

SUMMARY
The grey mould fungus Botrytis cinerea produces two major phytotoxins, the sesquiterpene botrydial, for which the biosynthesis gene cluster has been characterized previously, and the polyketide botcinic acid. We have identified two polyketide synthase (PKS) encoding genes, BcPKS6 and BcPKS9, that are up‐regulated during tomato leaf infection. Gene inactivation and analysis of the secondary metabolite spectra of several independent mutants demonstrated that both BcPKS6 and BcPKS9 are key enzymes for botcinic acid biosynthesis. We showed that BcPKS6 and BcPKS9 genes, renamed BcBOA6 and BcBO9 (for B. cinerea botcinic acid biosynthesis), are located at different genomic loci, each being adjacent to other putative botcinic acid biosynthetic genes, named BcBOA1 to BcBOA17. Putative orthologues of BcBOA genes are present in the closely related fungus Sclerotinia sclerotiorum, but the cluster organization is not conserved between the two species. As for the botrydial biosynthesis genes, the expression of BcBOA genes is co‐regulated by the Gα subunit BCG1 during both in vitro and in planta growth. The loss of botcinic acid production does not affect virulence on bean and tomato leaves. However, double mutants that do not produce botcinic acid or botrydial (bcpks6Δbcbot2Δ) exhibit markedly reduced virulence. Hence, a redundant role of botrydial and botcinic acid in the virulence of B. cinerea has been demonstrated.
INTRODUCTION
Fungi produce a great diversity of secondary metabolites including some that are of great interest in pharmacology, such as antibiotics (e.g. penicillin, cephalosporin) or immunosuppressants (e.g. cyclosporin), but also phytohormones (e.g. abscisic acid and gibberellins) and mycotoxins [e.g. trichothecenes (TRIs), fumonisins, aflatoxin]. These natural products can be classified as: (i) polyketides and fatty acid‐derived compounds; (ii) nonribosomal peptides and amino acid‐derived compounds; and (iii) terpenes (Hoffmeister and Keller, 2007). In contrast with the genes involved in fungal primary metabolism, genes that contribute to the biosynthesis of the same secondary metabolite are usually clustered at one genomic locus. Clustering provides an evolutionary advantage during horizontal gene transfer (HGT) as it facilitates the transmission of a whole pathway (Walton, 2000). Recently, such HGT of several clustered genes has been proven to occur between distant fungi (Khaldi et al., 2008). Clustering may also be maintained because of the advantage it confers for co‐regulation processes linked to chromatin structure (Bok et al., 2009; Palmer and Keller, 2010). Recent sequencing projects have revealed that some ascomycete genomes contain putative secondary metabolism gene clusters for the biosynthesis of more than 40 different metabolites (Soanes et al., 2007). The role of most of the corresponding metabolites remains elusive, but they may contribute to fungal fitness and protection against biotic and abiotic stresses in different biotopes (Fox and Howlett, 2008). In phytopathogenic species, some metabolites play crucial roles in interactions with host plants. Among them are some host‐selective toxins, such as the HC‐toxin of the maize pathogen Cochliobolus carbonum (Walton, 2006), nonhost‐selective toxins, such as the TRIs of Fusarium spp. (Desjardins et al., 1996), and a fungal polyketide which allows rice to recognize its pathogen Magnaporthe grisea (Collemare et al., 2008).
Botrytis cinerea (sexual form: Botryotinia fuckeliana) is the causal agent of grey mould disease that affects more than 200 ornamental and agriculturally important plant species. This necrotrophic fungus displays the capacity to kill host cells through the production of toxins, reactive oxygen species and the induction of a plant‐produced oxidative burst (Choquer et al., 2007; Williamson et al., 2007). Two groups of nonspecific phytotoxins have been identified, i.e. the sesquiterpene botrydial and related compounds (Colmenares et al., 2002) and botcinic acid and its botcinin derivatives (2005, 2006). Botrydial is produced during plant infection (Deighton et al., 2001) and induces chlorosis and cell collapse (Colmenares et al., 2002). Botcinic acid and derivatives have also been shown to induce chlorosis and necrosis (Cutler et al., 1996), but also have antifungal activities (Sakuno et al., 2007). The botrydial biosynthetic gene cluster consists of five genes (BcBOT1 to BcBOT5) that are co‐regulated by the Ca2+/calcineurin signal transduction pathway, which is under the control of the α subunit BCG1 of a heterotrimeric G protein (Pinedo et al., 2008; 2008a, 2008b). Gene inactivation has provided evidence that BcBOT2 is a sesquiterpene cyclase (presilphiperfolan‐8β‐ol synthase; Pinedo et al., 2008; Wang et al., 2009) responsible for the first step of botrydial synthesis, whereas BcBOT1 is a P450 monooxygenase that acts in a later step of biosynthesis (Siewers et al., 2005). Mutants blocked in botrydial production were generated in different genetic backgrounds, revealing a strain‐dependent impact of botrydial on virulence. Botrydial mutants in a T4 strain background presented a reduced virulence, whereas the same mutation in the B05.10 or SAS56 strains did not affect virulence. This strain‐dependent effect may be explained by the fact that the strain T4 does not produce either botcinic acid or any of its derivatives, whereas, in B05.10 and SAS56 strains, the absence of botrydial could be compensated for by the production of the second toxin botcinic acid (Siewers et al., 2005). This hypothesis is further supported by the fact that B05.10 bcbot2 mutants produce levels of botcinic acid that are markedly higher than those produced by the wild‐type (WT) strain (Pinedo et al., 2008). The identification of the genetic basis of botcinic acid biosynthesis is a prerequisite for studying the role of this toxin and the proposed redundant effect with botrydial during pathogenesis. The biosynthetic origin of the botcinic acid skeleton has been investigated previously by feeding B. cinerea with 13C‐ and 2H‐labelled precursors. This study demonstrated that botcinic acid and botcinins are acetate‐derived polyketides (Reino et al., 2006). Consequently, at least one polyketide synthase (PKS) is expected to be involved in the biosynthesis of these compounds.
The aim of this study was to identify the botcinic acid biosynthesis genes and to evaluate the role of this toxin in virulence. The availability of the genome sequence provided 20 candidate genes that were predicted to encode PKSs (Kroken et al., 2003). By using transcriptomic, reverse genetics and chemical approaches, we showed that two PKS‐encoding genes, located in two separate secondary metabolism gene clusters, are required for botcinic acid production. Mutants deficient in botcinic acid production and mutants that are additionally defective in botrydial production reveal the redundant role of the two toxins in virulence.
RESULTS
BcPKS6 and BcPKS9, which are predicted to encode reducing PKS, are up‐regulated during tomato leaf infection
Botcinic acid and botcinin lactone derivatives are eight‐carbon polyketides consisting of a highly substituted tetrahydropyran ring linked by an ester bound at carbon atom 7 (C‐7) to a chain of 4‐hydroxy‐2‐octenoic acid (Fig. 1). We therefore predicted that at least one of the enzymes involved in its biosynthesis should be a PKS. In addition, we proposed the hypothesis that the genes involved in phytotoxin production would be expressed during plant infection. In order to identify candidate genes, we used an expression approach based on reverse‐Northern blotting. The expression of the 20 BcPKS genes predicted from the genome sequence of strain B05.10 (Kroken et al., 2003) was studied at different physiological stages. The 3′ exon of each of the BcPKS genes was amplified by polymerase chain reaction (PCR) (see Experimental procedures) and the resulting fragments were spotted on Nylon filters (Fig. 2). The WT strain B05.10 was cultivated on rich medium or inoculated on tomato leaves. Total RNA was extracted after 3 days of growth (in vitro conditions) and 3 days post‐inoculation (dpi), respectively, labelled with radioactive 32P and hybridized to the membranes. When the fungus was grown on rich medium, 18 of the 20 BcPKS genes exhibited low levels of expression compared with the actin‐encoding gene used as control (Gioti et al., 2006). By contrast, during infection, only BcPKS6 and BcPKS9 exhibited higher levels of expression than the actin gene. Both experiments were repeated three times with similar results, indicating that BcPKS6 and BcPKS9 were significantly up‐regulated in planta (3 dpi) and that they may play a role in virulence.
Figure 1.
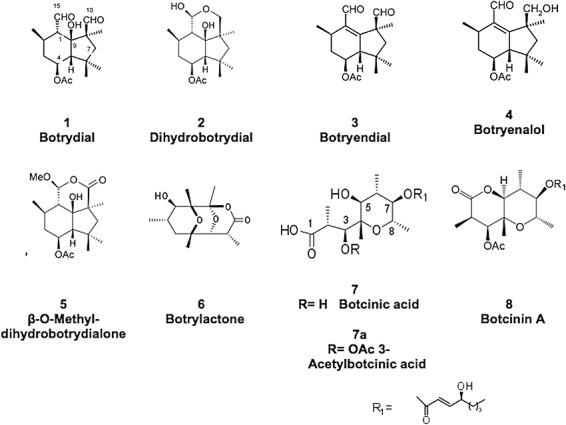
Chemical structures of the compounds secreted by Botrytis cinerea. The quantities isolated from the wild‐type (WT) strain and the mutants are indicated in Table 1.
Figure 2.
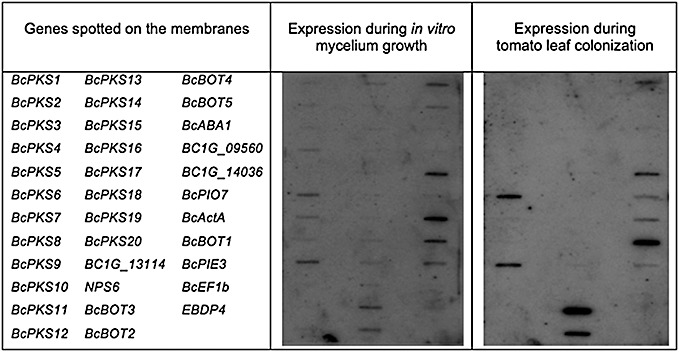
Expression of the polyketide synthase (PKS)‐encoding genes revealed by reverse‐Northern analysis. Polymerase chain reaction (PCR) fragments of BcPKS genes predicted by Kroken et al. (2003) and other Botrytis cinerea genes were spotted onto Nylon membranes. BcActA and EF1b are the actin‐ and elongation factor‐encoding genes used as constitutively expressed genes (Gioti et al., 2006). BcBOT genes are the botrydial biosynthesis genes described in Pinedo et al. (2008). All gene and primer information is given in Table S1. The B05.10 strain was cultivated on grape juice medium and inoculated on tomato leaves. Three days post‐inoculation, total RNA was extracted, labelled with 32P and hybridized to the membranes.
In their phylogenetic analysis of fungal PKSs, Kroken et al. (2003) classified BcPKS6 as a reducing PKS with similarities to PKS–NRPS hybrids (NRPS, nonribosomal peptide synthetases), whereas BcPKS9 was included in another clade of reducing PKSs. Using protein domain search tools [Protein Families (PFAM) and National Center for Biotechnology Information (NCBI) conserved domain search], functional domains have been predicted to be present in BcPKS6 and BcPKS9 protein sequences. Both proteins contain the three essential domains of PKSs, i.e. the ketoacyl synthase (KS), acyl transferase (AT) and phosphopantetheine (PP) domains. In addition, analysis of the protein sequences revealed some additional domains, i.e. the dehydratase (DH), methyltransferase (MT), enoyl reductase (ER) and ketoreductase (KR) domains. Although DH and KR domains are found in both PKS enzymes, the MT domain is present in BcPKS6 only, and the ER domain is present in BcPKS9 only (Fig. 3). In conclusion, BcPKS6 and BcPKS9 are predicted to encode reducing PKSs and are strongly up‐regulated during infection, thereby being candidate genes for botcinic acid biosynthesis.
Figure 3.
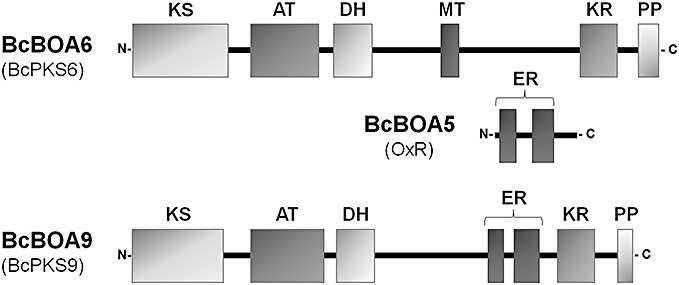
Domain organization of the polyketide synthases BcBOA6 (BcPKS6) and BcBOA9 (BcPKS9), and BcBOA5 (OxR), an enzyme presumably acting in concert with the polyketide synthase BcBOA6. Protein enzymatic domains are as follows: AT, acyl transferase; DH, dehydratase; ER, enoyl reductase; KR, ketoreductase; KS, β‐ketoacyl synthase; MT, methyltransferase; PP, phosphopantetheine attachment site. Domains were predicted by Protein Families (PFAM) and National Center for Biotechnology Information (NCBI) domain search (see Table 2).
Inactivation of BcPKS6‐ and BcPKS9‐encoding genes abolishes botcinic acid production
The BcPKS6 and BcPKS9 candidate genes were inactivated in order to test whether they were involved in botcinic acid biosynthesis. Gene replacement vectors pBcPKS6Δ (conferring resistance to hygromycin) and pBcPKS9Δ (conferring resistance to nourseothricin) were constructed [see Experimental procedures and Fig. S1 (Supporting Information)], and protoplasts of strain B05.10 were transformed with the BcPKS6 and BcPKS9 replacement fragments. Protoplast regeneration and further purification on selective medium led to the isolation of six BcPKS6 and 15 BcPKS9 transformants. PCR amplifications with one primer located upstream of the 5′ region of BcPKS6 and one primer located inside the hygromycin B phosphotransferase gene (hph) (see Experimental procedures) confirmed that the expected gene replacement event had taken place in three transformants, named bcpks6Δ‐1, bcpks6Δ‐4 and bcpks6Δ‐5. Similarly, PCR amplifications with one primer located upstream of the 5′ region of the BcPKS9 gene and one primer located inside the nourseothricin resistance gene (nat1) confirmed that the expected gene replacement event had taken place in six transformants, including bcpks9Δ‐3, bcpks9Δ‐14 and bcpks9Δ‐16. For all selected bcpks6Δ and bcpks9Δ mutants, Southern blot hybridization patterns confirmed that they were the result of a single integration event at the targeted locus (Fig. S1).
To investigate the production of botcinic acid and its derivatives, two bcpks9Δ mutants and the WT strain were cultivated on both solid malt medium and liquid‐modified Czapek–Dox medium. The corresponding culture media were extracted with ethyl acetate and analysed by extensive spectroscopic analysis [1H and 13C nuclear magnetic resonance (NMR)] and subsequent high‐performance liquid chromatography (HPLC) purification (see Experimental procedures) to detect the presence of toxins.
The production of metabolites on solid malt medium was higher and the extracts cleaner than those produced by surface culture fermentations. All data in Fig. 1 and Table 1 were obtained from cultivation on solid malt medium. The results indicate that none of the mutants were able to produce botcinic acid or its botcinin A derivative (Table 1, Fig. 1), thus demonstrating that both BcPKS6 and BcPKS9 are necessary for botcinic acid and botcinin production. Interestingly, a substantial amount (about 30 mg) of botrylactone (compound 6, Fig. 1) was isolated from the cultures of the bcpks9Δ mutants, whereas less than 1 mg was detected in WT culture. This unique metabolite with an interesting nine‐carbon polyketide lactone skeleton bearing two oxirane bridges has been isolated previously from B. cinerea and described as a strong antibiotic (Welmar et al., 1979). The isolation of botrylactone together with botcinins (Reino et al., 2006), and the similarity of their spectroscopic data, suggest that these two compounds have a common biosynthetic origin. In addition, a recent structural and biosynthetic study (I. G. Collado, unpublished results) has shown that the stereochemistry of botrylactone is different from that described previously (Bruns et al., 1995; Welmar et al., 1979) and identical to that proposed for botcinin. The isolation of botrylactone from bcpks9Δ and the absence of this metabolite in extracts from the bcpks6Δ mutants are consistent with a common biosynthetic pathway.
Table 1.
Metabolites identified in Botrytis cinerea wild‐type strain B05.10 and mutants (numbers correspond to compounds in Fig. 1).
| Culture | Botryanes (mg) | Botcinic acid and derivatives, botrylactone (mg) |
|---|---|---|
| B05.10 wild‐type | 1 (5), 2 (3.5), 3 (2), 4 (3), 5 (3) | 8 (6) |
| bcpks6Δ‐1 | 1 (9), 2 (4.5), 3 (4), 4 (3) | None |
| bcpks6Δ‐4 | 1 (7), 2 (6), 3 (6), 4 (2) | None |
| bcpks9Δ‐3 | 1 (12), 2 (12.5), 3 (4.5), 5 (5.5) | 6 (31) |
| bcpks9Δ‐14 | 1 (9), 2 (10), 3 (4), 5 (6) | 6 (28) |
| bcbot2Δ | None | 7a (57), 8 (3) |
| bcpks6Δ bcbot2Δ | None | None |
Quantities of purified compounds are indicated in milligrams per culture (see Experimental procedures).
To confirm that the loss of botcinic acid production is a result of the deletion of BcPKS6 and BcPKS9, complementation of bcpks6Δ and bcpks9Δ mutants with the corresponding WT gene copies was initiated by co‐transformation of B. cinerea, as described in Doehlemann et al. (2006). Complementation vectors containing about 10‐kb genomic fragments of BcPKS6 or BcPKS9, including the promoter regions, were generated. Several attempts were made to co‐transform each null mutant with its complementation vector, together with a vector containing the nourseothricin or hygromycin resistance gene. All resulting transformants showed integration of the resistance marker gene, but not the complementation vector, probably because of the large size of the gene (data not shown). As the complementation failed, we decided to use three bcpks6Δ and three bcpks9Δ mutants for all further phenotypic tests.
BcPKS6 and BcPKS9 are both part of secondary metabolite gene clusters
In a previous study, BcPKS6 has been characterized as part of a BCG1/calcineurin‐controlled gene cluster that is responsible for the biosynthesis of an as yet unknown polyketide (Schumacher et al., 2008a). Adjacent genes encode two cytochrome P450 monooxygenases (P450‐1 and P450‐2), a FAD‐binding monooxygenase (MO1), an oxidoreductase (OxR) and an NmrA‐like protein (ORF1) (Table 2; Fig. 4).
Table 2.
The putative botcinic acid biosynthetic genes (BcBOA).
| Gene name | Bc B05.10 annotation* | Homologue in Ss 1980† (BlastX, e‐value) | Predicted protein function‡ (domain search: hit, e‐value) | Open reading frame | Protein size (amino acid, aa) | Revision of automatic gene prediction (GenBank accession number, references) |
|---|---|---|---|---|---|---|
| BcBOA1 | — | Ss1g_12301, 2e‐97 | PF05368_NmrA‐like family, 6.9e‐16 | 1148 bp, 3 introns | 300 | AM930230 (ORF1); Schumacher et al. (2008a) |
| BcBOA2 | Bc1g_16083 | Ss1g_12300, 0.0 | PF00743_Flavin‐binding monooxygenase‐like, 1.1e‐07 | 1971 bp, 6 introns | 530 | AM930229 (MO1); Schumacher et al. (2008a) |
| BcBOA3 | Bc1g_16084 | Ss1g_09234, 0.0 | PF00067_Cytochrome P450, 1.1e‐66 | 1809 bp, 5 introns | 506 | AM930228 (P450‐2); Schumacher et al. (2008a) |
| BcBOA4 | Bc1g_16085 | Ss1g_09235, 0.0 | PF00067_Cytochrome P450, 6.9e‐43 | 1768 bp, 5 introns | 495 | AM930227 (P450‐1); Schumacher et al. (2008a) |
| BcBOA5 | — | Ss1g_09236, 9e‐148 | PF08240_Alcohol dehydrogenase GroES‐like domain, 1.5e‐06 | 1128 bp, no introns | 375 | AM930231 (OxR); Schumacher et al. (2008a) |
| BcBOA6 | Bc1g_16086 | Ss1g_09237, 0.0 | Reducing polyketide synthase, clade II | 7563 bp, 3 introns | 2460 | AAR90242 (PKS6); Kroken et al. (2003) AM930232 (PKS6); Schumacher et al. (2008a) |
| Bc1g_16087 | PF00109_β‐Ketoacyl synthase, N‐terminal domain, 1e‐72 | |||||
| PF02801_β‐Ketoacyl synthase, C‐terminal domain, 3.7e‐33 | ||||||
| PF00698_Acyl transferase domain, 1.8e‐52 | ||||||
| Smart00826_PKS_Dehydratase, 2e‐14 | ||||||
| PF08242_Methyltransferase domain, 4.9e‐17 | ||||||
| PF08659_Ketoacyl reductase domain, 1.4e‐45 | ||||||
| PF00550_Phosphopantetheine attachment site, 0.00053 | ||||||
| BcBOA7 | — | Ss1g_09238, 0.0 | PF00067_Cytochrome P450, 7.9e‐28 | 1780 bp, 4 introns | 517 | FR717895; this study |
| BcBOA8 | Bc1g_15836 | Ss1g_09239, 0.0 | PF01494_FAD‐binding domain, 3.4e‐12 | 2749 bp, 5 introns | 779 | FR718877; this study |
| BcBOA9 | Bc1g_15837 | Ss1g_09240, 0.0 | Polyketide synthase, clade IV | 7401 bp, 9 introns | 2294 | AAR90245 (PKS9); Kroken et al. (2003) |
| Bc1g_15838 | PF00109_β‐Ketoacyl synthase, N‐terminal domain, 4.4e‐73 | FR718878; this study | ||||
| Bc1g_15839 | PF02801_β‐Ketoacyl synthase, C‐terminal domain, 1.2e‐35 | |||||
| PF00698_Acyl transferase domain, 7e‐52 | ||||||
| Smart00826_PKS_Dehydratase, 9e‐22 | ||||||
| PF08240_Alcohol dehydrogenase GroES‐like domain, 1.4e‐08 | ||||||
| PF00107_Zinc‐binding dehydrogenase, 4.9e‐18 | ||||||
| PF08659_Ketoacyl reductase domain, 1.1e‐42 | ||||||
| PF00550_Phosphopantetheine attachment site, 0.0006 | ||||||
| BcBOA10 | Bc1g_15840 | Ss1g_09241, 2e‐66 | PF00975_ Thioesterase domain, 2.7e‐17 | 984 bp, 3 introns | 270 | FR718880; this study |
| BcBOA11 | Bc1g_15841 | Ss1g_09242, 0.0 | PF02458_Transferase family, 8.9e‐14 | 1413 bp, no introns | 470 | FR718882; this study |
| BcBOA12 | Bc1g_15842 | Ss1g_09243, 4e‐88 | Unknown function (no domains found) | 663 bp, no introns | 220 | FR718879; this study |
| BcBOA13 | Bc1g_15843 | Ss1g_09244, 2e‐134 | PF00172_Fungal Zn(2)‐Cys(6) binuclear cluster domain, 1e‐06 | 1901 bp, 3 introns | 568 | FR718881; this study |
| BcBOA14 | Bc1g_15845 | No hit | Unknown function (no domains found) | 171 bp, no introns | 56 | FR718883; this study |
| BcBOA15 | Bc1g_15846 | No significant hit | PF04082 _Fungal specific transcription factor domain, 3.3‐06 | 540 bp, no introns | 179 | FR718884; this study |
| BcBOA16 | Bc1g_15848 | No significant hit | PF01370_NAD‐dependent epimerase/dehydratase family, 0.015 | 477 bp, no introns | 158 | FR718885; this study |
| BcBOA17 | Bc1g_15849 | No significant hit | PF00106_Short‐chain dehydrogenase, 1.4e‐28 | 825 bp, no introns | 274 | FR718886; this study |
Botrytis cinerea Database (http://www.broadinstitute.org/annotation/genome/botrytis_cinerea/).
Sclerotinia sclerotiorum Database (http://www.broadinstitute.org/annotation/genome/sclerotinia_sclerotiorum/MultiHome.html), no genes are listed when the e‐value is 6e‐20 or lower.
The PFAM protein families database (http://pfam.sanger.ac.uk/search); National Center for Biotechnology Information (NCBI) conserved domain search (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Figure 4.
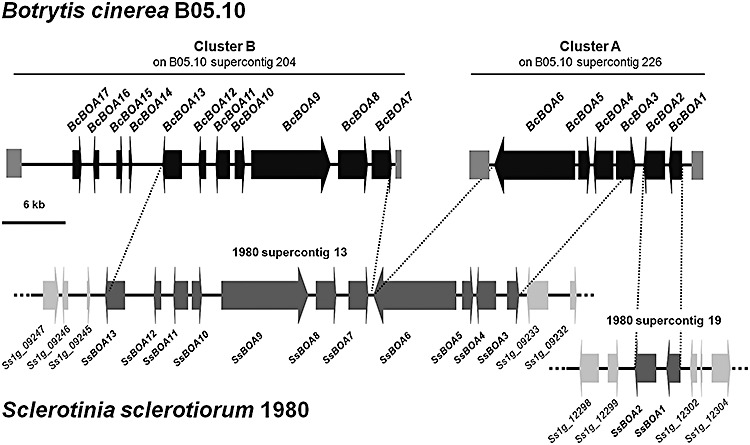
The putative botcinic acid biosynthetic genes (BcBOA) are separated into at least two clusters in the Botrytis cinerea wild‐type (WT) strain B05.10. Black arrows indicate genes that might be involved in botcinic acid biosynthesis in B. cinerea. Dark grey arrows in Sclerotinia sclerotiorum (strain 1980; Sclerotinia sclerotiorum Database, http://www.broadinstitute.org/annotation/genome/sclerotinia_sclerotiorum/MultiHome.html) indicate the genes that are the homologues of the B. cinerea BcBOA genes. Sclerotinia sclerotiorum genes that are probably not related to secondary metabolism and the botcinic acid cluster are indicated by light grey arrows. Grey boxes at the ends of the B. cinerea clusters indicate AT‐rich regions exhibiting AT contents from 70% to 90%. For more details on sequence assembly, see Experimental procedures; for predicted gene functions, see Table 2.
In this study, the genomic region adjacent to BcPKS9 was investigated using the available genome sequence of the B. cinerea strain B05.10 (B. cinerea Database; http://www.broadinstitute.org/annotation/genome/botrytis_cinerea/Home.html). The identified genome supercontig 204 comprises several putative open reading frames encoding proteins that might be involved in the biosynthesis of secondary metabolites. Automatic gene predictions by the Broad Institute were revised using cDNA sequences, and thermal asymmetric interlaced (TAIL)‐PCR approaches were applied to extend the sequences of both clusters (Table 2; for more details, see Experimental procedures). In summary, 11 genes are specified in the newly identified cluster, and their functions were predicted by searching conserved domains in the deduced protein sequences. A function could be predicted for nine, but the function of two proteins remains unknown as no conserved protein domains could be identified. Among the nine genes with predicted functions are two with similarities to specific transcription factors that might be involved in the regulation of cluster gene expression (BcBOA13 and BcBOA15).
The co‐expression of genes involved in the same biosynthetic pathway of a secondary metabolite is a common phenomenon (Hoffmeister and Keller, 2007). As described previously, the botrydial biosynthetic genes (BcBOT1–BcBOT5) are characterized by their dependence on different signalling components. Hence, the expression relies on the presence of the Gα subunit BCG1 and the active Ca2+‐regulated calcineurin phosphatase (Pinedo et al., 2008; Schumacher et al., 2008a; Viaud et al., 2003). The same expression pattern was found for the cluster genes located on supercontig 226 (Schumacher et al., 2008a). To determine whether the genes adjacent to BcPKS9 on supercontig 204 were subjected to the same mode of regulation as those on supercontig 226, the expression level of all genes was studied in WT strain B05.10 and the bcg1 deletion mutant by Northern blot analyses (Fig. 5). All expressed genes were similarly controlled by BCG1, as moderate expression levels were observed in the WT strain, but not in the bcg1 deletion mutant. However, for three genes (BcBOA14—BcBOA16), no hybridization signals could be detected, even though expression was expected on the basis of the cDNA fragments obtained. The co‐expression of most of the genes from both clusters suggests that the genes are functionally linked. Accordingly, the genes were named BcBOA1 to BcBOA17 (for B. cinerea botcinic acid biosynthesis).
Figure 5.
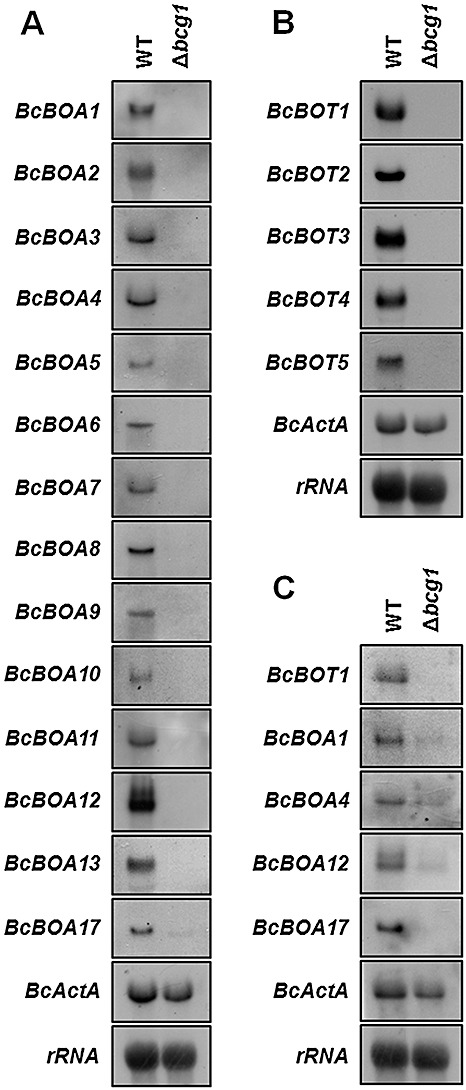
The expression of putative botcininc acid biosynthetic genes is co‐regulated and dependent on the presence of the α subunit BCG1 of a heterotrimeric G protein. (A) Expression of BcBOA genes in submerged culture. No hybridization signals for BcBOA14, BcBOA15 and BcBOA16 were detected (data not shown). (B) Expression of botrydial biosynthesis genes (BcBOT) in submerged culture. (C) Expression of several BcBOA and BcBOT genes in planta (48 h post‐inoculation). For cultivation conditions, see Experimental procedures. BcActA encoding actin and rRNA were used as loading controls.
Interestingly, Sclerotinia sclerotiorum, a close relative of B. cinerea (Fillinger et al., 2007), contains putative orthologues of the predicted BcBOA biosynthetic genes (SsBOA genes), as inferred from them being bidirectional best hits on nucleotide and protein levels. Accordingly, two supercontigs in the genome sequence of the S. sclerotiorum strain 1980 (http://www.broadinstitute.org/annotation/genome/sclerotinia_sclerotiorum/MultiHome.html) were identified comprising SsBOA1 to SsBOA13. For four genes (BcBOA14 to BcBOA17), no homologous genes could be found in the S. sclerotiorum database. Although SsBOA3 to SsBOA13 are located on supercontig 13, SsBOA1 and SsBOA2 are located on a different supercontig. In any case, the SsBOA genes are surrounded by genes whose products are probably not related to the biosynthesis of secondary metabolites (Fig. 4, Table S3). In contrast, the cluster sequences of B. cinerea are surrounded by regions that display AT contents from 70% to 90%. As these sequences hamper PCR and sequencing approaches, and subsequent sequence assembly, the neighbouring genes in B. cinerea have not yet been identified.
Strikingly, the PKS‐encoding genes are physically linked in S. sclerotiorum. As the sequence data downstream of BcBOA7 and BcBOA6 were missing, it could not be ruled out that both clusters were also physically linked in B. cinerea. PCR‐based approaches to test a physical linkage failed, and Southern blot analyses further supported the hypothesis that both clusters are separated in the genome of B. cinerea (data not shown).
Taken together, both PKS‐encoding genes in B. cinerea are adjacent to genes probably encoding enzymes of secondary metabolite pathways. In contrast with the genomic organization found in S. sclerotiorum, the PKS‐encoding genes and adjacent genes are separated into two different gene clusters. As the adjacent BcBOA genes exhibit the same expression pattern, it is assumed that the encoded enzymes might also be involved in botcinic acid biosynthesis.
Botcinic acid and botrydial have redundant functions
The availability of several mutants that do not produce botcinic acid provided the opportunity to test the importance of this toxin in virulence. On synthetic medium, botcinic acid‐deficient mutants (three bcpks6Δ and three bcpks9Δ) showed growth and conidiation rates similar to those of the WT strain (Fig. 6A, and data not shown). Pathogenicity tests were performed by inoculating plugs of 3‐day‐old mycelium onto 2‐week‐old bean leaves. As shown in Fig. 6B, C, the virulence of the three bcpks9Δ and three bcpks6Δ mutants on bean leaves was not significantly different from that of the WT. Similar data were obtained on tomato leaves (data not shown). Therefore, botcinic acid production is not essential for pathogenicity in the WT strain B05.10.
Figure 6.
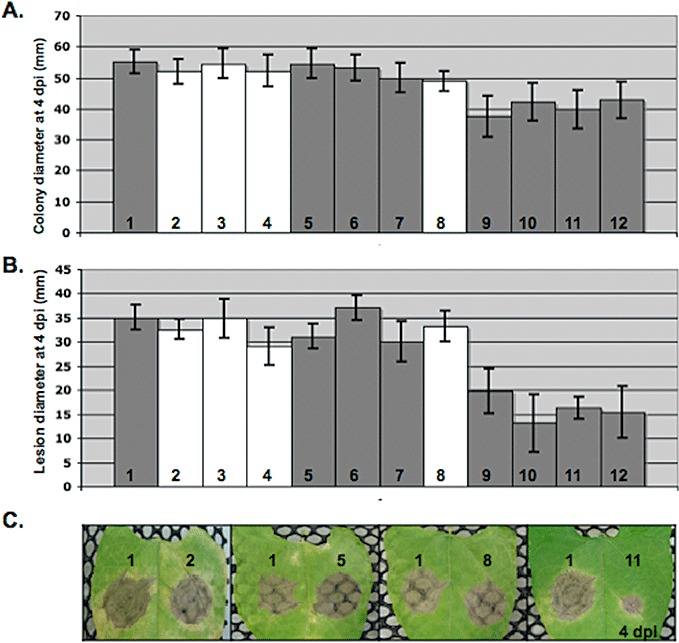
Growth and virulence of botcinic acid‐deficient mutants, botrydial‐deficient mutant and mutants deficient for the production of both toxins. (A) Growth on minimal medium: growth diameters were measured at 4 days post‐inoculation (dpi). (B) Virulence on bean leaves: plugs of young mycelium were inoculated on young leaves and lesion diameters were measured at 4 dpi. (C) Photographs of infected bean leaves taken at 4 dpi. The numbers indicate the strains as follows: B05.10 wild‐type strain (1), pks6Δ‐1 (2), pks6Δ‐4 (3), pks6Δ‐5 (4), pks9Δ‐3 (5), pks9Δ‐14 (6), pks9Δ‐16 (7), bcbot2Δ (8), bcbot2Δ pks6Δ−1 (9), bcbot2Δ pks6Δ‐2 (10), bcbot2Δ pks6Δ‐9 (11), bcbot2Δ pks6Δ‐14 (12).
In order to test the putative redundant role of botrydial and botcinic acid in infection, bcpks6Δ bcbot2Δ double mutants were generated. Protoplasts from the bcpks6Δ‐1 mutant were transformed with the pBcBOT2KO plasmid as described in Pinedo et al. (2008). Fifteen transformants were selected on gluphosinate selective medium. From these transformants, four double mutants were identified by PCR and confirmed by Southern blot (Fig. S1). Chemical analysis of one of these double mutants confirmed the absence of both botrydial and botcinic acid (Table 1). The in vitro growth and virulence of the four bcpks6Δ bcbot2Δ double mutants were compared with those of WT, the single botcinic acid mutants (bcpks6Δ and bcpks9Δ) and the single botrydial (bcbot2Δ) mutant obtained previously (Pinedo et al., 2008). As shown in Fig. 6A, the double mutants have a slightly slower growth rate than the WT strain and the single mutants. These phenotypes could suggest that botrydial and botcinic acid have an unexpected redundant role in saprophytic growth. As secondary metabolites may have a role in resistance to oxidative or ultraviolet (UV) stresses (Reverberi et al., 2010), we investigated these possibilities. The growth rates of simple and double mutants were monitored on minimal medium that contained menadione (250 and 500 µM) or H2O2 (10 and 25 mm). No strong differences were observed between the mutants and the WT strain. Sensitivity to UV was tested by growing the mutants in the dark, but no restoration of WT growth was observed. In conclusion, no role of the metabolites in oxidative or UV stress could be detected. Finally, the effect of the absence of both toxins was evaluated on virulence (Fig. 6B, C). All bcpks6Δ bcbot2Δ double mutants showed a reduced virulence on different plant tissues. The lesion size on bean leaves at 4 dpi was about 50% smaller than that on WT and the single mutants. These data clearly demonstrate that botrydial and botcinic acid have a redundant role in virulence.
DISCUSSION
Botcinic acid and derivatives produced by B. cinerea have been described as phytotoxins provoking chlorosis and necrosis (Cutler et al., 1996). In addition, the study of aggressiveness and toxin production in natural populations of B. cinerea suggested that strains that produce both botcinic acid and botrydial are more virulent than strains that produce botrydial only (Reino et al., 2004). Botcinic acid was first isolated from B. cinerea cultures by Cutler et al. (1993), who first named it botcinolide and described it as a nonane lactone ring with a fatty acid chain. The biosynthetic origin of the botcinolide skeleton later indicated its polyketidic nature (Reino et al., 2006). A careful reinvestigation of the spectroscopic data reported for botcinolide analogues allowed the revision of the structures of botcinolide derivatives into botcinic and botcineric acids and their ring‐closing derivatives, botcinins A–F (2005, 2006). Furthermore, the revised structures of this group of natural products have been unequivocally determined through their total synthesis (Fukui et al., 2008). Recently, we characterized botrylactone, previously described as an antibiotic (Welmar et al., 1979), as an additional related compound that has structural homology with botcinic acid, indicating that both compounds may be synthesized through a common pathway (Reino et al., 2006).
In this work, we have identified two PKS genes required for botcinic acid synthesis in B. cinerea. Our results indicate that the two PKSs encoded by BcBOA6 (formerly BcPKS6) and BcBOA9 (formerly BcPKS9) act in concert to synthesize botcinic acid. The iterative nature of fungal PKSs means that, in the majority of cases, there is only one PKS involved in the synthesis of one particular polyketide. However, some fungal polyketides are known to be assembled by the action of two PKSs. Two scenarios have been proposed for the biosynthesis. One possibility is that one PKS makes an advanced starter unit, which is passed on to a second PKS for further extension, as suggested for the biosynthesis of asperfuranone A in Aspergillus nidulans (Chiang et al., 2009), zearalenone in Gibberella zeae (Gaffoor and Trail, 2006) and T‐toxin in Cochliobolus heterostrophus (Baker et al., 2006). In the second scenario, two polyketides are produced independently by two PKSs and then attached together by means of an ester bound as suggested for lovastatin in Aspergillus terreus (Sutherland et al., 2001) and for the closely related compactin in Penicillium citrinum (Abe et al., 2002). Although the structure of botcinic acid, with two polyketide chains linked by an ester bond, is consistent with the second scenario, the accumulation of botrylactone as a metabolite obtained from the bcpks9/bcboa9Δ mutant on the one hand, and the absence of any intermediate in the bcpks6Δ/bcboa6Δ mutant on the other, is consistent with the first scenario: BcBOA6 would produce an advanced nonaketide yielding botrylactone as an intermediate; BcBOA9 would be responsible for the subsequent rearrangement of botrylactone and acylation, at the carbon 7 position of the tetrahydropyran ring of the 4‐hydroxyoctenoic acid chain, yielding the corresponding botcinic acid derivative. This proposal would explain the biosynthetic origin of the methyl group at carbon 8 of the botcinin skeleton, which proceeds from methyl‐methionine, instead of an acetate unit as expected in the starter units in polyketides (Reino et al., 2006). Further experiments of biotransformation of botrylactone to botcinin by the bcpks6Δ mutant are in progress to confirm this hypothesis (I. G. Collado et al., unpublished results). Interestingly, the observation that BcBOA6 is essential for the formation of the nonaketide ring structure of botrylactone fits well with the previous phylogenomic analysis of fungal PKSs (Baker et al., 2006; Kroken et al., 2003). Indeed, BcBOA6 is part of a clade including several PKSs responsible for the synthesis of cyclic polyketides: LovB (LNKS, nonaketide part of lovastatin; Sutherland et al., 2001), MlcA (nonaketide part of citrinin; Abe et al., 2002), EQS (equisetin; Sims et al., 2005) and FusS (fusarin; Song et al., 2004). By contrast, BcBOA9 is part of a clade including the FUM1 PKS responsible for the biosynthesis of fumonisin, a linear polyketide (Proctor et al., 2004).
Usually, genes that contribute to the biosynthesis of the same secondary metabolite are clustered at one genomic locus, but this is not the case for BcBOA6 and BcBOA9. They are at two distinct genomic loci and are both adjacent to other genes putatively involved in the biosynthesis of botcinic acid (Fig. 4). Similar to the situation in the lovastatin gene cluster of A. terreus, an ER‐encoding gene (BcBOA5) is located in close proximity to the PKS‐encoding gene (BcBOA6; Fig. 3). A close co‐operation of LovB (LNKS), possessing a nonfunctional ER domain, and LovC (ER) enzymes in lovastatin biosynthesis of A. terreus has been demonstrated by heterologous expression experiments in the nonproducing species A. nidulans. Only if LovB and LovC were co‐expressed was the correct nonaketide produced (Kennedy et al., 1999). As BcBOA6 lacks an ER domain, BcBOA5 may take over the ER function as described for LovB and LovC proteins.
Interestingly, a botcinic acid‐like gene cluster containing putative orthologues of most BcBOA genes has been identified in the closely related white mould fungus S. sclerotiorum. The metabolite produced by this cluster remains unknown. To our knowledge, no botcinic acid has ever been detected in S. sclerotiorum cultures, but other polyketides have been reported (Pedras and Ahiahonu, 2004). The B. cinerea and S. sclerotiorum BOA gene clusters share 13 common genes. In both species, the known BOA genes are organized at two different loci, but their repartitions are different (Fig. 4). In B. cinerea, BcBOA1 to BcBOA6 are located at one locus, whereas BcBOA7 to BcBOA17 are located at another locus. In S. sclerotiorum, SsBOA1 and SsBOA2 are located at one locus distinct from the main cluster (SsBOA3 to SsBOA13). Apart from these differences, the co‐localized genes are in the same order and orientation in the two species, suggesting that the genomic rearrangements that occur in this secondary metabolism cluster are mainly fissions or fusions. The most parsimonious hypothesis would be that BOA genes were originally clustered in the common ancestor of Botrytis and Sclerotinia and that different fission events occurred in the two genera. Fission may have occurred in Botrytis because of a DNA transposition event, as the AT‐rich region downstream of BcBOA6 contains transposon relics exhibiting similarities to Fot and Pot transposable elements from Fusarium oxysporum and M. grisea, respectively. In S. sclerotiorum, a different fission of the native cluster probably occurred, separating SsBOA1 and SsBOA2 from the main cluster. In addition, this rearrangement might be a result of a DNA transposition, as the predicted gene Ss1g_09233 downstream of the SsBOA3 gene (Fig. 4) encodes a putative transposase with the characteristic DDE superfamily endonuclease domain (Table S4 and Fig. S3, see Supporting Information). In addition to these hypothetical fission events, gene loss or gain may also have occurred on the native BOA cluster as homologues of BcBOA14 to BcBOA17 genes are lacking in the S. sclerotinium genome. Finally, the S. sclerotiorum and B. cinerea BOA clusters are located in different regions of their respective genomes (Table S4 and Fig. S3). Taken together, these results show that, even though B. cinerea and S. sclerotinium usually show a high degree of synteny for most parts of the genome because of their close phylogenetic relationship [Fillinger et al., 2007; J. Amselem (INRA, Versailles, France) et al., unpublished], the putative ancestral BOA gene cluster has been submitted to a rapid evolution in both species. The requirement of additional yet unknown genes for botcinic acid biosynthesis in a third genomic locus in B. cinerea cannot be excluded.
Comparative genomics in fungi have revealed that many rearrangements, such as gene duplications, translocations and losses, occur in secondary metabolism gene clusters (Bömke et al., 2008). In addition, there are several examples in which secondary metabolite gene clusters are split into two or more parts. Thus, in Fusarium graminearum and Fusarium sporotrichioides, the TRI biosynthetic genes are located at three loci: a 12‐gene TRI core cluster and two smaller TRI loci that consist of one or two genes. These three TRI loci have a complex evolutionary history that has included loss, nonfunctionalization and rearrangement of genes, as well as trans‐species polymorphism (Proctor et al., 2009). Two clusters of genes were also identified for lolitrem biosynthesis in the mutualistic endophyte of perennial ryegrass, Neotyphodium lolii (Young et al., 2006). In B. cinerea, the core gene cluster for abscisic acid biosynthesis has been identified (Siewers et al., 2006), but the gene encoding the key enzyme, a sesquiterpene synthase, is missing at this locus and must be elsewhere in the genome.
Remarkably, both gene clusters of B. cinerea that are responsible for botcinic acid biosynthesis are surrounded by AT‐rich (70%–90%) regions. Similar observations have been made for the 16‐kb botrydial gene cluster (Pinedo et al., 2008). In both cases, the AT‐rich repetitive sequences have complicated further cloning and sequencing of regions upstream and downstream of the clusters. Hence, the three gene clusters are isolated as, to date, they have not been connected to the genomic backbone, and also their chromosomal location is still unknown. However, these AT‐rich regions may have a function for the recognition of ‘border sequences’ of the clusters. Secondary metabolite gene clusters are often flanked by repetitive elements composed of transposable elements or transposon relics, and it has been proposed that they are involved in chromatin‐mediated transcriptional control of the cluster genes (Palmer and Keller, 2010).
Clustering of secondary metabolite genes might confer an advantage for the co‐regulation of the genes by ‘narrow‐’ or ‘broad’‐domain transcription factors, as well as by epigenetic processes (Fox and Howlett, 2008; Palmer and Keller, 2010). Although nothing is known about the importance of ‘broad’‐domain transcription factors, such as AreA, PacC and CreA, for the expression of BcBOA genes, the involvement of at least one pathway‐specific regulator appears to be likely. Thus, BcBOA13 is predicted to encode a Zn(ii)2Cys6 zinc binuclear cluster transcription factor. Members of this fungal‐specific group of transcription factors are often located within secondary metabolite clusters, positively regulating the expression of the adjacent cluster genes (Keller et al., 2005). Furthermore, a putative relic of another fungal‐specific transcription factor is found in the botcinic acid cluster. The corresponding domain has been identified in the predicted sequence of BcBOA15, and the open reading frames of both BcBOA14 and BcBOA15 share significant similarity with a predicted transcription factor in Aspergillus clavatus. However, the transcription factor in B. cinerea might be nonfunctional as the transcription of both BcBOA14 and BcBOA15 could not be detected by Northern blot analyses. In addition, sequencing of reverse transcription‐polymerase chain reaction (RT‐PCR) fragments revealed internal stop codons. Therefore, two small independent genes have been predicted whose functionality remains questionable.
In addition to the mentioned transcription factor(s), another gene product of the cluster might be important for the regulation of botcinic acid biosynthesis. BcBOA1 encodes a protein of the NmrA‐like family. Members of this family are often found in secondary metabolite clusters, such as in the ergot alkaloid biosynthesis gene cluster of Claviceps purpurea (N. Lorenz and P. Tudzynski, University of Münster, Germany, personal communication) and in the bikaverin biosynthesis gene cluster of Fusarium fujikuroi (Wiemann et al., 2009). In the latter case, it was demonstrated that the NmrA‐like protein Bik4 is probably involved in the regulation of bikaverin biosynthesis via a yet unknown mechanism. A catalytic function of Bik4 in the biosynthetic pathway has been ruled out as the bik4 deletion mutant is still able to produce the end product bikaverin, although in significantly lower quantities.
Although we do not know by which environmental signals botcinic acid production is driven, we demonstrated that the transcription of both botcinic acid and botrydial biosynthetic genes was dependent on the presence of the Gα subunit BCG1, the activity of the Ca2+‐regulated calcineurin phosphatase and the calcineurin‐responsive transcription factor BcCRZ1, but not on cyclic AMP‐mediated signal transduction (this study; Pinedo et al., 2008; 2008a, 2008b, 2008c). Siewers et al. (2005) showed that the bcg1Δ mutant does not produce botrydial or botcinic acid in submerged culture. Accordingly, the corresponding BcBOT and BcBOA genes are not expressed in axenic culture or in planta. These results are in agreement with the observation that the bcg1Δ mutants are severely affected in virulence (Schulze Gronover et al., 2001). The observation that botcinic acid production is increased in mutants deficient in botrydial production (bcbot2Δ background) might be a result of either altered expression levels of BcBOA genes or alterations in primary metabolism of the mutant. As both sesquiterpene and polyketide biosyntheses include acetyl‐CoA as a primary precursor unit, the inhibition of botrydial biosynthesis may result in the availability of more precursors for polyketide synthesis and, consequently, in increased botcinic acid production. Independent of the mode of regulation, the increased production of botcinic acid in B05.10 botrydial‐deficient strains may have a compensatory effect on virulence (Pinedo et al., 2008).
The fact that B05.10 mutants affected in the production of either botrydial or botcinic acid are not impaired in virulence on bean plants reveals the redundant function of both toxins in virulence. However, the impact of total phytotoxin biosynthesis on pathogenesis has been demonstrated clearly: botrydial and botcinic acid are required for the killing of host cells and, consequently, for the colonization of plant tissue.
EXPERIMENTAL PROCEDURES
Fungal strains and culture conditions
Strain B05.10 of Botrytis cinerea Pers. Fr. [Botryotinia fuckeliana (de Bary) Whetz] is derived from a Vitis field isolate (Quidde et al., 1999). The bcbot2Δ botrydial mutant has been described previously (Pinedo et al., 2008). The B05.10‐derivative strain bcg1Δ is deleted in the respective Gα subunit‐encoding gene and was characterized by Schulze Gronover et al. (2001). Standard procedures for the culture and maintenance of B. cinerea WT strain and mutants were carried out on NY medium (2 g/L malt extract, 2 g/L yeast extract, 15 g/L agar) at 21 °C with 16 h of daylight per day. Growth and conidiation rates were measured from cultures on V8, potato dextrose agar (PDA) and minimal medium (Viaud et al., 2003).
Standard molecular methods
Genomic DNA was extracted from fungal mycelium using a Sarcosyl‐based protocol (Dellaporta et al., 1983). Gel electrophoresis, restriction enzyme digestion and Southern blot experiments were performed using standard protocols (Sambrook et al., 1989). DNA probes were labelled by the random primer method using the Q‐Biogen (Illkirch, France) Nonaprimer Kit and 20 µCi α‐32P‐dCTP, as described previously (Levis et al., 1997). PCRs were performed with the Silverstar Taq DNA polymerase (Eurogentec, Seraing, Belgium), except for the vector construction for which the Phusion high‐fidelity polymerase (Ozyme, Montigny, France) was used. For cDNA synthesis, total RNA (1 µg) was subjected to DNase I treatment (RQ1 RNase‐Free DNase; Promega Corporation, Madison, WI, USA) and subsequently used for cDNA synthesis employing the oligo(dT)12–18 primer and SuperScript II reverse‐transcriptase (Invitrogen, Leek, the Netherlands), according to the manufacturer's instructions. The amplification was performed by PCR using the BioTherm™ DNA polymerase (GeneCraft GmbH, Münster, Germany) and the following PCR programme: 94 °C for 4 min; 35 cycles of 94 °C for 1 min, 58 °C for1 min and 70 °C for 3 min; and a 10 min final extension at 70 °C. Fragments were cloned into the pCR®2.1 TOPO® vector for sequencing.
Reverse‐Northern blot analyses
Twenty‐three secondary metabolism and control genes were amplified from B05.10 genomic DNA. PCR primer pairs were designed on the basis of the exon sequences at the 3′ part of the open reading frame (Table S1), so that they generated PCR products of approximately 700 bp, corresponding to the 3′ exon of the gene. Aliquots of 5 ng of the PCR products were denatured in 4 m NaOH and spotted onto Nylon filters. Total RNA was isolated from fungal cultures and infected tomato leaves using TRIzol. Then, mRNA was labelled with 30 µCi of 32P‐dCTP, as described previously (Viaud et al., 2003), and hybridized to the filters.
Gene inactivation and complementation by protoplast transformation
For construction of the gene replacement vector pΔbcpks6, the plasmid pOliHP (Rolke et al., 2004), carrying the Escherichia coli hph gene under the control of the A. nidulans oliC promoter and trpC terminator, was used as a basal vector. An 870‐bp fragment from the 5′ region of bcpks6 was amplified using the primers Bcpks6‐SacI (5′‐GAGCTCGTCTCAATGTTGTCGATATACATC‐3′) and Bcpks6‐EcoRI (5′‐GAATTCCCTATTCGACATGATACTGCGTG‐3′) containing restriction for further cloning. A 900‐bp fragment was generated as second flank using the primers Bcpks6‐KpnI (5′‐GGTACCGCCCATCATAATTCCGTGCTG‐3′) and Bcpks6‐SalI (5′‐GTCGACCTCGCAGAGCAACGTCGCAAGCG‐3′). The PCR fragments were cloned into pCR®2.1‐TOPO®, sequenced and isolated with SacI‐EcoRI and KpnI‐SalI, respectively, and cloned into the corresponding restriction sites of pOliHP, creating pΔbcpks6. For transformation, the replacement cassette was isolated by restriction with KpnI and SacI.
The BcPKS9 KO replacement cassette was generated using the double‐joint PCR strategy described by Yu et al. (2004). The nourseothricin resistance gene nat1 was amplified from the plasmid pNR1 (Malonek et al., 2004) using the primers Nat1‐5 (5′‐ACAAAAGCTGGAGCTCCACC‐3′) and Nat1‐3 (5′‐CGATATCGAATTCCTGCAGG‐3′. The pairs of primers BD38 (5′‐CAGAACTTTGTGTCGCTGGA‐3′)/BD37 (5′‐GGTGGAGCTCCAGCTTTTGTAGATCTGTGGCAGGAACCAT‐3′) and BD36 (5′‐CCTGCAGGAATTCGATATCGACGCGGCTCAGATATTGATT‐3′)/BD35 (5′‐CGTGCGTGAGAAATGATGAC‐3′) were used to amplify regions of about 1 kb in 5′ and in 3′ of BcPKS9, respectively. The italic tails overlap the sequence of the Nat1‐3 and Nat1‐5 primers and allow linkage of the three PCR products together (5′ region of BcPKS9 to Nat1 to 3′ region of BcPKS9) by a second PCR (Yu et al., 2004).
Transformation of B. cinerea
Protoplasts from B05.10 were prepared and transformed as described previously (Levis et al., 1997) using 2 µg of linear DNA. Transformed protoplasts were plated in molten osmotically stabilized medium agar containing 100 µg/mL hygromycin (Invitrogen), 70 µg/mL nourseothricin (Werner, Jena, Germany) or 100 µg/mL bialaphos (gluphosinate ammonium, Dr Ehrenstorfer GmbH, Augsburg, Germany). Transformants were selected after 6–8 days at 23 °C, subcultured twice on selective medium and single‐spore cultures were made to obtain genetically pure transformants. The screening for the BcPKS6 gene inactivation event was performed by PCR using the primers PKS6‐verif‐5 (5′‐GCTTGCATGACTGAGATTGC‐3′), located upstream of the 5′ flanking region, and Hyg‐deb‐R (5′‐CCGAGGGCAAAGGAATAGAG‐3′), located inside the hph gene. The screening for the BcPKS9 gene inactivation event was performed by PCR using the primers BD39 (5′‐ACTGAAGCGCCTACTTGTGG‐3′), located upstream of the 5′ flanking region, and Nat1F‐verif (5′‐GACACCGCCCTGTACGAC‐3′), located inside the nat1 gene. Inactivation of the BcBot2 gene and screening for the homologous recombination events were performed as described in Pinedo et al. (2008). All gene inactivations were verified by Southern blot hybridization as described in Fig. S1.
Analysis of metabolite production
Botrytis cinerea WT strain and mutants were grown on malt agar medium (20 g/L d‐glucose, 10 g/L malt extract, 20 g/L agar, pH 6.5–7) at 25 °C to produce mycelium plugs (1 cm) that were further used to inoculate medium for metabolite production. All studied strains were fermented in both surface and solid agar malt cultures. For surface cultures, mycelia were grown in 1‐L Roux bottles containing 200 mL of modified Czapek–Dox medium (50 mg/L d‐glucose, 1 g/L yeast extract, 5 g/L KH2PO4, 2 g/L NaNO3, 0.5 g/L MgSO4.7H2O and 0.01 g/L FeSO4.7H2O, pH 6.5–7.0) at 25 °C. For each experiment, 10 Roux bottles were inoculated with six mycelium plugs per bottle and incubated for 9 days. After incubation under fluorescent light, the culture medium was filtered, saturated with NaCl, extracted with ethyl acetate (3 × 0.5 vol.) and washed with water (3 × 0.25 vol.). For solid medium cultures, 20 Petri plates of malt agar medium were inoculated with two mycelium plugs per plate and incubated for 12 days. Then, mycelia and conidia were removed from the culture with a spatula and the solid agar malt medium was extracted with ethyl acetate (3 × 0.5 vol.) using an ultrasonic bath. The organic extracts from both liquid and solid cultures were dried over Na2SO4 and concentrated to dryness.
1H and 13C NMR measurements of metabolites isolated from culture extracts were obtained on Varian (Madrid, Spain) Unity 400 and Varian Innova 600 NMR spectrometers with SiMe4 as the internal reference. Mass spectra were recorded on a GC‐MS Thermoquest Voyager spectrometer (Finnigan, Manchester, UK) and a VG Autospec‐Q spectrometer (Manchester , UK). HPLC was performed with a Hitachi/Merck (Barcelona, Spain) L‐6270 apparatus equipped with a UV–visible detector (L 6200) and a differential refractometer detector (RI‐71). Thin layer chromatography (TLC) was performed on Merck Kiesegel 60 F254 (thickness, 0.2 mm). Silica gel (Merck) was used for column chromatography. HPLC purification was accomplished with a silica gel column (Hibar 60; 7 m; width, 1 cm; length, 25 cm). Chemicals were products of Fluka or Aldrich (Madrid, Spain). All solvents were freshly distilled.
For metabolite isolation and characterization, the yellow oil extract obtained from both experiments was separated by means of column chromatography on silica gel with a mixture of ethyl acetate–hexane (10, 20, 40, 60, 80 and 100% ethyl acetate) and 20% methanol in ethyl acetate as solvent.
Extensive spectroscopic analysis by 1H and 13C NMR was used to detect the presence of the different metabolites in each fraction. Candidate fractions were further purified by HPLC with an increasing gradient of ethyl acetate to petroleum ether. The compound structures were analysed by spectroscopic methods and direct comparison with authentic samples, previously isolated from strains of B. cinerea (Collado et al., 2007). Semi‐preparative HPLC afforded compounds 1–8 from both WT strains and mutants of B. cinerea (Table 1 and Fig. 1).
Cloning the gene clusters—revision of the automatic gene prediction
Genomic sequences comprising the PKS‐encoding genes BcPKS6/BcBOA6 and BcPKS9/BcBOA9, as well as the corresponding expressed sequence tags (ESTs) derived from the two genome projects of B. cinerea (B05.10 and T4; Fillinger et al., 2007), were assembled using Seqman, a program of the Lasergene package (DNASTAR, Madison, USA). In B05.10, two supercontigs (supercontigs 1.204 and 1.226), with lengths of 36 974 bp and 16 578 bp, respectively, comprising discontinuous sequences, were found. In the genome project of strain T4, only smaller genome contigs were present, which have not been considered for further assembly and gene prediction (Fig. S2, see Supporting Information).
The gene cluster located on supercontig 226 (cluster A) has been described previously. Gaps in the B05.10 sequence have been filled by sequencing a phage derived from a genomic library of B. cinerea SAS56, and the exon–intron structures of the genes have been determined by sequencing cDNA clones (Schumacher et al., 2008a). In this study, the gene cluster located on supercontig 204 containing BcPKS9/BcBOA9 (cluster B) was further characterized. Three gaps in the B05.10 sequence were filled by generating overlapping PCR fragments for sequencing (labelled as A–C in Fig. S2 and Table S2, see Supporting Information). For the generation of fragment D, primers have been designed using the sequence of S. sclerotiorum 1980 (Fillinger et al., 2007). cDNA sequences to confirm the intron positions were generated by performing PCR using cDNA derived from strain B05.10 as template and the primers summarized in Table S2. As it was not possible to obtain a full‐length cDNA clone for BcPKS9/BcBOA9, the gene was re‐annotated with the aid of the FGENESH (HMM‐based gene structure) prediction (http://linux1.softberry.com/berry.phtml). More details and GenBank accession numbers are given in Table 2.
TAIL‐PCR was performed in order to recover DNA fragments adjacent to the known cluster sequences using a protocol modified after Liu and Whittier (1995) and Terauchi and Kahl (2000). For TAIL‐PCR, five nested primers binding in the known sequence were combined with random hexamers (sequences not shown) in three consecutive reactions. TAIL‐PCR approaches were performed to obtain new sequence information downstream of BcBOA17 and BcBOA7 in order to extend supercontig 204, and downstream of BcBOA6/BcPKS6 and BcBOA1 to extend supercontig 226. However, approaches failed with one exception. Two rounds of TAIL‐PCR downstream of BcBOA1 resulted in a new sequence of 1940 bp that is characterized by high AT content. As for the other parts of the cluster, the third round of TAIL‐PCR did not result in any fragments. Consequently, the two gene clusters are restricted by very AT‐rich regions that hamper PCR and sequencing strategies. Taken together, the identified BoA genes are situated in two separate clusters (renamed clusters A and B) that are located on the B05.10 supercontig 226 (the expanded sequence contains 22 940 bp and comprises the open reading frames of BcBOA1–BcBOA6) and supercontig 204 (the revised sequence contains 36 579 bp and comprises the open reading frames BcBOA7–BcBOA17).
Detection of gene expression by Northern blot analyses
In order to detect phytotoxin gene expression under axenic conditions, conidia of the WT strain B05.10 and the bcg1 deletion mutant were cultivated for 62 h in 100 mL of liquid medium (0.2% yeast extract, 1.0% glucose, 0.2% KH2PO4, 0.15% K2HPO4, 0.1% (NH4)2SO4, 0.05% MgSO4.7H2O) at 20 g and 20 °C. Then, the mycelia were filtered and transferred to 100 mL of the same medium minus the nitrogen sources [yeast extract and (NH4)2SO4]. After an additional 7 h of cultivation, mycelia were harvested and used for RNA extraction. To study the gene expression in planta, conidia derived from the two strains were resuspended in Gamborg's B5 medium (DUCHEFA Biochemie B.V., Haarlem, the Netherlands) supplemented with 2% (w/v) glucose and 10 mm KH2PO4/K2HPO4, pH 6.4, to a final concentration of 2 × 105 conidia/mL. Droplets of the conidial suspensions (7.5 µL) were used to inoculate leaves of French bean (Phaseolus vulgaris). The plants were incubated in a plastic propagator box at 20 °C under natural illumination conditions. Primary lesions were harvested after 48 h of incubation and were subsequently employed for RNA isolation using TRIzol® reagent (Invitrogen). For Northern blot analyses, samples (25 µg of total RNA) were transferred to Hybond‐N+ membranes after electrophoresis on a 1% agarose gel containing formaldehyde, according to Sambrook et al. (1989). Blot hybridizations were carried out in 0.6 m NaCl, 0.16 m Na2HPO4, 0.06 m ethylenediaminetetraacetic acid (EDTA), 1% N‐lauroylsarcosine (Sigma‐Aldrich, Taufkirchen, Germany), 10% dextran sulphate (Eppendorf AG, Hamburg, Germany), 0.01% salmon sperm DNA, pH 6.2, at 65 °C in the presence of a random‐primed α‐32P‐dCTP‐labelled probe.
Infection assays
Infection assays of B. cinerea WT strain and mutants were performed on French bean (P. vulgaris) by the inoculation of detached leaves with young nonsporulating mycelium or conidial suspensions from cultures on NY medium. Bean plants (Caruso cultivar) were grown under glasshouse conditions. Leaves were harvested from 2‐week‐old plants and placed in a transparent plastic box lined with tissue moistened with sterile water. Leaves were inoculated with 1.8‐mm‐diameter plugs of 3‐day‐old mycelium. Alternatively, conidia were collected from 10‐day‐old plates and suspended in sucrose phosphate buffer (10 mm sucrose, 10 mm KH2PO4) to a final concentration of 105 conidia/mL. Droplets of 10 µL were applied to the leaves. Storage boxes containing inoculated leaves were incubated in a growth cabinet at 21 °C with 16 h of daylight. Disease development on leaves was recorded daily as the radial spread from the inoculation point to the lesion margin. Pathogenicity assays on leaves were repeated three times using at least five leaves per assay.
Supporting information
Fig. S1 Gene inactivation of BcPKS6 (A, B), BcPKS9 (C, D) and BcBOT2 (E, F) by homologous recombination. Target genes were inactivated by transforming the B05‐10 WT strain with the KO cassettes presented in (A), (C) and (E) (see Experimental procedures for more details). The letters C and H indicate ClaI and HpaI restriction sites, respectively. For Southern blots, genomic DNA was digested by ClaI (B, F) or HpaI (D). The numbers indicate the strains as follows: B05.10 WT strain (1), pks6Δ‐1 (2), pks6Δ‐4 (3), pks6Δ‐5 (4), pks9Δ‐3 (5), pks9Δ‐14 (6), pks9Δ‐16 (7), bcbot2Δ (8), bcbot2Δ pks6Δ −1 (9), bcbot2Δ pks6Δ‐2 (10), bcbot2Δ pks6Δ‐9 (11), bcbot2Δ pks6Δ‐14 (12).
Fig. S2 Physical maps of the characterized botcinic acid gene clusters of Botrytis cinerea. B05.10 genome supercontigs 1.204 and 1.226 (B. cinerea B05.10 Database; http://www.broadinstitute.org/annotation/genome/botrytis_cinerea/Home.html) and corresponding T4 genome contigs referred to as Bt4exctg (T4 genome project; http://urgi.versailles.inra.fr/index.php/urgi/Species/Botrytis) are indicated by black lines. Discontinuous sequences of B05.10 were corrected and expanded by generating polymerase chain reaction (PCR) fragments for sequencing (A–D and TAIL, respectively). For more details, see Experimental procedures.
Fig. S3 Physical maps of the Sclerotinia sclerotiorum strain 1980 genome supercontigs containing homologues of Botrytis cinerea BcBOA genes.
Table S1 Selected genes and designed primers used in the reverse‐Northern strategy.
Table S2 Primers used for the sequencing strategies.
Table S3 Predicted genes adjacent to SsBOA1 and SsBOA2 in Sclerotinia sclerotiorum strain 1980 (on supercontig 19).
Table S4 Predicted genes adjacent to SsBOA3 to SsBOA13 in Sclerotinia sclerotiorum strain 1980 (on supercontig13).
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGEMENTS
We thank Marine Vautier and Trang Le Thuy for technical support. IGC thanks the Ministerio de Educación y Ciencia (MEC), Spain for grant AGL2009‐13359‐C02‐01.
REFERENCES
- Abe, Y. , Suzuki, T. , Ono, C. , Iwamoto, K. , Hosobuchi, M. and Yoshikawa, H. (2002) Molecular cloning and characterization of an ML‐236B (compactin) biosynthetic gene cluster in Penicillium citrinum . Mol. Genet. Genomics, 267, 636–646. [DOI] [PubMed] [Google Scholar]
- Baker, S.E. , Kroken, S. , Inderbitzin, P. , Asvarak, T. , Li, B.Y. , Shi, L. , Yoder, O.C. and Turgeon, B.G. (2006) Two polyketide synthase‐encoding genes are required for biosynthesis of the polyketide virulence factor, T‐toxin, by Cochliobolus heterostrophus . Mol. Plant–Microbe Interact. 19, 139–149. [DOI] [PubMed] [Google Scholar]
- Bok, J.W. , Chiang, Y.M. , Szewczyk, E. , Reyes‐Dominguez, Y. , Davidson, A.D. , Sanchez, J.F. , Lo, H.C. , Watanabe, K. , Strauss, J. , Oakley, B.R. , Wang, C.C. and Keller, N.P. (2009) Chromatin‐level regulation of biosynthetic gene clusters. Nat. Chem. Biol. 5, 462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bömke, C. , Rojas, M.C. , Hedden, P. and Tudzynski, B. (2008) Loss of gibberellin production in Fusarium verticillioides (Gibberella fujikuroi MP‐A) is due to a deletion in the gibberellic acid gene cluster. Appl. Environ. Microbiol. 74, 7790–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns, W. , Horns, S. and Redlich, H. (1995) Botrylactone synthesis and structure revision: a convergent approach from d‐glucose. Synthesis, 3, 335–342. [Google Scholar]
- Chiang, Y.M. , Szewczyk, E. , Davidson, A.D. , Keller, N. , Oakley, B.R. and Wang, C.C.C. (2009) A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans . J. Am. Chem. Soc. 131, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquer, M. , Fournier, E. , Kunz, C. , Levis, C. , Pradier, J.‐M. , Simon, A. and Viaud, M. (2007) Botrytis cinerea virulence factors: new insights into a necrotrophic and polyphagous pathogen. FEMS Microbiol. Lett. 277, 1–10. [DOI] [PubMed] [Google Scholar]
- Collado, I.G. , Sanchez, A.J. and Hanson, J.R. (2007) Fungal terpene metabolites: biosynthetic relationships and the control of the phytopathogenic fungus Botrytis cinerea . Nat. Prod. Rep. 24, 674–686. [DOI] [PubMed] [Google Scholar]
- Collemare, J. , Pianfetti, M. , Houlle, A.E. , Morin, D. , Camborde, L. , Gagey, M.J. , Barbisan, C. , Fudal, I. , Lebrun, M.‐H. and Bohnert, H.U. (2008) Magnaporthe grisea avirulence gene ACE1 belongs to an infection‐specific gene cluster involved in secondary metabolism. New Phytol. 179, 196–208. [DOI] [PubMed] [Google Scholar]
- Colmenares, A.J. , Aleu, J. , Duran‐Patron, R. , Collado, I.G. and Hernandez‐Galan, R. (2002) The putative role of botrydial and related metabolites in the infection mechanism of Botrytis cinerea . J. Chem. Ecol. 28, 997–1005. [DOI] [PubMed] [Google Scholar]
- Cutler, H.G. , Jacyno, J.M. , Harwood, J.S. , Dulik, D.M. , Goodrich, P.D. and Roberts, R.G. (1993) Botcinolide: a biologically active natural product from Botrytis cinerea . Biosci. Biotechnol. Biochem. 57, 1980–1982. [Google Scholar]
- Cutler, H.G. , Parker, S.R. , Ross, S.A. , Crumley, F.G. and Schreiner, P.R. (1996) Homobotcinolide: a biologically active natural homolog of botcinolide from Botrytis cinerea . Biosci. Biotechnol. Biochem. 60, 656–658. [DOI] [PubMed] [Google Scholar]
- Deighton, N. , Muckenschnabel, I. , Colmenares, A.J. , Collado, I.G. and Williamson, B. (2001) Botrydial is produced in plant tissues infected by Botrytis cinerea . Phytochemistry, 57, 689–692. [DOI] [PubMed] [Google Scholar]
- Dellaporta, S.L. , Wood, J. and Hicks, B. (1983) A plant DNA minipreparation: version II. Plant Mol. Biol. Rep. 1, 19–21. [Google Scholar]
- Desjardins, A.E. , Proctor, R.H. , Bai, G. , McCormick, S.P. , Shaner, G. , Buechley, G. and Hohn, T.M. (1996) Reduced virulence of trichothecene‐nonproducing mutants of Gibberella zeae in wheat field test. Mol. Plant–Microbe Interact. 9, 775–781. [Google Scholar]
- Doehlemann, G. , Berndt, P. and Hahn, M. (2006) Different signalling pathways involving Gα protein, cAMP and a MAP kinase control germination of Botrytis cinerea conidia. Mol. Microbiol. 59, 821–835. [DOI] [PubMed] [Google Scholar]
- Fillinger, S. , Amselem, J. , Artiguenave, F. , Billault, A. , Choquer, M. , Couloux, A. , Cuomo, C. , Dickman, M. , Fournier, E. , Gioti, A. , Giraud, C. , Kodira, C. , Kohn, L. , Legeai, F. , Levis, C. , Mauceli, E. , Pommier, C. , Pradier, J.‐M. , Quévillon, E. , Rollins, J. , Ségurens, B. , Simon, A. , Viaud, M. , Weissenbach, J. , Wincker, P. and Lebrun, M.‐H. (2007) The genome projects of the plant pathogenic fungi Botrytis cinerea and Sclerotinia sclerotiorum In: Macromolecules of Grape and Wines (Jeandet P., Clément C. and Conreux A., eds), pp. 125–133. Paris: Editions TEC & DOC, Lavoisier. [Google Scholar]
- Fox, E.M. and Howlett, B.J. (2008) Secondary metabolism: regulation and role in fungal biology. Curr. Opin. Microbiol. 11, 1–7. [DOI] [PubMed] [Google Scholar]
- Fukui, H. , Hitomi, S. , Suzuki, R.S. , Ikeda, T. , Umezaki, Y. , Tsuji, K. and Shiina, I. (2008) Asymmetric total synthesis of botcinic acid and its derivatives: synthetic revision of the structure of botcinolides. Tetrahedron Lett. 49, 6514–6517. [Google Scholar]
- Gaffoor, I. and Trail, F. (2006) Characterization of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae . Appl. Environ. Microbiol. 72, 1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioti, A. , Simon, A. , Le Pêcheur, P. , Giraud, C. , Pradier, J.‐M. , Viaud, M. and Levis, C. (2006) Expression profiling of Botrytis cinerea genes identifies three patterns of up‐regulation in planta and an FKBP12 protein affecting pathogenicity. J. Mol. Biol. 358, 372–386. [DOI] [PubMed] [Google Scholar]
- Hoffmeister, D. and Keller, N.P. (2007) Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat. Prod. Rep. 24, 393–416. [DOI] [PubMed] [Google Scholar]
- Keller, N.P. , Turner, G. and Bennett, J.W. (2005) Fungal secondary metabolism—from biochemistry to genomics. Nat. Rev. Microbiol. 3, 937–947. [DOI] [PubMed] [Google Scholar]
- Kennedy, J. , Auclair, K. , Kendrew, S.G. , Park, C. , Vederas, J.C. and Hutchinson, C.R. (1999) Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science, 284, 1368–1372. [DOI] [PubMed] [Google Scholar]
- Khaldi, N. , Collemare, J. , Lebrun, M.‐H. and Wolfe, K.H. (2008) Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 9, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroken, S. , Glass, N.L. , Taylor, J.W. , Yoder, O.C. and Turgeon, B.G. (2003) Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. USA, 100, 15 670–15 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis, C. , Fortini, D. and Brygoo, Y. (1997) Transformation of Botrytis cinerea with the nitrate reductase gene (niaD) shows a high frequency of homologous recombination. Curr. Genet. 32, 157–162. [DOI] [PubMed] [Google Scholar]
- Liu, Y.G. and Whittier, R.F. (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics, 25, 674–681. [DOI] [PubMed] [Google Scholar]
- Malonek, S. , Rojas, M.C. , Hedden, P. , Gaskin, P. , Hopkins, P. and Tudzynski, B. (2004) The NADPH‐cytochrome P450 reductase gene from Gibberella fujikuroi is essential for gibberellin biosynthesis. J. Biol. Chem. 279, 25 075–25 084. [DOI] [PubMed] [Google Scholar]
- Palmer, J.M. and Keller, N.P. (2010) Secondary metabolism in fungi: does chromosomal location matter? Curr. Opin. Microbiol. 13, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedras, M.S. and Ahiahonu, P.W. (2004) Phytotoxin production and phytoalexin elicitation by the phytopathogenic fungus Sclerotinia sclerotiorum . J. Chem. Ecol. 30, 2163–2179. [DOI] [PubMed] [Google Scholar]
- Pinedo, C. , Wang, C.M. , Pradier, J.‐M. , Dalmais, B. , Choquer, M. , Le Pêcheur, P. , Morgant, G. , Collado, I.G. , Cane, D.E. and Viaud, M. (2008) Sesquiterpene synthase from the botrydial biosynthetic gene cluster of the phytopathogen Botrytis cinerea . ACS Chem. Biol. 3, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor, R.H. , Plattner, R.D. , Brown, D.W. , Seo, J.A. and Lee, Y.W. (2004) Discontinuous distribution of fumonisin biosynthetic genes in the Gibberella fujikuroi species complex. Mycol. Res. 108, 815–822. [DOI] [PubMed] [Google Scholar]
- Proctor, R.H. , McCormick, S.P. , Alexander, N.J. and Desjardins, A.E. (2009) Evidence that a secondary metabolic biosynthetic gene cluster has grown by gene relocation during evolution of the filamentous fungus Fusarium . Mol. Microbiol. 74, 1128–1142. [DOI] [PubMed] [Google Scholar]
- Quidde, T. , Büttner, P. and Tudzynski, P. (1999) Evidence for three different specific saponin‐detoxifying activities in Botrytis cinerea and cloning and functional analysis of a gene coding for a putative avenacinase. Eur. J. Plant Pathol. 105, 273–283. [Google Scholar]
- Reino, J.L. , Hernandez‐Galan, R. , Duran‐Patron, R. and Collado, I.G. (2004) Virulence–toxin production relationship in isolates of the plant pathogenic fungus Botrytis cinerea . J. Phytopathol. 152, 563–566. [Google Scholar]
- Reino, J.L. , Duran‐Patron, R.M. , Daoubi, M. , Collado, I.G. and Hernandez‐Galan, R. (2006) Biosynthetic studies on the botcinolide skeleton: new hydroxylated lactones from Botrytis cinerea . J. Org. Chem. 71, 562–565. [DOI] [PubMed] [Google Scholar]
- Reverberi, M. , Ricelli, A. , Zjalic, S. , Fabbri, A.A. and Fanelli, C. (2010) Natural functions of mycotoxins and control of their biosynthesis in fungi. Appl. Microbiol. Biotechnol. 87, 899–911. [DOI] [PubMed] [Google Scholar]
- Rolke, Y. , Liu, S. , Quidde, T. , Williamson, B. , Schouten, A. , Weltring, K.M. , Siewers, V. , Tenberge, K.B. , Tudzynski, B. and Tudzynski, P. (2004) Functional analysis of H2O2‐generating systems in Botrytis cinerea: the major Cu–Zn‐superoxide dismutase (BCSOD1) contributes to virulence on French bean, whereas a glucose oxidase (BCGOD1) is dispensable. Mol. Plant Pathol. 5, 17–27. [DOI] [PubMed] [Google Scholar]
- Sakuno, E. , Tani, H. and Nakajima, H. (2007) 2‐Epi‐botcinin A and 3‐O‐acetylbotcineric acid from Botrytis cinerea . Biosci. Biotechnol. Biochem. 71, 2592–2595. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory. [Google Scholar]
- Schulze Gronover, C. , Kasulke, D. , Tudzynski, P. and Tudzynski, B. (2001) The role of G protein alpha subunits in the infection process of the gray mold fungus Botrytis cinerea . Mol. Plant–Microbe Interact. 14, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Schumacher, J. , Viaud, M. , Simon, A. and Tudzynski, B. (2008a) The Gα subunit BCG1, the phospholipase C (BcPLC1) and the calcineurin phosphatase co‐ordinately regulate gene expression in the grey mould fungus Botrytis cinerea . Mol. Microbiol. 67, 1027–1050. [DOI] [PubMed] [Google Scholar]
- Schumacher, J. , de Larrinoa, I.F. and Tudzynski, B. (2008b) Calcineurin‐responsive zinc finger transcription factor CRZ1 of Botrytis cinerea is required for growth, development, and full virulence on bean plants. Eukaryot. Cell. 7, 584–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, J. , Kokkelink, L. , Huesmann, C. , Jimenez‐Teja, D. , Collado, I.G. , Barakat, R. , Tudzynski, P. and Tudzynski, B. (2008c) The cAMP‐dependent signaling pathway and its role in conidial germination, growth, and virulence of the gray mold Botrytis cinerea . Mol. Plant–Microbe Interact. 21, 1443–1459. [DOI] [PubMed] [Google Scholar]
- Siewers, V. , Viaud, M. , Jimenez‐Teja, D. , Collado, I.G. , Gronover, C.S. , Pradier, J.‐M. , Tudzynski, B. and Tudzynski, P. (2005) Functional analysis of the cytochrome P450 monooxygenase gene bcbot1 of Botrytis cinerea indicates that botrydial is a strain‐specific virulence factor. Mol. Plant–Microbe Interact. 18, 602–612. [DOI] [PubMed] [Google Scholar]
- Siewers, V. , Kokkelink, L. , Smedsgaard, J. and Tudzynski, P. (2006) Identification of an abscisic acid gene cluster in the grey mold Botrytis cinerea . Appl. Environ. Microbiol. 72, 4619–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims, J.W. , Fillmore, J.P. , Warner, D.D. and Schmidt, E.W. (2005) Equisetin biosynthesis in Fusarium heterosporum . Chem. Commun. 14, 186–188. [DOI] [PubMed] [Google Scholar]
- Soanes, D. , Richards, T.A. and Talbot, N.J. (2007) Insights from sequencing fungal and oomycete genomes: what can we learn about plant disease and the evolution of pathogenicity? Plant Cell, 19, 3318–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Z. , Cox, R.J. , Lazarus, C.M. and Simpson, T.J. (2004) Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum . Chembiochem. 5, 1196–1203. [DOI] [PubMed] [Google Scholar]
- Sutherland, A. , Auclair, K. and Vederas, J.C. (2001) Recent advances in the biosynthetic studies of lovastatin. Curr. Opin. Drug Discov. Devel. 4, 229–236. [PubMed] [Google Scholar]
- Tani, H. , Koshino, H. , Sakuno, E. and Nakajima, H. (2005) Botcinins A, B, C, and D, metabolites produced by Botrytis cinerea, and their antifungal activity against Magnaporthe grisea, a pathogen of rice blast disease. J. Nat. Prod. 68, 1768–1772. [DOI] [PubMed] [Google Scholar]
- Tani, H. , Koshino, H. , Sakuno, E. , Cutler, H.G. and Nakajima, H. (2006) Botcinins E and F and botcinolide from Botrytis cinerea and structural revision of botcinolides. J. Nat. Prod. 69, 722–725. [DOI] [PubMed] [Google Scholar]
- Terauchi, R. and Kahl, G. (2000) Rapid isolation of promoter sequences by TAIL‐PCR: the 5′‐flanking regions of Pal and Pgi genes from yams (Dioscorea). Mol. Gen. Genet. 263, 554–560. [DOI] [PubMed] [Google Scholar]
- Viaud, M. , Brunet‐Simon, A. , Brygoo, Y. , Pradier, J.‐M. and Levis, C. (2003) Cyclophilin A and calcineurin functions investigated by gene inactivation, cyclosporin A inhibition and cDNA arrays approaches in the phytopathogenic fungus Botrytis cinerea . Mol. Microbiol. 50, 1451–1465. [DOI] [PubMed] [Google Scholar]
- Walton, J.D. (2000) Horizontal gene transfer and the evolution of secondary metabolite gene clusters in fungi: an hypothesis. Fungal Genet. Biol. 30, 167–171. [DOI] [PubMed] [Google Scholar]
- Walton, J.D. (2006) HC‐toxin. Phytochemistry, 67, 1406–1413. [DOI] [PubMed] [Google Scholar]
- Wang, C.M. , Hopson, R. , Lin, X. and Cane, D.E. (2009) Biosynthesis of the sesquiterpene botrydial in Botrytis cinerea. Mechanism and stereochemistry of the enzymatic formation of presilphiperfolan‐8beta‐ol. J. Am. Chem. Soc. 131, 8360–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welmar, K. , Tschesche, R. and Breitmaier, E. (1979) Botrylactone, a new antibiotic from the culture medium of the fungus Botrytis cinerea . Chem. Ber. 112, 3598–3602. [Google Scholar]
- Wiemann, P. , Willmann, A. , Straeten, M. , Kleigrewe, K. , Beyer, M. , Humpf, H.‐U. and Tudzynski, B. (2009) Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Mol. Microbiol. 72, 931–946. [DOI] [PubMed] [Google Scholar]
- Williamson, B. , Tudzynski, B. , Tudzynski, P. and van Kan, J.A. (2007) Botrytis cinerea: the cause of grey mould disease. Mol. Plant Pathol. 8, 561–580. [DOI] [PubMed] [Google Scholar]
- Young, C.A. , Felitti, S. , Shields, K. , Spangenberg, G. , Johnson, R.D. , Bryan, G.T. , Saikia, S. and Scott, B. (2006) A complex gene cluster for indole‐diterpene biosynthesis in the grass endophyte Neotyphodium lolii . Fungal Genet. Biol. 43, 679–693. [DOI] [PubMed] [Google Scholar]
- Yu, J.‐H. , Hamari, Z. , Han, K.‐H. , Seo, J.‐A. , Reyes‐Dominguez, Y. and Scazzocchio, C. (2004) Double‐joint PCR: a PCR‐based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 41, 973–981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Gene inactivation of BcPKS6 (A, B), BcPKS9 (C, D) and BcBOT2 (E, F) by homologous recombination. Target genes were inactivated by transforming the B05‐10 WT strain with the KO cassettes presented in (A), (C) and (E) (see Experimental procedures for more details). The letters C and H indicate ClaI and HpaI restriction sites, respectively. For Southern blots, genomic DNA was digested by ClaI (B, F) or HpaI (D). The numbers indicate the strains as follows: B05.10 WT strain (1), pks6Δ‐1 (2), pks6Δ‐4 (3), pks6Δ‐5 (4), pks9Δ‐3 (5), pks9Δ‐14 (6), pks9Δ‐16 (7), bcbot2Δ (8), bcbot2Δ pks6Δ −1 (9), bcbot2Δ pks6Δ‐2 (10), bcbot2Δ pks6Δ‐9 (11), bcbot2Δ pks6Δ‐14 (12).
Fig. S2 Physical maps of the characterized botcinic acid gene clusters of Botrytis cinerea. B05.10 genome supercontigs 1.204 and 1.226 (B. cinerea B05.10 Database; http://www.broadinstitute.org/annotation/genome/botrytis_cinerea/Home.html) and corresponding T4 genome contigs referred to as Bt4exctg (T4 genome project; http://urgi.versailles.inra.fr/index.php/urgi/Species/Botrytis) are indicated by black lines. Discontinuous sequences of B05.10 were corrected and expanded by generating polymerase chain reaction (PCR) fragments for sequencing (A–D and TAIL, respectively). For more details, see Experimental procedures.
Fig. S3 Physical maps of the Sclerotinia sclerotiorum strain 1980 genome supercontigs containing homologues of Botrytis cinerea BcBOA genes.
Table S1 Selected genes and designed primers used in the reverse‐Northern strategy.
Table S2 Primers used for the sequencing strategies.
Table S3 Predicted genes adjacent to SsBOA1 and SsBOA2 in Sclerotinia sclerotiorum strain 1980 (on supercontig 19).
Table S4 Predicted genes adjacent to SsBOA3 to SsBOA13 in Sclerotinia sclerotiorum strain 1980 (on supercontig13).
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item