

SUMMARY
Rhizomania is one of the most devastating sugar beet diseases. It is caused by Beet necrotic yellow vein virus (BNYVV), which induces abnormal rootlet proliferation. To understand better the physiological and molecular basis of the disorder, transcriptome analysis was performed by restriction fragment differential display polymerase chain reaction (RFDD‐PCR), which provided differential gene expression profiles between non‐infected and infected sugar beet roots. Two distinct viral isolates were used to detect specific or general virus‐induced genes. Differentially expressed genes were selected and identified by sequence analysis, followed by reverse Northern and reverse transcriptase PCR experiments. These latter analyses of different plants (Beta vulgaris and Beta macrocarpa) infected under distinct standardized conditions revealed specific and variable expressions. Candidate genes were linked to cell development, metabolism, defence signalling and oxidative stress. In addition, the expression of already characterized genes linked to defence response (pathogenesis‐related protein genes), auxin signalling and cell elongation was also studied to further examine some aspects of the disease. Differential expression was retrieved in both B. vulgaris and B. macrocarpa. However, some candidate genes were found to be deregulated in only one plant species, suggesting differential response to BNYVV or specific responses to the BNYVV vector.
INTRODUCTION
Comparative transcript profiling provides essential information that can be used to explain a large range of biological differences between organisms, tissues, developmental processes (Whitelaw et al., 2000), cell fate (Whitelaw et al., 2002) and pathological situations (Liang and Pardee, 2003). A large range of methods can be used for transcript comparisons, from subtractive hybridization (Diatchenko et al., 1996) to produce expression libraries, to DNA microarrays to profile global changes (Duggan et al., 1999). The latter approach is applied in particular to completely sequenced organisms but is quite restrictive when applied to partially characterized genomes. Tools have been developed to assess comparative analysis of as yet uncharacterized genomes. Within such methods, restriction fragment differential display polymerase chain reaction (RFDD‐PCR) is of particular interest because this method permits the comparison of more than two samples and allows the identification of up‐ and down‐regulated genes (Gravesen et al., 2000). It is derived from the RNA differential display method (Liang and Pardee, 1992; Liang et al., 1992) but is more informative as it mainly provides nucleotide sequences of mRNA coding regions rather than just 3′ untranslated regions.
Rhizomania is a devastating viral disease of sugar beet (Beta vulgaris) and is present almost worldwide. The disease is induced by the phytovirus Beet necrotic yellow vein virus (BNYVV), which is transmitted by the soil‐borne vector Polymyxa betae in a persistent manner because of resting spores retaining the virus. Rhizomania studies have been extensively focused on the characterization of the function of virally encoded genes (Chiba et al., 2008; Klein et al., 2007; Link et al., 2005; Richards and Tamada, 1992; Tamada, 1999; Vetter et al., 2004) but little is known about the cellular activities that are modulated during the disease and how root proliferation is initiated. Histological characterization (Pollini and Giunchedi, 1989) and hormonal changes (Pollini et al., 1990) have been reported without information about the intimate molecular mechanisms present at the basis of such physiological alterations. Analyses of sugar beet infected by fungi or viruses have permitted the characterization of the pathogenesis‐related protein 2 gene (PR‐2) as well as the PR‐3 gene, which are induced during sugar beet infections or stresses (Burketova et al., 2003; Coutts et al., 1994; Fleming et al., 1991; Nielsen et al., 1993, 1996, 1994). As sugar beet is far from being a model plant, complete characterization of its genome will not be available for many years. Therefore, we used a differential display strategy to compare non‐infected and BNYVV‐infected root systems from susceptible B. vulgaris plants and characterize some differentially expressed genes. We also used Beta macrocarpa in our studies. Unlike B. vulgaris, B. macrocarpa leaves can be rubinoculated with BNYVV, thereby avoiding variation associated with the presence of the vector P. betae. B‐type and P‐type isolates that respectively contain four and five RNA species were used to infect plants. Subsequent gene expression profiling of selected genes was performed using reverse Northern and reverse transcriptase PCR (RT‐PCR) on distinct plant materials. These analyses were also performed to visualize the expression of other well‐characterized genes linked to defence response (e.g. PR‐protein genes) or related to sugar beet hormonal changes (e.g. auxin signalling) and root elongation (e.g. extensin gene).
RESULTS
Identification of differentially expressed genes by RFDD‐PCR
In order to study B. vulgaris root transcript profiles in response to typically BNYVV natural infections mediated by the P. betae vector, healthy plants (H) cultivated in soil mixtures containing aviruliferous P. betae were compared with infected plants grown in soil with viruliferous P. betae strains harbouring B‐type (A2) or P‐type (A4) BNYVV isolates. Two distinct viral isolates were used in this study in order to discriminate between genes generally induced in response to all types of BNYVV and genes specifically modulated by one viral isolate. Characteristic symptoms of BNYVV infection were detected on leaves (chlorosis and dwarfism) and on roots (root system disorders and rootlet proliferation) after 8 weeks. Total RNAs from roots of infected and healthy plants were extracted and first analysed by Northern blotting for the presence of viral RNA 1, 2, 3 and 5 (Fig. 1). Total RNAs from Chenopodium quinoa leaves mechanically inoculated with a BNYVV P‐type isolate was used as a viral control (C, Fig. 1). RNA 4 detection was omitted to better distinguish RNA 5, as RNA 4 length (1467 nt) is very close to RNA 5 length (1350 nt). RNA 4 detection was without interest here as this RNA species is present in all naturally infected plant because it is involved in virus transmission and pathogenesis (Rahim et al., 2007; Tamada and Abe, 1989). Our results confirmed that A2 and A4 sets of roots were respectively infected by B‐type and P‐type BNYVV isolates (A2, A4, Fig. 1). No BNYVV infection was detected on plants grown in aviruliferous soil (H, Fig. 1). Identical amounts of total RNAs from healthy (H) and infected (A2 and A4) root samples were analysed by RFDD‐PCR in duplicates with nine tested primer sets (0‐extension primers and displayPROBE primers). Although the majority of bands revealed no differential expression profiles between non‐infected and BNYVV‐infected plants, discriminative bands were also found and selected (Fig. 2). Reproducibility was estimated by comparison of the duplicated experiments. In total, 53 differentially expressed bands were originally isolated from RFDD‐PCR gels, and 42 bands were successfully re‐amplified and yielded colonies after transformation. The 42 selected bands were sequenced, 14 of which consisted of BNYVV viral RNA sequence (e.g. 2b, 4b, 6b, 10b, 14b, Fig. 2), thus leaving 28 bands of interest providing 50 sequences valid for further analysis. The 50 sequences were compared against non‐redundant databases (NCBI) using tBlastx and against the Arabidopsis thaliana database (TIGR) using WU‐Blast 2.0. On the basis of sequence identity, putative function and GO annotations were assigned to each sequenced cDNA. The results are summarized in Table 1.
Figure 1.
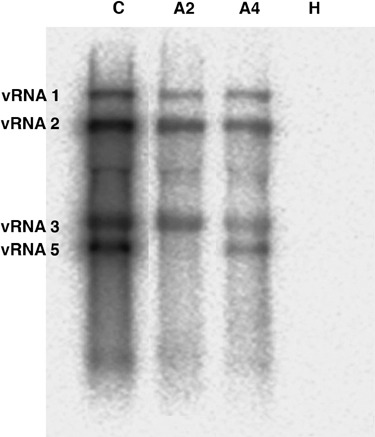
BNYVV RNA detection within total RNAs from Beta vulgaris roots infected by B‐type (A2) or P‐type (A4) BNYVV viral isolates. Total RNAs from non‐infected roots (H) were also tested. Viral RNA 1, 2, 3 and 5 were detected by Northern blotting with specific riboprobes. RNA 4 probe was omitted to allow the visualization of the RNA 5 species. Positions of viral RNAs were determined with the detection of viral RNAs within total RNAs from Chenopodium quinoa leaf lesions induced by a P‐type BNYVV isolate (C). Positions of viral species are indicated on the left.
Figure 2.

Autoradiogram obtained from RFDD‐PCR analysis showing the amplification products of RNAs from Beta vulgaris healthy roots (H) used as reference, and from roots infected by a B‐type BNYVV isolate (A2) or a P‐type BNYVV isolate (A4). Amplification reactions were made in duplicates (nos. 1 and 2) using the labelled 0‐extension primer in combination with one of the three displayPROBE primers (EU25, EU26 and EU27) tested in this example. Differentially expressed bands (detected here in infected samples) are indicated by dots and FDD name reported in Table 1. Names in italic correspond to BNYVV amplified sequences. Numbers on the left refer to the size of PCR fragments (base pairs).
Table 1.
Candidate transcripts detected by RFDD‐PCR analyses and differentially expressed between healthy (H) and BNYVV‐infected (A2 or A4) sugar beet roots
| No.* | FDD† | H‡ | A2 | A4 | Name§ | Size (bp)¶ | Predicted function (Organism)** | Accession no.†† | AT locus‡‡ | GO terms§§ |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1a | x | x | 18V15 | 584 | No significant similarity | — | |||
| 2 | 2a | x | 18V21 | 464 | Cytosolic heat shock 70 protein (Spinacia oleracea) | AF034618 | At3g12580 | Pro, RS, Rox | ||
| 3 | 18V25 | 455 | Phospholipase D alpha 1 (Arabidopsis thaliana) | NM_112443 | At3g15730 | Met, ST | ||||
| 4 | 5a | x | x | 18V56 | 262 | Glutathione S transferase (Arabidopsis thaliana) | AF349527 | At4g02520 | RS, Rox | |
| 5 | 6a | x | 18V61 | 185 | 26S rRNA (Chenopodium rubrum) | AM263434 | ||||
| 6 | 18V62 | 181 | Zinc‐containing alcohol dehydrogenase (Ralstonia eutropha) | YP_297879 | ||||||
| 7 | 18V63 | 181 | 23S rRNA (Pseudomonas stutzeri) | X87289 | ||||||
| 8 | 7a | x | 19S12 | 233 | Large‐subunit rRNA (Thaumatomonas sp. TMT002) | DQ980477 | ||||
| 9 | 19S13 | 874 | Large‐subunit rRNA (Thaumatomonas sp. TMT002) | DQ980477 | ||||||
| 10 | 8a | x | x | 19V12 | 61 | No significant similarity | — | |||
| 11 | 13a | x | x | 19V65 | 266 | Ubiquitin UBQ10 (Arabidopsis thaliana) | DQ793132 | At4g05320 | Pro, RS | |
| 12 | 14a | x | 19V72 | 196 | No significant similarity | — | ||||
| 13 | 17a | x | x | 20V16 | 576 | 23S rRNA gene (Rhizobium leguminosarum) | CP000133 | |||
| 14 | 18a | x | 20V21 | 441 | Amino acid transporter AAP4 (Arabidopsis thaliana) | NM_125780 | At5g63850 | Trans, RS | ||
| 15 | 20V26 | 443 | Genomic DNA, Os12g0190400 gene (Oryza sativa) | AP008218 | ||||||
| 16 | 19a | x | 20V34 | 369 | Unknown protein (Arabidopsis thaliana) | NM_105087 | At1g64140 | Un | ||
| 17 | 20V36 | 369 | Plasma membrane H+‐ATPase (Triticum aestivum) | AY829002 | At2g18960 | Trans, RS, ST | ||||
| 18 | 20a | x | x | 20V46 | 260 | No significant similarity | — | |||
| 19 | 5b | x | 25V51 | 313 | Carbamoyl‐phosphate synthase (Arabidopsis thaliana) | NM_102730 | At1g29900 | Met | ||
| 20 | 25V55 | 313 | FLS2 Leu‐rich repeat protein kinase (Arabidopsis thaliana) | NM_124003 | At5g46330 | ST, RS | ||||
| 21 | 7b | x | x | 25V75 | 213 | 2‐Dehydro‐3‐deoxyphosphoheptonate aldolase (Arabidopsis thaliana) | NM_120162 | At4g39980 | Met, RS | |
| 22 | 9b | x | 26V12 | 469 | Germin‐like protein (Mesembryanthemum crystallinum) | M93041 | At1g09560 | RS | ||
| 23 | 26V13 | 483 | LBVV‐pol gene for L protein (Lettuce big‐vein virus) | AB075039 | ||||||
| 24 | 11b | x | 26V31 | 365 | Large‐subunit rRNA (Thaumatomonas sp. TMT002) | DQ980477 | ||||
| 25 | 12b | x | x | 26V46 | 255 | Glucose‐6‐phosphate 1‐dehydrogenase (Arabidopsis thaliana) | AK227918 | At5g40760 | Met | |
| 26 | 13b | x | 26V52 | 235 | 26S ribosomal RNA (Maytenus fournieri) | AF222393 | ||||
| 27 | 16b | x | 27V21 | 89 | Genomic DNA (Staphylococcus aureus phage phi13) | AF424783 | ||||
| 28 | 27V24 | 700 | 23S rRNA (Acidovorax avenae subsp. citrulli) | CP000512 | ||||||
| 29 | 27V26 | 700 | Peroxidase, per2 gene (Beta vulgaris) | AJ276131 | At5g19890 | RS, Rox | ||||
| 30 | 17b | x | x | 27V31 | 585 | Microtubule‐associated protein (Trypanosoma cruzi) | AF158722 | |||
| 31 | 27V33 | 585 | 16S rRNA (Streptomyces glomeroaurantiacus) | AB249983 | ||||||
| 32 | 27V36 | 585 | 16S rRNA (Streptomyces sp. 4_C7_41) | EF540496 | ||||||
| 33 | 18b | x | x | 27V42 | 575 | 16S rRNA (Streptomyces sp. 4_C7_41) | EF540496 | |||
| 34 | 27V44 | 575 | 16S rRNA (Streptomyces sp. 4_C7_41) | EF540496 | ||||||
| 35 | 19b | x | 27V51 | 414 | 26S rRNA (Afrostyrax sp. Cheek 5007) | AF479115 | ||||
| 36 | 27V52 | 414 | Chloroplast DNA (Cyanidioschyzon merolae strain DBV201) | AY286123 | ||||||
| 37 | 20b | x | 27V61 | 302 | 26S rRNA (Medusagyne oppositifolia) | AF479120 | ||||
| 38 | 27V62 | 302 | l‐allo‐threonine aldolase (Malus x domestica) | AY742299 | At1g08630 | Met | ||||
| 39 | 1c | x | 22V12 | 368 | 18S ribosomal RNA (Allantion sp. ATCC 50734) | AF411265 | ||||
| 40 | 3c | x | x | 23V11 | 594 | TIP1 Palmitoyltransferase (Arabidopsis thaliana) | NM_122042 | At5g20350 | Cell, RS | |
| 41 | 23V13 | 52 | No significant similarity | — | ||||||
| 42 | 23V16 | 584 | 16S rRNA (Streptomyces sp. 4_C7_41) | EF540496 | ||||||
| 43 | 4c | x | x | 23V21 | 489 | No significant similarity | — | |||
| 44 | 23V22 | 494 | Phenylalanine ammonia lyase (Beta vulgaris) | AJ810175 | At2g37040 | Met, RS, Rox | ||||
| 45 | 23V24 | 492 | U2 snRNP auxiliary factor large subunit (Arabidopsis thaliana) | NM_104771 | At1g60900 | Rna | ||||
| 46 | 12c | x | 24S21 | 406 | 26S rRNA (Portulaca grandiflora) | AF479093 | ||||
| 47 | 24S22 | 406 | BvMS1 Methionine synthase (Beta vulgaris) | AB221011 | At5g17920 | Met | ||||
| 48 | 24S23 | 406 | No significant similarity | — | ||||||
| 49 | 24S24 | 338 | 26S rRNA (Nicotiana tabacum) | AF479172 | ||||||
| 50 | 10c | x | 24V31 | 134 | Mitochondrial genomic DNA (Beta vulgaris) | BA000024 |
Identifier of each cDNA.
Number of the band detected on denaturing gels.
Expression profile: x in the H column refers to a repressed gene during the infection, and ‘x’ in the A2 and/or A4 columns refers to up‐regulated genes.
Name of the pGEM‐T cloned sequence.
Size of the cloned cDNA.
The best Tblastx result with E value below 1 e−3.
Accession number of the Tblastx homologue.
The best‐matched Arabidopsis thaliana (AT) orthologue.
If the AT orthologue is identified, global gene function as described by the Gene Ontology Consortium. GO terms: response to abiotic or biotic stimulus (RS), response to oxidative stress (Rox), RNA binding and processing (Rna), protein binding, folding and modification (Pro), metabolism (Met), cell organization and biogenesis (Cell), signal transduction (ST), transport (Trans), unknown process (Un).
B. vulgaris candidate gene expression patterns
Differential display assays are known to produce bands consisting of more than one sequence, thus providing false‐positive clones among true differentially expressed candidate genes. The 50 cDNAs isolated previously were analysed by reverse Northern procedures both to validate the result of the RFDD‐PCR experiment and to distinguish between differentially expressed sequences and contaminating sequences (Fig. 3). This method permitted the multiple screening of candidates, which requires small amounts of RNA to produce only two labelled cDNA probes. PCR products were transferred in duplicate onto nylon membranes, one of which was hybridized with cDNA probes obtained from healthy roots (H panels, Fig. 3), and the other with cDNA probes obtained from P‐type BNYVV‐infected roots (A4 panels, Fig. 3). The detection of the viral control only with A4 cDNA probes validated the experiment (V, Fig. 3). Signals were integrated and compared between healthy and infected tissues. Using such a method for the confirmation of transcript profiling, one needs to consider the possible absence of detection due to a low abundance of some mRNAs and/or the low representation of full‐length radiolabelled cDNA that may not hybridize on the sequence amplified by RFDD‐PCR. Twenty clones were not or were only poorly detected (nos. 1, 6, 8, 12, 13, 14, 20, 26, 31–34, 36, 40–42, 45–47, 49; Fig. 3, see also Table 1 for details) whereas five clones presented no differential transcript accumulation (nos. 3, 25, 43, 44, 50). For most of the remaining clones (18/25) the nature of the variation (increase, decrease) was identical between cDNA‐AFLP and reverse Northern blotting. Transcript profiling was confirmed for cDNAs encoding a ubiquitin (no. 11), a plasma membrane H+‐ATPase (no. 17), a germin‐like (GLP, no. 22), a peroxidase (no. 29) and metabolism genes (nos. 19, 38), as well as sequences with unknown function (nos. 10, 16, 18), many ribosomal RNAs (nos. 5, 7, 24, 28, 35, 37) and most intriguingly virus, phage or protozoan sequences (nos. 23, 27, 30). For the last clones (7/25), the nature of the variation differed between the two procedures. Differences between RFDD‐PCR results and reverse Northern analysis were detected for genes encoding a heat shock 70 protein (HSC70, no. 2), a glutathione S transferase (GST, no. 4), a 2‐dehydro‐3‐deoxyphosphoheptonate aldolase (no. 21), unknown function sequences (nos. 15, 48) and ribosomal RNAs (nos. 9, 39). RNAs from prokaryotes or unrelated species that may differ within the soils were cloned as well. A Varicosavirus rhabdoviral‐related sequence (no. 23) might be explained by the presence of Olpidium brassicae in the soil, but no infection was detected by Northern blots (data not shown). Such sequences with no plant sequence identities (A. thaliana Locus, Table 1) reflected plant RNA contaminations with soil microorganism RNAs.
Figure 3.
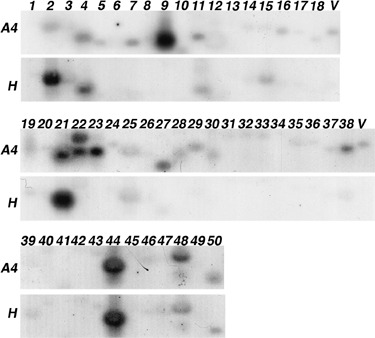
Expression patterns of genes selected after the initial characterization. Reverse Northern analysis consisted of replicate membranes harbouring PCR products of cDNA clones hybridized with 32P‐labelled cDNA probes derived from Beta vulgaris healthy (H panels) or infected (A4 panels) root mRNAs. The identifier of each cDNA indicated on the top is also reported in Table 1. A viral sequence (V) was used as a positive control for hybridizations.
Expression patterns of candidate genes in response to early BNYVV infection
To analyse the early expression of candidate cDNAs in response to premature BNYVV root infection, new complementary approaches were performed on the cultivated sugar beets (B. vulgaris ssp. vulgaris) and on the related wild species (Beta macrocarpa). In the first experiment, B. vulgaris healthy plantlets (H) cultivated in sand mixed with aviruliferous P. betae soil were compared with infected plantlets (B) grown in sand mixed with viruliferous P. betae soil harbouring B‐type BNYVV isolates. This bioassay, developed in the SESVanderHave laboratory, was the method of choice to provide standardized and rapid infections of young root systems. TAS‐ELISA assays were performed to quantify BNYVV multiplication in roots 7, 14, 21 and 28 days after infection (Fig. 4a). This analysis allowed us to detect an initial infection as soon as 7 days post‐infection (dpi), and an optimal infection at 14 dpi (B, Fig. 4a). These two time points were selected for further transcript analyses. In the second experiment, B. macrocarpa control plantlets (H) were compared with systemic infected plantlets (I) obtained after leaf infection by a B‐type BNYVV inoculum. Indeed, B. macrocarpa was chosen as it can be systemically infected after leaf rub inoculation without the need of the vector P. betae. Hence, the B. macrocarpa/BNYVV system allows for verification of our findings and for the selection of genes that are specifically associated with BNYVV infection from those that may be associated with the presence of P. betae or other microbes that were present in BNYVV/P. betae‐exposed versus non‐exposed B. vulgaris. BNYVV leaf rub inoculation of B. macrocarpa led to successful infection of the roots (Fig. 5a). These infected roots were harvested as soon as visible systemic symptoms (leaf blisters and chlorosis) appeared onto non‐inoculated leaves (approximately 13 dpi). Entire root systems from infected and non‐infected plants were submitted to immunoprinting to detect and precisely localize BNYVV infection within roots (Fig. 5a). This method distinguished between infected tissues (i) and non‐infected tissues (h) on the unique early‐infected root system (I, Fig. 5a). The control plant served only to determine the threshold chemiluminescence signal level (H, Fig. 5a). These specific tissues were extracted from the corresponding frozen roots and total RNAs were prepared for subsequent analyses. This method allowed us to study gene expression by comparison of infected and non‐infected samples from the same individual.
Figure 4.
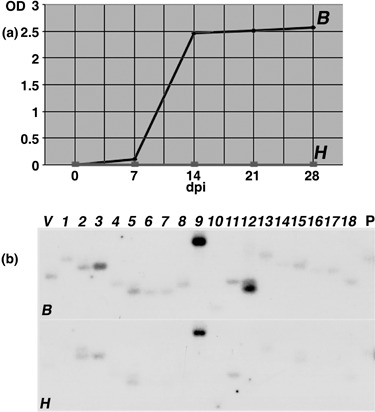
(a) BNYVV detection in Beta vulgaris roots infected by a B‐type BNYVV isolate (B) using standardized conditions (bioassay, SESVanderHave laboratory) or in control roots (H). Viral contents were monitored by ELISA analysis for BNYVV CP protein detection. Results are expressed as an optical density value (OD). (b) Differential expression of RFDD‐PCR candidate genes after B. vulgaris standardized infections. Reverse Northern hybridizations were performed with 32P‐labelled cDNA probes derived from B. vulgaris healthy (H) or infected (B) root RNAs extracted 14 days after treatment. The identifier of each cDNA indicated on the top is also reported in Table 1. A viral sequence (V) and a B. vulgaris beta‐1,3‐glucanase (accession no. BQ591809) PR‐2 sequence (P) were used as controls.
Figure 5.

(a) BNYVV localization in Beta macrocarpa roots after plant systemic infection. Viral contents of B. macrocarpa roots harvested 13 days after infection (I) or control roots (H) were monitored by immunoprinting and permitted the localization of infected tissues (i) and non‐infected tissues (h) selected for subsequent total RNA extraction. Background on control roots (H) corresponds to the processing of chemiluminescent reagent by cellular peroxidases. (b) Differential expression of RFDD‐PCR genes after B. macrocarpa systemic infection. Reverse Northern hybridizations were performed with cDNA probes derived from B. macrocarpa healthy (h) or infected (i) root RNAs. Identifiers of cDNA clones are indicated on the top. A viral sequence (V) was used as a positive control, as well as a B. vulgaris beta‐1,3‐glucanase PR‐2 (P) sequence (accession no. BQ591809) and a ubiquitin (U) sequence (accession no. BQ583989).
Expression profiles performed by reverse Northern blots were restricted to the set of 18 cDNA candidates (nos. 1–18, Table 1), due to the low amounts of total RNAs. Reverse Northern hybridizations consisted of comparisons between non‐infected and infected roots of B. vulgaris (Fig. 4b) and B. macrocarpa (Fig. 5b) RNA extracts harvested respectively at 14 and 13 dpi. The detection of the viral control only with infected cDNA probes validated the experiment (V, 4, 5). In addition, a B. vulgaris beta‐1,3‐glucanase sequence (PR‐2, accession no. BQ591809), known to be induced by plant stress and infection, was added (P, 4, 5), and also the B. vulgaris housekeeping ubiquitin sequence (accession no. BQ583989) to confirm ubiquitin gene activation (no. 11, Table 1) in B. macrocarpa (U, Fig. 5B). Signals were integrated and compared between healthy and infected tissues, and between new infection procedures and previous B. vulgaris root infections (3, 4, 5). Reverse Northern experiments confirmed RFDD‐PCR analysis based on the induction in both infected species of 23S rRNAs (nos. 7 and 13), unknown protein‐encoding genes (nos. 10, 12, 15, 16 and 18), and genes encoding a ubiquitin (no. 11), an amino acid permease (AAP, no. 14) and an H+‐ATPase (no. 17). The GST (no. 4) and unknown plant function sequences (nos. 1 and 6) were found to be induced only in the B. vulgaris bioassay experiment. For the remaining clones, the nature of the variation differed between the two species, showing up‐regulation in B. vulgaris (Fig. 4b) but repression in B. macrocarpa (Fig. 5b). This expression pattern was observed for HSC70 (no. 2), the phospholipase D gene (PLD, no. 3) and ribosomal RNAs (nos. 5, 8 and 9).
We extended some of our analyses only for cDNAs with putative function of interest. Semi‐quantitative RT‐PCR was performed on total RNAs from infected (B) and control (H) B. vulgaris rootlets collected at 7 dpi and from infected (i) and healthy (h) tissues extracted on the same B. macrocarpa plant at 13 dpi (Fig. 6). Signals were quantified and expression ratio calculated between infected and non‐infected values (ratio B/H and ratio i/h, Fig. 6). We analysed the early expression of the well‐studied B. vulgaris beta‐1,3‐glucanase gene (PR‐2, accession no. BQ591809) and the basic class IV chitinase gene (PR‐3, accession no. CF542771) previously detected in rhizomania‐infected sugar beet roots (Burketova et al., 2003). These two genes were found to be activated in both infected species (2.5‐ and 3.5‐fold increase for PR‐2; 2.7‐ and 4.0‐fold increase for PR‐3, Fig. 6). Among the previously identified cDNAs, some of them were found to be activated in both infected species. Up‐regulation was confirmed for genes encoding a ubiquitin (no. 11), an H+‐ATPase (no. 17) and a glucose‐6‐phosphate 1‐dehydrogenase (G6PDH, no. 25). However, GLP (no. 22) was found activated in B. vulgaris but repressed in B. macrocarpa. Finally, two genes, HSC70 (no. 2) and AAP (no. 14), did not reveal a significant difference.
Figure 6.
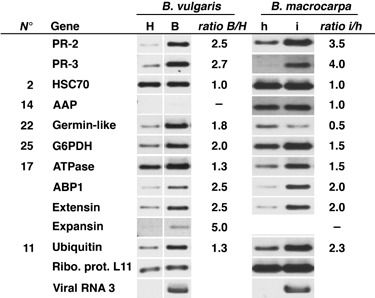
Expression patterns of RFDD‐PCR candidate genes and other genes of interest in Beta vulgaris and Beta macrocarpa roots upon response to early BNYVV infection. Analyses were conducted on healthy (H) or infected (B) B. vulgaris root total RNAs extracted after 7 days of bioassay treatment and on healthy (h) or infected (i) B. macrocarpa root total RNAs extracted after 13 days of plant systemic infection. Semi‐quantitative RT‐PCR was standardized with the L11 ribosomal protein gene (accession no. BE590348) and the BNYVV RNA 3 sequence was used as a viral control. Signals were quantified with ImageJ 1.38w software and transcript accumulation was expressed as a ratio between infected value and non‐infected value (ratio I/H or i/h). Identifiers (no.) of RFDD‐PCR cDNAs are those indicated in Table 1.
These RT‐PCR experiments also provided the opportunity to analyse other genes of interest. The auxin binding protein 1 gene (ABP1, accession no. BQ 584486) was analysed as auxin hormonal changes were previously reported in rhizomania syndrome (Pollini et al., 1990). Moreover, the extensin gene (accession no. BQ583860) was also studied. This gene belongs to the hydroxyproline‐rich glycoprotein (HRGP) family, key regulatory elements of cell‐wall development (Cassab, 1998) that can be induced by auxin, or biotic or abiotic stresses (Merkouropoulos and Shirsat, 2003). ABP1 and the extensin gene were activated in both infected species (2.5‐fold increase in B. vulgaris and 2.0‐fold increase in B. macrocarpa, Fig. 6). Finally, leaf blisters observed on systemically infected B. macrocarpa leaves were similar to the phenotypes observed on tomato leaves infected by Potato spindle tuber viroid (Qi and Ding, 2003). This led us to analyse expression variations of an expansin gene (accession no. BQ586143), a member of a gene family involved in root initiation and elongation and modulated by hormonal changes (Cho and Cosgrove, 2002). This expansin gene was found to be up‐regulated in infected B. vulgaris roots (5.0‐fold increase, Fig. 6) but it was not detected in B. macrocarpa root tissues.
DISCUSSION
To study the molecular mechanisms responsible for rhizomania syndrome of sugar beet, which results in root proliferation, we initiated RFDD‐PCR analyses to compare the root RNA contents of infected plants and healthy plants. We have reproduced the natural infection process, growing the plants in soil containing the aviruliferous and viruliferous P. betae vector to obtain healthy and infected plants, respectively. Viral isolates containing four and five RNA components were used to obtain B‐type (A2) and P‐type (A4) BNYVV‐infected plants, respectively (Fig. 1). Differential display patterns were then produced from infected and healthy plants (e.g. Fig. 2) and 53 bands were selected. Among the 42 bands successfully cloned and sequenced, 14 were assigned to BNYVV viral sequences (names in italic, Fig. 2), thus leaving 28 bands of interest (FDD names, Table 1) providing 50 sequences that were further characterized (Table 1). Many sequences corresponded to rRNA or DNA sequences either from unicellular eukaryotes (nos. 8, 9, 24, 30, 36, 39, Table 1) or prokaryotes (nos. 6, 7, 13, 27, 28, 31–34, 42) or unrelated virus (no. 23). The presence of soil‐dependent differential profiles was confirmed by the detection of prokaryotic RNA species with no plant sequence identities (A. thaliana Locus, Table 1). Therefore, to avoid such uncontrolled variations among total RNA samples, we performed similar reverse Northern analyses or semi‐quantitative RT‐PCR amplifications with plant rootlets naturally infected under standardized conditions (Fig. 4) or root portions selected on the same systemically infected plant (Fig. 5), which allowed us to focus only on genes mainly deregulated by the BNYVV infection and not by the vector or soil microorganisms.
Plant rRNAs were also identified (nos. 5, 26, 35, 37, 46, 49, Table 1). Within such rRNA sequences, differential expression of only three sequences (nos. 5, 35, and 37) was slightly visualized (Fig. 3) and was therefore not considered as significant. No signal or differential expression was found for unknown candidate nos. 41, 43, 48 and 50 as well as for sequences with plant putative function, i.e. nos. 20, 36, 40, 44, 45 and 47 (3, 4), which may be considered either as contaminating fragments during gel recovery or as false positive amplicons generated by RFDD‐PCR. Differential expression of unknown function sequences nos. 10, 12, 15, 16 and 18 as well as genes encoding for ubiquitin (no. 11, 3, 4, 5, 6), H+‐ATPase (no. 17, 3, 4, 5, 6), GLP (no. 22, Fig. 3; B. vulgaris, Fig. 6), AAP (no. 14, 4, 5) was detected by at least two approaches (reverse Northern and/or semi‐quantitative RT‐PCR) and corresponded to gene activation in response to BNYVV infection. Genes corresponding to HSC70 (no. 2, Fig. 4B), PLD (no. 3, Fig. 4B), GST (no. 4, Fig. 4B), G6PDH (no. 25, Fig. 6) and genes encoding a carbamoyl‐phosphate synthase (no. 19, Fig. 3), a peroxidase (no. 29, Fig. 3), and the l‐allo‐threonine aldolase (no. 38, Fig. 3) were confirmed by only a single experiment (reverse Northern or semi‐quantitative RT‐PCR). To explain such contradictions between experiments, it should be recalled that large gene families exist in plant genomes, and we could not exclude the possibility that different members of the same gene family with distinct expression profiles were cross‐hybridized by reverse Northern and alternatively amplified by RFDD‐PCR or RT‐PCR (e.g. HSC70). Therefore, the reverse Northern results may also reflect the differential regulation of members of a gene family rather than one specific gene. Further Southern blot and Northern blot experiments will be performed to identify the differentially regulated gene(s).
Comparison of B. vulgaris and B. macrocarpa differential expression patterns permitted the identification of genes that were differentially expressed because of the presence of BNYVV versus those genes that were differentially expressed possibly because of the presence of the vector P. betae. For example, one could argue that the GLP gene is induced by P. betae, as this gene was confirmed to be up‐regulated in the B. vulgaris experimental system (no. 22, 3, 6) but not in that of B. macrocarpa (no. 22, Fig. 6). An alternative explanation is that B. vulgaris and B. macrocarpa differ in their responses to BNYVV.
Differential expression of the ubiquitin gene was found to be induced in the three different reverse Northern analyses and was also confirmed by RT‐PCR. In maize kernels grown in vitro, the Ubi‐1 promoter is stimulated by the plant hormone indole‐acetic acid but not by pathogen infection (Muhitch and Shatters, 1998). Ubiquination plays major roles in the regulation of plant defence and plant signalling pathways (Devoto et al., 2003; Dreher and Callis, 2007). Until now, it has not been known whether virus‐mediated over‐expression of ubiquitin mRNA favours viral infection or plant defence, as both scenarios are possible (Angot et al., 2007). Transcriptome analyses of lesions induced after pathogen inoculation of wild‐type and ‘lesion mimics’spl11 mutant revealed the up‐regulation of several genes in which GST and GLP, as well as ATPase, expansin, extensin, phenylalanine ammonia‐lyase (PAL) and PR‐protein encoding genes are retrieved (Zeng et al., 2006). In our RFDD‐PCR approach, many stress‐response genes were found to be activated as well (the PLD, GST, PAL and peroxidase genes).
ABP1 was activated (Fig. 6), reinforcing the role of auxin hormonal signalling during B. vulgaris root infections. The ABP1 protein mediates cell expansion (Timpte, 2001). Auxin is also known to induce the expression of expansins that act together with H+‐ATPases to extend the plant cell wall but also to participate in cellular processes involved in growth and development (Choi et al., 2006). In the present study, a member of the H+‐ATPases gene was of particular interest: it was found to be induced by RFDD‐PCR and its differential expression was confirmed by the three reverse Northern analyses and by RT‐PCR. Acidification is required for cellular expansion of cell walls by expansin proteins. Interestingly, one expansin gene tested here was also found to be induced in B. vulgaris‐infected roots (Fig. 6). Recently, auxin has been shown to promote disease susceptibility (Navarro et al., 2006) and many pathogens use alternative strategies to take advantages of this property, from tobacco mosaic virus (Padmanabhan et al., 2005, 2008) to Pseudomonas syringae (Chen et al., 2007). Such deregulation can, however, be counterbalanced as it has been recently demonstrated that IAA‐amino synthetase GH3‐8 allows plants to inhibit expansin expression and promotes basal immunity to reduce pathogen invasion (Ding et al., 2008).
The extensin gene was shown to be induced during root initiation (Keller and Lamb, 1989) as well as by biotic and abiotic stresses (Merkouropoulos and Shirsat, 2003; Merkouropoulos et al., 1999). The hormones auxin, salicylic acid and methyl jasmonate activate the extensin gene (Merkouropoulos and Shirsat, 2003), expression of which has also been reported in programmed cell death (Asai et al., 2000). In our experiment, the extensin gene was found to be activated in roots of both species tested (Fig. 6).
Other putative up‐regulated cDNA candidates were also linked to auxin signalling as well as to cellular differentiation, responses to oxidative stress or defence signalling as revealed by Gene Ontology Annotations (Table 1). Among them, G6PDH and the carbamoyl‐phosphate synthase encoding gene are of particular interest. Indeed, during the infection process, G6PDH gene induction could benefit the virus as the enzyme could participate in the reduction of redox‐stress‐induced apoptosis (Fico et al., 2004). Moreover, carbamoyl‐phosphate synthase participates within arginine and pyrimidine biosynthesis (Kolloffel and Verkerk, 1982; O’Neal and Naylor, 1976; Slocum, 2005). Such compounds can lead to the production of nitric oxide and cGMP second messenger (Delledonne, 2005). Both molecules are involved in plant development and defence response and act together with auxin for root morphogenesis (Jacobi et al., 2006; Pagnussat et al., 2003, 2004). Further experiments will be required to confirm the differential expression of these two genes upon BNYVV root infection.
It is clear that our transcriptome analyses of Beta species did not provide consistent results. For comparisons performed by the use of natural infection methods, soils were different and brought significant variability. Using infected and non‐infected tissues from the same plant (i.e. B. macrocarpa), soil variability was resolved but amounts of RNA were not sufficient to conduct complete analyses. However, some candidate genes whose differential expression was well and truly confirmed highlighted the predominant role of auxin in root disease development. New approaches using transgenic plants expressing inducible BNYVV proteins will permit more efficient studies of initial and late events occurring in rhizomania syndrome.
EXPERIMENTAL PROCEDURES
Plant growth and infections
For B. vulgaris ssp. vulgaris root infections, susceptible plants were cultivated in soil mixtures containing viruliferous P. betae harbouring a B‐type BNYVV isolate (A2) or P‐type BNYVV isolate (A4) originating from soils collected from fields at Amifontaine (France) or Yèvre‐la‐Ville (France), respectively. Control plants were grown in virus‐free P. betae soil (H) coming from Rothamsted (UK). P. betae‐containing soils were mixed with sterilized compost (1:10) and ten sugar beet plants were grown in each compartment to allow the multiplication of P. betae. After this amplification step, plants were removed, 20 new seeds were sown and only ten plantlets of similar size were conserved after germination and allowed to grow for 8 weeks (six‐ to eight‐leaf stage) until visible symptoms appeared on infected plants. Plants were harvested, roots were carefully washed and pooled prior to being weighed and subsequently ground in liquid nitrogen and stored at −80 °C.
For standardized B. vulgaris infections (bioassay developed in SESVanderHave laboratory, Tienen, Belgium), five sets of 35 sugar beet seedlings were grown in individual tubes containing coarse sand mixed with 10% of viruliferous P. betae‐containing soil from Germany with a B‐type BNYVV isolate (B). Five other groups of 35 control seedlings were grown in sand mixed with aviruliferous P. betae‐containing soil (H). Roots of 35 plantlets were harvested at 0, 7, 14, 21 and 28 days after treatment, immediately frozen in liquid nitrogen and conserved at −80 °C. For each time point, 50 mg of fresh root tissue was reserved for virus quantification by a TAS‐ELISA at 405 nm using primary (H3) and monoclonal (MAFF 9) antibodies (Neogeneurope) directed against BNYVV coat protein (CP).
For B. macrocarpa systemic infections, one mature leaf of a 3‐week‐old seedling was rub inoculated with Stras1234, a B‐type BNYVV inoculum (B), as described previously (Lauber et al., 1998). Plants were harvested and roots collected as soon as visible systemic symptoms appeared on leaves (approximately 13 days after inoculation). Control plantlets were mock inoculated with sterile water (H). Entire root systems from infected and non‐infected plants were put between a set of nitrocellulose filter and a plastic sheet and then submitted to a pressure tissue print. Roots were then frozen and stored at −80 °C on the plastic sheet until RNA extraction; meanwhile, the nitrocellulose membrane was submitted to an immunodetection procedure using specific BNYVV CP antisera.
Total RNA isolation, mRNA purification and Northern blot hybridization
RNAs were extracted from 100 mg of frozen material using an RNeasy® Plant Mini Kit (Qiagen), according to the manufacturer's recommendations (including DNase treatment). Plant mRNAs were purified with the NucleoTrap® mRNA Mini Kit (Macherey‐Nagel) as described by the supplier's protocol. Northern blot analyses were performed using 7 µg of total RNAs from B. vulgaris non‐infected or infected roots or 2 µg of total RNAs from Chenopodium quinoa leaf lesions induced by a P‐type BNYVV isolate. Viral RNAs were detected with specific riboprobes complementary to viral RNA 1, 2, 3 and 5 as described previously (Lauber et al., 1998; Link et al., 2005).
RFDD‐PCR
The analysis was carried out as described in detail in the protocol from the displayPROFILETM kit (Qbiogene). Double‐stranded cDNAs were produced in duplicate using 1 µg of total RNAs and T25V (T25A, G or C) primer. Purified DNA was digested with TaqI endonuclease for 2 h at 65 °C and completed by the ligation of adaptor mixture containing an extension protection group for 3 h at 37 °C using T4 DNA ligase. The template was PCR amplified using a 0‐extension primer 33P‐labelled complementary to the EP adaptor in combination with a 3‐extension displayPROBE primer recognizing the standard adaptor and the three nucleotides adjacent to the TaqI site. For each template, PCR reactions using the labelled 0‐extension primer in combination with one of the nine displayPROBE primers tested (EU18, EU19, EU20, EU22, EU23, EU24, EU25, EU26 and EU27) were performed as follows: ten cycles of 94 °C for 30 s, 60 °C (−0.5 °C per cycle) for 30 s, and 72 °C for 1 min, followed by 15 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 1 min. PCR products were denatured and size fractionated in a 6% polyacrylamide gel (acryl/bis: 19:1) containing 8 m urea run for 3 h at 60 W. RFDD‐PCR gels were vacuum blotted and dried on Whatman 3MM paper for 1 h and subsequently exposed to X‐ray films. Bands of interest were selected and cut from the blotted gel on paper. The small paper cuttings were heated in distilled water for 15 min at 95 °C to elute the DNA from the paper. Samples were then amplified again by PCR and purified. After phosphorylation of the DNA 5′ ends, amplicons were cloned into pGEM®‐T vector (Promega) and six positive clones were sequenced for each pGEM®‐T construction. Sequences were compared against all sequences, using the tBlastx program with the NCBI database (http://www.ncbi.nlm.nih.gov/blast/) and the WU‐Blast 2.0 program with the TIGR A. thaliana database (http://tigrblast.tigr.org/er‐blast/). Candidates with appropriate A. thaliana orthologues were kept for further analyses. A biological function was then assigned, according to the Gene Ontology Annotations (GO terms) provided by the TAIR Search tools (http://www.arabidopsis.org/tools/bulk/go/) for each AT Locus identifier.
Reverse Northern hybridizations
Inserts of sequenced cDNA clones were amplified by PCR with SP6 and T7 primers that were complementary to pGEM®‐T vector sequences flanking both sides of cDNA inserts. One hundred nanograms of purified PCR products was analysed in duplicate by electrophoresis on a 2% agarose gel and transferred onto a nylon membrane. Five micrograms of total RNAs or purified mRNA from infected or non‐infected roots was used to prepare complex probes by reverse transcription in the presence of [α‐32P] dCTP. Half of the membrane was hybridized with control probes, the other half with probes corresponding to infected roots. Signals were quantified using a Fuji MAS1000 BioAnalyser and MacBAS software.
Semi‐quantitative RT‐PCR
cDNA were synthesized using 1 µg of total RNAs and oligo(dT)25 primer with SuperscriptTM II reverse transcriptase (Invitrogen) according to the manufacturer's recommendations. PCR amplifications were performed on 1–9 µL of the 10× diluted cDNA solution using Taq DNA Polymerase (Invitrogen) and consisted of 28 cycles of 94 °C for 15 s, 50 °C for 30 s and 72 °C for 1 min. Control reactions to normalize RT‐PCR amplification were run with primers derived from the L11 ribosomal protein gene (accession no. BE590348, G. Weyens, personal communication). PCR products were separated onto a 1.5% agarose gel stained with EtBr and image processing was carried out with a Bio‐Rad GelDoc apparatus. Signals were analysed with Quantity One software (Bio‐Rad) and quantified with ImageJ 1.38w software (http://rsb.info.nih.gov/ij/).
ACKNOWLEDGEMENTS
We thank Malek Alioua for sequence analysis and Danièle Scheidecker for logistical support. We are grateful to journal referees whose help to improve the manuscript. L.S. was supported by SESVanderHave under a CIFRE programme.
REFERENCES
- Angot, A. , Vergunst, A. , Genin, S. and Peeters, N. (2007) Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog. 3, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai, T. , Stone, J.M. , Heard, J.E. , Kovtun, Y. , Yorgey, P. , Sheen, J. and Ausubel, F. (2000) Fumonisin B1‐induced cell death in arabidopsis protoplasts requires jasmonate‐, ethylene‐, and salicylate‐dependent signaling pathways. Plant Cell, 12, 1823–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burketova, L. , Stillerova, K. and Feltlova, M. (2003) Immunohistological localization of chitinase and beta‐1,3‐glucanase in rhizomania‐diseased and benzothiadiazole treated sugar beet roots. Physiol. Mol. Plant Pathol. 63, 47–54. [Google Scholar]
- Cassab, G.I. (1998) Plant cell wall proteins. Annu. Rev. Plant Physiol. Plant Mol. Biol. 49, 281–309. [DOI] [PubMed] [Google Scholar]
- Chen, Z. , Agnew, J.L. , Cohen, J.D. , He, P. , Shan, L. , Sheen, J. and Kunkel, B.N. (2007) Pseudomonas syringae type III effector AvrRpt2 alters Arabidopsis thaliana auxin physiology. Proc. Natl. Acad. Sci. USA, 104, 20131–20136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba, S. , Miyanishi, M. , Andika, I.B. , Kondo, H. and Tamada, T. (2008) Identification of amino acids of the beet necrotic yellow vein virus p25 protein required for induction of the resistance response in leaves of Beta vulgaris plants J. Gen. Virol. 89, 1314–1323. [DOI] [PubMed] [Google Scholar]
- Cho, H.T. and Cosgrove, D.J. (2002) Regulation of root hair initiation and expansin gene expression in Arabidopsis. Plant Cell, 14, 3237–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, D. , Cho, H.‐T. and Lee, Y. (2006) Expansins: expanding importance in plant growth and development. Physiol. Plant 126, 511–518. [Google Scholar]
- Coutts, R.H. , Linton, D.J. and Bolwell, G.P. (1994) Patterns of protein synthesis in infected and stressed sugar beet (Beta vulgaris L.). J. Phytopathol. 142, 274–282. [Google Scholar]
- Delledonne, M. (2005) NO news is good news for plants. Curr. Opin. Plant Biol. 8, 390–396. [DOI] [PubMed] [Google Scholar]
- Devoto, A. , Muskett, P.R. and Shirasu, K. (2003) Role of ubiquitination in the regulation of plant defence against pathogens. Curr. Opin. Plant Biol. 6, 307–311. [DOI] [PubMed] [Google Scholar]
- Diatchenko, L. , Lau, Y.F. , Campbell, A.P. , Chenchik, A. , Moqadam, F. , Huang, B. , Lukyanov, S. , Lukyanov, K. , Gurskaya, N. , Sverdlov, E.D. and Siebert, P.D. (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue‐specific cDNA probes and libraries. Proc. Natl. Acad. Sci. USA, 93, 6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, X. , Cao, Y. , Huang, L. , Zhao, J. , Xu, C. , Li, X. and Wang, S. (2008) Activation of the indole‐3‐acetic acid‐amido synthetase GH3‐8 suppresses expansin expression and promotes salicylate‐ and jasmonate‐independent basal immunity in rice. Plant Cell, 20, 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher, K. and Callis, J. (2007) Ubiquitin, hormones and biotic stress in plants. Ann. Bot. 99, 787–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan, D.J. , Bittner, M. , Chen, Y. , Meltzer, P. and Trent, J.M. (1999) Expression profiling using cDNA microarrays. Nat. Genet. 21, 10–14. [DOI] [PubMed] [Google Scholar]
- Fico, A. , Paglialunga, F. , Cigliano, L. , Abrescia, P. , Verde, P. , Martini, G. , Iaccarino, I. and Filosa, S. (2004) Glucose‐6‐phosphate dehydrogenase plays a crucial role in protection from redox‐stress‐induced apoptosis. Cell Death Differ. 11, 823–831. [DOI] [PubMed] [Google Scholar]
- Fleming, T.M. , McCarthy, D.A. , White, R.F. , Antoniw, J.F. and Mikkelsen, J.D. (1991) Induction and characterization of some of the pathogenesis‐related proteins in sugar beet. Physiol. Mol. Plant Pathol. 39, 147–160. [Google Scholar]
- Gravesen, A. , Warthoe, P. , Knochel, S. and Thirstrup, K. (2000) Restriction fragment differential display of pediocin‐resistant Listeria monocytogenes 412 mutants shows consistent overexpression of a putative beta‐glucoside‐specific PTS system. Microbiology, 146, 1381–1389. [DOI] [PubMed] [Google Scholar]
- Jacobi, J. , Elmer, J. , Russell, K. , Soundur, R. and Portfield, D.M. (2006) Nitric oxide and cGMP dependent signaling in arabidopsis root growth. Gravit. Space Biol. 19, 157–158. [Google Scholar]
- Keller, B. and Lamb, C.J. (1989) Specific expression of a novel cell wall hydroxyproline‐rich glycoprotein gene in lateral root initiation. Genes Dev. 3, 1639–1646. [DOI] [PubMed] [Google Scholar]
- Klein, E. , Link, D. , Schirmer, A. , Erhardt, M. and Gilmer, D. (2007) Sequence variation within Beet necrotic yellow vein virus p25 protein influences its oligomerization and isolate pathogenicity on Tetragonia expansa. Virus. Res. 126, 53–61. [DOI] [PubMed] [Google Scholar]
- Kolloffel, C. and Verkerk, B.C. (1982) Carbamoyl phosphate synthetase activity from the cotyledons of developing and germinating pea seeds. Plant Physiol. 69, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber, E. , Guilley, H. , Tamada, T. , Richards, K.E. and Jonard, G. (1998) Vascular movement of beet necrotic yellow vein virus in Beta macrocarpa is probably dependent on an RNA 3 sequence domain rather than a gene product. J. Gen. Virol. 79, 385–393. [DOI] [PubMed] [Google Scholar]
- Liang, P. and Pardee, A.B. (1992) Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science, 257, 967–971. [DOI] [PubMed] [Google Scholar]
- Liang, P. and Pardee, A.B. (2003) Analysing differential gene expression in cancer. Nat. Rev. Cancer, 3, 869–876. [DOI] [PubMed] [Google Scholar]
- Liang, P. , Averboukh, L. , Keyomarsi, K. , Sager, R. and Pardee, A.B. (1992) Differential display and cloning of messenger RNAs from human breast cancer versus mammary epithelial cells. Cancer Res. 52, 6966–6968. [PubMed] [Google Scholar]
- Link, D. , Schmidlin, L. , Schirmer, A. , Klein, E. , Erhardt, M. , Geldreich, A. , Lemaire and Gilmer, D. (2005) Functional characterization of the Beet necrotic yellow vein virus RNA‐5‐encoded p26 protein: evidence for structural pathogenicity determinants. J. Gen. Virol. 86, 2115–2125. [DOI] [PubMed] [Google Scholar]
- Merkouropoulos, G. and Shirsat, A. (2003) The unusual Arabidopsis extensin gene atExt1 is expressed throughout plant development and is induced by a variety of biotic and abiotic stresses. Planta, 217, 356–366. [DOI] [PubMed] [Google Scholar]
- Merkouropoulos, G. , Barnett, D.C. and Shirsat, A.H. (1999) The Arabidopsis extensin gene is developmentally regulated, is induced by wounding, methyl jasmonate, abscisic and salicylic acid, and codes for a protein with unusual motifs. Planta, 208, 212–219. [DOI] [PubMed] [Google Scholar]
- Muhitch, M.J. and Shatters, R.G. (1998) Regulation of the maize ubiquitin (Ubi‐1) promoter in developing maize (Zea mays L.) seeds examined using transient gene expression in kernels grown in vitro. Plant Cell Rep. 17, 476–481. [DOI] [PubMed] [Google Scholar]
- Navarro, L. , Dunoyer, P. , Jay, F. , Arnold, B. , Dharmasiri, N. , Estelle, M. , Voinnet, O. and Jones, J.D. (2006) A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science, 312, 436–439. [DOI] [PubMed] [Google Scholar]
- Nielsen, J.E. , Nielsen, K.K. and Mikkelsen, J.D. (1996) Immunohistological localization of a basic class IV chitinase in Beta vulagris leaves after infection with Cercospora beticola . Plant Sci. 119, 191–202. [Google Scholar]
- Nielsen, K.K. , Mikkelsen, J.D. , Kragh, K.M. and Bojsen, K. (1993) An acidic class III chitinase in sugar beet: induction by Cercospora beticola, characterization, and expression in transgenic tobacco plants. Mol. Plant–Microbe. Interact. 6, 495–506. [DOI] [PubMed] [Google Scholar]
- Nielsen, K.K. , Bojsen, K. , Collinge, D.B. and Mikkelsen, J.D. (1994) Induced resistance in sugar beet against Cercospora beticola– induction by dichloroisonicotinic acid is dependent of chitinase and beta‐1,3‐glucanase transcript accumulation. Physiol. Mol. Plant Pathol. 45, 89–99. [Google Scholar]
- O’Neal T.D. and Naylor, A.W. (1976) Some regulatory properties of pea leaf carbamoyl phosphate synthetase. Plant Physiol. 57, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan, M.S. , Goregaoker, S.P. , Golem, S. , Shiferaw, H. and Culver, J.N. (2005) Interaction of the tobacco mosaic virus replicase protein with the Aux/IAA Protein PAP1/IAA26 is associated with disease development. J. Virol. 79, 2549–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan, M.S. , Kramer, S.R. , Wang, X. and Culver, J.N. (2008) Tobacco mosaic virus replicase‐auxin/indole acetic acid protein interactions: reprogramming the auxin response pathway to enhance virus infection. J. Virol. 82, 2477–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnussat, G.C. , Lanteri, M.L. and Lamattina, L. (2003) Nitric oxide and cyclic GMP are messengers in the indole acetic acid‐induced adventitious rooting process. Plant Physiol. 132, 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnussat, G.C. , Lanteri, M.L. , Lombardo, M.C. and Lamattina, L. (2004) Nitric oxide mediates the indole acetic acid induction activation of a mitogen‐activated protein kinase cascade involved in adventitious root development. Plant Physiol. 135, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollini, C.P. and Giunchedi, L. (1989) Comparative histopathology of sugar beets that are susceptible and partially resistant to rhizomania. Phytopath. Medit. 28, 16–21. [Google Scholar]
- Pollini, C.P. , Masia, A. and Giunchedi, L. (1990) Free indole‐3‐acetic acid in sugar‐beet root of rhizomania‐susceptible and moderately resistant cultivars. Phytopath. Medit. 29, 191–195. [Google Scholar]
- Qi, Y. and Ding, B. (2003) Inhibition of cell growth and shoot development by a specific nucleotide sequence in a noncoding viroid RNA. Plant Cell, 15, 1360–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim, M.D. , Andika, I.B. , Han, C. , Kondo, H. and Tamada, T. (2007) RNA4‐encoded p31 of beet necrotic yellow vein virus is involved in efficient vector transmission, symptom severity and silencing suppression in roots. J. Gen. Virol. 88, 1611–1619. [DOI] [PubMed] [Google Scholar]
- Richards, K. and Tamada, T. (1992) Mapping functions on the multipartite genome of beet necrotic yelow vein virus. Annu. Rev. Phytopathol. 30, 291–313. [Google Scholar]
- Slocum, R.D. (2005) Genes, enzymes and regulation of arginine biosynthesis in plants. Plant Physiol. Biochem. 43, 729–745. [DOI] [PubMed] [Google Scholar]
- Tamada, T. (1999) Benyviruses In: Encyclopedia of Virology, 2nd edn (Webster R. and Granoff A., eds.). New York, N.Y.: Academic Press, pp. 154–160. [Google Scholar]
- Tamada, T. and Abe, H. (1989) Evidence that beet necrotic yellow vein virus RNA‐4 is essential for transmission by the fungus Polymyxa betae . J. Gen. Virol. 70, 3391–3398. [DOI] [PubMed] [Google Scholar]
- Timpte, C. (2001) Auxin binding protein: curiouser and curiouser. Trends Plant Sci. 6, 586–590. [DOI] [PubMed] [Google Scholar]
- Vetter, G. , Hily, J.M. , Klein, E. , Schmidlin, L. , Haas, M. , Merkle, T. and Gilmer, D. (2004) Nucleo‐cytoplasmic shuttling of the beet necrotic yellow vein virus RNA‐3‐encoded p25 protein. J. Gen. Virol. 85, 2459–2469. [DOI] [PubMed] [Google Scholar]
- Whitelaw, C.A. , Ruperti, B. and Roberts, J.A. (2000) Differential display. Analysis of gene expression during plant development. Methods Mol. Biol. 141, 19–32. [DOI] [PubMed] [Google Scholar]
- Whitelaw, C.A. , Ruperti, B. and Roberts, J.A. (2002) Differential display: analysis of gene expression during plant cell separation processes. Mol. Biotechnol. 21, 251–258. [DOI] [PubMed] [Google Scholar]
- Zeng, L.‐R. , Vega‐Sanchez, M.E. , Zhu, T. and Wang, G.‐L. (2006) Ubiquitination‐mediated protein degradation and modification: an emerging theme in plant–microbe interactions. Cell Res. 16, 413–426. [DOI] [PubMed] [Google Scholar]