

SUMMARY
Plant pathogenic bacteria possess a large number of genes that allow them to grow and cause disease on plants. In planta gene expression analysis is important to understand the impact of these genes on bacterial virulence. A new mRNA‐based approach using multiplexed Northern hybridization was developed. High‐quality bacterial and plant total RNA was successfully isolated from leaf tissue infiltrated with Pseudomonas syringae. The procedure employs a new extraction buffer formulation containing glycine, sodium dodecylsulphate, cetyltrimethylammonium bromide, high‐molecular‐weight polyethylene glycol and β‐mercaptoethanol. Cell lysis and classical acid–phenol extraction steps followed by LiCl precipitation yielded large amounts of total RNA of high purity and integrity. Multiplexing of DIG and chemically fluorescently labelled RNA probes was developed and expression data were normalized using the 23S rRNA gene as reference. The method was validated by studying in planta expression of the P. syringae genes mucD, cmaA, cfl, corR, corS and corP comprising a selection of highly expressed biosynthetic and low‐expressed regulatory genes. The method was assessed regarding its sensitivity and might by useful for studying a variety of plant–microbe interactions.
INTRODUCTION
Analysis of in planta gene expression can significantly contribute to our understanding of the interaction of plant pathogenic bacteria with their plant hosts. For the plant pathogenic bacterium Pseudomonas syringae, thus far classical reporter genes such egfp or uidA have been utilized to measure gene expression inside infected host tissue (Keith et al., 2003; Weingart et al., 2004). Differential gene expression of P. syringae on plant surfaces was studied with gfp, lux or inaZ reporter gene fusions (Cirvilleri and Lindow, 1994; Joyner and Lindow, 2000; Marco et al., 2005; Waterhouse et al., 1993). The activity of reporter genes is easy to measure but each investigated gene requires an elaborate study design and introduction of a resistance marker‐carrying reporter construct into the bacterial test strain. Expression data can be biased by altered mRNA processing due to reporter gene fusion or the stability of the reporter protein.
Direct measurement of mRNA levels with quantitative RT‐PCR or Northern hybridization would be suitable alternatives to gain accurate and reliable in planta gene expression data. An important prerequisite is the extraction of high‐quality total RNA from infected leaf tissue. Plants produce secondary metabolites, which can strongly interfere with the RNA extraction and enzymatic downstream applications such as RT‐PCR. Bacterial mRNA has a shorter half‐life and is more unstable than that of eukaryotes because eukaryotic mRNA is capped and polyadenylated. There are several plant RNA extraction protocols available that address the problems of plant tissue rich in polysaccharides or secondary metabolites. The detergent cetyltrimethylammonium bromide (CTAB) was successfully used to separate nucleic acids from polysaccharides (Chang et al., 1993). High‐molecular‐weight polyethylene glycol (HMW‐PEG) was found to eliminate large amounts of phenolic compounds and polysaccharides (Gehrig et al., 2000). The reducing agent β‐mercaptoethanol can be used to prevent the oxidation of phenolic compounds (Jaakola et al., 2001) and to inactivate RNases. Extraction protocols for bacterial total RNA use rapid cell lysis under heat with the detergent sodium dodecylsulphate (SDS) followed by repeated organic extractions with acid‐phenol (Majumdar et al., 1991; Sambrook et al., 1989).
RNA isolated from soybean leaf tissue infected with P. syringae represents a mixture of bacterial and plant total RNA at an unknown ratio. Therefore, normalization of expression data becomes an important issue, especially if generated by Northern hybridization, where equal amounts of total RNA from to‐be‐compared samples are usually required. For quantitative RT‐PCR analysis, normalization of data against reference genes is indispensable. Normalization of Northern hybridization data with a reference gene would require two probes, one for the gene of interest and one for a reference gene.
To our knowledge, there are no reports dealing with the simultaneous extraction of bacterial and plant total RNA suitable for Northern hybridization and quantitative RT‐PCR experiments. Here we report for the first time the simultaneous extraction of high‐quality bacterial and plant total RNA from infected leaf tissue and a refined method of bacterial in planta gene expression quantification based on classical and simple non‐radioactive, fluorescent Northern hybridization, as demonstrated on selected genes of interest.
In this study, two P. syringae pv. glycinea strains were used which differ in the presence of the functional alternative sigma factor AlgT, PG4180 (algT −) and PG4180.muc (algT +). As a result, PG4180.muc is able to produce large amounts of the exopolysaccharide alginate, whereas alginate production is negligible in PG4180 (Schenk et al., 2006). We investigated the in vitro and in planta gene expression of mucD, coding for a periplasmic serine protease previously implicated in alginate regulation (Schenk et al., 2006), expression of lsc, coding for levansucrase required for synthesis of the exopolysaccharide levan (Li et al., 2001), expression of cmaA and cfl, genes involved in the biosynthesis of the phytotoxin coronatine (Budde et al., 1998), and expression of corR, corS and corP, genes coding for regulators of coronatine biosynthetic genes (Weingart et al., 2004). Several of these genes were previously reported to exhibit a thermoresponsive pattern of expression in vitro (Budde et al., 1998). Consequently, two temperatures, 18 and 28 °C, were used. Alginate and levan, and coronatine are important virulence and fitness factors of P. syringae, respectively. An appropriate and sensitive method to analyse their potential plant inducibility consequently is the key to a better understanding of their role(s) in the plant–microbe interaction.
RESULTS
Extraction of total RNA from infected leaf tissue
The alginate‐producing P. syringae pv. glycinea strain PG4180.muc and the non‐alginate‐producing strain PG4180 were inoculated into soybean leaves by infiltration. Infected and uninfected control plants were kept in growth chambers set to 18 or 28 °C. Three days after inoculation, infiltrated leaf regions showed typical symptoms and in planta samples were taken. For in vitro samples, PG4180.muc and PG4180 were grown in HSC medium at 18 and 28 °C to an OD600 of 1.0. Total RNA was extracted using the extraction buffer and method described herein. Yields of total RNA were in the range 75–125 µg per plant tissue sample (150–200 mg plant tissue) and 75–100 µg per bacterial cell pellet (5 × 109 CFU). A260/A280 ratios ranged between 2.1 and 2.2. Determined A260/A230 ratios above 1.6 indicated high purity and absence of protein and polysaccharide contaminations. Aliquots of the preparations were further analysed using the Agilent 2100 Bioanalyzer (Fig. 1). The isolated RNA was of high quality and integrity, as demonstrated by sharp ribosomal RNA peaks.
Figure 1.
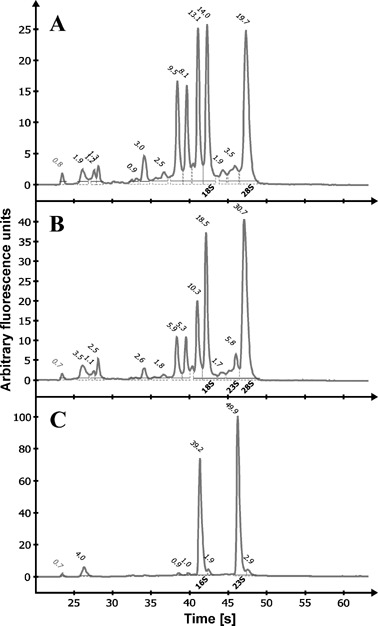
Electropherograms of total RNA extractions. Total RNA from uninfected soybean leaf tissue (A), soybean leaf tissue infected with P. syringae pv. glycinea (B) and in vitro grown P. syringae pv. glycinea (C) was analysed using a Agilent 2100 Bioanalyzer. Main ribosomal RNA peaks of bacteria (16S, 23S) and plant (18S, 28S) are indicated. Numbers above the peaks show their quantitative percentage within the entire sample.
23S rRNA probe for normalization of bacterial in planta gene expression data
Northern or Spot blot expression data of bacterial total RNA can be directly generated from hybridization signals, as each line or spot contains a defined amount of bacterial RNA. Samples isolated from infected leaf tissues consist of a mixture of bacterial and plant total RNA and might vary in their bacterial total RNA content. Consequently, normalization of hybridization signals is required to generate bacterial in planta expression data. The high abundance and relatively constant expression of the 16S and 23S bacterial ribosomal RNAs make them a suitable target for reference. Potential probe binding sites in the 23S rRNA were identified by alignment of 23S rDNA gene sequences from Pseudomonas syringae, Pseudomonas putida, Pseudomonas fluorescens, Pseudomonas aeruginosa, Arabidopsis thaliana chloroplast, and Nicotiana tabacum chloroplast. Two short regions, one located at the beginning (23S#1) and one located at the end (23S#2) of the 2903‐bp 23S rDNA gene from P. syringae, showed sufficient sequence uniqueness (data not shown). DIG‐labelled RNA probes 23S#1 and 23S#2 were generated and analysed by Northern hybridization with plant total RNA, total RNA isolated from infected tissue and bacterial total RNA (Fig. 2). Both probes showed strong signals at the size of the 23S rRNA in the total RNA isolated from infected tissue and bacterial total RNA samples. No cross‐hybridization with total RNA derived from uninfected plants was detectable.
Figure 2.
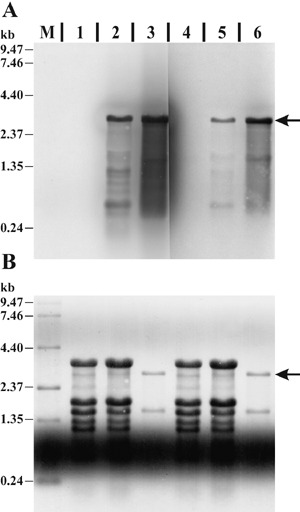
Evaluation of two 23S rRNA‐specific probes. (A) Northern blot membrane hybridized with DIG‐labelled probe 23S#1 (lanes M, 1–3) or 23S#2 (lanes 4–6). Lanes contain total RNA extracted from uninfected soybean leaf tissue (1, 4; 5 µg), soybean leaf tissue infected with P. syringae pv. glycinea (2, 5; 5 µg), in vitro cultured P. syringae pv. glycinea (3, 6; 0.5 µg) and an RNA size standard (M; 3.0 µg). Hybridization signals specific to the 23S rRNA are indicated by the arrow. (B) Methylene‐blue‐stained membrane prior to Northern hybridization for size estimation, control of total RNA quantity and successful Northern transfer. The same region of the membrane as in A is shown.
Multiplexed fluorescent Northern hybridization
Bacterial gene‐specific and 23S rRNA signals were analysed using a new multiplexed fluorescent Northern hybridization approach. A DIG‐labeled gene‐specific and an Oyster‐645 fluorescence‐labelled 23S#1 RNA probe were multiplexed in a single Northern hybridization reaction. Northern and Spot blotting techniques were evaluated for their usability in our multiplexed hybridization approach. For mucD analysis, total RNA samples of PG4180.muc and PG4180 grown in vitro and in planta at 18 and 28 °C and total RNA from uninfected plants as control were separated by denaturating RNA gel electrophoresis, followed by Northern transfer and hybridization (Fig. 3). The mucD Northern blot showed a transcript at a size of ~1.75 kb (Fig. 3A) and sharp bands for the 23S rRNA at ~2.9 kb (Fig. 3B). Neither the mucD nor the 23S#1 probe showed cross‐hybridization with uninfected plant total RNA controls. Cross‐talk of the two fluorescent dyes ECF and Oyster‐645 was not observed, as determined by scanning the membrane sequentially on the FLA‐3000 phosphoimager with appropriate laser and filter combinations. Signal intensities were always in the linear range of detection. The coronatine genes, cmaA, cfl, corR, corS and corP, were analysed with PG4180 in vitro and in planta total RNA samples. Samples were spotted in triplicate and hybridized (Fig. 4). The Spot blots showed signals for cmaA (Fig. 4A), cfl, corR, corS, corP and the 23S#1 probe (Fig. 4B). Signals obtained for cmaA and cfl biosynthetic genes were stronger than those for the regulatory genes corR, corS and corP. No cross‐hybridizations with the plant total RNA controls were observed.
Figure 3.
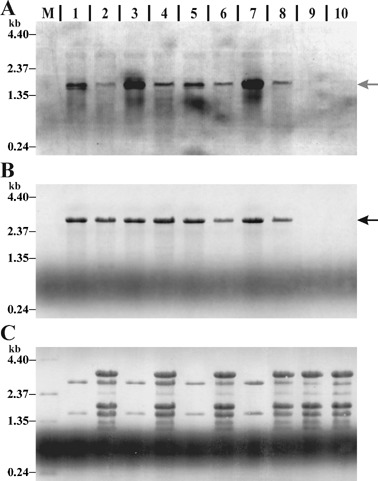
Multiplexed fluorescent Northern blot hybridization. Northern blot membrane hybridized with a DIG‐labelled mucD probe (A) multiplexed with an Oyster‐645‐labelled 23S#1 probe (B). Lanes contain total RNA extracted from in vitro cultured PG4180.muc at 18 (1) or 28 °C (3), PG4180.muc‐infected soybean tissue at 18 (2) or 28 °C (4), in vitro cultured PG4180 at 18 (5) or 28 °C (7), PG4180‐infected soybean tissue at 18 (6) or 28 °C (8), uninfected soybean leaf tissue at 18 (9) or 28 °C (10), and an RNA size standard (M). Arrows indicate probe‐specific signals. For in vitro samples 1.5 µg and for in planta samples 6 µg RNA was loaded. (C) Methylene‐blue‐stained membrane prior Northern hybridization. A, B and C show the same region of the membrane.
Figure 4.

Multiplexed fluorescent Spot blot hybridization. Example of a Spot blot membrane hybridized with a DIG‐labelled cmaA probe (A) multiplexed with an Oyster‐645‐labelled 23S#1 probe (B). Spots contain total RNA extracted from in vitro cultured PG4180 at 18 (1) or 28 °C (3), PG4180‐infected soybean tissue at 18 (2) or 28 °C (4), and uninfected soybean leaf tissue at 18 (5) or 28 °C (6). For in vitro samples 0.2 µg and for in planta samples 1 µg RNA was spotted. (C) Methylene‐blue‐stained membrane prior Northern hybridization. A, B and C show the same region of the membrane.
Quantitative data were acquired for Northern and Spot blot signals using the AIDA V4.00 software package (Raytest). For each hybridization signal, a total volume and local background value was obtained, which led to background‐corrected volume values after subtraction. Corrected volume values were normalized to their corresponding 23S rRNA signals by division and, when possible, standard deviations were calculated. For each data set the highest value was set to 100% relative expression and other data were related to this. Figure 5 summarizes the normalized data. Expression of mucD was slightly higher in vitro than in planta and about two‐fold higher at 28 than at 18 °C, whereas in planta this situation was almost reversed. Both investigated strains exhibited similar expression patterns with slightly lower signals for PG4180.muc than for PG4180. The in vitro expression of cmaA and cfl in PG410 showed a clear temperature‐dependent phenotype with 12‐fold higher expression at 18 than at 28 °C. The temperature phenotype was confirmed in planta for both strains but at a 50‐fold lower overall gene expression level than in vitro. PG4180 corR, corS and corP regulatory genes could be quantified in vitro and in planta with expression values varying independent of temperature and environmental background up to three‐fold.
Figure 5.
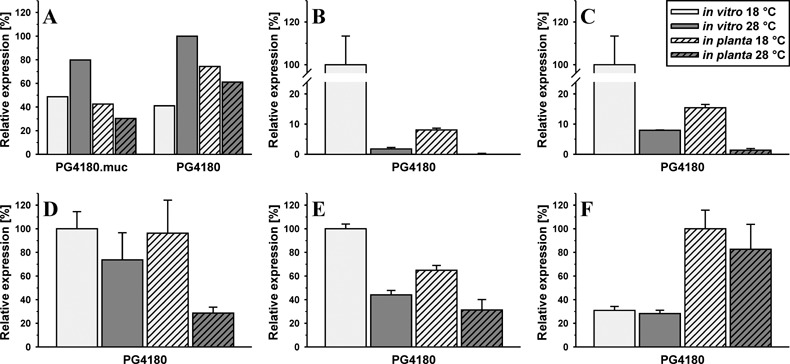
Quantitative in vitro and in planta gene expression. Relative abundance of P. syringae pv. glycinea mucD (A), cmaA (B), cfl (C), corR (D), corS (E) and corP (F) mRNA in vitro and in planta at 18 and 28 °C. Data were generated by quantification of gene‐specific signals and normalization to the corresponding 23S rRNA signals. Bars in B–F represent the mean of three replica spots and their standard deviation.
In order to assess the sensitivity of the method described herein, in planta detection limits were determined for the moderately expressed bacterial lsc gene coding for levansucrase (2001, 2006) (Fig. 6). For this, a very low inoculation density (OD of 0.001) and sampling after 2, 4, 7 and 10 days were used. No signal could be detected with 5.2 × 102 CFU per leaf disc. For the 23S rRNA probe a signal was observed with 5 × 104 CFU per leaf disc (Fig. 6A) and for lsc 2.5 × 106 CFU per leaf disc was required to obtain a signal. The data suggested that expression of genes from ~106 cells per sample can be monitored by this method.
Figure 6.
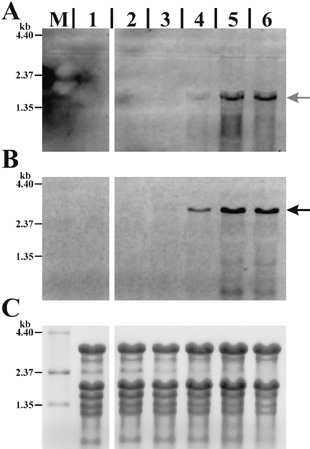
Evaluation of in planta sensitivity of multiplexed fluorescent Northern blot hybridization. Membrane hybridized with a DIG‐labelled lsc probe (A) multiplexed with an Oyster‐645‐labeled 23S#1 probe (B). Lanes contain 10 µg total RNA extracted from uninfected soybean leaf tissue (1), soybean leaf tissue infected with P. syringae pv. glycinea PG4180 (2–6) and 3 µg of RNA size standard (M). Soybeans were infiltrated with a bacterial suspension of OD 0.001 and leaf discs were sampled for CFU determination and RNA extraction at different time points. Lane 2, 2 days (5.2 × 102 CFU per leaf disc); Lane 3, 4 days (5.0 × 104 CFU per leaf disc); Lane 4, 7 days (2.5 × 106 CFU per leaf disc); Lane 5, 10 days (1.3 × 107 CFU per leaf disc); and Lane 6, 16 days (2.2 × 107 CFU per leaf disc). Arrows indicate probe‐specific signals. (C) Methylene‐blue‐stained membrane prior to Northern hybridization. A, B and C show the same region of the membrane.
DISCUSSION
Prerequisite for any gene expression experiment at the RNA level is reliable starting material. For in planta experiments, quality and yield are far more important than sample throughput. Therefore, we developed a new extraction buffer formulation and method for the isolation of total RNA from leaf tissue infected with P. syringae, yielding consistently large amounts of high‐quality total RNA. A combination of SDS and β‐mercaptoethanol in the extraction buffer with a short hot lysis step facilitated cell‐wall dissolution and recovery of RNA. Addition of HMW‐PEG improved integrity and purity of RNA by removing phenolic compounds (Gehrig et al., 2000). Successive organic extraction with acid‐phenol and addition of CTAB detergent in the last extraction step greatly improved separation of RNA from proteins and polysaccharides (Chang et al., 1993; Jaakola et al., 2001; Majumdar et al., 1991). Finally, selective RNA precipitation using LiCl further increased RNA purity. Spectrophotometric and gel electrophoretic analysis demonstrated the high quality of the bacterial and plant total RNA. This RNA was applicable for enzymatic downstream applications such as cDNA synthesis and RT‐PCR.
With a yield of high‐quality total RNA, we evaluated the usability of well‐established, non‐expensive, low‐tech Northern and Spot blot hybridization methods for bacterial in planta gene expression analysis, although the more expensive quantitative RT‐PCR method needs to be kept as a sensitive and high‐throughput option. For RT‐PCR it is essential to have demonstrated in vitro that the probe is specific to a transcript of a given size. However, that technique might not detect variable transcript sizes in high‐throughput experiments in planta. Such variations in transcript sizes might arise from differential regulation in the natural setting. The method described herein circumvents this often neglected potential problem. The quality of the RNA obtained by the extraction procedure described herein is suitable for RT‐PCR as determined in a previous study (Schenk et al., 2006).
As total RNA samples isolated from infected leaf tissue represent a mixture in which the bacterial RNA portion is unknown, all expression data had to be normalized to a reference. We took advantage of the 23S rRNA as a target of high abundance and relatively constant expression. Two RNA probes, directed against 23S rRNA, were developed and yielded clear hybridization signals highly specific for P. syringae 23S rRNA. To increase data accuracy and to reduce the amount of blots and chemicals, we successfully developed multiplexing of a gene‐specific and a 23S rRNA probe. The total RNA samples isolated from infected leaf tissue contained only a low proportion of bacterial mRNA. Sensitivity of gene‐specific probes was increased by labelling them with the DIG system. This has the advantage of producing an amplified ECF‐fluorescence signal mediated by an antibody binding cascade and alkaline phosphatase action. In contrast to gene‐specific mRNAs, the abundance of 23S rRNA in total RNA samples is high. Therefore, the 23S rRNA probe was directly labelled by chemical linkage of an activated fluorescent dye. Taking into account that a higher number of potential probe targets demands more RNA probe, large amounts of in vitro synthesized probe were generated by large‐scale in vitro transcription. Development and quantification of the ECF and Oyster‐645 fluorescence signals was successfully carried out on a phosphoimager system, utilizing two different laser and filter settings. Cross‐talk or fluorescence resonance energy transfer (FRET) between the two fluorophors used was not detectable as excitation and emission spectra of the dyes did not show a significant overlap.
Validation of the method was addressed by in planta gene expression analysis of the P. syringae genes mucD, cmaA, cfl, corR, corS and corP, which we had investigated in previous studies (Budde et al., 1998; Schenk et al., 2006; Weingart et al., 2004). Sensitivity was assessed by monitoring the moderately expressed lsc gene, indicating that gene expression from ~106 CFU per leaf disc can be detected. In vitro data for mucD obtained from Northern blots confirmed our previously reported data (Schenk et al., 2006). In planta data showed that mucD transcription in P. syringae is not plant‐inducible and supported our previous finding that it is not dependent on the sigma factor AlgT, as mucD expression was not elevated in PG4180.muc (algT +) as compared with PG4180 (algT −). The two‐fold induction observed in vitro at 28 versus 18 °C was not observed in planta. In vitro and in planta data obtained for the biosynthetic genes involved in coronatine biosynthesis, cmaA and cfl, confirmed our previous findings, which showed a strong temperature dependence but no plant induction in P. syringae strain PG4180, in contrast to the situation in strain DC3000 (Weingart et al., 2004). In our previous study, cmaA expression was measured using egfp as reporter gene. In the present study, we observed about 50‐fold lower in planta expression than in vitro. This can be explained by growth phase differences because in planta samples are likely to represent the mid‐stationary phase whereas in vitro samples correspond to the early exponential growth phase. Highest EGFP accumulation in vitro occurred in the stationary phase, whereas highest levels for the cmaA transcript were found in the early exponential phase and declined in the stationary phase (Weingart et al., 2004). Reporter gene fusions measure promoter activity over time by accumulation of the reporter protein, whereas mRNA‐based methods provide snapshots of mRNA levels at a defined time point.
Sensitivity of the multiplexed fluorescent Northern hybridization is also applicable for in planta gene expression analysis of regulatory genes, as demonstrated for corR, corS and corP. The method described herein can provide further insight into bacterial and plant gene expression during their natural pathogenic or symbiotic interaction(s).
EXPERIMENTAL PROCEDURES
Bacterial strains, growth conditions and harvesting of bacteria
P. syringae was routinely maintained at 28 °C on mannitol‐glutamate (MG) medium (Keane et al., 1970). For liquid cultures at 18 or 28 °C, bacteria were grown in Hoitink‐Sinden minimal medium (HSC) (Palmer and Bender, 1993) and growth was continuously monitored by measuring the optical density at 600 nm (OD600). For three RNA extractions, a culture equivalent of 15 mL OD600 of 1.0 (c. 15 × 109 CFU) was harvested at a defined OD600 by transfer into a falcon tube filled with 15 mL ice‐cold killing buffer [20 mm Tris‐HCl (pH 7.5); 20 mm NaN3) and centrifuged for 15 min at 3200 g and 4 °C. The supernatant was discarded, the pellet resuspended in 3 mL ice‐cold killing buffer, distributed to three 1.5‐mL centrifuge tubes and centrifuged for 5 min at 6600 g and 4 °C. The supernatants were carefully removed, the tubes immediately frozen in liquid nitrogen and stored at –80 °C for further RNA extraction.
Plant material, inoculation procedures and harvesting of plant material
Soybean seedlings [Glycine max (L.) Merr. cv. Maple Arrow] were grown in shelves equipped with fluorescent lamps at 22–25 °C, 55% humidity, with a 16‐h photoperiod (350 µE/m2/s). Two days prior to inoculation plants were transferred to growth chambers set to 18 or 28 °C, 55% humidity, with a 16‐h photoperiod (350 µE/m2/s). Bacteria were infiltrated into leaves by means of a needleless syringe at an OD600 of 0.1 (c. 1 × 108 CFU/mL) and plants were kept in growth chambers at 18 or 28 °C. Three days after inoculation 20 discs (7 mm diameter) containing infected leaf tissue (about 150–200 mg) were excised with a cork borer, transferred into a 2.0‐mL centrifuge tube, immediately frozen in liquid nitrogen and stored at –80 °C for further RNA extraction.
Extraction of total RNA from infected leaf tissue
The total RNA was isolated from infected leaf tissue by a combination of several described methods and reagents (Chang et al., 1993; Gehrig et al., 2000; Geraats et al., 2002; Jaakola et al., 2001; Majumdar et al., 1991; Sambrook et al., 1989). All solutions were prepared with 0.1% (v/v) diethylpyrocarbonate (DEPC)‐treated and autoclaved water. Prior to use, glassware, mortars, pestles and spatulas were wrapped in aluminium foil and backed at 200 °C for at least 6 h. Additional precautions were taken for isolating total RNA (Sambrook et al., 1989). Chemicals and reagents, if not otherwise stated, were obtained from AppliChem (Darmstadt, Germany) and, if available, at molecular biology grade. The bacteria plant extraction (BPEX) buffer [0.35 m glycine; 0.7 m NaCl; 2% (w/v) polyethylene glycol 20000 (Merck, Darmstadt, Germany); 40 mm EDTA (0.5 m EDTA stock solution, pH 8.0); 50 mm NaOH (10 m NaOH stock solution); 4% (w/v) SDS] and CTAB/NaCl solution [10% (w/v) CTAB; 0.7 m NaCl] was prepared under gentle warming and stirring. Aliquots of the BPEX buffer were supplemented with 100 mmβ‐mercaptoethanol shortly before use. Total RNA was extracted according to the steps as listed in Table 1.
Table 1.
Bacteria and/or plant total RNA extraction protocol.
| 1. | This step is only necessary for plant tissue, for bacterial cell pellet start at step 2. Grind the plant tissue sample (150–200 mg) to a fine powder in liquid nitrogen with a pre‐cooled pestle and mortar. Transfer the powder to a 2.0‐mL centrifuge tube with a pre‐cooled spatula. Keep it in liquid nitrogen until BPEX buffer is added. |
| 2. | Add 750 µL supplemented BPEX buffer to the grinded plant tissue or the bacterial cell pellet, mix completely by vortexing for 10 s (grinded plant tissue) or pipeting up and down (bacterial cell pellet) and incubate in a pre‐heated thermomixer at 900 r.p.m. and 95 °C for 90 s. |
| 3. | Add 750 µL water‐saturated, stabilized phenol/chloroform mix (5:1; pH 4.0). Shake in a mixer at room temperature for 5 min (speed has to be adjusted until an emulsion is formed). Centrifuge at 16 000 g and room temperature for 7 min. |
| 4. | Transfer 675 µL of the upper phase to a fresh 2.0‐mL centrifuge tube, preloaded with 675 µL water‐saturated, stabilized phenol/chloroform mix (5:1; pH 4.0). Shake in a mixer at room temperature for 5 min. Centrifuge at 16 000 g and room temperature for 7 min. |
| 5. | Transfer 575 µL of the upper phase to a fresh 2.0‐mL centrifuge tube, preloaded with 575 µL water‐saturated, stabilized phenol/chloroform/isoamyl alcohol mix (25:24:1; pH 4.0). Shake in a mixer at room temperature for 5 min. Centrifuge at 16 000 g and room temperature for 6 min. |
| 6. | Transfer 495 µL of the upper phase to a fresh 2.0‐mL centrifuge tube, preloaded with 550 µL water‐saturated, chloroform/isoamyl alcohol mix (24:1) and overlay with 55 µL CTAB/NaCl solution (prewarmed to 55 °C). Shake in a mixer at room temperature for 5 min. Centrifuge at 16 000 g and room temperature for 6 min. |
| 7. | Transfer 435 µL of the upper phase to a fresh 1.5‐mL centrifuge tube, preloaded with 145 µL 8 m LiCl solution. Mix by inverting the tube and precipitate RNA at –20 °C for 30 min. Centrifuge at 16 000 g and 4 °C for 20 min. |
| 8. | Decant the supernatant and dab the tube on a fresh paper towel. Wash the pellet by adding 1 mL ice‐cold 75% ethanol and inverting the tube several times. Centrifuge at 16 000 g and 4 °C for 10 min. |
| 9. | Optional: Repeat step 8 to remove any LiCl traces. |
| 10. | Decant the supernatant and collect ethanol remains by short centrifugation. Remove any ethanol remains by pipeting. Air‐dry the RNA pellet briefly at room temperature for 5 min. |
| 11. | Resuspend the RNA pellet in 50 µL RNase‐free water and store at –80 °C. |
RNA quantification and quality assessment
The purity and concentration of the purified RNA was determined spectrophotometrically. An aliquot of the total RNA was diluted 1:70 in 10 mm Tris/HCl buffer (pH 8.0), and RNA concentration was measured by its absorbance at 260 nm (A260) in a Microvolume cell and a Ultrospec 2100 pro UV/Visible Spectrophotometer (Amersham Biosciences, Freiburg, Germany). A260/A280 and A260/A230 ratios were determined to check for protein and polysaccharide contaminations (Rapley and Heptinstall, 1998). Quality and integrity of RNA was assessed by analysing total RNA aliquots in an RNA 6000 Nano LabChip® Kit on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
Denaturating RNA gel electrophoresis, Northern and spot blot
Aliquots of total RNA (1.5 µg bacterial or 6 µg plant/bacterial total RNA per lane) and an RNA size standard [2 µg 0.24–9.5 kb RNA Ladder (Invitrogen, Karlsruhe, Germany)] were separated by denaturing glyoxal RNA agarose gel electrophoresis as described by Burnett (1997) and transferred to a positively charged nylon membrane (Pall, Dreieich, Germany) as described by Ingelbrecht et al. (1998). For RNA spot blot analysis, aliquots of total RNA (200 ng bacterial or 1 µg plant/bacterial total RNA per spot) were transferred in 10× SSC (1.5 m NaCl, 0.15 m sodium citrate, adjusted with HCl to pH 7.0) to a positively charged nylon membrane (Pall) using the Minifold I Spot‐Blot System (Schleicher & Schuell BioScience, Dassel, Germany) according to the manufacturer's recommendations. Even and successful transfer of the RNA was verified prior to hybridization by reversible staining of the membrane with methylene blue (Herrin and Schmidt, 1988).
Generation of DIG and fluorescently labelled RNA hybridization probes
RNA probes were generated by in vitro transcription of T7‐modified PCR products. Gene‐specific primers (Table 2) were used to amplify PCR products from genomic DNA of P. syringae. The reverse PCR primers carry a T7 promoter sequence at their 5′‐end.
Table 2.
Oligonucleotide primers used in this study.
| Oligonucleotide primer | Nucleotide sequence (5′–3′)* |
|---|---|
| mucD_fwd | CGAATTTCTCGAGCGCAGCATGC |
| mucD_revT7 | TAATACGACTCACTATAGGGAGGGGAGCGGGTAAATATCTGCG |
| cmaA_fwd | TTTGAGTCGGTCTGCACGCA |
| cmaA_revT7 | TAATACGACTCACTATAGGGAGGGCTGTACGTTGTCTACTAG |
| cfl_fwd | ATGAGTCTGATTTCTGAGTTCCGCA |
| cfl_revT7 | TAATACGACTCACTATAGGGAGGTAGTTATTCCTGTGGTGC |
| corR_fwd | ATGCCGAGCTCTTCGATCTTGC |
| corR_revT7 | TAATACGACTCACTATAGGGAGGAGTATCGCCTGGACATGG |
| corS_fwd | AATACGGCGCGCTGTCAGTT |
| corS_revT7 | TAATACGACTCACTATAGGGAGGAATGGATGGCCTAATAGGCG |
| corP_fwd | ATCACGCCTTGTTCCGTT |
| corP_revT7 | TAATACGACTCACTATAGGGAGGTCATTTGCAAATCGAGCAAGATGAGATCG |
| 23SrRNA_fwd#1 | ACGTGGACCAGCCCTTAAGTTGTATTG |
| 23SrRNA_revT7#1 | TAATACGACTCACTATAGGGAGGCCCCCATATTCAGACAAG |
| 23SrRNA_fwd#2 | CTTGAGTTCCCTGAAGGGCCGTCGAAG |
| 23SrRNA_revT7#2 | TAATACGACTCACTATAGGGAGGTGGTCAAGCCTCACGGGC |
| lsc_fwd | GTCAGTGCGGACTTTCCGGTCATG |
| lsc_revT7 | TAATACGACTCACTATAGGGAGGGATCGCGAAAGTTCCAGCTC |
T7 RNA polymerase promoter sequences incorporated in primers are shown in italics.
DIG‐labelled RNA probes were synthesized by using the Strip‐EZ™ RNA T7 Kit (Applied Bioscience, Cambridgeshire, UK) and digoxigenin‐11‐UTP (Roche Diagnostics, Mannheim, Germany), yielding hybridization probes of the following sizes internal to the structural genes: for mucD, 511 nt (mucD_fwd, mucD_revT7); cmaA, 523 nt (cmaA_fwd, cmaA_revT7); cfl, 520 nt (clf_fwd, cfl_revT7); corR, 524 nt (corR_fwd, corR_revT7); corS, 608 nt (corS_fwd, corS_revT7); corP, 143 nt (corP_fwd, corP_revT7); lsc, 543 nt (lsc_fwd, lsc_revT7); 23S#1, 146 nt (23S_fwd#1, 23S_revT7#1); and 23S#2, 108 nt (23S_fwd#2, 23S_revT7#2).
Fluorescently labelled RNA probes were generated by incorporation of 5‐(3‐aminoallyl)‐UTP during in vitro transcription and a subsequent chemical linkage of an N‐hydroxysuccinimide‐activated fluorescent dye (Denovo Biolabels, Muenster, Germany). For a 40‐µL in vitro transcription reaction, 2 µg of T7‐modified PCR product adjusted to 20 µL with RNase‐free water, 8 µL 5× rNTP‐mix [20 mm ATP, 20 mm CTP, 20 mm GTP, 10 mm UTP (Fermentas, St. Leon‐Rot, Germany); 10 mm 5‐(3‐aminoallyl)‐UTP (Ambion Europe)], 4 µL 10× T7 RNA Polymerase transcription buffer (Ambion Europe), 2 µL 200 mm MgCl2, 2 µL inorganic Pyrophosphatase [0.3 U/µL, lyophilized inorganic Pyrophosphatase, S. cerevisiae (Sigma, Taufkirchen, Germany) dissolved in 10 mm Tris‐HCl (pH 7.5), 0.1 mm EDTA (pH 8.0), 50% (v/v) glycerol], 4 µL T7 RNA Polymerase‐Plus™[20 U/µL (Applied Bioscience Europe)] were combined in a 1.5‐mL centrifuge tube, mixed and incubated at 37 °C for 12 h. The PCR template was digested by addition of 2 µL DNase I (RNase‐free) [2 U/µL (Applied Bioscience)] and incubation at 37 °C for 15 min. After addition of 5 µL sodium acetate (pH 5.2) and 35 µL 2‐propanol, RNA was precipitated at –20 °C for 2 h followed by centrifugation at 16 000 g and 4 °C for 40 min. Supernatant was removed by pipeting and pellet was washed three times by addition of 1 mL 75% ice‐cold ethanol followed by centrifugation at 16 000 g and 4 °C for 5 min. RNA pellet was briefly air‐dried and resuspended in 41 µL RNase‐free water. RNA concentration in a 1‐µL aliquot was determined.
The in vitro synthesized RNA was then chemically labelled with a fluorescent dye. One hundred to 200 µg of the in vitro transcribed RNA, adjusted to 40 µL with RNase‐free water, was mixed with 40 µL 200 mm sodium bicarbonate buffer (pH 8.5). The Oyster®645‐NHS dye [0.2 mg N‐hydroxysuccinimide‐activated bifunctional pentamethine fluorophor (Denovo Biolabels, Münster, Germany)] was completely dissolved in 20 µL dimethyl sulfoxide by vortexing. The bicarbonate‐buffered RNA was immediately added to the dissolved dye and incubated under gentle shaking at room temperature for 45 min. Unbound dye was quenched by addition of 50 µL ethanolamine and gentle shaking at room temperature for 15 min. After addition of 300 µL RNase‐free water, 50 µL sodium acetate (pH 5.2) and 350 µL 2‐propanol, RNA was precipitated at –20 °C for 2 h followed by centrifugation at 16 000 g and 4 °C for 40 min. Supernatant was removed by pipeting and pellet was washed twice by addition of 1 mL 75% ice‐cold ethanol followed by centrifugation at 16 000 g and 4 °C for 5 min. RNA pellet was briefly air‐dried, resuspended in 50 µL RNase‐free water and stored at –20 °C until further use.
Multiplexed fluorescent Northern hybridization and imaging
The Northern or Spot blot membranes were incubated in hybridization solution [50% formamide; 7% SDS; 2% blocking reagent (Roche Diagnostics); 0.1% N‐laurylsarcosine; 5× SSC] in roller bottles (Hybaid, Middlesex, UK) at 68 °C for 1 h. The hybridization solution was then discarded and pre‐warmed (10 min at 95°C) hybridization solution containing Oyster‐645 and/or DIG‐labelled RNA probe was added. After hybridization at 68 °C for 16 h, the membranes were washed twice for 5 min at room temperature in 2× SSC/0.1% SDS, followed by two washes for 15 min in 0.2× SSC/0.1% SDS at 68 °C. Hybridization signals for DIG‐labelled probes were generated by incubation with anti‐digoxigenin‐AP Fab fragments (Roche Diagnostics) and ECF substrate (Amersham, Freiburg, Germany) according to the manufacturers’ instructions. Fluorescence signals were quantified using a FLA‐3000 phosphoimager (ECF: 473‐nm laser/520‐nm filter; Oyster‐645: 633‐nm laser/675‐nm filter) and the manufacturer's image analysis software package (Raytest, Straubenhardt, Germany).
ACKNOWLEDGEMENTS
A.S. thanks Dorotha Konopka and Leif Steil for support and stimulating discussions. This work was supported by grants of the Max‐Planck Society and the Deutsche Forschungsgemeinschaft.
REFERENCES
- Budde, I.P. , Rohde, B.H. , Bender, C.L. and Ullrich, M.S. (1998) Growth phase and temperature influence promoter activity, transcript abundance, and protein stability during biosynthesis of the Pseudomonas syringae phytotoxin coronatine. J. Bacteriol. 180, 1360–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett, W.V. (1997) Northern blotting of RNA denatured in glyoxal without buffer recirculation. Biotechniques, 22, 668–671. [DOI] [PubMed] [Google Scholar]
- Chang, S. , Puryear, J. and Cairney, J. (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116. [Google Scholar]
- Cirvilleri, G. and Lindow, S.E. (1994) Differential expression of genes of Pseudomonas syringae on leaves and in culture evaluated with random genomic lux fusions. Mol. Ecol. 3, 249–257. [Google Scholar]
- Gehrig, H.H. , Winter, K. , Cushman, J. , Borland, A. and Taybi, T. (2000) An improved RNA isolation method for succulent plant species rich in polyphenols and polysaccharides. Plant Mol. Biol. Rep. 18, 369–376. [Google Scholar]
- Geraats, B.P. , Bakker, P.A. and Van Loon, L.C. (2002) Ethylene insensitivity impairs resistance to soilborne pathogens in tobacco and Arabidopsis thaliana . Mol. Plant–Microbe Interact. 15, 1078–1085. [DOI] [PubMed] [Google Scholar]
- Herrin, D.L. and Schmidt, G.W. (1988) Rapid, reversible staining of northern blots prior to hybridization. Biotechniques, 6, 196–197, 199–200. [PubMed] [Google Scholar]
- Ingelbrecht, I.L. , Mandelbaum, C.I. and Mirkov, T.E. (1998) Highly sensitive northern hybridization using a rapid protocol for downward alkaline blotting of RNA. Biotechniques, 25, 420–423, 425–426. [DOI] [PubMed] [Google Scholar]
- Jaakola, L. , Pirttila, A.M. , Halonen, M. and Hohtola, A. (2001) Isolation of high quality RNA from bilberry (Vaccinium myrtillus L.) fruit. Mol. Biotechnol. 19, 201–203. [DOI] [PubMed] [Google Scholar]
- Joyner, D.C. and Lindow, S.E. (2000) Heterogeneity of iron bioavailability on plants assessed with a whole‐cell GFP‐based bacterial biosensor. Microbiology, 146, 2435–2445. [DOI] [PubMed] [Google Scholar]
- Keane, P.J. , Kerr, A. and New, P.B. (1970) Crown gall of stone fruit. II. Identification and nomenclature of Agrobacterium isolates. Aust. J. Biol. Sci. 23, 585–595. [Google Scholar]
- Keith, R.C. , Keith, L.M. , Hernandez‐Guzman, G. , Uppalapati, S.R. and Bender, C.L. (2003) Alginate gene expression by Pseudomonas syringae pv. tomato DC3000 in host and non‐host plants. Microbiology, 149, 1127–1138. [DOI] [PubMed] [Google Scholar]
- Li, H. and Ullrich, M.S. (2001) Characterization and mutational analysis of three allelic isc genes encoding levansucrase in Pseudomonas syringae . J. Bacteriology, 183, 3282–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Schenk, A. , Srivastava, A. , Zhurina, D. and Ullrich, M.S. (2006) Thermo‐responsive expression and differential secretion of the extracellular enzyme levansucrase in the plant pathogenic bacterium Pseudomonas syringae pv. glycinea. FEMS Microbial. Lett. 265, 178–185. [DOI] [PubMed] [Google Scholar]
- Majumdar, D. , Avissar, Y.J. and Wyche, J.H. (1991) Simultaneous and rapid isolation of bacterial and eukaryotic DNA and RNA: a new approach for isolating DNA. Biotechniques, 11, 94–101. [PubMed] [Google Scholar]
- Marco, M.L. , Legac, J. and Lindow, S.E. (2005) Pseudomonas syringae genes induced during colonization of leaf surfaces. Environ. Microbiol. 7, 1379–1391. [DOI] [PubMed] [Google Scholar]
- Palmer, D.A. and Bender, C.L. (1993) Effects of environmental and nutritional factors on production of the polyketide phytotoxin coronatine by Pseudomonas syringae pv. glycinea. Appl. Environ. Microbiol. 59, 1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapley, R. and Heptinstall, J. (1998) RNA Isolation and Characterization Protocols. Totowa, NJ: Humana Press. [Google Scholar]
- Sambrook, J. , Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Schenk, A. , Berger, M. , Keith, L.M. , Bender, C.L. , Muskhelishvili, G. and Ullrich, M.S. (2006) The algT gene of Pseudomonas syringae pv. glycinea and new insights into the transcriptional organization of the algT‐muc gene cluster. J. Bacteriol. 188, 8013–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, R.N. , Silcock, D.J. , White, H.L. , Buhariwalla, H.K. and Glover, L.A. (1993) The cloning and characterization of phage promoters, directing high expression of luciferase in Pseudomonas syringae pv. phaseolicola, allowing single cell and microcolony detection. Mol. Ecol. 2, 285–293. [DOI] [PubMed] [Google Scholar]
- Weingart, H. , Stubner, S. , Schenk, A. and Ullrich, M.S. (2004) Impact of temperature on in planta expression of genes involved in synthesis of the Pseudomonas syringae phytotoxin coronatine. Mol. Plant–Microbe Interact. 17, 1095–1102. [DOI] [PubMed] [Google Scholar]