

SUMMARY
Sequence analysis of five of the six endopolygalacturonase‐encoding genes (Bcpg1, Bcpg2, Bcpg3, Bcpg4, Bcpg5) from 32 strains of Botrytis cinerea showed marked gene to gene differences in the amount of among‐strains diversity. Bcpg4 was almost invariable in all strains; Bcpg3 and Bcpg5 showed a moderate variability, similar to that of non‐pathogenicity‐associated genes examined in other studies. Conversely, Bcpg1 and Bcpg2 were highly variable and were shown to be under positive selection based on the McDonald–Kreitman test and likelihood ratio test. The evolution of the five endopolygalacturonase genes is explained by their different ecophysiological role. Diversification and balancing selection, as detected in Bcpg1 and Bcpg2, can be used by the pathogen to escape recognition by the host and delay plant reaction in the early phases of infection. The analysis of the polymorphisms and the location of the sites with high probability of being positively selected highlighted the relevance of variability of the BcPG1 and BcPG2 proteins at their C‐terminal end. By contrast, the absence of variability in Bcpg4 suggests that the efficiency of the product of this gene is critical for B. cinerea growth in late phases of infection or during intraspecific competition, thus markedly affecting strain fitness.
INTRODUCTION
Botrytis cinerea Persoon: Fries [teleomorph: Botryotinia fuckeliana (de Bary) Whetzel] is a plant pathogenic fungus that causes fruit and leaf rot as well as flower blight in over 200 plant species (Barrie, 1994; Williamson et al., 2007). To colonize the host tissue, B. cinerea produces a battery of cell‐wall‐degrading enzymes. Among them, the endopolygalacturonases (endoPGs) cleave the linkages between d‐galacturonic acid residues in homogalacturonan and cause tissue maceration, resulting in soft‐rot symptoms. In B. cinerea up to 13 different endoPG isoforms were detected (van der Cruyssen et al., 1994) that are encoded by a family of at least six genes denoted as Bcpg1–6 (ten Have et al., 1998; Wubben et al., 1999).
Bcpg1 and Bcpg2 are required for full virulence and their inactivation by gene knockout produced mutants with a significant decrease in virulence (ten Have et al., 1998; Kars et al., 2005). However, the interplay of the different gene products in B. cinerea pathogenesis is not fully understood. Possibly, the redundancy of endoPG genes is a way of coping with the different environments that a highly polyphagous pathogen may encounter. Pathogen behaviour may differ significantly from one host species to the other and, on any given host, some strains may be more successful than others. The study of population structure and diversity is therefore fundamental for understanding the molecular dynamics of the interaction between B. cinerea and its hosts. Population structure and specialization in B. cinerea has been the subject of extensive studies in recent years. Giraud et al. (1997, 1999) postulated the occurrence of two sympatric sibling species, named vacuma and transposa, for B. cinerea on grape and other plants. The transposa strains were characterized by the presence in their genome of two transposable elements, Boty and Flipper, whereas strains of vacuma had neither. The frequencies of the markers Flipper and Boty were significantly different in strains collected from different host plants, leading to the conclusion that transposa and vacuma do not have the same ability to infect different hosts. This supposition was supported by an analysis of strains collected in Chile (Muñoz et al., 2002), showing that the isolates clustered together according to their host rather than their geographical origin. Later, Albertini et al. (2002) and Fournier et al. (2003, 2005) used a multiple‐genealogies approach based on four nuclear genes to show that B. cinerea isolates can be grouped into two genetically isolated subgroups, one including only vacuma strains (Group I), and the other both vacuma and transposa (Group II).
Due to their complex involvement in the interaction with the plant, genes coding endoPGs are excellent candidates for the investigation of possible host specialization in B. cinerea populations. Rowe and Kliebenstein (2007) reported the analysis of a collection of primarily Californian isolates for nucleotide sequence variations in Bcpg1, Bcpg2 and Bcpg3. No evidence of host specialization was found at the three loci, but the results indicated a notable difference in the degree of diversity. Bcpg1 and Bcpg2 showed a higher level of genetic diversity than Bcpg3 and neutral genes and statistics suggested that the former two genes were under balancing selection. In this work we confirm and expand the conclusions of Rowe and Kliebenstein (2007) by analysing 32 European B. cinerea strains for the diversity in Bcpg1, Bcpg2, Bcpg3 and in two additional genes, Bcpg4 and Bcpg5. We present evidence that some of these genes do not show a neutral evolution pattern and discuss this observation in relation to current views on the roles of the different gene products in the interaction with the host plant.
RESULTS
Sequence analysis
BLAST searches in the partially annotated genome of B. cinerea B05.10 recently released by the Broad Institute (http://www.broad.mit.edu/annotation/genome/botrytis_cinerea/Home.html) and in the as yet unpublished genome of strain T4 (J. A. L. van Kan, personal communication) provided no evidence of endoPG genes other than Bcpg1–6. Furthermore, using a PCR‐based screening approach with degenerate oligonucleotides no additional genes could be detected in B. cinerea SAS 405 and SAS56, and in B. fabae NCB1496. It is therefore likely that Bcpg1–6 and their homologues represent the entire endoPG gene family in B. cinerea and closely related species.
The endoPG coding gene sequences were studied in a collection comprising 32 strains of B. cinerea isolated primarily from Italy and Croatia and three strains of B. fabae (Table 1). The sequences of the Bcpg1/Bfpg1 and Bcpg2/Bfpg2 genes were determined for the entire collection of 35 strains. As our preliminary results (not published) suggested that some of the endoPG genes had limited diversity, we used a gel‐based approach to scan for polymorphisms in Bcpg3/Bfpg3, Bcpg4/Bfpg4 and Bcpg5/Bfpg5 prior to sequencing. For each locus, pairwise hybridization and digestion of heteroduplexes with a mismatch‐specific endonuclease enabled us to distinguish on gel between identical and non‐identical sequences, because each polymorphism generated two bands (Fig. 1). As a result, nine haplotypes in Bcpg3, four in Bcpg4 and nine in Bcpg5 were detected. Thus, the number of strains sequenced for all Bcpg3–5 was reduced to 16 strains, including all detected haplotypes. The sequences of strains SAS56, available from public databases, were also included in the sequence analysis.
Table 1.
List of the strains used in the present study; the fungal species is B. cinera where not otherwise specified.
| Isolate* | Host | Country |
|---|---|---|
| T4 (t) | Tomato | France |
| B05.10 (v) | unknown | Germany |
| BD90 (t) | Grapevine | France |
| C22 (t) | Grapevine (leaves) | Italy |
| PM10 (v) | Grapevine (leaves) | Italy |
| T23 (v) | Grapevine (shoot) | Italy |
| WS92 (t) | Grapevine (wine shoot) | Italy |
| 97 (t) | Grapevine (wine shoot) | Italy |
| G1 (t) | Grapevine (grape) | Italy |
| D1 (t) | Grapevine (grape) | Italy |
| DA1 (t) | Grapevine (grape) | Italy |
| RO18 (t) | Grapevine (grape) | Italy |
| SP37 (v) | Grapevine (grape) | Italy |
| RD1 (v) | Grapevine (grape) | Croatia |
| 134 (t) | Strawberry (flowers) | Croatia |
| 137(v) | Strawberry (flowers) | Croatia |
| 138 (t) | Strawberry (fruits) | Croatia |
| 139 (v) | Strawberry (fruits) | Croatia |
| 149 (v) | Strawberry (fruits) | Croatia |
| 154 (t) | Strawberry (fruits) | Croatia |
| CI199 (v) | Cyclamen (stem) | Italy |
| CI212 (t) | Cyclamen (leaves) | Italy |
| CI221 (v) | Cyclamen (flowers) | Italy |
| CI226 (t) | Cyclamen (leaves) | Italy |
| 193 (t) | Chrysanthemum (flowers) | Croatia |
| 194 (v) | Chrysanthemum (flowers) | Croatia |
| WS195 (v) | Carnation | Italy |
| VB24 (t) | Carnation | Italy |
| WS38 (t) | Strawberry | Italy |
| WS252 (t) | Strawberry | Switzerland |
| WS264 (v) | Strawberry | Portugal |
| 142649 (u) | Broad bean (leaves) | UK |
| SAS405 | WS55 x ? (parentals isolated from rose)† | |
| SAS56 | WS158 x ? (parentals isolated from grape)* | |
| B. fabae NCB1496 | Broad bean | Italy |
| B. fabae 145553 | Broad bean | UK |
| B. fabae 225852 | Broad bean | UK |
v = vacuma; t = transposa; u = uknown.
SAS56 and SAS405 are monoascosporic strains obtained from a spermatization of microconidi from different isolates, and thus one of the parental strains is unknown.
Figure 1.
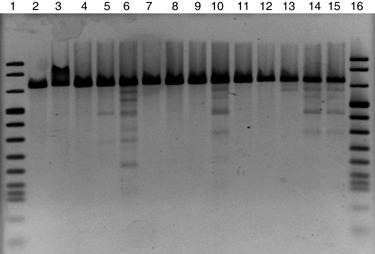
An example of polymorphism analysis of the DNA fragment PG5MD. Lanes 1 and 16, marker VI (Roche); Lane 2, G1‐WS252; Lane 3, G1‐149; Lane 4, G1‐RD1; Lane 5, G1‐RO18; Lane 6, G1‐PM10; Lane 7, G1‐138; Lane 8, G1‐WS92; Lane 9, G1‐B05.10; Lane 10, G1‐CI221; Lane 11, G1‐CI212; Lane 12, G1‐CI226; Lane 13, G1‐194; Lane 14, G1‐142649; Lane 15, G1‐T23. In lanes 5, 6, 10, 13, 14 and 15, the complex banding patterns are evidence of polymorphism.
The number of polymorphic sites was very different in the endoPG genes sequenced. Bcpg1 was the most diverse gene (Pi = 0.018), comprising 80 polymorphic sites (among which 40 were non‐synonymous sites, affecting 27 amino acid residues) carried by eight different haplotypes. Comparison with the results reported by Rowe and Kliebenstein (2007) showed that all but one of the polymorphisms scored in our European strain collection were present in their primarily Californian collection. Strong linkage disequilibrium (ZnS = 0.55) and absence of recombination (R per gene = 0.001; R between adjacent sites = 0) were estimated for Bcpg1, and each haplotype showed a distinct pattern of substitutions.
A similar variability was observed in the Bcpg2 gene sequence (Pi = 0.0097), with 62 polymorphic sites, including 30 non‐synonymous sites affecting 25 amino acids. There were 12 different haplotypes in Bcpg2, four of which were not present in the primarily Californian collection analysed by Rowe and Kliebenstein (2007), due to the presence of 14 additional polymorphic sites that were newly detected. Bcpg2 did not show the strong linkage disequilibrium and absence of recombination (ZnS = 0.16; R per gene = 2.7; R between adjacent sites = 0.0021) that were estimated for Bcpg1.
Bcpg3, Bcpg4 and Bcpg5 were substantially less variable than Bcpg1 and Bcpg2. For Bcpg3 we found the same diversity as reported by Rowe and Kliebenstein (2007), i.e. 11 polymorphic sites in nine haplotypes (Pi = 0.0016) for the European dataset vs. eight polymorphic sites in seven haplotypes (Pi = 0.0017) for the Californian one. Bcpg5 was more variable (Pi = 0.0041), with 28 polymorphic sites in nine haplotypes. Bcpg3 and Bcpg5 variability indices were comparable with those of the neutral evolving genes analysed by Fournier et al. (2005).
Conversely, sequence variation in the Bcpg4 gene among strains was extremely limited, with four haplotypes and an estimated Pi (= 0.0006) that was 30‐fold lower than for Bcpg1 (Table 2). None of the three polymorphisms detected affected the amino acid composition of the protein.
Table 2.
Population genetic indices.
| Locus | Length* | Pi† | Hd ‡ | CBI§ | ENC¶ | ZnS** | R per gene†† |
|---|---|---|---|---|---|---|---|
| Bcpg1 | 1055 | 0.018 | 0.555 | 0.682 | 30.8 | 0.55 | 0.001 |
| Bcpg2 | 1282 | 0.0097 | 0.784 | 0.422 | 50.1 | 0.16 | 2.7 |
| Bcpg3 | 1479 | 0.0016 | 0.800 | 0.493 | 42.03 | 0.10 | 16.1 |
| Bcpg4 | 1257 | 0.0006 | 0.654 | 0.552 | 37.91 | 0.05 | > 104 |
| Bcpg5 | 1370 | 0.0041 | 0.724 | 0.403 | 47.26 | 0.27 | 0.001 |
Nucleotides sequenced and analysed for each gene.
Polymorphism per polymorphic site.
Haplotype diversity.
Codon Bias Index.
Effective number of codons.
Statistic for linkage disequilibrium.
††Recombination parameter. See text for references.
Bcpg1 and Bcpg4 were also characterized by a codon bias higher than other endoPG genes (Table 2).
Preliminary investigations by sequencing fragments from randomly selected strains showed that Bcpg6 has a moderate variability, i.e. similar to Bcpg5 (unpublished results).
With reference to the genes that were analysed in both the populations sampled in Rowe and Kliebenstein (2007) and in this work, the F ST statistic was used to compare polymorphism levels. The low F ST values calculated for Bcpg1 (0.0836), Bcpg2 (0.0856) and Bcpg3 (0.00) indicated that there was not a significant population structure separating the Californian and the European populations.
Selection analysis
The three most variable endoPG genes were analysed for positive selection by comparison of synonymous (silent) and non‐synonymous (amino acid‐altering) substitutions, a common method for studying the models of DNA sequence evolution (Gillespie, 1991; Kimura, 1983; Ohta, 1993; Yang et al., 2000).
Bcpg1, Bcpg2 and Bcpg5 were tested for positive selection with the McDonald–Kreitman test, which is not affected by population demography (Nielsen, 2005). According to the results of this test, Bcpg1 and Bcpg2 have undergone significant (P < 0.05) diversifying selection whereas Bcpg5 has not (Table 3). Further support for the hypothesis of positive selection in these two endoPG genes was provided by the likelihood ratio test (LRT) with the Codeml program implemented in the Paml 3.15 package (Yang, 1997). This approach requires the evolutionary relationships between taxa to be represented by phylogenetic trees, and therefore recombination may alter the results, but our data indicated that recombination is absent in Bcpg1 and very modest in Bcpg2. As shown in Table 4, in both M1a vs. M2a and M7 vs. M8 model comparisons the 2Δl value for both genes resulted higher than χ2, thus supporting the hypothesis that Bcpg1 and Bcpg2 were under positive selection.
Table 3.
Results from the McDonald–Kreitman test.
| endoPG* | Substitutions | Fixed differences | Polymorphic sites | G value† | P‐value |
|---|---|---|---|---|---|
| BcPG1 | Synonymous | 5 | 48 | 10.701 | 0.001 < P < 0.01 |
| Non Synonymous | 16 | 27 | |||
| BcPG2 | Synonymous | 22 | 23 | 6.572 | 0.01 < P < 0.05 |
| Non Synonymous | 8 | 29 | |||
| BcPG5 | Synonymous | 2 | 16 | 1.206 | not significant |
| Non Synonymous | 2 | 4 |
B. fabae was chosen as second species for BcPG1 and BcPG5, B. calthae for BcPG2.
G value with Williams’ correction.
Table 4.
Results of the LRT analysis for BcPG1 and BcPG2.
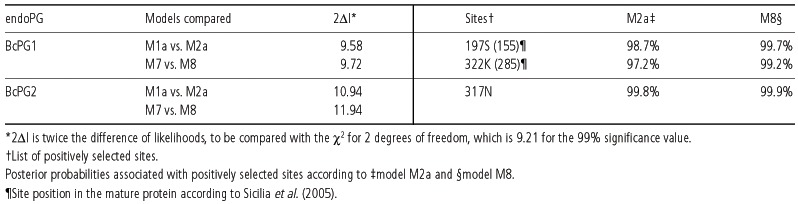
The sites under positive selection and their associated posterior probabilities, as calculated using the Bayes theorem, are reported in Table 4. According to the current protein structure model (Federici et al., 2001; Sicilia et al., 2005; van Santen et al., 1999), site 197S (corresponding to residue 155 in the model of Sicilia et al., 2005) of BcPG1 is located in a loop that forms the boundaries of the active site cleft. This site is 3 amino acids apart from the conserved HNTD motif, and that in the polygalacturonase of Fusarium moniliforme (FmPG) has been shown to play a major role in both substrate binding and recognition by PvPGIP2, the Polygalacturonase Inhibiting Protein 2 of Phaseolus vulgaris (Federici et al., 2001; Sicilia et al., 2005). A docking simulation predicted that the N‐terminal of BcPG1 is interacting with PvPGIP2 (Sicilia et al., 2005), and the active site is partially buried by the inhibitor. Therefore, in the hypothetical generalization of the FmPG‐PvPGIP2 interaction model, residue 197S of Bcpg1 would be in contact with the PGIP, while site 322 K (285), located at the C‐terminal end of the protein, would not.
No protein structure model is presently available for BcPG2, although a similar structure can be assumed, given the high similarity between BcPG1 and BcPG2 (72% identity, according to Wubben et al., 1999). With this further assumption, the positively selected site 317N, located at the C‐terminal end of the enzyme, would not play any role in the interaction with the substrate or the inhibitor protein.
The high similarity between BcPG1 and BcPG2 allowed their unambiguous alignment, revealing that the polymorphic sites do not occur randomly in these two proteins, but are preferably localized in particular regions. Figure 2a, reporting the result of a sliding window analysis of the co‐occurrence of polymorphisms in the two proteins, shows that such regions are most frequently and consistently found at the C‐terminal end. The alignment of Fig. 2b, relative to the C‐terminal half of the proteins, shows that in six instances the same position was found to be polymorphic in both proteins, and that the two positively selected sites in the C‐terminal of the protein are located only three positions apart from each other in their respective sequences. Moreover, BcPG5 also showed variability in this region, as a further indication of the significance of variability in this domain. Several other sites in BcPG2 were found with BEB posterior probabilities > 80%, although not statistically significant (site 277N, P = 83.3%; site 279K, P = 91%; site 283S, P = 87.3%; site 326M, P = 92.4%).
Figure 2.
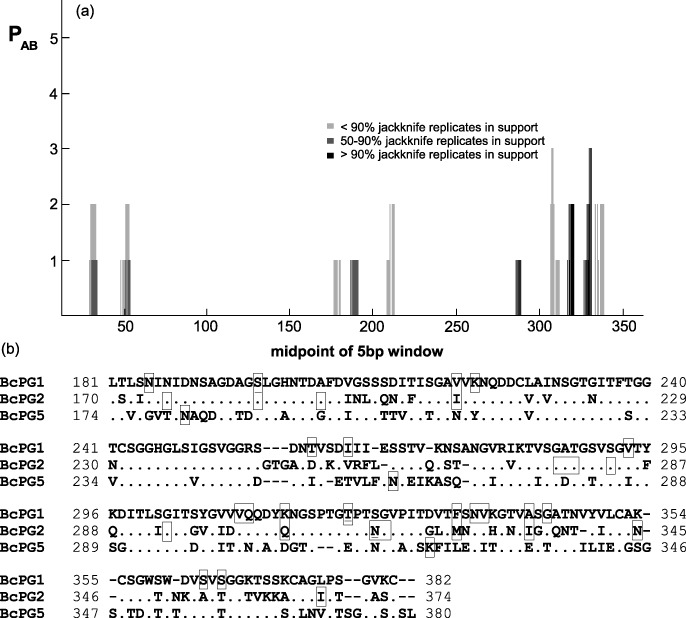
(a) Plot of a sliding window analysis of the co‐occurrence of polymorphic amino acid sites in BcPG1 and BcPG2. The darker the bar, the higher the percent of jackknife replicates in support. (b) Alignment of BcPG1, BcPG2 and BcPG5 from the proximity of AGSLG loop to the end. Boxes with filled lines represent polymorphic sites. Underlined residues indicate sites under positive selection.
The analysis of a dataset comprising the sequences obtained in this work and those obtained by Rowe and Kliebenstein (2007) confirmed the above results and pointed up site 53T in Bcpg2 as a further putative site under positive selection.
Phylogenetic analysis with Bcpg genes
The maximum parsimony trees derived from the analysis of Bcpg1–5 genes (3, 4) provided no obvious evidence of population partition according to the host of isolation, transposon content or geographical origin. In general, the genealogies were poorly congruent, revealing that the five endoPG genes are not linked.
Figure 3.
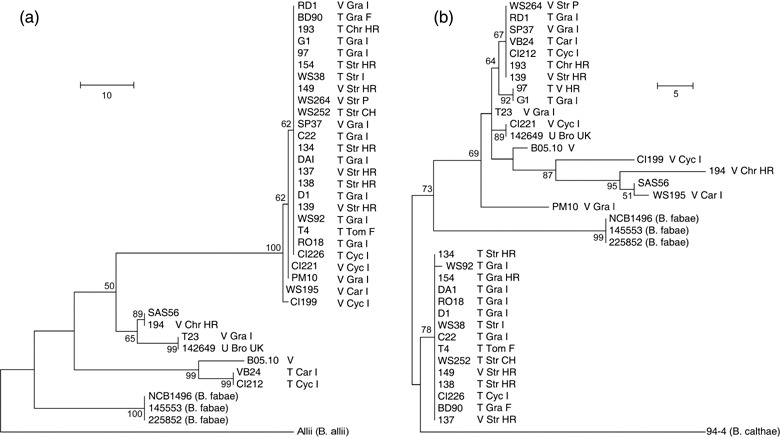
Maximum parsimony trees with bootstrap significance values supported by > 50% for PG1 (a) and PG2 (b) coding genes. Abbreviations used in the strain name labels are as follows. Transposon content: V = vacuma, T = transposa isolates. Host of isolation: Bro = broad bean, Car = carnation, Chr = chrysanthemum, Cyc = cyclamen, Gra = grapevine, Str = strawberry. Geographical origin: CH = Switzerland, F = France, HR = Croatia, I = Italy, P = Portugal and UK = United Kindom. Sequences for isolates SAS56, 94‐4 and Allii were retrieved from databases. The trees in (a) and (b) were rooted using B. allii strain Allii and B. calthae strain 94‐4, respectively, as outgroups.
Figure 4.
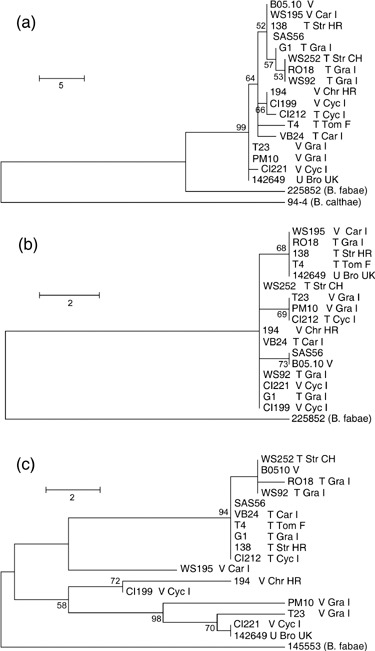
Maximum parsimony trees with bootstrap significance values supported by > 50% for minimum dataset. (a) PG3 coding genes, rooted using B. calthae 94‐4 as an outgroup; (b) PG4 coding genes, rooted using B. fabae 225852 as an outgroup; (c) PG5 coding genes, rooted using B. fabae 145553 as an outgroup. See the legend to Fig. 3 for abbreviations. Sequences for isolates SAS56 and 94‐4 were retrieved from databases.
As the selection models presumed for Bcpg1, Bcpg2 and Bcpg4 were not neutral, and the number of nucleic acid polymorphisms in Bcpg3 was small, the phylogenies based on these genes are expected to be inaccurate (Taylor et al., 2000). Conversely, the maximum parsimony analysis of Bcpg5 was informative and highlighted a clade of closely related strains that included strain T4 and all other transposa strains. This was reminiscent of the results of Fournier et al. (2005), who hypothesized two subpopulations in B. cinerea, named Group I and Group II, which include only vacuma strains or both vacuma and transposa, respectively. As strain T4 was the only strain in common between this study and that of Fournier et al. (2005), an assignment of our strains to either Group I or Group II was not possible. However, for a subjective subdivision into one group including T4 and relatives (WS252, B05.10, RO18, WS92, SAS56, VB24, T4, G1, 138, CI212, WS195) and another including the remaining strains (194, CI199, PM10, T23, CI221, 142649), the estimated values of F ST (0.64) and Nm (0.27) for Bcpg5 were similar to those calculated for Cyp51 (F ST 0.749 and Nm 0.17) by Fournier et al. (2005). Other genetic differentiation estimates such as Nei's χ2 (17, P < 0.05), H S (0.752, P < 0.05), K S* (1.180, P < 0.0001) and Z* (3.465, P < 0.001) were all significant, indicating that there was differentiation between groups. Thus, the results of the analysis of the genealogy derived from Bcpg5 were similar to those reported by Fournier et al. (2005) for Cyp51.
DISCUSSION
In the last 10 years, the work of Giraud et al. (1997, 1999), Albertini et al. (2002) and Fournier et al. (2003, 2005) has shown that B. cinerea is not a single, largely clonal population as was once thought. According to population genetics, two reproductively isolated groups (Group I and Group II) are distinct phylogenetic species. Attempts to use population genetics to establish possible host specificity or preference of B. cinerea subpopulations gave contrasting results, as the hypothesis of population subdivision has been corroborated by results obtained in Chile (Muñoz et al., 2002), but not by those obtained in California (Ma and Michailides, 2005) and Spain (Moyano et al., 2003).
In the present study, we carried out a sequence analysis of five pathogenicity‐associated genes coding for endoPGs in a collection of strains isolated primarily in Italy and Croatia, and found no congruent association between genetic diversity and either host of isolation, transposon content or geographical origin. However, most of the genes examined were either not evolving according to a neutral model or poorly variable, and therefore Bcpg5 was the only endoPG coding gene that could be used for phylogenetic inference. Its sequence analysis supported the latest results of Fournier et al. (2003, 2005) that B. cinerea comprises two reproductively isolated subpopulations, one of which was invaded by the transposons Boty and Flipper.
Despite the modest contribution to the clarification of the population structure in B. cinerea, analysis of the other genes highlighted a large difference in the frequency of nucleotide substitutions in the different endoPG genes, which ranged from very low to very high polymorphism, in the order Bcpg4, Bcpg3, Bcpg5, Bcpg2 and Bcpg1.
A major result of the work reported here is that Bcpg4 showed no diversification at the protein level, and almost negligible nucleotide variability. The exceptionally low rate of variation in the nucleotide sequence of Bcpg4 is consistent with background selection due to prolonged purifying selection against deleterious mutations. This is known to cause a reduction in the amount of genetic variation in the surrounding neutral sites, producing a region of low overall variability (Charlesworth et al., 1993). The strong selection against variation, and the codon bias that was detected in Bcpg4 but not in neutrally evolving endoPG genes support the notion that the product of this gene has a relevant function in the life cycle of B. cinerea.
Several studies provided evidence that the different endoPGs of B. cinerea differ in many important characteristics, such as specific enzymatic activity, optimum pH, processivity and necrotizing activity (Kars et al., 2005). By evaluating in vitro expression, it has been shown (Wubben et al., 2000) that Bcpg4 is induced by monogalacturonic acid, while other genes (with the exception of Bcpg6) are not; moreover Bcpg4 is expressed to high levels in planta during the late phases of infection (ten Have et al., 2001). Taken together with the results of our sequence study, this suggests that the enzymatic activity of BcPG4 is a relevant component of the complex mechanism that determines the fitness of this pathogen.
In contrast, balancing selection is the model that better explains the evolution of Bcpg1 and Bcpg2. The linkage disequilibrium and divergence of the haplotypes and the results of the McDonald–Kreitman test are consistent with this model. Balancing selection increases nucleotide diversity not only in selected loci but also in the surrounding genomic regions (Charlesworth, 2006), and this may explain the high polymorphism along the length of the entire genes. Evidence of balancing selection in these genes has already been pointed out by Rowe and Kliebenstein (2007).
Diversifying selection is characteristic of key genes which regulate the interaction of organisms with their environment. It also enables pathogens to maintain genetic diversity within their populations, often driven by selection imposed by host defence systems (Guttman et al., 2006). Cases have been reported in the past of surface‐exposed domains of parasites and immunogenic proteins of animal pathogens (Jiggins et al., 2002; Urwin et al., 2002).
Here we report for the pathogenicity‐associated genes Bcpg1 and Bcpg2 of B. cinerea the evidence of maintenance of high genotypic diversity for orthologous endoPG genes among strains of the same species. The hypothesis that the diversification of Bcpg1 and Bcpg2 is related to the involvement of these genes in the relationship with the host is in agreement with the evidence that both genes were shown to be required for full virulence by gene replacement studies (ten Have et al., 1998; Kars et al., 2005), while Bcpg3–6 were not (Kars, 2007).
The putative host component driving BcPG1 and BcPG2 adaptive selection may be an antagonistically interacting protein and in particular the polygalacturonase inhibiting protein (PGIP), which has been shown to bind directly to pathogen endoPGs. Stotz et al. (2000) provided evidence of positive selection for several PGIPs and highlighted amino acid residues in PGIP that should sustain adaptive substitutions. A similar positive selection acts in the counterpart of the arms race, the pathogen. Stotz et al. (2000) compared specific domains of non‐orthologous genes and identified evolutionarily selected sites in different regions of the protein.
In this work we show that two regions in particular are kept variable within specific populations. The location of only one of the sites with high probability of being positively selected in a region of putative interaction with PGIP suggests that the adaptive evolution of Bcpg1–2 is driven, beside the interaction with PGIP, by other mechanisms, such as the interaction with the enzyme substrate or other plant proteins.
The comparison of the analysis of variation in Bcpg1 and Bcpg2 in two independent populations (this work; Rowe and Kliebenstein, 2007) sheds further light on the evolution of these genes. Considering the independent sampling of the two populations, the uniform geographical distribution of the polymorphisms in Bcpg1 is striking. Moreover, the presence of ‘footprints’ of polymorphic silent sites near the selected ones and of conserved substitution patterns in B. fabae and B. alii suggest long‐term maintenance of variation due to host–pathogen coevolution (Charlesworth, 2006). These facts suggest that the highly diversified haplotypes in Bcpg1, originally evolved by diversifying positive selection, were maintained over time by long‐term balancing selection.
Similar sequence diversity was found in the analysis of Bcpg2, although the European population appeared to be more variable than the Californian one. The combined analysis of the sequences obtained in this work with those of Rowe and Kliebenstein (2007) showed for Bcpg2 less polymorphic sites but more haplotypes when compared with Bcpg1, with some geographical partition, and less pronounced linkage disequilibrium. A possible interpretation of these observations is that Bcpg1 is subject to a stronger selection pressure than Bcpg2. The notion that Bcpg1 is constitutively expressed (ten Have et al., 2001; Wubben et al., 2000) is consistent with this interpretation.
In conclusion, the information gathered in this work about the nucleotide diversity of B. cinerea endoPG genes supports the hypothesis of differential function of the genes. Accordingly, balancing selection in Bcpg1 and Bcpg2 aids the diversification of the gene products as a means to delay host recognition and reaction during the initial phases of the infection. BcPG4 acts in a later or different stage, when the full activity of the endopolygalacturonase machinery of the pathogen is required for efficient maceration. In this regard, the high conservation of the Bcpg4 nucleotide sequence detected in this study indicates a selection pressure that minimizes mutations that may compromise full enzyme activity. Thu,s on the one hand the pathogen appears to have an arsenal of PG genes that are conveniently used at the strain level modulating gene expression with time or environmental conditions, while, on the other, there is an arsenal of Bcpg1 and Bcpg2 variants that are available at the population level and used to optimize the confrontation with the host. In such a context, polyphagy may play a major role in maintaining diversity and may be regarded as the key to pathogen success.
EXPERIMENTAL PROCEDURES
Fungal growth and DNA extraction
The strains of Botrytis cinerea and B. fabae, listed in Table 1, were provided either by Drs F. Faretra and S. Pollastro (Università di Bari, Italy) or by CABI Bioscience (Egham, UK). Strains from Bari had been previously characterized for the presence of transposons (De Miccolis Angelini et al., 2003). For nucleic acid preparation, strains were grown for 7–8 days in Petri dishes of malt extract agar (Fluka) on a nitrocellulose membrane (Biorad Laboratories) at 25 °C in the dark. Extraction of total DNA from 0.2–0.3 g of mycelium was carried out according to the protocol described by Lecellier and Silar (1994). DNA samples were resuspended in 50 µL of sterile distilled water.
PCR amplifications and sequencing
Amplification of the nearly‐complete DNA sequence of Bcpg1/Bfpg1, Bcpg2/Bfpg2, Bcpg3/Bfpg3, Bcpg4/Bfpg4 and Bcpg5/Bfpg5 was carried out with the primers listed in Table 5, designed with the program Primer3 (Rozen and Skaletsky, 2000). The genes were sequenced to 91.9, 100, 91.75, 95.70 and 100 of their coding sequence, respectively. Three overlapping fragments were independently amplified for each Bcpg1/Bfpg1 and Bcpg2/Bfpg2 and two for Bcpg3/Bfpg3, Bcpg4/Bfpg4 and Bcpg5/Bfpg5. The PCR product sizes were 632 bp for PG1I, 363 bp for PG1III and 616 bp for PG1II; 560 bp for PG2II, 702 bp for PG2 and 297 bp for PG2III; 873 bp for PG3MD and 764 bp for PG3MD1; 851 bp for PG4MD and 687 bp for PG4MD1; and 746 bp for PG5MD and 777 bp for PG5MD1. The PCR reaction for fragment amplification prior to sequencing contained 1× buffer (Roche S.p.A., Monza, Italy), 400 ng of each primer, 80 µm dNTPs (Roche) and 2 U Taq Polymerase in a standard reaction of 100 µL. When the products had to be used for both screening of polymorphism and sequencing, PCRs were carried out in a 50‐µL reaction containing 1× buffer and 2 mm MgSO4 (Transgenomic Ltd, Hillington, Glasgow, UK), 20 pmol of each primer, 160 µm dNTPs (Roche) and 2.5 U Optimase Polymerase (Transgenomic).
Table 5.
Primers used for gene amplifications.
| Primer | Sequence 5′–3′ | Position* |
|---|---|---|
| Bcpg1/Bfpg1 | ||
| PG1IF | GCCAATATGGTTCAACTTCTC | 861–891 |
| PG1IR | GAACCAACATCGAAAGCATC | 1473–1492 |
| PG1IIIF | GGAAAGACCAAGCCAAAGTT | 1290–1309 |
| PG1IIIR | GGAGACAGTGTTGTCGGAAC | 1633–1652 |
| PG1IIF | CATTGACAACTCTGCTGGAG | 1427–1445 |
| PG1IIR | GGTTTGATGGTCAAGGTGTT | 2023–2042 |
| Bcpg2/Bfpg2 | ||
| PG2IIF | GCTCATCAATCAGCACAGTC | 263–283 |
| PG2IIR | TTCCGGCAGAAACATTAGAT | 804–823 |
| PG2F | GTGCATCCGGAAACAAGATT | 749–768 |
| PG2R | GTCAATCCGGTGATTGGAAC | 1440–1459 |
| PG2IIIF | GAGTTGGCATTGATGTTCA | 1375–1392 |
| PG2IIIR | TTATCAACCATTGTCAGTCG | 1652–1671 |
| Bcpg3/Bfpg3 | ||
| PG3MDF | CTTTCTGTGCTACCTACACCCAAG | 625–648 |
| PG3MDR | GTAAGTCCAGAGACGGTAAGACCT | 1475–1498 |
| PG3MD1F | GGATGTTGTCAACGGTGTTATCTC | 1397–1420 |
| PG3MD1R | GAAACTTTCCATGCCTCAGTTC | 2140–2161 |
| Bcpg4/Bfpg4 | ||
| PG4MDF | CCAGGCTCTCTTCTACCACTTGTTCT | 1396–1420 |
| PG4MDR | GCAGTCATCTTGGTTGTAGACAGT | 2224–2247 |
| PG4MD1F | CCTACATCACTGTTGATGCTTCC | 2068–2090 |
| PG4MD1R | GAGTATGTCTTACCACCAGTGACAG | 2740–2755 |
| Bcpg5/Bfpg5 | ||
| PG5MDF | TACACTCTCATCTCTTGGCTCATC | 1059–1082 |
| PG5MDR | CCACGCAGTCATCTTGATTGTA | 1784–1805 |
| PG5MD1F | GACACTGACAGTCTTGGTGCTAAC | 1703–1726 |
| PG5MD1R | CGCGAGTCTTACTTGAAAGTGTCTAC | 2455–2480 |
| PG5MD1R | CGCGAGTCTTACTTGAAAGTGTCTAC | 2455–2480 |
PCR conditions for Bcpg1/Bfpg1 and Bcpg2/Bfpg2 were as follows: 2 min at 95 °C; 35 cycles of 30 s for PG1III and PG2II (1 min for the others) at 95 °C, 1 min at the annealing temperature (54 °C for PG1III and PG2II; 56 °C for PG1I, PG1II, PG2III, PG2) and 1 min (1.5 min for PG1III and PG2II) at 72 °C; and finally 10 min at 72 °C. For the other genes cycling parameters were: 2 min at 94 °C; 30 cycles of 30 s at 94 °C, 45 s at the annealing temperature (59 °C for PG3MD1 and PG5MD; 61 °C for PG4MD and PG4MD1; 62 °C for PG3MD and PG5MD1) and 1.5 min at 72 °C; and finally 5 min at 72 °C.
Amplified DNA was routinely electrophoresed on 1% (w/v) agarose gels at 100 V for 1 h in Tris‐acetate buffer, stained with EtBr (0.5 µg/mL) and photographed under UV light.
PCR products were purified in vacuum filter plates (MANU 030 PCR, Millipore) and sequenced with a 96‐capillary sequencer (3730 DNA Analyzer, Applied Biosystems) according to standard procedures. Accession numbers of the sequences obtained in this work are AM491602, AM491603 (Bfpg1); AM491885–AM491916, AM941436 (Bcpg1); AM696308–AM696310 (Bfpg2); AM697717–AM697748 (Bcpg2); AM941434 (Bfpg3); AM697607–AM697609, AM697611–AM697613, AM697615–AM697619, AM697624, AM697627, AM697632‐AM697633, AM941433 (Bcpg3); AM941428 (Bfpg4) AM941411–AM941427 (Bcpg4); AM490853 (Bfpg5), AM491491, AM491495–AM491498, AM491500–AM491501, AM491508, AM491512, AM491514, AM491517–AM491521, AM941435 (Bcpg5). Additional sequences obtained from Genbank and used in this work were EF195810 (B. allii strain allii gene for PG1), EF195879 and EF195915 (B. calthae 94‐4 genes for PG2 and PG3), and U68715, U68716, U68717, U68719 and U68721 (B. cinerea strain SAS56 genes Bcpg1–6).
Screening for additional Bcpg genes
Possible missing members of the endoPG gene family were searched with the aid of the GenomeWalker kit (Clontech Laboratories, Palo Alto, CA), as detailed below. Four separate fungal DNA aliquots from each of the three strains B. cinerea SAS56, B. cinerea SAS 405 and B. fabae NCB1496 were thoroughly digested with four different restriction enzymes (EcoRV, DraI, PvuII, StuI) leaving blunt ends. Following digestion, each pool of DNA fragments was ligated to adaptors. For each fragment library, primary PCR amplifications were carried out using an adaptor primer provided in the kit and outer, gene‐specific primers [5′‐TG(CT)TC(ACT)GG(AT)GG(CT)CA(CT)GGTCT‐3′]. The primary PCR product was then diluted and used as a template for a secondary PCR amplification using a nested adaptor primer and nested gene‐specific primers [5′‐CA(CT)GGTCT(CT)TC(AC)(AG)T(CT)GG(AT)TC(CT)GT(CT)GG‐3′]. The resulting DNA amplimers were cloned. A total of 20 colonies were randomly picked and sequenced.
Polymorphism detection analysis
The Surveyor Mutation Detection Kit for Standard Gel Electrophoresis (Transgenomic Ltd) was used to scan polymorphisms among fungal strains. The kit is based on the amplification of DNA fragments from a reference and a test sample, their hybridization to create heteroduplexes with mismatches due to insertion/deletion or base substitution, digestion with a mismatch‐specific DNA endonuclease that cuts both strands of a DNA heteroduplex at the mismatch site, and analysis of the digestion products by agarose gel electrophoresis. The amplification protocols were as described above. Hybridization of at least 200 ng of each amplified sample was performed with a thermocycler with the following steps: 95 °C for 2 min, 95–85°C with a decrease of 2 °C/s, 85–25 °C with a decrease of 0.1 °C/s and hold at 4 °C. Two hundred to 400 ng of hybridized DNA was digested with Surveyor Nuclease S and Enhancer S (ratio 1:1) at 42 °C for 20 min and the reaction arrested with 1/10 of stop solution. Digestion products were analysed on 1% (w/v) agarose MS‐6 Metagel (Laboratories Conda, Torrejon de Ardoz, Madrid, Spain) gels at 75 V for 2.5 h in Tris‐borate buffer, stained and photographed as described.
Analysis of DNA sequences
Sequence manipulation and alignment were carried out using ClustalX version 1.83 (Thompson et al., 1997) and Bioedit version 7.0.5.3 (Hall, 2005). Sequence quality was checked thoroughly by inspection of all chromatograms with Finch Tv 1.4 (http://www.geospiza.com/finchtv/).
The program Mega version 3.1 (Kumar et al., 2004) was used to translate the nucleotide into amino acid sequences and to reconstruct phylogenies via maximum parsimony, using Close Neighbor Interchange with random addition trees for tree selection and a bootstrap test with 1000 replicates. DnaSP 4.10 (Rozas et al., 2003) was used to calculate genetic indices, estimated for the entire dataset of 32 strains, such as Hd (haplotype diversity) and Pi (polymorphism per polymorphic site) (Nei, 1987), the ZnS statistic for linkage disequilibrium (Kelly, 1997), recombination per gene (with R = 4Nr where N is the effective population size and r is the recombination rate per gene sequence; Hudson, 1987), CBI (codon bias index; Morton, 1993), ENC (effective number of codons; Wright, 1990) and McDonald–Kreitman test (McDonald and Kreitman, 1991). When comparing the results of this work with those presented by Rowe and Kliebenstein (2007), the genetic indices were newly estimated from a dataset comprising the regions in all sequences determined by Rowe and Kliebenstein (2007) common to our dataset.
Positive selection was tested by the McDonald–Kreitman test in coding regions only. This test is based on a comparison of synonymous and non‐synonymous (replacement) variation within and between species. In the absence of diversifying selection, the ratio of replacement to synonymous fixed substitutions (differences) between species should be the same as the ratio of non‐synonymous to synonymous polymorphisms within species. A G‐test of independence with Williams's correction is used to test the null hypothesis. To minimize the number of multiple mutations at single nucleotide sites we have chosen for comparison the related species B. fabae and B. calthae. The maximum‐likelihood implementation in the CODEML program, contained in the PAML 3.15 package (Yang, 1997), was used to corroborate the results of the McDonald–Kreitman test and to identify sites under positive selection. The program evaluates the ω ratio (ω = dN/dS, where dN and dS are defined as the number of non‐synonymous and synonymous substitutions per site, respectively; a value of ω > 1 means positive selection) which is an important indicator of selective pressure at the protein level. It specifies site models that allow ω to vary among sites, compares null models with models that assume adaptive selection, then computes the difference of their likelihoods and produces a significance value to be compared with a χ2 distribution. From UPGMA trees obtained from the analysis of Bcpg1 and Bcpg2 sequences, we ran several site models (M0, M1a, M2a, M3, M7 and M8), in accordance with the suggestion of the authors (Nielsen and Yang, 1998; Yang et al., 2000). Moreover, the Bayes empirical Bayes (BEB) calculation of posterior probabilities for site classes was implemented for models M2a and M8 (Yang et al., 2005). M1a and M2a were used to construct an LRT, M7 and M8 to construct another LRT, and M2a and M8 to identify sites under positive selection with BEB.
For analysis of the common occurrence of polymorphisms of BcPG1 and BcPG2 in the same regions, a sliding window approach was applied using a RUBY script (not published). Starting from an alignment of the amino acid sequences, for each window of five amino acids the index PAB = PA × PB was calculated, where PA and PB were the number of sites in BcPG1 and BcPG2, respectively, that were polymorphic. Thus, PAB had a non‐zero value only when both proteins showed polymorphism(s) in the window. The calculation was repeated for 100 half‐deleted jackknife replicate sets.
To compare the differentiation of groups defined by the phylogenetic analysis of Bcpg5 (this work) and of Cyp51 (Fournier et al., 2005), Nei's χ2 test (Nei, 1987) and H S, K*S and Z* statistics (Hudson et al., 1992a) were evaluated. Gene flow between groups was tested by estimating Nm (N is the effective population size and m the fraction of migrants per generation) based on F ST estimates (Hudson et al., 1992b).
ACKNOWLEDGEMENTS
We are grateful to Drs Franco Faretra and Stefania Pollastro for providing most of the strains used for this work, Dr Jan A. L. van Kan for sharing unpublished data, and Dr Francesco Favaron for helpful discussions. We acknowledge the Italian Ministry of University and Research for funding.
REFERENCES
- Albertini, C. , Thebaud, G. , Fournier, E. and Leroux, P. (2002) Eburicol 14 alpha‐demethylase gene (CYP51) polymorphism and speciation in Botrytis cinerea . Mycol. Res. 106, 1171–1178. [Google Scholar]
- Barrie, A. (1994) The importance of Botrytis cinerea as a storage rot of apple cv. Cox and pear. J. Agr. Sci. S17, 383–389. [Google Scholar]
- Charlesworth, B. , Morgan, M.T. and Charlesworth, D. (1993) The effect of deleterious mutations on neutral molecular variation. Genetics, 134, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D. (2006) Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Cruyssen, G. , De Meester, E. and Kamoen, O. (1994) Expression of polygalacturonases of Botrytis cinerea in vitro and in vivo. Med. Fac. Landbouwwet. Rijksuniv. Gent. 59, 895–905. [Google Scholar]
- De Miccolis Angelini, R.M. , Milicevic, T. , Natale, P. , Lepore, A. , De Guido, M.A. , Pollastro, S. , Cvjetkovic, B. and Faretra, F. (2003) Botryotinia fuckeliana isolates carrying different transposons show differential response to fungicides and localization on hosts plants. J. Plant Pathol. 85, 285. [Google Scholar]
- Federici, L. , Caprari, C. , Mattei, B. , Savino, C. , Di Matteo, A. , De Lorenzo, G. , Cervone, F. and Tsernoglou, D. (2001) Structural requirements of endopolygalacturonase for the interaction with PGIP (polygalacturonase‐inhibiting protein). Proc. Natl Acad. Sci. USA, 98, 13425–13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier, E. , Levis, C. , Fortini, D. , Leroux, P. , Giraud, T. and Brygoo, Y. (2003) Characterization of Bc‐hch, the Botrytis cinerea homolog of the Neurospora crassa het‐c vegetative incompatibility locus, and its use as a population marker. Mycologia, 95, 251–261. [PubMed] [Google Scholar]
- Fournier, E. , Giraud, T. , Albertini, C. and Brygoo, Y. (2005) Partition of the Botrytis cinerea complex in France using multiple gene genealogies. Mycologia, 97, 1251–1267. [DOI] [PubMed] [Google Scholar]
- Gillespie, J.H. (1991) The Causes of Molecular Evolution. Oxford: Oxford University Press. [Google Scholar]
- Giraud, T. , Fortini, D. , Levis, C. , Leroux, P. and Brygoo, Y. (1997) RFLP markers show genetic recombination in Botryotinia fuckeliana (Botrytis cinerea) and transposable elements reveal two sympatric species. Mol. Biol. Evol. 14, 1177–85. [DOI] [PubMed] [Google Scholar]
- Giraud, T. , Fortini, D. , Levis, C. , Lamarque, C. , Leroux, P. and Brygoo, K.L.a.Y. (1999) Two sibling species of the Botrytis cinerea complex, transposa and vacuma, are found in sympatry on numerous host plants. Phytopathology, 89, 967–973. [DOI] [PubMed] [Google Scholar]
- Guttman, D.S. , Gropp, S.J. , Morgan, R.L. and Wang, P.W. (2006) Diversifying selection drives the evolution of the type III secretion system pilus of Pseudomonas syringae . Mol. Biol. Evol. 23, 2342–2354. [DOI] [PubMed] [Google Scholar]
- Hall, T. (2005) BioEdit, http://www.mbio.ncsu.edu/BioEdit/bioedit.html.
- Ten Have, A. , Mulder, W. , Visser, J. and Van Kan, J.A.L. (1998) The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea . Mol. Plant–Microbe Interact. 11, 1009–1016. [DOI] [PubMed] [Google Scholar]
- Ten Have, A. , Breuil W.O., Wubben J.P., Visser, J. and Van Kan, J.A.L. (2001) Botrytis cinerea endopolygalacturonase genes are differentially expressed in various plant tissues. Fungal Genet Biol. 33, 97–105. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R. (1987) Estimating the recombination parameter of a finite population model without selection. Genet. Res. 50, 245–250. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R. , Boos, D.D. and Kaplan, N.L. (1992a) A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9, 198–151. [DOI] [PubMed] [Google Scholar]
- Hudson, S.S. , Slaktin, M. and Maddison, W.P. (1992b) Estimation of levels of gene flow from DNA sequence data. Genetics, 132, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins, F.M. , Hurst, G.D.D. and Yang, Z.H. (2002) Host‐symbiont conflicts: Positive selection on an outer membrane protein of parasitic but not mutualistic Rickettsiaceae. Mol. Biol. Evol. 19, 1341–1349. [DOI] [PubMed] [Google Scholar]
- Kars, I. (2007) The role of pectin degradation in pathogenesis of Botrytis cinerea . PhD Thesis, Wageningen University. [Google Scholar]
- Kars, I. , Krooshof, G.H. , Wagemakers, L. , Joosten, R. , Benen, J.A. and Van Kan, J.A.L. (2005) Necrotizing activity of five Botrytis cinerea endopolygalacturonases produced in Pichia pastoris . Plant J. 43, 213–225. [DOI] [PubMed] [Google Scholar]
- Kelly, J.K. (1997) A test of neutrality based on interlocus associations. Genetics, 146, 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M. (1983) The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press. [Google Scholar]
- Kumar, S. , Tamura, K. and Nei, M. (2004) MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 5, 150–163. [DOI] [PubMed] [Google Scholar]
- Lecellier, G. and Silar, P. (1994) Rapid methods for nucleic acids extraction from Petri dish‐grown mycelia. Curr. Genet. 25, 122–123. [DOI] [PubMed] [Google Scholar]
- Ma, Z. and Michailides, T.J. (2005) Genetic structure of Botrytis cinerea from different host plants in California. Plant Dis. 89, 1083–1089. [DOI] [PubMed] [Google Scholar]
- McDonald, J.H. and Kreitman, M. (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature, 351, 652–654. [DOI] [PubMed] [Google Scholar]
- Morton, B.R. (1993) Chloroplast DNA codon use: Evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 37, 273–280. [DOI] [PubMed] [Google Scholar]
- Moyano, C. , Alfonso, C. , Gallego, J. , Raposo, R. and Melgarejo, P. (2003) Comparison of RAPD and AFLP marker analysis as a means to study the genetic structure of Botrytis cinerea populations. Eur. J. Plant Pathol. 109, 515–522. [Google Scholar]
- Muñoz, G. , Hinrichsen, P. , Brygoo, Y. and Giraud, T. (2002) Genetic characterisation of Botrytis cinerea populations in Chile. Mycol. Res. 106, 594–601. [Google Scholar]
- Nei, M. (1987) Molecular Evolutionary Genetics. New York: Columbia University Press. [Google Scholar]
- Nielsen, R. (2005) Molecular signatures of natural selection. Ann. Rev. Genet. 39, 197–218. [DOI] [PubMed] [Google Scholar]
- Nielsen, R. and Yang, Z.H. (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV‐1 envelope gene. Genetics 148, 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T. (1993) The nearly neutral theory of molecular evolution. Ann. Rev. Ecol. Syst. 23, 263–286. [Google Scholar]
- Rowe, H.C. and Kliebenstein, D.J. (2007) Elevated genetic variation within virulence‐associated Botrytis cinerea polygalacturonase loci. Mol. Plant–Microbe Interact. 20, 1126–1137. [DOI] [PubMed] [Google Scholar]
- Rozas, J. , Sanchez‐DelBarrio, J.C. , Messeguer, X. and Rozas, R. (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 19, 2496–2497. [DOI] [PubMed] [Google Scholar]
- Rozen, S. and Skaletsky, H.J. (2000) Primer3 on the WWW for general users and for biologist programmers In: Bioinformatics Methods and Protocols: Methods in Molecular Biology, Vol. (Krawetz S. and Misener S., eds), pp. 365–386. Totowa, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Van Santen, Y. , Benen, J.A.E. , Schroter, K.H. , Kalk, K.H. , Armand, S. , Visser, J. and Dijkstra, B.W. (1999) 1.68‐angstrom crystal structure of endopolygalacturonase II from Aspergillus niger and identification of active site residues by site‐directed mutagenesis. J. Biol. Chem. 274, 30474–30480. [DOI] [PubMed] [Google Scholar]
- Sicilia, F. , Fernandez‐Recio, J. , Caprari, C. , De Lorenzo, G. , Tsernoglou, D. , Cervone, F. and Federici, L. (2005) The polygalacturonase‐inhibiting protein PGIP2 of Phaseolus vulgaris has evolved a mixed mode of inhibition of endopolygalacturonase PG1 of Botrytis cinerea . Plant Physiol. 139, 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz, H.U. , Bishop, J.G. , Bergmann, C.W. , Koch, M. , Albersheim, P. , Darvill, A.G. and Labavitch, J.M. (2000) Identification of target amino acids that affect interactions of fungal polygalacturonases and their plant inhibitors. Physiol. Mol. Plant Pathol. 56, 117–130. [Google Scholar]
- Taylor, J.W. , Jacobson, D.J. , Kroken, S. , Kasuga, T. , Geiser, D.M. , Hibbett, D.S. and Fisher, M.C. (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 31, 21–32. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D. , Gibson, T.J. , Plewniak, F. , Jeanmougin, F. and Higgins, D.G. (1997) The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin, R. , Holmes, E.C. , Fox, A.J. , Derrick, J.P. and Maiden, M.C.J. (2002) Phylogenetic evidence for frequent positive selection and recombination in the meningococcal surface antigen PorB. Mol. Biol. Evol. 19, 1686–1694. [DOI] [PubMed] [Google Scholar]
- Williamson, B. , Tudzynski, B. , Tudzynski, P. , Van Kan, J.A.L. (2007) Botrytis cinerea: the cause of grey mould disease. Mol. Plant Pathol. 8, 561–580. [DOI] [PubMed] [Google Scholar]
- Wright, F. (1990) The ‘effective number of codons’ used in a gene. Gene, 87, 23–29. [DOI] [PubMed] [Google Scholar]
- Wubben, J.P. , Mulder, W. , Ten Have, A. , Van Kan, J.A.L. and Visser, J. (1999) Cloning and partial characterization of endopolygalacturonase genes from Botrytis cinerea . Appl. Environ. Microbiol. 65, 1596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wubben, J.P. , Ten Have, A. , Van Kan, J.A.L. and Visser, J. (2000) Regulation of endopolygalacturonase gene expression in Botrytis cinerea by galacturonic acid, ambient pH and carbon catabolite repression. Curr. Genet. 37, 152–157. [DOI] [PubMed] [Google Scholar]
- Yang, Z. (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556. [DOI] [PubMed] [Google Scholar]
- Yang, Z.H. , Nielsen, R. , Goldman, N. and Pedersen, A.M.K. (2000) Codon‐substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.H. , Wong, W.S.W. and Nielsen, R. (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. [DOI] [PubMed] [Google Scholar]