

SUMMARY
A novel, unbiased approach to plant viral disease diagnosis has been developed which requires no a priori knowledge of the host or pathogen. Next‐generation sequencing coupled with metagenomic analysis was used to produce large quantities of cDNA sequence in a model system of tomato infected with Pepino mosaic virus. The method was then applied to a sample of Gomphrena globosa infected with an unknown pathogen originally isolated from the flowering plant Liatris spicata. This plant was found to contain a new cucumovirus, for which we suggest the name ‘Gayfeather mild mottle virus’. In both cases, the full viral genome was sequenced. This method expedites the entire process of novel virus discovery, identification, viral genome sequencing and, subsequently, the development of more routine assays for new viral pathogens.
INTRODUCTION
The detection and identification of new viruses currently rely on a large range of techniques, both traditional and modern. Typically, the process starts by screening for a range of suspected ‘known’ viruses, using a panel of specific tests, based on either serological [e.g. enzyme‐linked immunosorbent assay (ELISA)] or molecular [e.g. polymerase chain reaction (PCR) or nucleic acid spot hybridization)] methods. Subsequently, if pathogens are not detected, more ‘investigational’ techniques are applied, such as electron microscopy, host plant inoculation and, if available, PCR using degenerate primers (Gibbs and Mackenzie, 1997; James et al., 2006). More recently, microarray techniques have been developed that offer a more flexible approach to viral detection, allowing a large number of target pathogens to be tested for simultaneously (Boonham et al., 2007; Mumford et al., 2006). Although often successful, these techniques suffer from several significant drawbacks, especially when trying to identify ‘unknown’ agents, e.g. either a pathogen infecting a new host or a previously uncharacterized pathogen. Firstly, specific assays require a prediction of the infective agent, based on a reliable knowledge of which pathogens infect which hosts. However, these data are frequently incomplete or even totally absent, especially for many of the more obscure or novel hosts, such as ornamentals. Second, specific tests, including microarrays, utilize reagents (DNA primers, probes or antibodies) with a finite specificity covering a particular strain, individual species or small group of pathogens. These reagents are often incapable of detecting variants, such as new strains, that can arise unexpectedly. As a result, these can evade detection and spread rapidly. Third, nonspecific methods (e.g. host plant inoculation or electron microscopy) can be useful for the detection of the presence of a viral pathogen and, in some cases, for the identification of the family or genus of viruses to which it belongs. However, further analysis is then required to reach a definitive identification. These techniques are also not truly universal, as many viruses are either not readily transmissible to indicator hosts or easily visualized using standard electron microscopic procedures, again allowing certain agents to remain undetected. The final significant issue is the length of time needed for the identification of a new disease. Often as a result of the requirement for the performance of multiple parallel tests, even the identification of a previously characterized virus in a new host can take weeks or months. The identification of a novel pathogen can take considerably longer; for example, the identification of Blackcurrant reversion virus, the causal agent of reversion disease in Ribes, took years using traditional virological and molecular methods (Susi, 2004). Given these drawbacks, virologists have continued to look for novel approaches that will improve the identification of the causes of new or unusual diseases. One such promising approach is the use of metagenomics.
Metagenomics is an approach for the study of microbial populations in a sample by analysing the nucleotide sequence content. It has been applied to a wide range of environmental samples, including bacterial metagenomes from deep mines (Edwards et al., 2006) and the sea (Sogin et al., 2006; Venter et al., 2004), viral metagenomes from the human gut (Breitbart et al., 2003; Zhang et al., 2006), sea water (Angly et al., 2006; Williamson et al., 2008) and fresh water (Breitbart et al., 2008), and bacteria, archaea, fungi and viruses in soil (Fierer et al., 2007). Early metagenomic projects used Sanger sequencing and were costly, but the advent of next‐generation sequencing technology, such as Roche's GS‐FLX Genome Sequencer (Roche Diagnostics Ltd., Burgess Hill, West Sussex, UK), has made large‐scale metagenomic studies more practical and cost‐effective. A metagenomic approach to diagnostic plant virology offers the possibility of overcoming the problems of pathogen prediction associated with parallel screening methods and the nonspecificity associated with traditional investigational techniques. Sequences produced from an infected plant will include sequences from any pathogens present. The extraction of RNA from the infected plant, the production of cDNA with a random priming method and, finally, sequencing will produce sequences from a large range of potential pathogens. RNA viruses, viroids and the RNA stages of actively replicating DNA viruses can be directly sequenced. This approach should also produce sequences of mRNA and rRNA from any phytoplasma, bacteria or fungi present in the sample. By sequencing cDNA and not genomic DNA, only active host genes and ribosomes will be sequenced, avoiding the large amounts of untranscribed genomic DNA found in higher plants, and also avoiding integrated genomes of some plant viruses, such as badnaviruses.
To date, there have been three published accounts of the combination of next‐generation sequencing and metagenomics in disease diagnostics. Cox‐Foster et al. (2007) sequenced cDNA from a series of beehives with symptoms of colony collapse disorder and identified the presence of Israeli acute paralysis virus. Palacios et al. (2008) sequenced cDNA from samples of tissue taken from three transplant patients, who had died after receiving transplants from the same donor. Using this approach, a new arenavirus was identified in all three samples. Nakamura et al. (2009) applied next‐generation sequencing techniques to human nasal and faecal samples, and were able to type and obtain genomic information on the viral infections present.
In this article, we describe the development of a metagenomic diagnostic technique utilizing next‐generation sequencing, and its application for the detection of plant viruses, with two specific examples presented: the first using a model system, Pepino mosaic virus infecting tomato; and the second using a real diagnostic sample, consisting of a previously uncharacterized virus infecting an ornamental host.
RESULTS
Pepino mosaic virus (PepMV)
Sequencing of cDNA prepared from total RNA extracted from a tomato plant infected with PepMV produced a total of 65 691 individual sequences with a mean read length of 252 bp, giving a total of 16 590 395 bp of DNA sequence. Contig assembly reduced this to 387 contigs and 6963 unassembled sequences. Examination of the blast results revealed that 20.1% (13 183) of the sequences were PepMV. Of the remaining sequences, approximately 70% were plant ribosomal or chloroplast in origin.
Of the contigs produced, seven corresponded to PepMV, covering 97% of the viral genome. A small 200‐nucleotide region at the 5′ end of the genome remained unconstructed until additional PepMV sequences were extracted from the unassembled sequence dataset. Using a published PepMV genome (accession number AJ606361) as a scaffold, the extracted PepMV sequences were aligned and full coverage was achieved. Figure 1a shows a histogram of read coverage along the length of the genome sequence. A minimum of 200‐fold sequencing coverage was achieved for the whole virus genome. The isolate of PepMV was found to have a genome of 6382 nucleotides in length excluding the polyA tail. The closest relative (accession number EF408821), with a sequence homology of 98%, was from Poland and was described as a US2 strain (Hasiow et al., 2008). Five genes were found in the genome sequence: RNA‐dependent RNA polymerase, three small proteins known collectively as the ‘triple gene block’ and the coat protein. The sequence was submitted to GenBank (accession number FJ212288).
Figure 1.

Sequence coverage and gene positions along the lengths of the viral genomes (5′–3′): (A) Pepino mosaic virus; (B) Gayfeather mild mottle virus (GMMV) RNA 1; (C) GMMV RNA 2; (D) GMMV RNA 3.
Metagenomic analysis using megan (Huson et al., 2007) revealed that the most common sequence origin was tomato (Solanum lycopersicum) and that the majority of the sequences were homologous to dicotyledons. PepMV was also identified. In addition to the expected host plant and PepMV assignments, sequences with significant homology to human, chimpanzee, bacterial and phage sequences were also identified, but accounted for less than 0.001% of the sequences obtained.
The high levels of sequence originating from the host plant in the PepMV model system suggested that a modification to the protocol would be required if viral sequence was to be obtained from lower titre infections. A subtractive hybridization approach was chosen. This method was initially carried out on the cDNA from the PepMV‐infected tomato subtracted with cDNA synthesized from uninfected tomato. The resulting cDNA was cloned and sequenced using conventional techniques. A significant proportion (60%) of the clones contained PepMV DNA, three times more than in the unsubtracted sample.
Gayfeather mild mottle virus (GMMV)
Sap from a Liatris spicata plant showing symptoms of mild mottling was mechanically inoculated onto a series of indicator plants, which produced a range of symptoms (see Table 1). Photographs of these symptoms are included as Supporting Information (Figs S1–S8).
Table 1.
Symptoms produced by Gayfeather mild mosaic virus (GMMV) in host plant Liatris and in mechanically inoculated indicator plants.
| Indicator plant | Symptoms | |
|---|---|---|
| Local | Systemic | |
| Liatris spicata | Mild mottling | |
| Amarantus caudatus | Chlorotic local lesions becoming necrotic with time | |
| Atriplex hortensis | Chlorotic local lesions, grey necrotic spots | |
| Celosia argentea | Local necrotic vein banding, ringspots | |
| Chenopodium amaranticolor | Necrotic small local lesions | |
| C. ambrosioides | Chlorotic local light green ringspots | |
| C. murale | Chlorotic local lesions | |
| Cucumis sativus | Chlorotic ringspots | |
| Gomphrena globosa | Local necrotic lesions, that become clear paper thin | Leaf distortion, necrotic etching on leaf base |
| Nicotiana alata | Local chlorotic diffuse spots with necrotic etched borders | Leaf necrosis turning into stem necrosis |
| N. occidentalis | Local necrotic ringspots followed by vein necrosis | Severe leaf distortion, plant stunting |
| N. rustica | Systemic mottling | |
| N. glutinosa | Local chlorotic lesions | Mottling on the base of leaves, leaf distortion, light green mottling |
| N. debneyi | Large necrotic lesions | Mottling and severe leaf distortion |
| N. tabacum | Local etched ringspots | |
Using the subtractive sample preparation methodology, a Gomphrena globosa plant infected with the L. spicata virus was sequenced, and a total of 71 146 individual sequences were produced with a mean read length of 227 bp, giving a total of 16 149 138 bp of DNA sequence. Contig assembly produced 738 contigs with 8556 sequences left unassembled. Examination of the blast results revealed that 40.9% (29 095) of the sequences were related to, but distinct from, the cucumoviruses Cucumber mosaic virus (CMV), Peanut stunt virus (PSV) and Tomato aspermy virus (TAV), and approximately 47% of the sequences were ribosomal or chloroplast in origin. The tripartite genome of the new Cucumovirus, named ‘Gayfeather mild mottle virus’, did not require additional assembly with reference genomes and was constructed entirely from the contigs. RNA 1 is 3350 bp in length and codes for only one protein, 1a (replicase). RNA 2 is 2935 bp in length and codes for two proteins, 2a (RNA‐dependent RNA polymerase) and 2b (unknown function). RNA 3 is 2214 bp in length and codes for two proteins, 3a (movement protein) and 3b (coat protein). Figure 1 shows the coverage for all three RNA segments, together with gene locations. No correlation between the level of coverage and open reading frame location was observed. The minimum coverage was onefold for the very beginning of RNA 3, with a minimum of sevenfold for the rest of the genomes. The average coverage was over 800‐fold. A fourth (subgenomic) RNA was not detected, nor were there any satellite RNAs.
A neighbour‐joining tree was constructed from the 1a protein sequences from all known viruses within the family Bromoviridae (Fig. 2) to confirm GMMV's position within the family. GMMV clusters with the other cucumoviruses, as suggested by the initial blast results. To clarify whether GMMV was a new virus or a new strain of a known Cucumovirus, the three full‐length RNAs were combined end‐to‐end and aligned with Cucumovirus reference sequences from GenBank and a Bromovirus outgroup (Broadbean mottle virus). The full‐genome nucleotide alignment was used to produce a second phylogenetic tree (Fig. 3) and percentage nucleotide differences (Table 2). The tree and percentage nucleotide differences observed strongly suggested that GMMV was a new virus within the genus Cucumovirus. The three RNA segments of the GMMV genome were submitted to GenBank with the following accessions: FM881899, FM881900 and FM881901.
Figure 2.
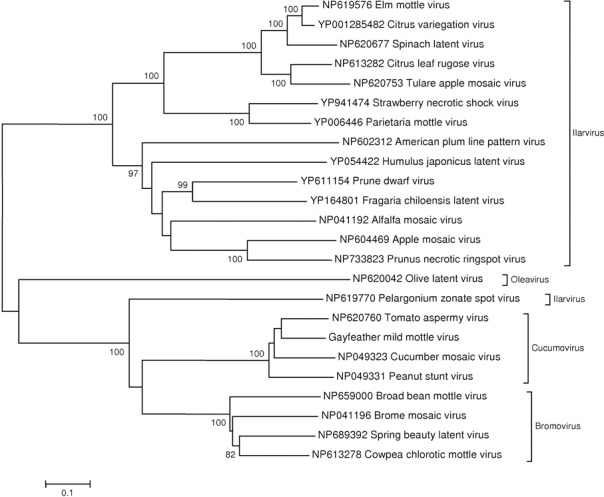
Bootstrapped neighbour‐joining tree (1000 replicates) constructed from an alignment of 1a protein sequences from the Bromoviridae family. Gayfeather mild mottle virus (GMMV) is placed within the Cucumovirus genus.
Figure 3.

Bootstrapped neighbour‐joining tree (1000 replicates) constructed using uncorrected P distances derived from an alignment of the full reference genomes of all known cucumoviruses [Cucumber mosaic virus (CMV), Peanut stunt virus (PSV) and Tomato aspermy virus (TAV)], Gayfeather mild mosaic virus and a Bromovirus, Broad bean mottle virus (BBMV). Reference genome accession numbers: PSV (NC_002038, NC_002039, NC_002040); CMV (NC_002035, NC_002034, NC_001440); TAV (NC_003837, NC_003838, NC_003836); BBMV (NC_004008, NC_004007, NC_004006).
Table 2.
Percentage nucleotide differences between the whole genomes of the currently recognized cucumoviruses [Cucumber mosaic virus (CMV), Peanut stunt virus (PSV) and Tomato aspermy virus (TAV)], Gayfeather mild mottle virus (GMMV) and a bromovirus, Broad bean mottle virus (BBMV).
| GMMV | PSV | TAV | CMV | |
|---|---|---|---|---|
| PSV | 43.19% | — | ||
| TAV | 40.06% | 35.88% | — | |
| CMV | 41.19% | 34.97% | 35.16% | — |
| BBMV | 55.50% | 55.19% | 54.24% | 54.26% |
megan analysis of this sample identified contigs with homology to the viruses CMV, PSV and TAV. These contigs were shown to be the complete genome of GMMV, as described above. Also identified were a large number of plant sequences, as well as small numbers of sequences from bacteria, fungi and the common glasshouse pest western flower thrips (Frankliniella occidentalis), which was present in the glasshouse when the plant was being grown.
DISCUSSION
In this article, we describe the development and use of a metagenomic diagnostic technique utilizing next‐generation sequencing for several plant viruses. Unlike traditional techniques, such as ELISA, PCR or hybridization methods, this method requires no a priori knowledge of the suspected pathogen. The technique does not utilize any virus‐specific reagents, such as antibodies or primers/probes, and, in this sense, the method developed has more in common with investigational virology techniques, such as electron microscopy. The method was initially developed using the well‐characterized PepMV as a model system, and subsequently applied to an uncharacterized virus disease of the ornamental flowering plant Gayfeather or Blazing star (L. spicata).
Using tomato plants infected with the high‐titre PepMV as a model system, we were able to identify a total of 13 183 viral sequences amongst the 65 691 fragments of primarily plant host sequences generated in a single run. Using a published PepMV genome as a scaffold, we were able to assemble the sequence fragments into a full genome (6382 nucleotides excluding the poly‐A tail) for the virus, with an average nucleotide coverage of 200.
The uncharacterized virus from L. spicata was isolated into indicator hosts and subjected to a subtractive hybridization method to enrich for viral cDNA above the background of redundant host cDNA (primarily ribosomal in origin) produced. The subtraction method developed has the advantage that the cDNA amplification requires considerably less starting material than the direct double‐stranded cDNA synthesis approach used for the PepMV sample. From the 71 146 fragments of sequence generated, 29 095 were identified as having similarity to published viruses based on blast searching. By generating contiguous sequence from the fragments with viral homology, each of the three fragments of the viral tripartite genome were assembled, without the need for a scaffold sequence. Using the subtractive approach, an average nucleotide coverage of 800 was achieved across the entire genome.
During initial characterization work, the new virus only weakly cross‐reacted with CMV antibodies in ELISA (data not shown). Cluster analysis from an alignment of 1a replicase protein sequences from the Bromoviridae family reliably placed the new virus within the Cucumovirus genus, and whole‐genome comparisons showed the most related virus to be TAV (59.04% nucleotide identity). These sequence differences were further supported by biological differences, following mechanical transmission to a range of indicator hosts. The host range was again most similar to TAV (Brunt et al., 1996), although the new virus was able to systemically infect and produce symptoms in Cucumis sativus, whereas TAV produces small local chlorotic spot symptoms. The symptoms produced were distinct from those described for all other members of the Cucumovirus genus. Based on the criteria detailed in the 8th Report of the International Committee on Taxonomy of Viruses (Fauquet et al., 2005), the uncharacterized virus was sufficiently distinct from all other sequenced cucumoviruses to warrant the status of a new virus species. As a result, we suggested the name ‘Gayfeather mild mottle virus’, and placed the virus as a new species within the genus Cucumovirus.
The results show that, even without the use of existing viral genomes as a scaffold, we were able to assemble a complete genome sequence for a previously uncharacterized virus. The sample preparation took 2 days and, at the time of writing, a 1/16 partition of a GS‐FLX 454 sequencing plate (the smallest possible partition) produced approximately 10 000 sequences, probably enough to attain a full‐genome sequence of GMMV with more than 100‐fold coverage using the described method. In most diagnostic situations, a full‐genome sequence is not a necessity, and it is probable that, as shown by Cox‐Foster et al. (2007) and Palacios et al. (2008), at least some viral sequences would be recovered from even low‐titre infections (especially using the subtractive technique), and greater sensitivity could be achieved by increasing the amount of sequencing performed. At present, the analysis costs approximately £1000 per sample, but this sum is likely to be reduced considerably in the future.
The identification of the new virus using this approach proved to be extremely rapid. The generation of sequence in this way would enable the development of routine diagnostics (e.g. PCR methods) very rapidly after initial identification, allowing timely epidemiological work to be completed and potentially enabling control or eradication strategies to take place.
In this case, only one viral sequence was recovered from the infected plant and the virus was successfully transmitted from the initial host to a range of indicator hosts, giving virus symptoms in each case. Although technically not completing Koch's postulates, in the absence of any other virus being recovered, it seems probable from the work completed that the new Cucumovirus was the cause of disease in the G. globosa plant, and probably the original L. spicata plant. As none of the original infected L. spicata plant material is available, it is now impossible for this to be confirmed. In addition, although in the examples presented only one virus was found to be present, it seems likely that, if multiple viruses had been present as part of a mixed infection, they would have been identified and characterized correctly, as we were easily able to correctly assemble the three distinct genome segments of GMMV without reference to published genome sequences of related viruses.
As exemplified by other accounts of a metagenomic approach to the identification of unknown agents (Cox‐Foster et al., 2007; Palacios et al., 2008), recovering a virus sequence does not provide a direct link to the cause of the disease. However, the use of methods of this kind has potential benefits when compared with other commonly accepted methods in this regard. The described method generates sequence in an unbiased fashion; therefore, if multiple viruses are present in a sample, all are likely to be sequenced and further investigations can be carried out as appropriate based on this information. Traditional approaches (e.g. electron microscopy followed by virus purification and sequencing) could easily generate misleading results, especially with mixed infections. For example, if a sample contained a mixed infection of an easy to identify rod‐shaped virus (e.g. potyvirus) as well as a difficult to identify spherical virus (e.g. nepovirus), subsequent virus purification and sequencing would be biased to the further characterization of the rod‐shaped virus. Koch's postulates could be erroneously completed for the rod‐shaped virus, and yet it may be difficult to identify the cryptic spherical virus causing the disease.
Looking forward to the broader use of these techniques, employing the software package megan, we were able to identify host plant and viral sequences (including phage), as well as bacterial, fungal, insect and human (presumably contamination from the operator) RNA. The identification of bacteria and fungi suggests that this technique may be useful in identifying unknown pathogens other than viruses.
We have developed a generic unbiased tool which allows the identification and genome sequencing of novel viral infections in plants using metagenomics coupled with pyrosequencing. The technique should prove to be a valuable tool for the rapid identification of suspect viral infections, and is also likely to be useful for suspect infections of other pathogens, such as fungi and bacteria.
EXPERIMENTAL PROCEDURES
Virus maintenance
An isolate of PepMV (Potexvirus, Flexiviridae) was maintained on a tomato plant (variety Moneymaker) in a glasshouse at 18–20 °C. Leaves were taken for RNA extraction 3 weeks post‐inoculation.
A sample of L. spicata showing mild mottling on the leaves was brought from Poland to the Experimental Station of Field Floriculture in Vilnius, Lithuania, in 2005. The virus was mechanically inoculated onto a series of indicator plants to check for symptoms, and then maintained in G. globosa in the glasshouse at 18–20 °C. Leaves were taken for RNA extraction 3 weeks post‐inoculation.
RNA isolation
RNA was isolated from 300 mg of fresh leaf material using a modification of the cetyltrimethylammonium bromide (CTAB) method described in Chang et al. (1993). The leaves were frozen in liquid nitrogen, ground to a powder and resuspended in 2 mL of buffer containing 100 mm tris(hydroxymethyl)aminomethane (Tris)‐HCl (pH 8), 2% CTAB, 20 mm ethylenediaminetetraacetic acid (EDTA), 1.5 m NaCl, 2% polyvinylpyrrolidone (PVP) and 1% Na2SO3. After incubation at 65 °C for 10 min, the samples were mixed with an equal volume of chloroform–isoamyl alcohol (24 : 1) and centrifuged for 10 min at 12 000 g. The aqueous phase was then mixed with an equal volume of 4 m LiCl and incubated at –20 °C for 1 h. The samples were then centrifuged for 25 min at 12 000 g, and the pellets were washed in 70% ethanol, air dried and resuspended in 100 µL of water. The samples were then further purified using RNAeasy columns according to the manufacturer's instructions (Qiagen, Crawley, West Sussex, UK). The RNAse‐free DNAse set was used to remove any contaminating DNA from the samples. The RNA was eluted from the columns in 100 µL of water and quantified using a Nanodrop ND‐1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
cDNA synthesis
Double‐stranded cDNA was synthesized from the tomato sample using the Superscript double‐stranded cDNA synthesis kit (Invitrogen, Paisley, Renfrewshire, UK) following the manufacturer's protocol. Total RNA (100 µg) was processed in two separate reactions, one primed with random hexameric primers and one with oligo‐dT primer. The two samples were combined and yielded 500 ng of double‐stranded DNA, as determined by the Quant‐iT dsDNA BR assay kit (Invitrogen).
Using G. globosa plants, both healthy and infected with the suspected new virus, cDNA was produced using a modification of the methods described in Cox‐Foster et al. (2007). First‐strand cDNA was synthesized from 5 µg of total RNA using Tag dT (GTTTCCCAGTAGGTCTCTTTTTTTTTTTTTTT) and Tag random (GTTTCCCAGTAGGTCTCNNNNNNNN) primers. This cDNA was amplified using tag random and tag (CGCCGTTTCCCAGTAGGTCTC) primers in a ratio of 1 : 10. A proof‐reading taq, Advantage II (Clontech, Mountain View, CA, USA), was used for amplification. The initial five cycles were carried out with an annealing temperature of 25 °C and a further 10 cycles with an annealing temperature of 55 °C. The cDNA from the uninfected plant was amplified using a nucleotide mixture in which half of the usual dTTP was replaced with biotin‐16‐dUTP (Roche Diagnostics Ltd.). cDNA from the infected plant was amplified using unlabelled nucleotides. The production of double‐stranded DNA was determined using the Quant‐iT dsDNA BR assay kit (Invitrogen).
PCR cloning and Sanger sequencing
PCR products were cloned into pGEMTeasy (Promega, Southampton, Hampshire, UK) and sequenced using cycle sequencing on an ABI 3130XL (Applied Biosystems, Warrington, Cheshire, UK).
Subtractive hybridization
Subtractive hybridization was carried out using a modification of the methods of Pradel et al. (2002). Amplified cDNA from uninfected and infected plants, in 50 µL Tris‐EDT (TE), was mixed in a ratio of 10 : 1 uninfected : infected and heated at 95 °C for 4 min. The samples were hybridized following the addition of 12 µL of 5 m NaCl and incubation at 65 °C for 18 h. EEN buffer (100 µL of 1 mm EDTA, 500 mm NaCl) was added and the sample was mixed with 100 µL of 1% Streptavidin Dynabeads (Invitrogen) resuspended in 100 µL EEN. After 20 min, a magnet was used to collect the beads, which were washed twice in 100 µL EEN. All EEN washes were collected, precipitated with 2.5 vol of ethanol and centrifuged at 14 000 g for 30 min. The resulting DNA pellet was resuspended in 20 µL of water and digested with 100 U S1 nuclease (Fermentas, York, UK) for 30 min at 37 °C to remove any single‐stranded DNA. Finally, 1 µL of 20 mm EDTA was added and the sample was incubated at 70 °C for 10 min to inactivate the enzyme.
cDNA was amplified using tag primer with an annealing temperature of 55 °C for 25 cycles. To inactivate the polymerase, 2 U proteinase K (Fermentas) was added to 50 µL of PCR product and incubated for 1 h at 45 °C. Proteinase K was then inactivated by heating the sample to 90 °C for 10 min. The DNA was finally blunt ended to remove any overhangs by the addition of 15 U T4 DNA polymerase (Fermentas) and incubated at 16 °C for 30 min. Heating the sample to 70 °C for 10 min then inactivated this polymerase. Finally, the sample was cleaned using a Qiaquick column (Qiagen), and the resulting DNA was suspended in 100 µL of water prior to sequencing.
Sequencing
Samples were sequenced using one‐quarter of a plate from a GS‐FLX Genome Sequencer (Advanced Genomics Facility, Liverpool University, Liverpool, UK).
Analysis
Contig assembly was carried out using the CLC Genomics Workbench (CLC Bio, Aarhus, Denmark). blast analysis was performed locally using blastn version 2.2.18 (Altschul et al., 1997) against the GenBank nucleotide database (Benson et al., 2008). Unassembled sequences showing homology to PepMV following blast analysis were extracted from the main dataset using a custom Perl script. Assembly of the consensus PepMV genome sequence was carried out in SeqMan (DNAStar, Madison, WI, USA) by combining the contigs already produced and the PepMV sequences extracted from the unassembled dataset. GMMV genome sequences did not require a scaffold for construction and were assembled de novo using the CLC Genomics Workbench.
Metagenomic analysis was performed using megan version 2 beta 14 (Huson et al., 2007). megan uses the results of a blast analysis of the metagenomic sequences and assigns each sequence to a taxon using the National Center for Biotechnology Information (NCBI) taxonomic database. The taxonomic level of the assignment depends on the degree of sequence homology. megan was set to only use blast results with high homology by setting the minimum score as 200. The read coverage of the viral genomes was calculated for each 5 bp of genome sequence, and these values were plotted as a histogram (Fig. 1). Phylogenetic trees were constructed in mega 4.1 (Tamura et al., 2007) and all alignments were created with ClustalW within mega.
Supporting information
Fig. S1 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Latris spicata.
Fig. S2 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Amaranthus caudatus.
Fig. S3 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Chenopodium murale.
Fig. S4 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Gomphrena globosa.
Fig. S5 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Gomphrena globosa.
Fig. S6 Gayfeather mild mottle virus (GMMV) symptoms on a stem of Nicotiana alata.
Fig. S7 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Nicotiana debneyi.
Fig. S8 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Nicotiana occidentalis.
Fig. S9 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Nicotiana rustica.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
ACKNOWLEDGEMENTS
We would like to thank Margaret Hughes from the Liverpool Advanced Genomics Facility for carrying out the sequencing.
This work was funded by the Plant Health Division of Defra (project PH0424) and Defra Seedcorn.
REFERENCES
- Altschul, S.F. , Madden, T.L. , Schaffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly, F.E. , Felts, B. , Breitbart, M. , Salamon, P. , Edwards, R.A. , Carlson, C. , Chan, A.M. , Haynes, M. , Kelley, S. , Liu, H. , Mahaffy, J.M. , Mueller, J.E. , Nulton, J. , Olson, R. , Parsons, R. , Rayhawk, S. , Suttle, C.A. and Rohwer, F. (2006) The marine viromes of four oceanic regions. PLoS Biol. 4, e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, D.A. , Karsch‐Mizrachi, I. , Lipman, D.J. , Ostell, J. and Wheeler, D.L. (2008) GenBank. Nucleic Acids Res. 36, D25–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonham, N. , Tomlinson, J. and Mumford, R. (2007) Microarrays for rapid identification of plant viruses. Annu. Rev. Phytopathol. 45, 307–328. [DOI] [PubMed] [Google Scholar]
- Breitbart, M. , Hewson, I. , Felts, B. , Mahaffy, J.M. , Nulton, J. , Salamon, P. and Rohwer, F. (2003) Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 185, 6220–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart, M. , Hoare, A. , Nitti, A. , Siefert, J. , Haynes, M. , Dinsdale, E. et al. (2008) Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Cienegas, Mexico. Environ. Microbiol. 11, 16–34. [DOI] [PubMed] [Google Scholar]
- Brunt A.A., Crabtree K., Dallwitz M.J., Gibbs A.J., Watson L. and Zurcher E.J. (eds) (1996 onwards) Plant Viruses Online: Descriptions and Lists from the VIDE Database . Version: 15th January 2007. URL http://biology.anu.edu.au/Groups/MES/vide/ [accessed on 10th January 2009].
- Chang, S. , Puryear, J. and Cairney, J. (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116. [Google Scholar]
- Cox‐Foster, D.L. , Conlan, S. , Holmes, E.C. , Palacios, G. , Evans, J.D. , Moran, N.A. , Quan, P.L. , Briese, T. , Hornig, M. , Geiser, D.M. , Martinson, V. , VanEngelsdorp, D. , Kalkstein, A.L. , Drysdale, A. , Hui, J. , Zhai, J.H. , Cui, L.W. , Hutchison, S.K. , Simons, J.F. , Egholm, M. , Pettis, J.S. and Lipkin, W.I. (2007) A metagenomic survey of microbes in honey bee colony collapse disorder. Science, 318, 283–287. [DOI] [PubMed] [Google Scholar]
- Edwards, R.A. , Rodriguez‐Brito, B. , Wegley, L. , Haynes, M. , Breitbart, M. , Peterson, D.M. , Saar, M.O. , Alexander, S. , Alexander, E.C., Jr. and Rohwer, F. (2006) Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics, 7, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauquet C.M., Mayo M.A., Maniloff J., Desselberger U. and Ball L.A. (eds) (2005) 8th Report of the International Committee on Taxonomy of Viruses. San Diego, CA: Academic Press. [Google Scholar]
- Fierer, N. , Breitbart, M. , Nulton, J. , Salamon, P. , Lozupone, C. , Jones, R. , Robeson, M. , Edwards, R.A. , Felts, B. , Rayhawk, S. , Knight, R. , Rohwer, F. and Jackson, R.B. (2007) Metagenomic and small‐subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 73, 7059–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, A. and Mackenzie, A. (1997) A primer pair for amplifying part of the genome of all potyvirids by RT‐PCR. J. Virol. Methods, 63, 9–16. [DOI] [PubMed] [Google Scholar]
- Hasiow, B. , Borodynko, N. and Pospieszny, H. (2008) Complete genomic RNA sequence of the Polish Pepino mosaic virus isolate belonging to the US2 strain. Virus Genes, 36, 209–214. [DOI] [PubMed] [Google Scholar]
- Huson, D.H. , Auch, A.F. , Qi, J. and Schuster, S.C. (2007) MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, D. , Varga, A. , Pallas, V. and Candresse, T. (2006) Strategies for simultaneous detection of multiple plant viruses. Can. J. Plant Pathol. 28, 16–29. [Google Scholar]
- Mumford, R.A. , Jarvis, B. , Harju, V. , Boonham, N. and Skelton, A. (2006) The first report of Broad bean wilt virus 2 in the UK: findings in foxglove and salvia. Plant Pathol. 55, 819. [Google Scholar]
- Nakamura, S. , Yang, C.S. , Sakon, N. , Ueda, M. , Tougan, T. , Yamashita, A. , Goto, N. , Takahashi, K. , Yasunaga, T. , Ikuta, K. , Mizutani, T. , Okamoto, Y. , Tagami, M. , Morita, R. , Maeda, N. , Kawai, J. , Hayashizaki, Y. , Nagai, Y. , Horii, T. , Iida, T. and Nakaya, T. (2009) Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high‐throughput sequencing approach. PLoS ONE, 4, e4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios, G. , Druce, J. , Du, L. , Tran, T. , Birch, C. , Briese, T. , Conlan, S. , Quan, P.L. , Hui, J. , Marshall, J. , Simons, J.F. , Egholm, M. , Paddock, C.D. , Shieh, W.J. , Goldsmith, C.S. , Zaki, S.R. , Catton, M. and Lipkin, W.I. (2008) A new arenavirus in a cluster of fatal transplant‐associated diseases. N. Engl. J. Med. 358, 991–998. [DOI] [PubMed] [Google Scholar]
- Pradel, N. , Leroy‐Setrin, S. , Joly, B. and Livrelli, V. (2002) Genomic subtraction to identify and characterize sequences of Shiga toxin‐producing Escherichia coli O91:H21. Appl. Environ. Microbiol. 68, 2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin, M.L. , Morrison, H.G. , Huber, J.A. , Mark, W.D. , Huse, S.M. , Neal, P.R. , Arrieta, J.M. and Herndl, G.J. (2006) Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc. Natl. Acad. Sci. USA, 103, 12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susi, P. (2004). Black currant reversion virus, a mite‐transmitted nepovirus. Mol. Plant Pathol. 5, 167–173. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Dudley, J. , Nei, M. and Kumar, S. (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. [DOI] [PubMed] [Google Scholar]
- Venter, J.C. , Remington, K. , Heidelberg, J.F. , Halpern, A.L. , Rusch, D. , Eisen, J.A. , Wu, D. , Paulsen, I. , Nelson, K.E. , Nelson, W. , Fouts, D.E. , Levy, S. , Knap, A.H. , Lomas, M.W. , Nealson, K. , White, O. , Peterson, J. , Hoffman, J. , Parsons, R. , Baden‐Tillson, H. , Pfannkoch, C. , Rogers, Y.H. and Smith, H.O. (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science, 304, 66–74. [DOI] [PubMed] [Google Scholar]
- Williamson, S.J. , Rusch, D.B. , Yooseph, S. , Halpern, A.L. , Heidelberg, K.B. , Glass, J.I. , Andrews‐Pfannkoch, C. , Fadrosh, D. , Miller, C.S. , Sutton, G. , Frazier, M. and Venter, J.C. (2008) The Sorcerer II Global Ocean Sampling Expedition: metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE, 3, e1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Breitbart, M. , Lee, W.H. , Run, J.Q. , Wei, C.L. , Soh, S.W. , Hibberd, M.L. , Liu, E.T. , Rohwer, F. and Ruan, Y. (2006) RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol. 4, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Latris spicata.
Fig. S2 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Amaranthus caudatus.
Fig. S3 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Chenopodium murale.
Fig. S4 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Gomphrena globosa.
Fig. S5 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Gomphrena globosa.
Fig. S6 Gayfeather mild mottle virus (GMMV) symptoms on a stem of Nicotiana alata.
Fig. S7 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Nicotiana debneyi.
Fig. S8 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Nicotiana occidentalis.
Fig. S9 Gayfeather mild mottle virus (GMMV) symptoms on a leaf of Nicotiana rustica.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item