato mosaic virus (ToMV) coat protein (CP) in Nicotiana benthamiana protoplasts over‐expressing KELP or KELP‐DsRed. Protoplasts were prepared from N. benthamiana leaves infiltrated with untransformed agrobacteria (lanes 1 and 2) or agrobacteria transformed with pART27‐KELP (lane 3) or pART27‐KELP‐DsRed (lane 4). The freshly prepared protoplasts were treated with 10 mm phosphate buffer (lane 1) or inoculated with virion‐derived ToMV RNA (lanes 2–4). Western blot analysis was performed 24 h after inoculation to detect ToMV CP (top), KELP (middle) and KELP‐DsRed (middle). To estimate the amount of loaded proteins, the 55‐kDa ribulose‐1,5‐bisphosphate carboxylase/oxygenase (Rubisco) large subunit (LS) on the sodium dodecylsulphate‐polyacrylamide gel, stained by Coomassie Brilliant Blue, is shown (bottom). The positions of ToMV CP (18 kDa), KELP (19 kDa), KELP‐DsRed (48 kDa) and Rubisco LS (55 kDa) are indicated on the right. The positions of molecular size markers are shown on the left.
DISCUSSION
KELP has been identified as a putative transcriptional coactivator (Cormack et al., 1998), and has been shown to interact biochemically with ToMV MP, CMV MP and the MP of a wasabi strain of a crucifer tobamovirus (Matsushita et al., 2001). In this study, we examined the effects of the transient over‐expression of KELP in leaf tissue on ToMV infection. As a result, it was found that the movement of the GFP‐expressing ToMV from cells over‐expressing KELP was inhibited. In these cells, the virus was allowed to amplify and produce the reporter GFP at detectable levels. In addition, the over‐expression of KELP in protoplasts did not affect the amplification of wild‐type ToMV. These data suggest that the over‐expression of KELP may have an inhibitory effect on the cell‐to‐cell movement of the virus genome, but not on its amplification in individual cells.
Although the over‐expression of KELP‐DsRed, KELPdC‐DsRed and native KELP clearly inhibited the cell‐to‐cell movement of ToMV, in comparison with the case for free DsRed, the inhibitory effect of native KELP was apparently lower than that of KELP‐DsRed and KELPdC‐DsRed. Because the over‐expression of untagged, native KELP could not be confirmed in any bombarded cells, the protein could be absent or could accumulate at very low levels in some of the bombarded cells, leading to an underestimation of its inhibitory effect. Alternatively, these results may suggest that the fusion of KELP and KELPdC to DsRed can somehow augment their inherent ability to block virus movement. Possibly, fused KELP may interact more effectively than native KELP with MP.
In addition to the inhibitory effect on virus movement, the over‐expression of KELP interfered with the association of ToMV MP with punctate structures along the cell periphery. The association of MPs with peripheral punctate structures represents their localization at plasmodesmata (Blackman et al., 1998; Oparka et al., 1997). The plasmodesmal localization of MPs is assumed to be involved in the structural modification of plasmodesmata for virus movement (McLean et al., 1993; Tremblay et al., 2005). In the case of TMV, MP accumulates at plasmodesmata during virus infection (Heinlein et al., 1998) and functions in expanding the size exclusion limit of plasmodesmata (Oparka et al., 1997), allowing virus RNA–MP complexes to pass through plasmodesmata. Thus, the interference with the plasmodesmal localization of ToMV MP by over‐expressed KELP may result in inefficient transport of the virus genome to neighbouring cells through plasmodesmata.
The observed interference with the plasmodesmal localization of ToMV MP was incomplete, which is an apparent discrepancy with the nearly complete inhibition of the movement of the GFP‐expressing ToMV by the over‐expression of KELP‐DsRed. In intracellular localization experiments, KELP and MP were expressed presumably at comparable levels under the control of the constitutively active 35S promoter, which possibly allowed a certain amount of MP to escape from the inhibitory effect of KELP with a certain frequency. However, in the case of GFP‐expressing ToMV, MP is translated from subgenomic RNA during virus replication. It has been shown that the production of tobamovirus MP from subgenomic RNA is temporal and limited, depending on the infection cycle (Watanabe et al., 1984). Moreover, the transcription of the virus genome from the 35S promoter may not be efficient because of its large size (more than 7 kb). Thus, it is possible that KELP is produced more rapidly and efficiently than MP in our co‐expression experiments, and that the excess amount of KELP over MP during early infection of the virus may almost completely prevent MP from accumulating and functioning at plasmodesmata for virus movement.
The decrease in the plasmodesmal localization of ToMV MP was accompanied by a corresponding increase in aggregation and filament‐associated localization. In the non‐plasmodesmal localization patterns, KELP was predominantly co‐localized with ToMV MP in irregular or filamentous structures and in the nucleus. These results strongly imply that KELP interacts with MP in vivo as well as in vitro (Matsushita et al., 2001), and that the interaction results in changes in the intracellular localization patterns of MP. As KELP is localized predominantly in the cytoplasm and the nucleus when transiently expressed alone, and MP is not targeted to the nucleus in the absence of over‐expressed KELP, the two proteins probably encounter and interact in the cytoplasm. Possibly, the interaction prevents MP from associating with the host factors necessary for its plasmodesmal localization, and thereby keeps the two proteins in the cytoplasm. Alternatively, it may facilitate their rearrangement to particular sites, such as irregular and filamentous structures and the nucleus.
We have demonstrated that KELP is localized in the nucleus and the cytoplasm, and that the nuclear localization signal resides in the N‐terminal half of the protein. These results support the possibility that KELP functions in the nucleus as a transcriptional coactivator (Cormack et al., 1998). Based on the hypothesis that KELP may be involved in as yet unknown host defence gene expression against pathogen attacks (Cormack et al., 1998), we have previously discussed a viral strategy in which the interaction between ToMV MP and KELP in the cytoplasm may block the translocation of KELP to the nucleus and interfere with host defence gene expression (Matsushita et al., 2001). It is possible that the over‐expression of KELP may abrogate the viral strategy by providing the nucleus with sufficient KELP to induce the host defence system. In this study, we have demonstrated that the N‐terminal half of KELP is involved in and sufficient for its inhibitory effects on MP function for virus movement. Amino acid sequences with high similarities to known transcriptional coactivators are located in the C‐terminal half rather than the N‐terminal half of KELP (Matsushita et al., 2001). Thus, it seems unlikely that the putative host defence‐related function is implicated in the inhibitory effects of the N‐terminal half of KELP.
Wild‐type A. thaliana allows the local and systemic spread of ToMV (Ohshima et al., 1998), although the endogenous KELP mRNA is produced in leaves and other organs at detectable levels (Cormack et al., 1998). It is possible that the amount of endogenous KELP may not be sufficient to exert its potential inhibitory effects on virus movement. Alternatively, an insufficient amount of KELP may favour viral escape from the host defence system discussed above. To elucidate an inherent role of KELP in virus movement, biological analyses using A. thaliana mutants lacking the functional KELP gene are in progress.
A database search revealed KELP homologues in various plants, including tobacco (accession no. EB681766), tomato (accession no. BM411434) and rice (accession no. NM_001050549), in addition to that in B. campestris (accession no. AB050390), which was originally identified as a cDNA of a ToMV MP‐interacting protein (Matsushita et al., 2001). This indicates that KELP‐related proteins may be distributed in plant species and have similar functions. It would be of interest to examine whether these proteins can interact with ToMV MP and other MPs, and whether their over‐expression can interfere with virus movement as observed in the current study. In a preliminary co‐bombardment experiment, we have found that the transient over‐expression of the N‐terminal half of B. campestris KELP efficiently blocks the movement of GFP‐expressing ToMV (N. Sasaki et al., unpublished data). Further investigation is required to obtain an understanding of the inhibitory mechanisms of such host factors, which may lead us to generate novel virus‐resistant plants using genetic resources of plant origin.
EXPERIMENTAL PROCEDURES
Plants and growth conditions
Nicotiana benthamiana plants were grown on Rock Fiber blocks (Nittobo, Tokyo, Japan) for 2 weeks at 25 °C with a 16‐h light/8‐h dark photoperiod. Seedlings on the Rock Fiber blocks were transferred to pots filled with vermiculite and grown under the same conditions with 0.1% Hyponex solution (Hyponex Japan, Osaka, Japan), which was given once a week. For all the experiments, 5–6‐week‐old plants were used.
Plasmid construction
The plasmids described below were constructed by standard cloning techniques (Sambrook and Russell, 2001) or the Gateway cloning technique, according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). Briefly, in Gateway cloning, the first polymerase chain reaction (PCR) was performed with template DNA and appropriate primers containing partial attB1 and attB2 recombination sites. After purification of the PCR fragment, the second PCR was carried out with the attB1 adaptor primer (5′‐GGGGACAAGTTTGTACAAAAAAGCAGGCT‐3′) and the attB2 adaptor primer (5′‐GGGGACCACTTTGTACAAGAAAGCTGGGT‐3′). The PCR product with complete attB1 and attB2 recombination sites was cloned into the entry vector pDONR221 (Invitrogen) using BP Clonase enzyme (Invitrogen), and then transferred to a destination vector by LR Clonase enzyme (Invitrogen). PCR‐derived coding sequences in the plasmids were confirmed by sequencing.
piL.erG3L
piL.erG3L is a derivative of piL.erG3 containing an infectious clone of a modified ToMV, in which part of the CP gene is replaced by the GFP gene (Tamai et al., 2003). To construct piL.erG3L, the BstEII‐SpeI fragment of the CP gene was obtained from pTLW3 containing a wild‐type ToMV cDNA (Kawakami et al., 1999) and inserted between the BstEII and SacI sites of piL.erG3.
pTH2‐KELP‐GFP and pTH2‐KELP‐DsRed
To construct pTH2‐KELP‐GFP, PCR was carried out with pGEX‐P‐KELP (Matsushita et al., 2001) as a template and KELP‐F02 (5′‐ATATGTCGACATGGAGAAAGAGACGAAG‐3′) and KELP‐R02 (5′‐ATATCCATGGCAGAACCACCTGTACCACCGACACGCGATTCCATTTT‐3′) as primers. The resultant PCR product was digested with SalI and NcoI, and ligated to the SalI‐NcoI large fragment from the 35‐sGFP(S65T) plasmid (Chiu et al., 1996). To construct pTH2‐KELP‐DsRed, pDsRed‐Monomer (BD Biosciences Clontech, Palo Alto, CA, USA) was digested with NcoI and NotI. The fragment containing the DsRed gene was ligated to the NcoI‐NotI large fragment from pTH2‐KELP‐GFP. The expressed proteins KELP‐GFP and KELP‐DsRed contain extra amino acids (Gly‐Gly‐Tyr‐Gly‐Gly‐Ser‐Ala) between KELP and each tag protein.
pART7‐KELP
The DNA fragment with the coding sequence of the KELP gene was amplified from an A. thaliana cDNA library through PCR using primers KELP‐F01 and KELP‐R01, as described previously (Matsushita et al., 2001). These were digested with EcoRI and XhoI, and ligated to the EcoRI‐XhoI large fragment from the pCR2.1 vector (Invitrogen) to generate pCR‐KELP. To add a BamHI site after the stop codon of the KELP cDNA, PCR was carried out with pCR‐KELP as a template and KELP‐F01 and KELP/BamHI/R (5′‐TAGGATCCTCAGACACGCGATTC‐3′) as primers. The resultant PCR product was digested with EcoRI and BamHI, and ligated to the EcoRI‐BamHI large fragment from the pART7 vector (Gleave, 1992).
pART7‐DsRed
pDsRed‐Monomer was digested with BamHI and SpeI. The fragment containing the DsRed gene was ligated to the BamHI‐XbaI large fragment from the pART7 vector.
pART7‐MP‐YFP
cDNA of the MP gene was amplified by PCR with pTLW3 as a template and pTLW3F01 (5′‐GCGGTACCATGGCTCTAGTTGTTAAA‐3′) and pTLW3R01 (5′‐GCTCTAGATTAATACGAATCAGAATC‐3′) as primers, and digested with KpnI and XhoI. The KpnI‐XhoI fragment was ligated to the KpnI‐XhoI large fragment from pART7 to generate the plasmid pART7‐30K. To remove the stop codon of MP cDNA, PCR was carried out with pART7‐30K as template and 35S (5′‐CCTTCGCAAGACCCTTCCTC‐3′) and pTLW3R02 (5′‐GCGGATCCGCATACGAATCAGAATCCGC‐3′) as primers. The PCR product was digested with XhoI and BamHI, and ligated to the BamHI‐XhoI large fragment from pART7 to generate the plasmid pART7‐30KdTAA. pART7‐30KdTAA and pEYFP (BD Biosciences Clontech) were digested with XbaI and NotI, respectively, and blunt‐ended with the Klenow fragment of DNA polymerase I. Following the subsequent digestion of both plasmids with BamHI, the fragment containing the EYFP sequence was ligated to the large fragment from pART7‐30KdTAA to generate pART7‐MP‐YFP. The expressed protein MP‐YFP contains extra amino acids (Ala‐Asp‐Pro‐Arg‐Val‐Pro‐Val‐Ala‐Thr) between MP and YFP.
pART7‐KELPdC‐YFP and pART7‐KELPdN‐YFP
First, pART7‐YFP was constructed by ligating the XhoI‐SmaI large fragment from pART7 to the fragment containing the EYFP coding sequence, which was prepared from pEYFP by digestion with NotI, blunt‐ending and second digestion with SalI. The Gateway conversion cassette (reading frame C.1, Invitrogen) was introduced to the SmaI site of pART7‐YFP to generate a Gateway destination vector pART7‐GWC‐YFP. To obtain an entry plasmid for KELPdC (designated as pDONR‐KELPdC), the first PCR was performed with pART7‐KELP as a template and attB1/KELP/F (5′‐AAAAAGCAGGCTACCATGGAGAAAGAGACGAAG‐3′) and attB2/KELPdC/R (5′‐AGAAAGCTGGGTTTCCTTTACCACAATCCTT‐3′) as primers. After cloning of the second PCR product to pDONR221, the insert in pDONR‐KELPdC was recombined into pART7‐GWC‐YFP to generate pART7‐KELPdC‐YFP. pART7‐KELPdN‐YFP was constructed in the same way as pART7‐KELPdC‐YFP, except that a primer set of attB1/KELPdN/F (5′‐AAAAAGCAGGCTACCATGAACAAAGAGTTTGAT‐3′) and attB2/KELP/R (5′‐AGAAAGCTGGGTTGACACGCGATTCCATTTT‐3′) was used in the first PCR to obtain an entry plasmid for KELPdN (designated as pDONR‐KELPdN). The expressed proteins KELPdC‐YFP and KELPdN‐YFP contain extra amino acids (Asn‐Pro‐Ala‐Phe‐Leu‐Tyr‐Lys‐Val‐Val‐Arg‐Typ‐Val‐Pro‐Val‐Ala‐Thr) between KELPdC or KELPdN and YFP.
pART7‐KELPdC‐DsRed
The Gateway conversion cassette (reading frame C.1, Invitrogen) was first introduced to the SmaI site of pDsRed‐Monomer to construct pDsRed‐Monomer‐GWC. The SpeI‐HindIII fragment of pDsRed‐Monomer‐GWC, which contains the cassette and the DsRed coding sequence, was ligated to the XbaI‐HindIII large fragment from pART7 to construct a Gateway destination vector pART7‐GWC‐DsRed. The entry plasmid pDONR‐KELPdC was used for recombination with the destination vector pART7‐GWC‐DsRed to generate pART7‐KELPdC‐DsRed. The expressed protein KELPdC‐DsRed contains extra amino acids between KELPdC and DsRed, which are the same as those in KELPdC‐YFP (see above).
pART27‐KELP and pART27‐KELP‐DsRed
A Gateway destination vector pART27‐35Sa‐GWB was constructed by inserting the expression cassette from pART7 into the NotI site of the binary vector pART27 (Gleave, 1992) and introducing the Gateway conversion cassette (reading frame B, Invitrogen) at the SmaI site in the expression cassette. To obtain an entry plasmid for KELP (designated as pDONR‐KELP), the first PCR was performed with pCR‐KELP as a template and attB1/KELP/F and attB2/KELP/R (5′‐AGAAAGCTGGGTTGACACGCGATTCCATTTT‐3′) as primers. After cloning of the second PCR product to pDONR221, the entry plasmid pDONR‐KELP was used for recombination with the destination vector pART27‐35Sa‐GWB to generate pART27‐KELP. pART27‐KELP‐DsRed was constructed in the same way as pART27‐KELP, except that a template plasmid pTH2‐KELP‐DsRed and a primer set of attB1/KELP/F and attB2/DsRed‐Mono/Rev (5′‐AGAAAGCTGGGTGCTACTGGGAGCCGGAGTGGC‐3′) were used in the first PCR to obtain an entry plasmid for KELP‐DsRed (designated as pDONR‐KELP‐DsRed). The KELP‐DsRed protein encoded by pART27‐KELP‐DsRed contains the same linker amino acids as mentioned above for pTH2‐KELP‐DsRed.
Particle bombardment
Particle bombardment of N. benthamiana leaves was carried out using the Biolistic PDS‐1000/He Particle Delivery System (Bio‐Rad Laboratories, Hercules, CA, USA). The 1.0‐µm gold particles (Bio‐Rad Laboratories) were washed vigorously twice with 100% ethanol, once with 75% ethanol and three times with distilled water. The gold particles were resuspended in 50% glycerol at a final density of 60 mg/mL. To the glycerol suspension containing 3 mg of gold particles, 2.5 µg of a single plasmid or 5.0 µg of a combination of two plasmids (2.5 µg each) were used. The gold particles and plasmid DNA were mixed well by vortexing in the presence of 1.14 m CaCl2 and 45.5 mm spermidine, kept for 1 min at room temperature and centrifuged briefly. The DNA‐coated gold particles were washed once with 70% ethanol and twice with 100% ethanol, and resuspended in an appropriate volume of 100% ethanol. Young leaves (5–6 cm in length) were excised and used for bombardment. The DNA‐coated gold particles were fired into the leaves at a distance of 6 cm and a pressure of 900 MPa. The bombarded leaves were kept on wet filter paper in Petri dishes at 25 °C and observed 1 day and 2 days after bombardment.
Agroinfiltration
Agrobacterium tumefaciens strain GV3101 (pMP90) was transformed with pART27‐KELP or pART27‐KELP‐DsRed. Transformation was performed using a Gene Pulse II Electroporation System (Bio‐Rad Laboratories), according to the manufacturer's protocol. The agrobacteria were grown by shaking overnight at 28 °C in Luria–Bertani (LB) medium containing rifampicin (50 µg/mL), kanamycin (20 µg/mL) and streptomycin (20 µg/mL). An aliquot of the overnight culture was transferred to new LB medium containing 10 mm 2‐(N‐morpholino)ethanesulphonic acid (MES) and antibiotics, and grown until the optical density at 600 nm (OD600) reached ~1.0. The bacteria were collected by centrifugation for 10 min at 4900 g at 4 °C, resuspended in an infiltration buffer containing 10 mm MES (pH 5.7), 10 mm MgCl2 and 150 µm acetosyringone to adjust the OD600 value to 0.4–0.6, and kept at room temperature for 2–3 h prior to infiltration. Young leaves (5–6 cm in length) attached on plants were injured with plastic tips to produce holes on the leaves, and infiltrated with the agrobacteria through plastic syringes without needles. The plants were kept for 2 days at 25 °C before protoplast preparation.
Protoplast preparation and viral RNA inoculation
After agroinfiltration, N. benthamiana leaves were treated for 2–3 h at room temperature with 1% Cellulase Onozuka R‐10 (Yakuruto Biochemicals, Tokyo, Japan) and 0.05% Macerozyme R‐10 (Yakuruto Biochemicals) in washing buffer (0.6 m mannitol, 10 mm CaCl2 and 5 mm MES, pH 5.6). Protoplasts were separated from tissue debris by filtration through four layers of cheesecloth, and centrifuged for 3 min at 1600 g. The purified protoplasts were washed at least twice with washing buffer (pH 6.5). In all, 5 × 105 protoplasts were inoculated with 3 µg of virion‐derived ToMV RNA by the polyethylene glycol method (Kroner and Ahlquist, 1992), and incubated for 24 h at 25 °C in incubation buffer (0.6 m mannitol, 0.2 mg/mL chloramphenicol, 5 mm MES, 0.2 mm KH2PO4, 1 mm KNO3, 1 mm MgSO4, 1 µm KI, 0.1 µm CuSO4, 10 mm CaCl2, pH 6.5).
Purification of ToMV virion and preparation of virion RNA
Nicotiana benthamiana leaves at 2 weeks after inoculation with ToMV were homogenized in 100 mm sodium phosphate buffer (pH 7.0) containing 0.1% 2‐mercaptoethanol (extraction buffer). After the homogenate had been passed through four layers of cheesecloth, the filtrate was mixed vigorously with the same volume of chloroform and centrifuged at 9000 gfor 10 min at 4 °C. The supernatant was centrifuged at 20 000 gfor 20 min at 4 °C and layered on the extraction buffer containing 25% sucrose, which was followed by centrifugation at 78 000 gfor 60 min at 4 °C. The pellets were resuspended in 10 mm phosphate buffer and centrifuged at 9500 gfor 20 min at 4 °C. The supernatant was collected as the virus suspension. Virion RNA was prepared from the suspension by a first extraction with phenol–chloroform–isoamylalcohol (25 : 24 : 1), a second extraction with chloroform, and precipitation with 100% ethanol in the presence of 0.3 m sodium acetate. The concentration of virion RNA was determined using a NanoDrop ND 1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA).
Protein analysis
Protoplasts inoculated with ToMV were resuspended and boiled in 1 × Lane Marker Reducing Sample Buffer (Pierce Biotechnology, Rockford, IL, USA). Proteins were separated by electrophoresis on 15% polyacrylamide gels containing 0.1% sodium dodecylsulphate (SDS) and stained using CBB Stain One (Nacalai Tesque, Inc., Kyoto, Japan). Western blotting on polyvinylidene difluoride (PVDF) membrane was carried out using 1 × Tris‐glycine buffer (Bio‐Rad Laboratories). The blots were treated with EzBlock blocking buffer (ATTO, Tokyo, Japan) and incubated in blocking buffer with appropriate antisera. ToMV CP was detected with a rabbit polyclonal antiserum against ToMV CP (Japan Plant Protection Association, Tokyo, Japan) and a goat anti‐rabbit immunoglobulin G (IgG) conjugated with alkaline phosphatase (Promega, Madison, WI, USA). A rabbit polyclonal antiserum raised against a recombinant KELP protein (Sawady Technology, Tokyo, Japan) and a goat anti‐rabbit IgG conjugated with peroxidase (Pierce Biotechnology) were used to detect KELP and KELP‐DsRed. To obtain the recombinant KELP protein, a glutathione transferase (GST)‐KELP fusion protein in Escherichia coli carrying pGEX‐P‐KELP was produced (Matsushita et al., 2001), and digested with PreScission Protease (Amersham Pharmacia Biotech, Piscataway, NJ, USA) to remove the GST portion. 5‐Bromo‐4‐chloroindol‐3‐yl phosphate/nitroblue tetrazolium (BCIP/NBT) Color Development Substrate (Promega) and Super Signal West Dura Extended Duration Substrate (Pierce Biotechnology) were used as substrates for alkaline phosphatase and peroxidase, respectively. Colour images were analysed on LAS‐3000 (Fujifilm, Tokyo, Japan).
DAPI staining
For the staining of the nucleus, leaves at 24 h after bombardment were incubated for 1 h in DAPI solution (10 µg/mL) and subjected to fluorescence microscopy.
Imaging of fluorescent proteins
Bombarded leaf tissues were observed under a BX50 epifluorescence microscope using the filter units UMWU for DAPI, UMWIBA/GFP for GFP and U‐MRFPHQ for DsRed (Olympus, Tokyo, Japan), or with a TCS‐NT laser scanning confocal microscope system equipped with the FITC/TRITC filter set (version 1.6.587, Leica, Heidelberg, Germany) to detect GFP, YFP and DsRed.
Supporting information
Fig. S1 Co‐localization of movement protein‐yellow fluorescent protein (MP‐YFP) and KELP‐DsRed in Nicotiana benthamiana epidermal cells. Epidermal cells expressing MP‐YFP and KELP‐DsRed were observed under a confocal microscope 24 h after bombardment. Green and red colours indicate fluorescence captured by detection units for green fluorescent protein (GFP) and DsRed, respectively. C, F, I and L are merged images of A and B, D and E, G and H, and J and K, respectively. Co‐localization of MP‐YFP and KELP‐DsRed in the nucleus (A–C) and irregular (D–F) or filamentous (G–I) structures is shown. No fluorescence of DsRed was detected as punctate structures (J–L). Scale bars, 10 µm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
ACKNOWLEDGEMENTS
We thank Dr Ken‐ichi Konagaya for technical advice on virus purification. We also thank Yuko Takahashi for technical assistance. We acknowledge Dr Yasuo Niwa for providing the 35Ω‐sGFP(S65T) plasmid. This work was supported in part by a Grant‐in‐Aid for Encouragement of Young Scientists (to Y.M., no. 13760035), a Grant‐in‐Aid for Scientific Research (C) (to Y.M., nos. 15580031 and 19580045), a Grant‐in‐Aid for Scientific Research on Priority Areas (to H.N., nos. 13039007 and 15028206) and a Grant‐in‐Aid for Young Scientists (B) (to N.S., no. 20780030) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a Grant‐in‐Aid for Young Scientists (Start‐up) (to N.S., no. 18880010) from the Japan Society for the Promotion of Science. This work was also supported by the Research for the Future Program of the Japan Society for the Promotion of Science.
REFERENCES
- Blackman, L.M. , Boevink, P. , Santa Cruz, S. , Palukaitis, P. and Oparka, K.J. (1998) The movement protein of cucumber mosaic virus traffics into sieve elements in minor veins of Nicotiana clevelandii . Plant Cell, 10, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.‐H. and Citovsky, V. (2003) Systemic movement of a tobamovirus requires host cell pectin methylesterase. Plant J. 35, 386–392. [DOI] [PubMed] [Google Scholar]
- Chen, M.‐H. , Tian, G.W. , Gafni, Y. and Citovsky, V. (2005) Effects of calreticulin on viral cell‐to‐cell movement. Plant Physiol. 138, 1866–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, W. , Niwa, Y. , Zeng, W. , Hirano, T. , Kobayashi, H. and Sheen, J. (1996) Engineered GFP as a vital reporter in plants. Curr. Biol. 6, 325–330. [DOI] [PubMed] [Google Scholar]
- Cormack, R.S. , Hahlbrock, K. and Somssich, I.E. (1998) Isolation of putative plant transcriptional coactivators using a modified two‐hybrid system incorporating a GFP reporter gene. Plant J. 14, 685–692. [DOI] [PubMed] [Google Scholar]
- Curin, M. , Ojangu, E.L. , Trutnyeva, K. , Ilau, B. , Truve, E. and Waigmann, E. (2007) MPB2C, a microtubule‐associated plant factor, is required for microtubular accumulation of tobacco mosaic virus movement protein in plants. Plant Physiol. 143, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave, A.P. (1992) A versatile binary vector system with a T‐DNA organisational structure conducive to efficient integration of cloned DNA into the plant genome. Plant Mol. Biol. 20, 1203–1207. [DOI] [PubMed] [Google Scholar]
- Haseloff, J. , Siemering, K.R. , Prasher, D.C. , and Hodge, A. (1997) Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc. Natl. Acad. Sci. USA, 94, 2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinlein, M. , Padgett, H.S. , Gens, J.S. , Pickard, B.G. , Casper, S.J. , Epel, B.L. and Beachy, R.N. (1998) Changing patterns of localization of the tobacco mosaic virus movement protein and replicase to the endoplasmic reticulum and microtubules during infection. Plant Cell, 10, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami, S. , Padgett, H.S. , Hosokawa, D. , Okada, Y. , Beachy, R.N. and Watanabe, Y. (1999) Phosphorylation and/or presence of serine 37 in the movement protein of tomato mosaic tobamovirus is essential for intracellular localization and stability in vivo. J. Virol. 73, 6831–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragler, F. , Curin, M. , Trutnyeva, K. , Gansch, A. and Waigmann, E. (2003) MPB2C, a microtubule‐associated plant protein binds to and interferes with cell‐to‐cell transport of tobacco mosaic virus movement protein. Plant Physiol. 132, 1870–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner, P. and Ahlquist, P. (1992) RNA‐based viruses In: Molecular Plant Pathology. A Practical Approach, Vol. I (Gurr S.J., McPherson M.J. and Bowles D.J., eds), pp. 23–34. Oxford: IRL Press. [Google Scholar]
- Lee, J.Y. , Taoka, K. , Yoo, B.C. , Ben‐Nissan, G. , Kim, D.J. and Lucas, W.J. (2005) Plasmodesmal‐associated protein kinase in tobacco and Arabidopsis recognizes a subset of non‐cell‐autonomous proteins. Plant Cell, 17, 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, W.J. (2006) Plant viral movement proteins: agents for cell‐to‐cell trafficking of viral genomes. Virology, 344, 169–184. [DOI] [PubMed] [Google Scholar]
- Matsushita, Y. , Deguchi, M. , Youda, M. , Nishiguchi, M. and Nyunoya, H. (2001) The tomato mosaic tobamovirus movement protein interacts with a putative transcriptional coactivator KELP. Mol. Cells 12, 57–66. [PubMed] [Google Scholar]
- Matsushita, Y. , Hanazawa, K. , Yoshioka, K. , Oguchi, T. , Kawakami, S. , Watanabe, Y. , Nishiguchi, M. and Nyunoya, H. (2000) In vitro phosphorylation of the movement protein of tomato mosaic tobamovirus by a cellular kinase. J. Gen. Virol. 81, 2095–2102. [DOI] [PubMed] [Google Scholar]
- Matsushita, Y. , Miyakawa, O. , Deguchi, M. , Nishiguchi, M. and Nyunoya, H. (2002) Cloning of a tobacco cDNA coding for a putative transcriptional coactivator MBF1 that interacts with the tomato mosaic virus movement protein. J. Exp. Bot. 53, 1531–1532. [PubMed] [Google Scholar]
- Matsushita, Y. , Ohshima, M. , Yoshioka, K. , Nishiguchi, M. and Nyunoya, H. (2003) The catalytic subunit of protein kinase CK2 phosphorylates in vitro the movement protein of Tomato mosaic virus. J. Gen. Virol. 84, 497–505. [DOI] [PubMed] [Google Scholar]
- McLean, B.G. , Waigmann, E. , Citovsky, V. and Zambryski, P. (1993) Cell‐to‐cell movement of plant viruses. Trends Microbiol. 1, 105–109. [DOI] [PubMed] [Google Scholar]
- McLean, B.G. , Zupan, J. and Zambryski, P.C. (1995) Tobacco mosaic virus movement protein associates with the cytoskeleton in tobacco cells. Plant Cell, 7, 2101–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima, K. , Taniyama, T. , Yamanaka, T. , Ishikawa, M. and Naito, S. (1998) Isolation of a mutant of Arabidopsis thaliana carrying two simultaneous mutations affecting tobacco mosaic virus multiplication within a single cell. Virology, 243, 472–481. [DOI] [PubMed] [Google Scholar]
- Okada, Y. (1999) Historical overview of research on the tobacco mosaic virus genome: genome organization, infectivity and gene manipulation. Philos. Trans. R. Soc. London, B: Biol. Sci. 354, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oparka, K.J. , Prior, D.A. , Santa Cruz, S. , Padgett, H.S. and Beachy, R.N. (1997) Gating of epidermal plasmodesmata is restricted to the leading edge of expanding infection sites of tobacco mosaic virus (TMV). Plant J. 12, 781–789. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. and Russell, D.W. (2001) Molecular Cloning. A Laboratory Manual, 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Tagami, Y. and Watanabe, Y. (2007) Effects of brefeldin A on the localization of Tobamovirus movement protein and cell‐to‐cell movement of the virus. Virology, 361, 133–140. [DOI] [PubMed] [Google Scholar]
- Tamai, A. , Kubota, K. , Nagano, H. , Yoshii, M. , Ishikawa, M. , Mise, K. and Meshi, T. (2003) Cucumovirus‐ and bromovirus‐encoded movement functions potentiate cell‐to‐cell movement of tobamo‐ and potexviruses. Virology, 315, 56–67. [DOI] [PubMed] [Google Scholar]
- Tomenius, K. , Clapham, D. and Meshi, T. (1987) Localization by immunogold cytochemistry of the virus‐coded 30K protein in plasmodesmata of leaves infected with tobacco mosaic virus. Virology, 160, 363–371. [DOI] [PubMed] [Google Scholar]
- Tremblay, D. , Vaewhongs, A.A. , Turner, K.A. , Sit, T.L. and Lommel, S.A. (2005) Cell wall localization of Red clover necrotic mosaic virus movement protein is required for cell‐to‐cell movement. Virology, 333, 10–21. [DOI] [PubMed] [Google Scholar]
- Trutnyeva, K. , Bachmaier, R. and Waigmann, E. (2005) Mimicking carboxyterminal phosphorylation differentially affects subcellular distribution and cell‐to‐cell movement of Tobacco mosaic virus movement protein. Virology, 332, 563–577. [DOI] [PubMed] [Google Scholar]
- Waigmann, E. , Chen, M.H. , Bachmaier, R. , Ghoshroy, S. and Citovsky, V. (2000) Regulation of plasmodesmal transport by phosphorylation of tobacco mosaic virus cell‐to‐cell movement protein. EMBO J. 19, 4875–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waigmann, E. , Curin, M. and Heinlein, M. (2007) Tobacco mosaic virus—a model for macromolecular cell‐to‐cell spread In: Viral Transport in Plants, Plant Cell Monograph 7 (Waigmann E. and Heinlein M., eds), pp. 29–62. Heidelberg/Berlin: Springer‐Verlag. [Google Scholar]
- Watanabe, Y. , Emori, Y. , Ooshika, I. , Meshi, T. , Ohno, T. and Okada, Y. (1984) Synthesis of TMV‐specific RNAs and proteins at the early stage of infection in tobacco protoplasts: transient expression of the 30K protein and its mRNA. Virology, 133, 18–24. [DOI] [PubMed] [Google Scholar]
- Wolf, S. , Deom, C.M. , Beachy, R.N. and Lucas, W.J. (1989) Movement protein of tobacco mosaic virus modifies plasmodesmatal size exclusion limit. Science, 246, 377–379. [DOI] [PubMed] [Google Scholar]
- Yoshioka, K. , Matsushita, Y. , Kasahara, M. , Konagaya, K. and Nyunoya, H. (2004) Interaction of tomato mosaic virus movement protein with tobacco RIO kinase. Mol. Cells, 17, 223–229. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Co‐localization of movement protein‐yellow fluorescent protein (MP‐YFP) and KELP‐DsRed in Nicotiana benthamiana epidermal cells. Epidermal cells expressing MP‐YFP and KELP‐DsRed were observed under a confocal microscope 24 h after bombardment. Green and red colours indicate fluorescence captured by detection units for green fluorescent protein (GFP) and DsRed, respectively. C, F, I and L are merged images of A and B, D and E, G and H, and J and K, respectively. Co‐localization of MP‐YFP and KELP‐DsRed in the nucleus (A–C) and irregular (D–F) or filamentous (G–I) structures is shown. No fluorescence of DsRed was detected as punctate structures (J–L). Scale bars, 10 µm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item