

SUMMARY
Flow cytometers are probably the most multipurpose laboratory devices available. They can analyse a vast and very diverse range of cell parameters. This technique has left its mark on cancer, human immunodeficiency virus and immunology research, and is indispensable in routine clinical diagnostics. Flow cytometry (FCM) is also a well‐known tool for the detection and physiological status assessment of microorganisms in drinking water, marine environments, food and fermentation processes. However, flow cytometers are seldom used in plant pathology, despite FCM's major advantages as both a detection method and a research tool. Potential uses of FCM include the characterization of genome sizes of fungal and oomycete populations, multiplexed pathogen detection and the monitoring of the viability, culturability and gene expression of plant pathogens, and many others. This review provides an overview of the history, advantages and disadvantages of FCM, and focuses on the current applications and future possibilities of FCM in plant pathology.
INTRODUCTION
In plant pathology, detection and characterization, quantification and viability assessment of pathogens are crucial to the development or application of control measures. Fast detection methods are indispensable, as plant pathogen population levels often fluctuate rapidly. In spite of these benefits, the time‐consuming isolation and culture of microorganisms, based on the methods developed by Koch, Hesse and Petri in the early 1880s (Lopez et al., 2008), persist as the gold standard in many detection protocols. Alternatives such as enzyme‐linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR) were introduced in microbiology in 1971 (Engvall and Perlmann, 1971) and 1983 (Mullis et al., 1986), respectively. Very soon after their introduction, these methods were adopted in plant pathology (Deng and Hiruki, 1990; Dunez, 1977) and are now established routine detection methods and research tools (Palacio‐Bielsa et al., 2009). In addition to the advantage of faster characterization, both methods are culture independent and can be more specific than plate counts. One disadvantage is their inability to perform viability discrimination.
Flow cytometry (FCM) is an alternative method that can be used for both routine detection and research. FCM can give a very precise estimation of fungal and oomycete genome sizes or provide quantitative information on the presence and viability of cells, and a myriad of other parameters, e.g. size and shape, membrane potential or mitochondrial activity. FCM was introduced before PCR or ELISA, with the first commercial flow cytometer used in 1969 (Shapiro, 2003). It soon became an indispensable method in medical diagnosis and is a commonly used technique in food microbiology, veterinary research and water analysis. However, it remains rather unknown and unused in plant pathology. This review focuses on the current applications and future possibilities of FCM in plant pathology for research and routine detection.
WHAT IS FCM?
FCM is a technique for the measurement and counting of small particles in a fluid stream. A flow cytometer comprises three systems: fluidics, optics and electronics (Fig. 1). In essence, every single particle is excited by a light source and is finally displayed on a graph (Shapiro, 2003). Common flow cytometers detect multiple parameters: forward scatter (FSC), sideward scatter (SSC) and a number of fluorescent wavelengths (FL1, FL2, and so on), depending on the excitation source and the complexity of the instrument. FSC and SSC signals provide information about the size, shape and complexity of the cell. FSC is the narrow angle light scatter and is dependent on the size and refraction index of the cell (Longobardi, 2001). SSC is the right angle light scatter and depends on the external granularity, internal complexity and shape of the cell (Shapiro, 2003). The sensitivity of each photomultiplier tube (PMT) can be adjusted separately to suit the application. During the analysis, the instrument can be triggered on one of its parameters. Only if a particle is positive for the triggering parameter, its signal intensity for that parameter and all other parameters will be displayed on the outputs (Rehse et al., 1995).
Figure 1.
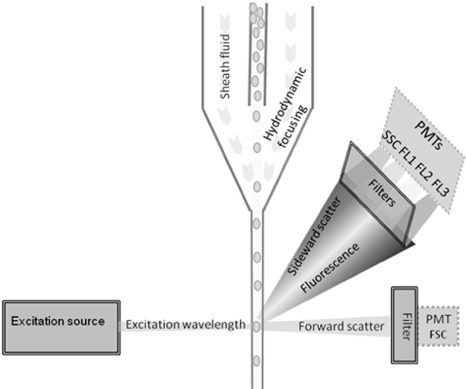
Conceptual figure of a flow cytometer, showing the fluidics system (solid lines), optical system (double lines) and electronics system (dotted lines). The fluidics system delivers the particles of the sample in a single file to the flow cuvette. This is done by injecting the sample into a sheath fluid, that narrows down the sample stream into a single cell line by hydrodynamic focusing. The optical system consists of one or more excitation sources (laser, lamp or light emitting diode) to excite the cells in the flow cuvette. A set of filters and mirrors deflects and passes certain wavelengths of the emitted fluorescence (FL1‐3) and scattered laser light (SSC and FSC). The key parts of the electronics system are the photomultiplier tubes (PMTs) that detect the incoming photons, multiply the current they produce and send this electric signal to the computer where it is displayed as single‐parameter histogram or two‐parameter dot plot.
Shapiro (2003) has authored the most comprehensive overview of FCM in all of its aspects. His book can be accessed at no cost on the Internet. Doležel et al. (2007b) focus on all plant‐related FCM topics.
HISTORY OF FCM
The first fluorescence‐based flow cytometer was developed in 1968 by Wolfgang Göhde and was commercialized a year later (Shapiro, 2003). It was soon adopted as a detection method for human immunodeficiency virus (HIV) (Shapiro, 2003), cancer (Barrett et al., 1976) and malaria (Jackson et al., 1977), but also for the detection of medically relevant viruses (Hercher et al., 1979) and bacteria (Steen, 2000). A detailed history of FCM can be found in Shapiro (2003) and on the websites of many flow cytometer manufacturers.
The basics of all major clinical FCM applications today were developed during the first 10 years of FCM. Currently, FCM is still mostly used for immunophenotyping: determination of blood type, transplant compatibility, detection of stem cell disorders, leukaemias and lymphomas, and immunological monitoring of HIV‐infected patients. These routine clinical practices rely on FCM and monoclonal antibodies (Brown and Wittwer, 2000; Tait et al., 2009). Since the 1980s, a major change in FCM has taken place. While the basic principles are still the same, technological advances have resulted in cheaper machines, more sensitive instruments and better fluorochromes, which, in turn, have resulted in higher speed and smaller volumes. FCM analyses at a rate of one sample or over 10 000 cells per second in multiwell plates have already become standard practice in many diagnostic laboratories (Krishhan et al., 2009). An up‐and‐coming technology in clinical FCM is high‐content flow cytometric screening: a combination of robotic fluid handling, flow cytometric instrumentation and bioinformatics software capable of screening a large number of samples in a short time (Naumann and Wand, 2009).
Although the earliest report on the FCM analysis of plant material was among the very first FCM publications (Heller, 1973), it took until 1990 for the first plant pathogen to be detected with FCM (Hardham and Suzaki, 1990). This huge gap persists between the applications of FCM and its evolution in medicine and plant pathology. For example, new developments in the medical sector strive to go beyond the limitations of detecting ‘only’ 17 fluorescent labels at once. Plant pathologists, in contrast, consider a three‐colour experiment to be exceptional.
The cost of a flow cytometer is often pinpointed as the major cause for this disparity. Clearly, flow cytometers are not cheap, but neither are the real‐time PCR machines used by plant pathologists. A well‐equipped flow cytometer capable of detecting four colours and two scatter parameters costs approximately €35 000–106 000. A real‐time PCR machine (detecting only one fluorescence parameter) costs €18 000–67 000. In the plant sciences, FCM is almost exclusively established in plant breeding (Doležel et al., 2007c), where it is used routinely for ploidy and genome size analysis. The cost or availability of flow cytometers thus cannot be the main cause of the lack of applications in plant pathology. The reason is more likely to be the complexity of the instrument and the lack of knowledge, training and support needed to operate it. Companies in this new field can make a breakthrough for the technique by providing automated equipment, ready‐to‐use kits and specialized training.
APPLICATIONS OF FCM IN PLANT PATHOLOGY
Flow cytometric applications in plant pathology can be divided into three groups: genome size measurement, detection and physiological status assessment (Fig. 2). Genome size estimation is based on the comparison of the amount of fluorescence emitted by DNA stained with an intercalating fluorochrome with that of a reference standard with a known genome size. Detection can be based on nonspecific staining of nucleic acids or the characterization of autofluorescence and scatter patterns. This helps to detect the presence of pathogens and to enumerate them, but does not allow discrimination between two morphologically similar organisms. Therefore, labelling with specific probes, such as antibodies or nucleic acid probes, is often required. The physiological status of an organism can be measured with FCM by quantifying the fluorescence intensity of one or more of the emitted wavelengths. The metabolic activity of cells can be measured on the basis of the fluorescence intensity. A few of the many examples are the uptake of a membrane integrity probe, the fluorescence of an esterase or mitochondrial activity probe or the amount of green fluorescent protein (GFP) expression.
Figure 2.
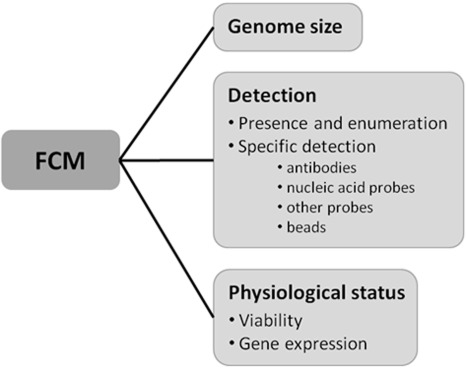
Schematic overview of the applications of flow cytometry in plant pathology.
Genome size
In the plant sector, FCM has become the method of choice for ploidy and genome size determination because it is fast, cheap and easy. FCM can also be performed in an early growth stage of the plant, or even on seeds (Doležel et al., 2007a). A 5‐min preparation by razor blade chopping (Galbraith et al., 1983) is sufficient to obtain a nuclear suspension that can be measured with a single‐parameter flow cytometer. The results are presented as a fluorescence intensity histogram with a peak (G1) and often a second peak (G2), correlated with the DNA content of the cell and mitotic cell (Kron et al., 2007). A comparison of the peak position of the sample with that of an external reference is often sufficient to detect large differences in DNA content between sample and reference, e.g. different ploidy levels. To obtain a more exact genome size estimate, an internal standard with known genome size is co‐chopped, stained and analysed with the sample. This is necessary, as some secondary metabolites, such as polyphenols, may cause small shifts in fluorescence peaks and give rise to small but significant variations between measurements (Greilhuber et al., 2007). When a biologically similar internal standard is used, both sample and standard peaks are influenced in the same way and the proportion between the peak positions stays constant (Suda and Leitch, 2010).
Genome size analysis has gained increasing attention over the past decade in both the plant and animal kingdoms, owing to more accurate and efficient quantification techniques (Gregory et al., 2007). Relationships between genome size and biological parameters, such as cell size, cell division rate and the ability of an organism to overcome selection pressure, have become more documented (Leitch and Bennet, 2007). In fungi, in general, variations in chromosome number and size seem to be the rule rather than the exception. Ploidy levels ranging from 1x to 50x and genome sizes in the range 1C= 0.007–0.81 pg have been found so far (Gregory et al., 2007). Variations in genome size of plant pathogens can cause variation in pathogenicity and complicate the control of a disease (Gregory et al., 2007). In particular plant pathogenic fungi and oomycetes are known for their high degree of genome plasticity. In these cases, it is extremely important to obtain information about the structure of the genome and to understand the dynamic forces which give rise to the high level of pathotype variation observed in the field (O'Sullivan et al., 1998).
As a result of the small (genome) size of bacteria, the relatively large amount of RNA and the absence of a distinct mitotic phase caused by the constant chromosome replication, bacterial genome size estimation is very difficult. Consequently, there are very few publications on the estimation of the bacterial genome size using FCM, and none involve plant pathogens (Button and Robertson, 2001; Steen, 2000).
Preparation and buffer systems applied for plants can also be used for plant pathogens (Kim et al., 2000). Commercial kits for the genome size determination of plants are available and allow easy sample preparation. They have also been successfully used on oomycetes (Si‐Ammour, 2002; Vercauteren, 2010). Figure 3 shows an example of genome size determination on Phytophthora ramorum taken from the work of Vercauteren et al. (2011). The positions of the G1 peaks of sample and reference are determined and the genome size of the sample is calculated as follows: C value of sample = (C value of reference × peak position of sample)/peak position of reference.
Figure 3.
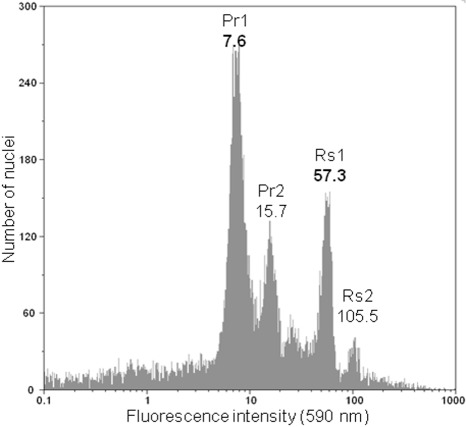
Flow cytometer histogram from the genome size determination on mycelium of Phytophthora ramorum European isolate ‘2299’ (A1 mating type) with Raphanus sativus‘Saxa’ (2C = 1.11 pg) leaf material as an internal standard (Doležel et al., 1992). Logarithmic histogram of orange fluorescence (590 nm) with P. ramorum G1 peak (Pr1) at position 7.6 and G2 peak (Pr2) consisting of dividing cells at position 15.7; the G1 peak of R. sativus (Rs1) appears at position 57.3, so the proportion of both peaks is 0.1326, resulting in a 2°C genome size of 0.147 pg.
The correct expression of the genome size of fungi and fungus‐like organisms is difficult, as they have complicated life cycles with different ploidy levels and the basic chromosome numbers are often not known. Therefore, genome sizes in this article were expressed as holoploid genome sizes or C values, defined as ‘the DNA content of the whole complement of chromosomes characteristic for the organism, irrespective of the degree of generative polyploidy, aneuploidies, etc.’ (Greilhuber et al., 2005). To avoid confusion, genome sizes were given as 1C or 2C values, reflecting the life stages in which they were measured. Life stages were specified with superscripts as described by Greilhuber and Doležel (2009). For example, Phytophthora mycelium, which is diplophasic, was indicated as 2d C, whereas haploid pycniospores, a monokaryon life stage, were indicated as 1Mk C, and spermatia, which are microgametes, as 1miG C.
A reliable and reproducible genome size estimate depends on standardization. Standardization methods for plant analysis and factors influencing genome size have been described by several authors (Bennett et al., 2003; Doležel and Bartos, 2005; Suda and Leitch, 2010). This review gives an overview on the standardization required for plant analysis, but such an overview is equally valid for all types of FCM genome size analysis.
The crucial factor in genome size estimation is the internal standard, which should: (i) be cytologically stable and uniform, without intraspecific variation; (ii) have a low level of secondary metabolites; (iii) be easily and readily available; (iv) have an appropriate and well‐defined genome size obtained by FCM (Bennett et al., 2003), preferably no more than three times larger/smaller than the sample (Doležel et al., 1992); (v) produce a well‐defined, high‐resolution G1 peak (Barow and Jovtchev, 2007); and (vi) be biologically similar to the sample, meaning that plants should be measured with plant standards, fungi with fungal standards, etc. (Suda and Leitch, 2010). Another important consideration is that only intercalating fluorochromes are suitable for total genome size determination (Doležel et al., 2007a). Other factors, such as buffer constitution, dye concentration and staining time, can also influence the outcome (Bainard et al., 2010).
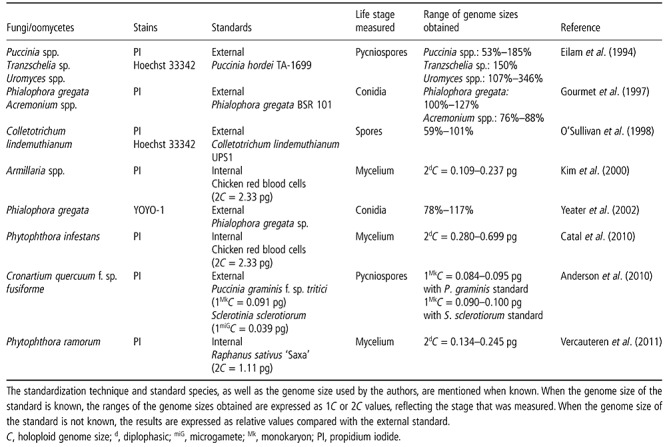
Standardization is still a problem for fungal and oomycete analysis (Kullman et al., 2005) because many standards used for fungal or oomycete analysis today: (i) are subject to intraspecific variations (Catal et al., 2010), chromosomal length polymorphisms (Kullman, 2000), variations in genome size as a result of the gain/loss of complete chromosomes (Zolan, 1995) or, as in chicken red blood cells, differences in sex chromosomes (Mendonca et al., 2010); (ii) contain secondary metabolites, as they are essential to their survival (Howlett, 2006); (iii) are obligate pathogens or are subject to strict biosafety regulations; (iv) have an unknown genome size, a genome size determined by sequencing (Bennett et al., 2003), a genome size that differs widely among studies or a genome size that has been calculated on the basis of an unreliable standard (Greilhuber et al., 2007; Kullman, 2000); (v) are heterokaryotic and hence produce several G1 peaks (Catal et al., 2010); or (vi) meet most of the other criteria, but are not biologically similar (Table 1).
Table 1.
Overview of DNA content measurements with flow cytometry (FCM) relevant to plant pathology.
Plant standards seem to be the best option currently available. They exist for a wide span of genome sizes, are easy to cultivate and some are well described as FCM standards (Loureiro et al., 2007). However, these have drawbacks as well. The genome size of plants is too large compared with most fungi or oomycetes, often necessitating logarithmic measurements, and the condition of biological similarity is not fulfilled. In short, there is a need for stable and well‐characterized fungal and oomycete standards. Regardless, internal standardization remains a necessity for FCM, even when no biologically similar standard is available.
An example of the importance of the use of adequate standards on the resulting genome size is illustrated by the results of Eilam et al. (1994), who estimated the DNA content of rust pycniospores, including Puccinia graminis f. sp. tritici, relative to P. hordei (Table 1). On the basis of these data, Leonard and Szabo (2005) later calculated the absolute genome sizes of these rust fungi, using the sequenced genome size for P. graminis f. sp. tritici (1Mk C= 0.069 pg; Backlund and Szabo, 1993). However, the latest sequenced genome size estimation for P. graminis f. sp. tritici is 1Mk C= 0.091 pg, which is still believed to be an underestimation of the true genome size (Anderson et al., 2010). While gaps remain at telomeres, nucleolus organizer regions (NORs) or centromeres, genome sizes obtained by sequencing will always underestimate the true DNA content as measured with FCM, and should therefore be avoided as standard values (Bennett et al., 2003).
When looking at the overview of DNA content measurements on plant pathogenic fungi and oomycetes in Table 1, it is apparent that intraspecific genome size differences up to 59% were found (Anderson et al., 2010; Kim et al., 2000; O'Sullivan et al., 1998). This is in contrast with plants, where intraspecific genome size variation is controversial (Greilhuber, 1998). Distinct differences in DNA content between fungi isolated from susceptible and resistant plants were reported by Yeater et al. (2002). FCM measurement of Phytophthora species revealed complex nuclear conditions, such as heterokaryosis and indications of aneuploidy (Catal et al., 2010; Vercauteren et al., 2011).
The genome size of plant pathogenic fungi was first estimated in 1980 using Feulgen microspectrophotometry (Typas and Heale, 1980; Voglmayr and Greilhuber, 1998). In general, there is a good correlation between Feulgen and FCM data, but FCM is often the method of choice as it is faster and more accurate (Greilhuber et al., 2007). Feulgen microspectrophotometry estimates the amount of DNA by measuring the amount of light absorbed by a stained nucleus. A more recent method of measuring the DNA amount in Feulgen‐stained nuclei is image analysis densitometry, which measures the staining intensity of a microscope image using a CCD camera and image analysis software (Hardie et al., 2002). Feulgen densitometry is performed on fixed cells on microscope slides. This requires only a very small number of cells and samples can be stored. The disadvantages to this technique are a time‐consuming fixation process and a loss of accuracy because of the small sample volume (Greilhuber, 2008).
Genome sizes of fungi and the method by which they were obtained can be found at http://www.zbi.ee/fungal‐genomesize.
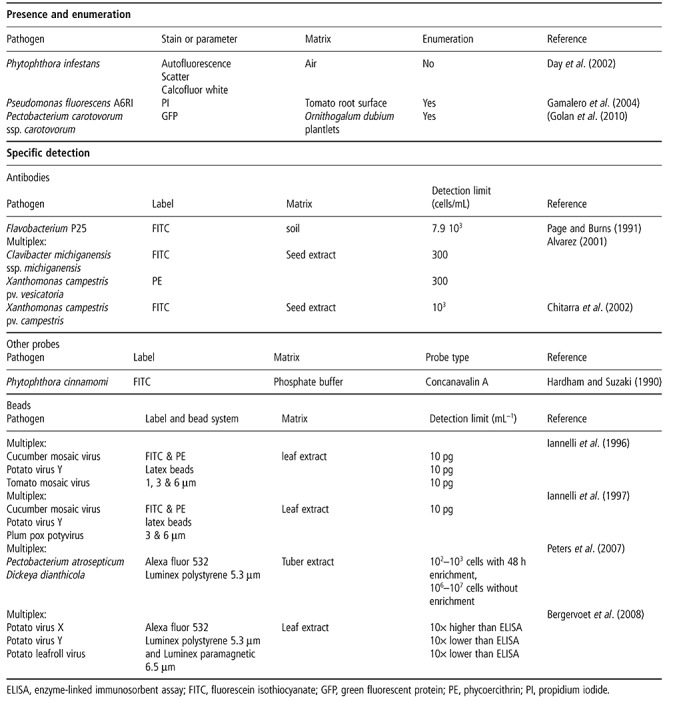
Detection
Presence and enumeration
The discrimination of microorganisms from background particles often depends on fluorescent staining. Extensive lists of fluorescent dyes and their properties are described in Tracy et al. (2010). For nonspecific staining of biological material, DNA stains, such as 4′,6‐diamidino‐2‐phenyl‐indole (DAPI), propidium iodide (PI) and ethidium bromide (EB), are most often used. Online fluorescence spectral viewers can help to select a stain with excitation and emission wavelengths that fit the instrument used. However, other criteria should also be considered, such as membrane permeability, photostability, pH, temperature sensitivity, etc. (Alvarez‐Barrientos et al., 2000; Hammes and Egli, 2010; Tracy et al., 2010).
Absolute cell counting is one of the most straightforward and useful functions of FCM, as it is much faster than microscopy. Total bacterial counts can be used as a quality parameter for water (Hammes and Egli, 2010), food or beverages (Comas‐Riu and Rius, 2009), or as a fast tool to detect microbial contamination in sterile matrices, such as in a cell culture medium (Mchugh and Tucker, 2007).
Applications for plant pathogens are given in Table 2. Day et al. (2002) tested FCM as a means to quickly detect and quantify airborne Phytophthora infestans sporangia based on scatter and autofluorescence, in order to better predict fungicide application times than with climatic models. Gamalero et al. (2004) used FCM and plate counts to quantify and study the evolution of culturable and nonculturable PI‐stained Pseudomonas fluorescens cells in different root zones. Golan et al. (2010) counted GFP‐tagged Pectobacterium carotovorum ssp. carotovorum cells in Ornithogalum dubium plantlets to screen for resistant cultivars in an early growth stage. All of the applications described above are based on nonspecific staining or are meant to study pure cultures or GFP‐tagged organisms. Although some of these applications have a certain degree of specificity, they are unsuitable for the detection of the presence of a specific organism in an environmental sample.
Table 2.
Overview of plant pathogens analysed with flow cytometry (FCM) for detection and enumeration.
Specific detection
Specific detection methods require specific labelling and are mostly based on immunofluorescence or fluorescent in situ hybridization (FISH); labelling of these specific probes can be performed by organic fluorophores, such as fluorescein isothiocyanate (FITC) and phycoercithrin (PE), or by inorganic components, such as quantum dots. Quantum dots have a very good photostability, a broad excitation spectrum and a narrow emission spectrum. However, for environmental samples, they do not always perform better than organic fluorophores (Ferrari and Bergquist, 2007).
Antibodies. Medically relevant fungi, yeasts and parasites are often detected with FCM and fluorescently labelled antibodies (Alvarez‐Barrientos et al., 2000). The detection of numerous bacterial species in a wide range of different body fluids can be accomplished in only 30 min from sample preparation to FCM output with a sensitivity of 100 cells/mL.
In plant pathology, the availability of specific antibodies is often problematic. Nevertheless, specific detection methods with antibodies and FCM have been successfully applied to plant pathogens (Table 2). Chitarra et al. (2002) used FITC‐labelled antibodies and FCM to detect 103 Xanthomonas campestris pv. campestris cells/mL in seed extracts of Brassica sp., even in the presence of nonpathogenic Xanthomonas campestris. Alvarez (2001) reported a detection limit of 300 Clavibacter michiganensis cells/mL in tomato seed extract, in the presence of a 1000 times larger background population. Simultaneous detection of C. michiganensis and X. campestris in the same matrix has also been reported (Alvarez, 2001).
Nucleic acid probes. Flow‐FISH is an alternative to immunoassays. This technique, which is similar to microscopy‐based FISH, uses short nucleic acid oligomers labelled with a fluorescent molecule and hybridized to the target RNA or DNA of the cells. Flow‐FISH can be used to rapidly screen a population or to identify and enumerate one specific organism. The prerequisite for successful FISH is a sufficiently strong signal for detection (Porter et al., 1997a; Vives‐Rego et al., 2000). Therefore, there are very few applications of this technique to date in microbiology in general (Alvarez‐Barrientos et al., 2000), and none in the field of plant pathology.
Other probes. Probes can also be specific to certain receptors or binding sites of a cell. A binding site‐specific FCM assay, using neither oligomers nor antibodies, was performed by Hardham and Suzaki (1990). They used FITC‐labelled concanavalin A (ConA) and soybean agglutinin to quantify the number of ConA binding sites on Phytophthora cinnamomi zoospores during encystment (Table 2).
Beads. Immunoassays in FCM are often combined with beads. Beads are spherical particles, usually with a diameter ranging from a few nanometres to a few micrometres. The beads act as a carrier of the probes that are suspended in the sample. The contact zone between probes and sample is hence much larger than in a well‐plate assay and results in faster binding kinetics. Although beads are most often coated with antibodies, they can also be used with nucleic acid probes or other ligands.
Most bead manufacturers provide beads that can be custom‐coated, such as carboxylated beads, streptavidin‐coated beads, anti‐IgG beads, etc. Although the coating process is technically straightforward in most cases, the optimization of antibody and buffer concentrations can take time, and small changes in the protocol can make an enormous difference.
The simultaneous use of different sizes or colours of antibody‐coated beads allows the simultaneous detection of multiple target cells (Dunbar et al., 2003). For example, 1996, 1997) used different sizes of latex beads for multiplex FCM detection of three different plant pathogenic viruses (Table 2).
A special case of bead‐based immunoassays uses paramagnetic beads and immunomagnetic separation (IMS). IMS allows the rapid and efficient recovery and concentration of target cells, whilst, at the same time, nontarget components are removed from the test material (Boschke et al., 2005). To this end, immunomagnetic beads are incubated with the sample and beads will adhere to the target cells upon collision. When the sample is subsequently placed on a magnetic separator, the beads, and thus target cells, will be drawn to the wall of the sample tube closest to the magnet. This allows the isolation, concentration and purification of target cells prior to analysis. IMS allows the enrichment of rare cells up to 10 000‐fold and is therefore common practice in medical immunology (Grutzkau and Radbruch, 2010). In other fields of study, including plant pathology, IMS is habitually applied as a pre‐enrichment technique prior to plate assays (de Leon et al., 2008), or is used to remove inhibiting components to increase PCR sensitivity (Grant et al., 2000; Walcott et al., 2002). The combination of IMS and FCM allows the fast and selective capture and concentration of target pathogens from complex matrices, combined with rapid quantitative analysis of fluorescently labelled or live/dead stained bacteria (Hibi et al., 2007). The only combination of IMS and FCM for plant pathogen detection was performed by Bergervoet et al. (2008), who used paramagnetic Luminex beads for the simultaneous detection of three potato viruses (Table 2). They found that the use of paramagnetic beads drastically increased the signal‐to‐noise ratio.
Luminex flow cytometers are instruments that are specially and solely designed for bead‐based applications; they use 5.3–6.5‐µm microspheres that are internally dyed with a certain proportion of red and infrared stains. The instruments have a green and red laser; the red laser identifies the bead and the green laser excites the reporter fluorochrome if present. Given the availability of 100 different shades of bead, theoretically 100 different tests can be performed in one analysis. Luminex offers easy‐to‐use kits and platforms for high‐throughput screening; many routine diagnostic tests in medicine are based on this technology (Krishhan et al., 2009; Tait et al., 2009).
For plant pathogens, five is the actual maximum number of pathogens that can be multiplexed with a Luminex kit, and supplies for the detection of three bacterial species and nine viruses are commercially available. Bergervoet et al. (2008) reported an immunoassay with paramagnetic beads for the simultaneous detection of three potato viruses (Table 2). Results comparable with enrichment ELISA and PCR were obtained by Peters et al. (2007), who developed an enrichment microsphere immunoassay for the simultaneous detection of two bacterial potato diseases on the Luminex platform.
Although the Luminex technology has proven its use for routine testing, it is less flexible for research purposes as it only works for bead applications.
Physiological status
Viability
Viability measurement of microorganisms with FCM is frequently used to monitor the efficiency of water treatment (Hammes and Egli, 2010) or to detect viable yeast cells in wine and bacterial contamination of milk (Comas‐Riu and Rius, 2009). In clinical settings, FCM is often the method of choice to test antibiotic, antifungal and antiparasitic drugs on a microbial population (Alvarez‐Barrientos et al., 2000). Although measurement of the PI uptake by FCM is a fast and accurate way to determine antifungal activity (Green et al., 1994), most viability studies performed with FCM involve bacteria. The following section thus focuses on bacterial viability.
The death of a microorganism has long been defined as the inability to grow to a visible colony on culture media (Berney et al., 2007). This definition of viability makes assessment simple: an organism is either alive or dead. However, since the first report of the viable but nonculturable (VBNC) state in bacteria in 1982 (Xu et al., 1982), more and more researchers have reported this third physiological state (Oliver, 2005). The increased use of fluorescent dyes, the growing application of culture‐independent methods and increasingly frequent reports of the VBNC state in bacteria have given rise to a discussion about what is ‘live’ and what is ‘dead’. Cell death is now characterized by parameters such as membrane permeability, deficient efflux pump activity, lack of enzymatic activity, loss of membrane potential, etc. (Joux and Lebaron, 2000). FCM allows the determination of up to seven different stages between living and dead (Joux et al., 1997; Nebe‐von Caron et al., 1998). Therefore, the comparison between live counts by FCM and plate counts may vary, especially for organisms under stress. Even microscopic counts of a live/dead stained population can differ from FCM counts of the same sample, as the human eye cannot dissect the emitted colour into separate wavelengths and operator bias can occur (Jenson et al., 1998).
Some commercial kits for viability assessment of bacteria, yeasts and fungi can be used with both microscopy and FCM. Nevertheless, correct staining should be tested for every new species, as some microorganisms show different staining patterns according to their growth stage (Shi et al., 2007). Viability staining can also be influenced by dye concentrations and combinations (Stocks, 2004), pH (Boulos et al., 1999), incubation time (Yu et al., 1995), temperature (Jernaes and Steen, 1994), salinity (Lebaron et al., 1998; Martens et al., 1981), the presence of soil particles (Pascaud et al., 2009), etc.
Most viability staining protocols used in FCM are based on membrane integrity, esterase activity or membrane potential (Chitarra and van den Bulk, 2003; Sträuber and Müller, 2010). When used correctly, viability staining in combination with FCM is a very fast and accurate tool to research viability and efficacy of treatments in plant pathology, as reviewed by Chitarra and van den Bulk (2003). One example comes from our own research on a Syto9/PI‐stained lettuce pathogen, Pseudomonas cichorii, before and after heat treatment (Fig. 4).
Figure 4.
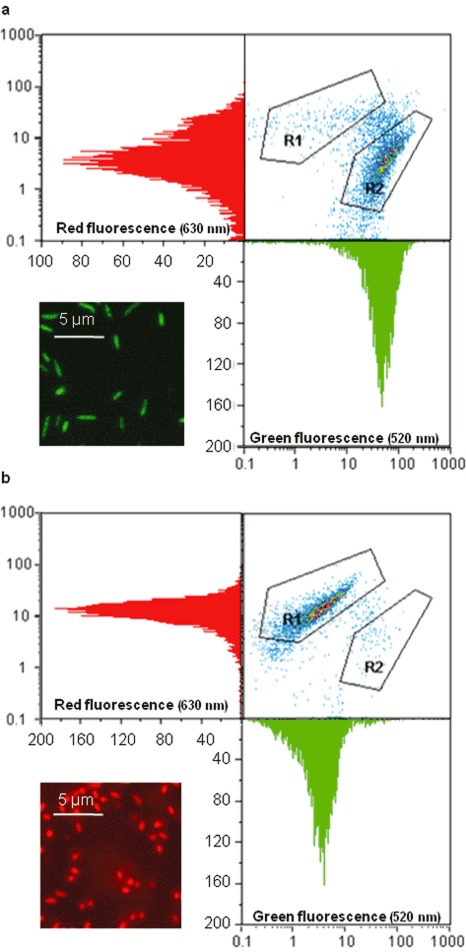
Flow cytometer output and fluorescence microscope images of Syto9/PI stained Pseudomonas cichorii before and after heat treatment (60°C, 10 min). (a) Living P. cichorii have a high green fluorescence intensity and a lower red fluorescence intensity and appear in gate R2 on the 520–630 nm dot plot, a minority of the bacteria are dead and appear in gate R1; microscopic observation shows green fluorescent bacteria. (b) Heat‐killed P. cichorii have a low green fluorescence intensity and a high red fluorescence intensity and appear in gate R1 on the 520–630 nm dot plot, a few bacteria survived and are still visible in gate R2; microscopic observation shows red fluorescent bacteria.
In plant pathology, the major uses of viability application with FCM are research related and often involve the induction of VBNC states. Table 3 gives an overview of viability studies with FCM related to plant pathology. Only Assaraf et al. (2002) used FCM on plant pathogenic fungi to determine stress and viability on conidia during and after heat treatment. Several authors have used FCM on plant pathogenic bacteria to compare different fluorochromes for viability assessment (Chitarra et al., 2006; Porter et al., 1997b), whereas others have tested survival under stress (Ordax et al., 2006; van Overbeek et al., 2004). Most of the authors that have compared FCM with plate counts detected more bacteria using FCM (Chitarra et al., 2006; Ordax et al., 2006; Porter et al., 1997b; van Overbeek et al., 2004); this discrepancy could be as high as 108/mL–1 for bacteria in the VBNC state (Ordax et al., 2006).
Table 3.
Physiological status assessments with flow cytometry (FCM) for plant pathology research.
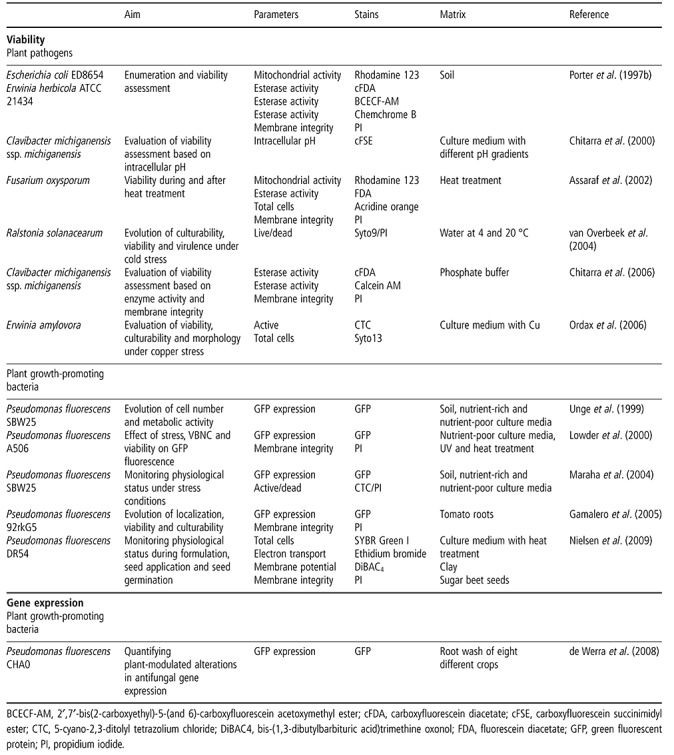
One very promising application of FCM in plant disease research is the monitoring of the physiological status of plant growth‐promoting bacteria after introduction into the soil. In order to optimize the survival and root colonization of microbial inoculants, information is needed about their physiological status in the environment and the influence of stress conditions encountered in the soil. Several authors have used FCM to study fluorescent pseudomonads. FCM assays revealed that almost all bacteria lose culturability, become VBNC or die less than 10 days after introduction into the soil (Table 3).
Gene expression
The expression of specific genes can be measured in cells using GFP‐based reporters (Ghim et al., 2010). As FCM allows for the quantification of fluorescence intensity and for the counting of the number of GFP‐expressing bacteria, the average gene expression per bacterium can be calculated. The only example of this being used in plant pathology is a quantitative FCM study of antifungal gene expression in Pseudomonas fluorescens CHA0 during root colonization (Table 3). Using FCM, significant differences in expression levels between plant species were found (de Werra et al., 2008).
PROS AND CONS OF FCM
FCM is a very fast technique, capable of the analysis of thousands of cells per second. It can thus generate enormous amounts of data. The wide variety of fluorescent markers and stains available makes it possible to screen for a vast range of physiological parameters and biochemical characteristics of cells. Technological advances have resulted in cheaper and more specialized instruments, ranging from small, simple and easy‐to‐operate flow cytometers for one specific application to seven‐laser instruments that allow the simultaneous detection of 32 parameters (Lorkowski and Cullen, 2003). Certain instruments allow volumetric counting, whereas others require a bead standard to determine the cell concentration and exact concentration of any subpopulation defined by the user. Many flow cytometers also have a sorting function, which allows the deflection of subpopulations in real time for culture or further analysis (Bergquist et al., 2009).
Of course, FCM also has disadvantages, some of which result directly from its potential. The first is related to the adaptability of the instrument to specific needs and experimental designs. The user thus needs to implement the adaptations required and to set up the instrument for the intended experiment. The sensitivity and detection threshold of each PMT must be found empirically to detect weak fluorescent signals, but still avoid noise. This requires the appropriate controls and standards; for multicolour experiments, compensation may be necessary to avoid spectral overlap between fluorochromes. Second, flow cytometric outputs still require interpretation. For some applications, this proves to be difficult even with the appropriate controls.
The diversity of plant pathogens—fungi, oomycetes, bacteria, viruses, viroids and phytoplasmas—implicates a wide variety in size, nucleic acid content, shape and structure. Although the first flow cytometers were not designed for the detection of small particles such as bacteria, some of the current instruments are capable of detecting 0.5‐µm particles solely based on scatter properties (Robert et al., 2008). When analysing microorganisms with FCM, it becomes clear that individual microorganisms, even those in ‘clonal’ populations, may differ widely from each other in terms of morphology, genetic composition, physiology or biochemistry (Davey and Kell, 1996). Because of this, FCM outputs of microorganisms often show more variation than expected. This can make it challenging to correctly characterize each group on the outputs.
However, FCM is culture independent. This makes the technique suitable for the analysis of environmental samples and obligate pathogens, as well as organisms in the VBNC state (Oliver, 2005). FCM is particularly valuable for plant pathology, because the number of VBNC reports is steadily increasing (Ordax et al., 2006). One of the major advantages of FCM is quantitative viability assessment. Culture‐independent live/dead assessment is now often evaluated with fluorescence microscopy, but can be performed more rapidly and more precisely with a flow cytometer.
Plant pathogens often have low infection thresholds and usually require concentration before they can be detected. In addition, the isolation or discrimination of the pathogen from its natural environment can also be problematic. Cells that cannot be dispersed into a single‐cell suspension cannot be measured; this can be a problem for biofilm‐forming bacteria or soil‐associated microorganisms. Plant pathogens are present in or on very diverse substrates, such as plant cells, seeds, soil, water, insects, pollen, etc. In general, matrix components, such as culture media, silica particles or chlorophyll, can influence the measurement by causing unwanted background fluorescence and light scattering, or even an extra group on the output. Every application thus requires an adapted protocol.
CONCLUSION
Flow cytometers are one of the most versatile laboratory instruments available, capable of yielding a great amount and wide variety of data, but therefore requiring highly skilled operators.
FCM has had—and is still having—a very significant impact on human cell biology (Steen, 2000). The potential of FCM is much larger for microbiology and, indeed, microbial applications have increased notably over the past few years (Hammes and Egli, 2010). Despite this trend and the hope expressed by some plant pathologists (Bergervoet et al., 2007; Chitarra and van den Bulk, 2003), FCM remains an unknown technique in plant pathology. Nevertheless, FCM is a very valuable tool for the study of fungi, oomycetes, bacteria, viruses and plant–microbe interactions.
One of the most straightforward FCM applications is genome size measurement in fungi and oomycetes. It can reveal a huge amount of information about non‐Mendelian inheritance, chromosomal aberrations, aneuploidy and other genetic processes that contribute to the adaptive process of plant pathogenic fungi and fungus‐like organisms. Fast and specific detection methods for bacteria and viruses will aid in phytosanitary decisions and reduce harvest losses. In particular, simultaneous bead‐based testing for multiple pathogens can speed up the certification of seed lots and be a more cost‐effective alternative for the routine testing of planting material (Bergervoet et al., 2008). Viability staining and the subsequent counting of living and dead cells is a fast and accurate way to identify factors causing stress, the induction of VBNC states and the effectiveness of control measures. Factors inducing the VBNC state in the environment and correct quantification of the number of living pathogens under VBNC‐inducing circumstances can be invaluable for correct risk assessment. The monitoring of rhizosphere colonization of biocontrol strains can provide valuable information on the conditions required for successful biological control strategies.
The lack of basic reagents, protocols and training in nonmedical cytometry presents a major obstacle to the establishment of FCM methods in phytopathology laboratories. As a result of the scarcity of commercially available methods and trained personnel, it is seldom cost‐effective to invest in a flow cytometer solely for phytopathological research. However, that need not be a constraint. Most FCM analyses worldwide are performed by flow core facilities: small groups of trained people operating a variety of machines for an entire hospital, university or company. Commercial services will probably be eager to broaden their horizon and measure plant pathogens. Plant pathologists can and should draw on the knowledge available in medicine and immunology, but there is also value in the machines and the knowledge present closer to home in the plant (breeding) sector.
The potential of FCM in plant pathology is huge, but is hampered by a lack of knowledge. Companies are interested in this new field, but will only provide specialized training and equipment when the market is sufficiently large. This will only happen when more people appreciate the potential of FCM and start to explore it, despite the fact that they have to start from scratch and develop new methods by trial and error. We are trapped in a vicious circle until more plant pathologists see the light and use it.
ACKNOWLEDGEMENTS
The first author would like to thank the Institute for Agricultural and Fisheries Research (ILVO) for the allocated scholarship.
REFERENCES
- Alvarez, A.M. (2001) Differentiation of bacterial populations in seed extracts by flow cytometry In: Plant Pathogenic Bacteria (De Boer S.H., ed.), pp. 393–396. Dordrecht: Kluwer Academic Publishers. [Google Scholar]
- Alvarez‐Barrientos, A. , Arroyo, J. , Canton, R. , Nombela, C. and Sanches‐Perez, M. (2000) Applications of flow cytometry to clinical microbiology. Clin. Microbiol. Rev. 13, 167–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, C.L. , Kubisiak, T.L. , Smith, J.A. and Davis, J.M. (2010) Genome size variation in the pine fusiform rust pathogen Cronartium quercuum f. sp. fusiforme as determined by flow cytometry. Mycologia, 102, 1295–1302. [DOI] [PubMed] [Google Scholar]
- Assaraf, M.P. , Ginzburg, C. and Katan, J. (2002) Weakening and delayed mortality of Fusarium oxysporum by heat treatment: flow cytometry and growth studies. Phytopathology, 92, 956–963. [DOI] [PubMed] [Google Scholar]
- Backlund, J.E. and Szabo, L.J. (1993) Physical characteristics of the genome of the phytopathogenic fungus Puccinia graminis . Curr. Genet. 24, 89–93. [DOI] [PubMed] [Google Scholar]
- Bainard, J.D. , Fazekas, A.J. and Newmaster, S.G. (2010) Methodology significantly affects genome size estimates: quantitative evidence using Bryophytes. Cytometry A, 77, 725–732. [DOI] [PubMed] [Google Scholar]
- Barow, M. and Jovtchev, G. (2007) Endopolyploidy in plants and its analysis by flow cytometry In: Flow Cytometry with Plant Cells (Doležel J., Greilhuber J. and Suda J., eds), pp. 349–372. Weinheim: Wiley‐VCH. [Google Scholar]
- Barrett, D.L. , King, E.B. , Jensen, R.H. and Merrill, J.T. (1976) Cytomorphology of gynecologic specimens analyzed and sorted by 2 parameter flow cytometry. Acta Cytol. 20, 585–586. [PubMed] [Google Scholar]
- Bennett, M.D. , Leitch, I.J. , Price, H.J. and Johnston, J.P. (2003) Comparisons with Caenorhabditis (100 Mb) and Drosophila (175 Mb) using flow cytometry show genome size in Arabidopsis to be 157 Mb and thus 25% larger than the Arabidopsis Genome Initiative estimate of 125 Mb. Ann. Bot. 91, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergervoet, J.H.W. , van der Wolf, J.M. and Peters, J. (2007) Detection and viability assessment of plant pathogenic microorganisms using flow cytometry In: Flow Cytometry with Plant Cells (Doležel J., Greilhuber J. and Suda J., eds), pp. 217–229. Weinheim: Wiley‐VCH. [Google Scholar]
- Bergervoet, J.H.W. , Peters, J. , van Beckhoven, J.R.C.M. , Van den Bovenkamp, G.W. , Jacobson, J.W. and van der Wolf, J.M. (2008) Multiplex microsphere immuno‐detection of potato virus Y, X and PLRV. J. Virol. Methods, 149, 63–68. [DOI] [PubMed] [Google Scholar]
- Bergquist, P.L. , Hardiman, E.M. , Ferrari, B.C. and Winsley, T. (2009) Applications of flow cytometry in environmental microbiology and biotechnology. Extremophiles, 13, 389–401. [DOI] [PubMed] [Google Scholar]
- Berney, M. , Hammes, F. , Bosshard, F. , Weilenmann, H.U. and Egli, T. (2007) Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight kit in combination with flow cytometry. Appl. Environ. Microbiol. 73, 3283–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschke, E. , Steingroewer, J. and Bley, T. (2005) Application of biomagnetic separation to the microbiological quality control of foods. Chem. Ing. Tech. 77, 912–919. [Google Scholar]
- Boulos, L. , Prevost, M. , Barbeau, B. , Coallier, J. and Desjardins, R. (1999) LIVE/DEAD BacLight: application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. J. Microbiol. Methods, 37, 77–86. [DOI] [PubMed] [Google Scholar]
- Brown, M. and Wittwer, C. (2000) Flow cytometry: principles and clinical applications in hematology. Clin. Chem. 46, 1221–1229. [PubMed] [Google Scholar]
- Button, D.K. and Robertson, B.R. (2001) Determination of DNA content of aquatic bacteria by flow cytometry. Appl. Environ. Microbiol. 67, 1636–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catal, M. , King, L. , Tumbalam, P. , Wiriyajitsomboon, P. , Kirk, W.W. and Adams, G.C. (2010) Heterokaryotic nuclear conditions and a heterogeneous nuclear population are observed by flow cytometry in Phytophthora infestans . Cytometry A, 77, 769–775. [DOI] [PubMed] [Google Scholar]
- Chitarra, L.G. and van den Bulk, R.W. (2003) The application of flow cytometry and fluorescent probe technology for detection and assessment of viability of plant pathogenic bacteria. Eur. J. Plant Pathol. 109, 407–417. [Google Scholar]
- Chitarra, L.G. , Breeuwer, P. , van den Bulk, R.W. and Abee, T. (2000) Rapid fluorescence assessment of intracellular pH as a viability indicator of Clavibacter michiganensis subsp michiganensis . J. Appl. Microbiol. 88, 809–816. [DOI] [PubMed] [Google Scholar]
- Chitarra, L.G. , Langerak, C.J. , Bergervoet, J.H.W. and van den Bulk, R.W. (2002) Detection of the plant pathogenic bacterium Xanthomonas campestris pv. campestris in seed extracts of Brassica sp. applying fluorescent antibodies and flow cytometry. Cytometry, 47, 118–126. [DOI] [PubMed] [Google Scholar]
- Chitarra, L.G. , Breeuwer, P. , Abee, T. and van den Bulk, R.W. (2006) The use of fluorescent probes to assess viability of the plant pathogenic bacterium Clavibacter michiganensis subsp. michiganensis by flow cytometry. Fitopat. Brasil. 31, 349–356. [Google Scholar]
- Comas‐Riu, J. and Rius, N. (2009) Flow cytometry applications in the food industry. J. Ind. Microbiol. Biotechnol. 36, 999–1011. [DOI] [PubMed] [Google Scholar]
- Davey, H.M. and Kell, D.B. (1996) Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single‐cell analyses. Microb. Rev. 60, 641–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, J.P. , Kell, D.B. and Griffith, G.W. (2002) Differentiation of Phytophthora infestans sporangia from other airborne biological particles by flow cytometry. Appl. Environ. Microbiol. 68, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, S.J. and Hiruki, C. (1990) Enhanced detection of a plant pathogenic Mycoplasma‐like organism by polymerase chain‐reaction. Proc. Jpn. Acad. B: Phys. Biol. Sci. 66, 140–144. [Google Scholar]
- Doležel, J. and Bartos, J. (2005) Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 95, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doležel, J. , Sgorbati, S. and Lucretti, S. (1992) Comparison of 3 DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant 85, 625–631. [Google Scholar]
- Doležel, J. , Greilhuber, J. and Suda, J. (2007a) Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244. [DOI] [PubMed] [Google Scholar]
- Doležel, J. , Greilhuber, J. and Suda, J. (2007b) Flow Cytometry with Plant Cells. Weinheim: Wiley‐VCH. [Google Scholar]
- Doležel, J. , Greilhuber, J. and Suda, J. (2007c) Flow cytometry with plants: an overview In: Flow Cytometry with Plant Cells (Doležel J., Greilhuber J. and Suda J., eds), pp. 41–66. Weinheim: Wiley‐VCH. [Google Scholar]
- Dunbar, S.A. , Vander Zee, C.A. , Oliver, K.G. , Karem, K.L. and Jacobson, J.W. (2003) Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP (TM) system. J. Microbiol. Methods, 53, 245–252. [DOI] [PubMed] [Google Scholar]
- Dunez, J. (1977) Application of immunoenzymatic techniques to detection of some plant‐viruses—ELISA technique (Enzyme Linked Immunosorbent Assay). Ann. Phytopathol. 9, 219–221. [Google Scholar]
- Eilam, T. , Bushnell, W.R. and Anikster, Y. (1994) Relative nuclear‐DNA content of rust fungi estimated by flow cytometry of propidium iodide‐stained pycniospores. Phytopathology, 84, 728–735. [Google Scholar]
- Engvall, E. and Perlmann, P. (1971) Enzyme‐linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G. Immunochemistry, 8, 871–874. [DOI] [PubMed] [Google Scholar]
- Ferrari, B.C. and Bergquist, P.L. (2007) Quantum dots as alternatives to organic fluorophores for Cryptosporidium detection using conventional flow cytometry and specific monoclonal antibodies: lessons learned. Cytometry A, 71, 265–271. [DOI] [PubMed] [Google Scholar]
- Galbraith, D.W. , Harkins, K.R. , Maddox, J.M. , Ayres, N.M. , Sharma, D.P. and Firoozabady, E. (1983) Rapid flow cytometric analysis of the cell‐cycle in intact plant‐tissues. Science, 220, 1049–1051. [DOI] [PubMed] [Google Scholar]
- Gamalero, E. , Lingua, G. , Capri, F.G. , Fusconi, A. , Berta, G. and Lemanceau, P. (2004) Colonization pattern of primary tomato roots by Pseudomonas fluorescens A6RI characterized by dilution plating, flow cytometry, fluorescence, confocal and scanning electron microscopy. FEMS Microbiol. Ecol. 48, 79–87. [DOI] [PubMed] [Google Scholar]
- Gamalero, E. , Lingua, G. , Tombolini, R. , Avidano, L. , Pivato, B. and Berta, G. (2005) Colonization of tomato root seedling by Pseudomonas fluorescens 92rkG5: spatio‐temporal dynamics, localization, organization, viability, and culturability. Microb. Ecol. 50, 289–297. [DOI] [PubMed] [Google Scholar]
- Ghim, C.M. , Lee, S.K. , Takayama, S. and Mitchell, R.J. (2010) The art of reporter proteins in science: past, present and future applications. Bmb Rep. 43, 451–460. [DOI] [PubMed] [Google Scholar]
- Golan, A. , Kerem, Z. , Tun, O.M. , Luzzatto, T. , Lipsky, A. and Yedidia, I. (2010) Combining flow cytometry and gfp reporter gene for quantitative evaluation of Pectobacterium carotovorum ssp. carotovorum in Ornithogalum dubium plantlets. J. Appl. Microbiol. 108, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Gourmet, C. , Gray, L.E. and Rayburn, A.L. (1997) Flow cytometric analysis of conidia of fungi isolated from soybean vascular tissue. J. Phytopathol. 145, 405–408. [Google Scholar]
- Grant, I.R. , Pope, C.M. , O'Riordan, L.M. , Ball, H.J. and Rowe, M.T. (2000) Improved detection of Mycobacterium avium subsp. paratuberculosis in milk by immunomagnetic PCR. Vet. Microbiol. 77, 369–378. [DOI] [PubMed] [Google Scholar]
- Green, L. , Petersen, B. , Steimel, L. , Haeber, P. and Current, W. (1994) Rapid determination of antifungal activity by flow‐cytometry. J. Clin. Microbiol. 32, 1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, R.T. , Nicol, J.A. , Tamm, H. , Kullman, B. , Kullman, K. , Leitch, I.J. , Murray, B.G. , Kapraun, D.F. , Greilhuber, J. and Bennet, M. (2007) Eukaryotic genome size databases. Nucleic Acids Res. 35, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber, J. (1998) Intraspecific variation in genome size: a critical reassessment. Ann. Bot. 82, 27–35. [Google Scholar]
- Greilhuber, J. (2008) Cytochemistry and C‐values: the less‐well‐known world of nuclear DNA amounts. Ann. Bot. 101, 791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber, J. and Doležel, J. (2009) 2C or not 2C: a closer look at cell nuclei and their DNA content. Chromosoma, 118, 391–400. [DOI] [PubMed] [Google Scholar]
- Greilhuber, J. , Doležel, J. , Lysak, M.A. and Bennett, M.D. (2005) The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C‐value’ to describe nuclear DNA contents. Ann. Bot. 95, 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber, J. , Temsch, E.M. and Loureiro, J.C.M. (2007) Nuclear DNA content measurement In: Flow Cytometry with Plant Cells (Doležel J., Greilhuber J. and Suda J., eds), pp. 67–101. Weinheim: Wiley‐VCH. [Google Scholar]
- Grutzkau, A. and Radbruch, A. (2010) Small but mighty: how the MACS technology based on nanosized superparamagnetic particles has helped to analyze the immune system within the last 20 years. Cytometry A, 77, 643–647. [DOI] [PubMed] [Google Scholar]
- Hammes, F. and Egli, T. (2010) Cytometric methods for measuring bacteria in water: advantages, pitfalls and applications. Anal. Bioanal. Chem. 397, 1083–1095. [DOI] [PubMed] [Google Scholar]
- Hardham, A.R. and Suzaki, E. (1990) Glycoconjugates on the surface of spores of the pathogenic fungus Phytophthora cinnamomi studied using fluorescence and electron microscopy and flow cytometry. Can. J. Microbiol. 36, 183–192. [Google Scholar]
- Hardie, D.C. , Gregory, T.R. and Hebert, P.D.N. (2002) From pixels to picograms: a beginners' guide to genome quantification by Feulgen image analysis densitometry. J. Histochem. Cytochem. 50, 735–749. [DOI] [PubMed] [Google Scholar]
- Heller, F.O. (1973) DNA measurement of Vicia faba L. with pulse cytophotometry. Ber. Deut. Bot. Ges. 86, 437–441. [Google Scholar]
- Hercher, M. , Mueller, W. and Shapiro, H.M. (1979) Detection and discrimination of individual viruses by flow cytometry. J. Histochem. Cytochem. 27, 350–352. [DOI] [PubMed] [Google Scholar]
- Hibi, K. , Mitsubayashi, K. , Fukuda, H. , Ushio, H. , Hayashi, T. , Ren, H. and Endo, H. (2007) Rapid direct determination using combined separation by prepared immunomagnetic and flow cytometry of Flavobacterium psychrophilum . Biosens. Bioelectron. 22, 1916–1919. [DOI] [PubMed] [Google Scholar]
- Howlett, B.J. (2006) Secondary metabolite toxins and nutrition of plant pathogenic fungi. Curr. Opin. Plant Biol. 9, 371–375. [DOI] [PubMed] [Google Scholar]
- Iannelli, D. , D'Apice, L. , Pasquini, G. , Capparelli, R. , Monti, L. , Parrella, G. , Scala, F. and Noviello, C. (1996) Cytofluorometric method for the detection of cucumber mosaic virus. Phytopathology, 86, 959–965. [Google Scholar]
- Iannelli, D. , D'Apice, L. , Cottone, C. , Viscardi, M. , Scala, F. , Zoina, A. , Del, S.G. , Spigno, P. and Capparelli, R. (1997) Simultaneous detection of cucumber mosaic virus, tomato mosaic virus and potato virus Y by flow cytometry. J. Virol. Methods, 69, 137–145. [DOI] [PubMed] [Google Scholar]
- Jackson, P.R. , Winkler, D.G. , Kimzey, S.L. and Fisher, F.M. (1977) Cytofluorograph detection of Plasmodium yoelii, Trypanosoma gambiense, and Trypanosoma equiperdum by laser‐excited fluorescence of stained rodent blood. J. Parasitol. 63, 593–598. [PubMed] [Google Scholar]
- Jenson, H.B. , Grant, G.M. , Ench, Y. , Heard, P. , Thomas, C.A. , Hilsenbeck, S.G. and Moyer, M.P. (1998) Immunofluorescence microscopy and flow cytometry characterization of chemical induction of latent Epstein–Barr virus. Clin. Diagn. Lab. Immunol. 5, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernaes, M.W. and Steen, H.B. (1994) Staining of Esherichia coli for flow cytometry: influx and efflux of ethidium bromide. Cytometry, 17, 302–309. [DOI] [PubMed] [Google Scholar]
- Joux, F. and Lebaron, P. (2000) Use of fluorescent probes to assess physiological functions of bacteria at single‐cell level. Microbes Infect. 2, 1523–1535. [DOI] [PubMed] [Google Scholar]
- Joux, F. , Lebaron, P. and Troussellier, M. (1997) Succession of cellular states in a Salmonella typhimurium population during starvation in artificial seawater microcosms. FEMS Microbiol. Ecol. 22, 65–76. [Google Scholar]
- Kim, M.S. , Klopfenstein, N.B. , McDonald, G.I. , Arumuganathan, K. and Vidaver, L.K. (2000) Characterization of North American Armillaria species by nuclear DNA content and RFLP analysis. Mycologia, 92, 874–883. [Google Scholar]
- Krishhan, V.V. , Khan, I.H. and Luciw, P.A. (2009) Multiplexed microbead immunoassays by flow cytometry for molecular profiling: basic concepts and proteomics applications. Crit. Rev. Biotechnol. 29, 29–43. [DOI] [PubMed] [Google Scholar]
- Kron, P. , Suda, J. and Husband, B.C. (2007) Applications of flow cytometry to evolutionary and population biology. Annu. Rev. Ecol. Evol. Syst. 38, 847–876. [Google Scholar]
- Kullman, B. (2000) Application of flow cytometry for measurement of nuclear DNA content in fungi. Folia Cryptog. Estonica, 36, 31–46. [Google Scholar]
- Kullman, B. , Tamm, H. and Kullman, K. (2005) Fungal genome size database Available at http://www.zbi.ee/fungal‐genomesize[accessed on 30 October 2010].
- Lebaron, P. , Parthuisot, N. and Catala, P. (1998) Comparison of blue nucleic acid dyes for flow cytometric enumeration of bacteria in aquatic systems. Appl. Environ. Microbiol. 64, 1725–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch, I.J. and Bennet, M.D. (2007) Genome size and its uses: the impact of flow cytometry In: Flow Cytometry with Plant Cells (Doležel J., Greilhuber J. and Suda J., eds), pp. 153–176. Weinheim: Wiley‐VCH. [Google Scholar]
- de Leon, L. , Rodriguez, A. , Lopez, M.M. and Siverio, F. (2008) Evaluation of the efficacy of immunomagnetic separation for the detection of Clavibacter michiganensis subsp michiganensis in tomato seeds. J. Appl. Microbiol. 104, 776–786. [DOI] [PubMed] [Google Scholar]
- Leonard, K.J. and Szabo, L.J. (2005) Stem rust of small grains and grasses caused by Puccinia graminis . Mol. Plant Pathol. 6, 99–111. [DOI] [PubMed] [Google Scholar]
- Longobardi, G.A. (2001) Flow Cytometry: First Principles. New York: Wiley‐Liss. [Google Scholar]
- Lopez, M.M. , Llop, P. , Olmos, A. , Marco‐Noales, E. , Cambra, M. and Bertolini, E. (2008) Are molecular tools solving the challenges posed by detection of plant pathogenic bacteria and viruses? Curr. Issues Mol. Biol. 11, 13–45. [PubMed] [Google Scholar]
- Lorkowski, S. and Cullen, P. (2003) Sample preparation and supplementary tools In: Analysing Gene Expression (Lorkowski S. and Cullen P., eds), pp. 97–162. Weinheim: Wiley‐VCH. [Google Scholar]
- Loureiro, J.C.M. , Suda, J. , Doležel, J. and Santos, C. (2007) FLOWer: a plant DNA flow cytometry database In: Flow Cytometry with Plant Cells (Doležel J., Greilhuber J. and Suda J., eds), pp. 423–438. Weinheim: Wiley‐VCH. [Google Scholar]
- Lowder, M. , Unge, A. , Maraha, N. , Jansson, J.K. , Swiggett, J. and Oliver, J.D. (2000) Effect of starvation and the viable‐but‐nonculturable state on green fluorescent protein (GFP) fluorescence in GFP‐tagged Pseudomonas fluorescens A506. Appl. Environ. Microbiol. 66, 3160–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraha, N. , Backman, A. and Jansson, J.K. (2004) Monitoring physiological status of GFP‐tagged Pseudomonas fluorescens SBW25 under different nutrient conditions and in soil by flow cytometry. FEMS Microbiol. Ecol. 51, 123–132. [DOI] [PubMed] [Google Scholar]
- Martens, A.C.M. , Vandenengh, G.J. and Hagenbeek, A. (1981) The fluorescence intensity of propidium iodide bound to DNA depends on the concentration of sodium chloride. Cytometry, 2, 24–25. [DOI] [PubMed] [Google Scholar]
- Mchugh, I.O.L. and Tucker, A.L. (2007) Flow cytometry for the rapid detection of bacteria in cell culture production medium. Cytometry A, 71, 1019–1026. [DOI] [PubMed] [Google Scholar]
- Mendonca, M.A.C. , Carvalho, C.R. and Clarindo, W.R. (2010) DNA content differences between male and female chicken (Gallus gallus domesticus) nuclei and Z and W chromosomes resolved by image cytometry. J. Histochem. Cytochem. 58, 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis, K. , Faloona, F. , Scharf, S. , Saiki, R. , Horn, G. and Erlich, H. (1986) Specific enzymatic amplification of DNA in vitro—the polymerase chain‐reaction. Cold Spring Harb. Symp. Quant. Biol. 51, 263–273. [DOI] [PubMed] [Google Scholar]
- Naumann, U. and Wand, M.P. (2009) Automation in high‐content flow cytometry screening. Cytometry A, 75, 789–797. [DOI] [PubMed] [Google Scholar]
- Nebe‐von Caron, G. , Stephens, P. and Badley, R.A. (1998) Assessment of bacterial viability status by flow cytometry and single cell sorting. J. Appl. Microbiol. 84, 988–998. [DOI] [PubMed] [Google Scholar]
- Nielsen, T.H. , Sjoholm, O.R. and Sorensen, J. (2009) Multiple physiological states of a Pseudomonas fluorescens DR54 biocontrol inoculant monitored by a new flow cytometry protocol. FEMS Microbiol. Ecol. 67, 479–490. [DOI] [PubMed] [Google Scholar]
- O'Sullivan, D. , Tosi, P. , Creusot, F. , Cooke, M. , Phan, T.H. , Dron, M. and Langin, T. (1998) Variation in genome organization of the plant pathogenic fungus Colletotrichum lindemuthianum . Curr. Genet. 33, 291–298. [DOI] [PubMed] [Google Scholar]
- Oliver, J.D. (2005) The viable but nonculturable state in bacteria. J. Microbiol. 43, 93–100. [PubMed] [Google Scholar]
- Ordax, M. , Marco‐Noales, E. , López, M.M. and Biosca, E.G. (2006) Survival strategy of Erwinia amylovora against copper: induction of the viable‐but‐nonculturable state. Appl. Environ. Microbiol. 72, 3482–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Overbeek, L.S. , Bergervoet, J.H.H. , Jacobs, F.H.H. and van Elsas, J.D. (2004) The low‐temperature‐induced viable‐but‐nonculturable state affects the virulence of Ralstonia solanacearum biovar 2. Phytopathology, 94, 463–469. [DOI] [PubMed] [Google Scholar]
- Page, S. and Burns, R.G. (1991) Flow cytometry as a means of enumerating bacteria introduced into soil. Soil Biol. Biochem. 23, 1025–1028. [Google Scholar]
- Palacio‐Bielsa, A. , Cambra, M.A. and Lopez, M.M. (2009) PCR detection and identification of plant pathogenic bacteria: updated review of protocols (1989–2007). J. Plant Pathol. 91, 249–297. [Google Scholar]
- Pascaud, A. , Amellal, S. , Soulas, M.L. and Soulas, G. (2009) A fluorescence‐based assay for measuring the viable cell concentration of mixed microbial communities in soil. J. Microbiol. Methods, 76, 81–87. [DOI] [PubMed] [Google Scholar]
- Porter, J. , Deere, D. , Hardman, M. , Edwards, C. and Pickup, R. (1997a) Go with the flow—use of flow cytometry in environmental microbiology. FEMS Microbiol. Ecol. 24, 93–101. [Google Scholar]
- Porter, J. , Pickup, R. and Edwards, C. (1997b) Evaluation of flow cytometric methods for the detection and viability assessment of bacteria from soil. Soil Biol. Biochem. 29, 91–100. [Google Scholar]
- Peters, J. , Sledz, W. , Bergervoet, J.H.W. and van der Wolf, J.M. (2007) An enrichment microsphere immunoassay for the detection of Pectobacterium atrosepticum and Dickeya dianthicola in potato tuber extracts. Eur. J. Plant Pathol. 117, 97–107. [Google Scholar]
- Rehse, M.A. , Corpuz, S. , Heimfeld, S. , Minie, M. and Yachimiak, D. (1995) Use of fluorescence threshold triggering and high‐speed flow cytometry for rare event detection. Cytometry, 22, 317–322. [DOI] [PubMed] [Google Scholar]
- Robert, S. , Poncelet, P. , Lacroix, R. , Arnaud, L. , Giraudo, L. , Hauchard, A. , Sampol, J. and Dignat‐George, F. (2008) Standardization of platelet‐derived microparticle counting using calibrated beads and a Cytomics FC500 routine flow cytometer: a first step towards multicenter studies? J. Thromb. Haemost. 7, 190–197. [DOI] [PubMed] [Google Scholar]
- Shapiro, H.M. (2003) Practical Flow Cytometry. Hoboken: John Wiley & Sons, Inc. [Google Scholar]
- Shi, L. , Günther, S. , Hübschmann, T. , Wick, L.Y. , Harms, H. and Müller, S. (2007) Limits of propidium iodide as a cell viability indicator for environmental bacteria. Cytometry A, 71, 592–598. [DOI] [PubMed] [Google Scholar]
- Si‐Ammour, A. (2002) Molecular analysis of the Arabidopsis–Phytopthora pathosystem. PhD Thesis. Fribourg: University of Fribourg.
- Steen, H.B. (2000) Flow cytometry of bacteria: glimpses from the past with a view to the future. J. Microbiol. Methods, 42, 65–74. [DOI] [PubMed] [Google Scholar]
- Stocks, S.M. (2004) Mechanism and use of the commercially available viability stain, BacLight. Cytometry A, 61, 189–195. [DOI] [PubMed] [Google Scholar]
- Sträuber, H. and Müller, S. (2010) Viability states of bacteria‐specific mechanisms of selected probes. Cytometry A, 77, 623–634. [DOI] [PubMed] [Google Scholar]
- Suda, J. and Leitch, I.J. (2010) The quest for suitable reference standards in genome size research. Cytometry A, 77, 717–720. [DOI] [PubMed] [Google Scholar]
- Tait, B.D. , Hudson, F. , Cantwell, L. , Brewin, G. , Holdsworth, R. , Bennett, G. and Jose, M. (2009) Review article: luminex technology for HLA antibody detection in organ transplantation. Nephrology, 14, 247–254. [DOI] [PubMed] [Google Scholar]
- Tracy, B.P. , Gaida, S.M. and Papoutsakis, E.T. (2010) Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr. Opin. Biotechnol. 21, 85–99. [DOI] [PubMed] [Google Scholar]
- Typas, M.A. and Heale, J.B. (1980) DNA content of germinating spores, individual hyphal cells and resting structure cells of Verticillium spp. measured by microdensitometry. J. Gen. Microbiol. 121, 231–242. [Google Scholar]
- Unge, A. , Tombolini, R. , Molbak, L. and Jansson, J.K. (1999) Simultaneous monitoring of cell number and metabolic activity of specific bacterial populations with a dual gfp‐luxAB marker system. Appl. Environ. Microbiol. 65, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercauteren, A. (2010) Genetic diversity, mating type, aneuploid sexual reproduction and survival of Phytopthora ramorum . PhD Thesis. Ghent: Ghent University.
- Vercauteren, A. , Boutet, X. , D'hondt, L. , Van Bockstaele, E. , Maes, M. , Leus, L. , Chandelier, A. and Heungens, K. (2011) Aberrant genome size and instability of Phytophthora ramorum oospore progenies. Fungal Genet. Biol. 48, 537–543. doi: 10.1016/j.fgb.2011.01.008 [DOI] [PubMed] [Google Scholar]
- Vives‐Rego, J. , Lebaron, P. and Nebe‐von Caron, G. (2000) Current and future applications of flow cytometry in aquatic microbiology. FEMS Microbiol. Rev. 24, 429–448. [DOI] [PubMed] [Google Scholar]
- Voglmayr, H. and Greilhuber, J. (1998) Genome size determination in Peronosporales (Oomycota) by Feulgen image analysis. Fungal Genet. Biol. 25, 181–195. [DOI] [PubMed] [Google Scholar]
- Walcott, R.R. , Gitaitis, R.D. , Castro, A.C. , Sanders, F.H. and az‐Perez, J.C. (2002) Natural infestation of onion seed by Pantoea ananatis, causal agent of center rot. Plant Dis. 86, 106–111. [DOI] [PubMed] [Google Scholar]
- de Werra, P. , Baehler, E. , Huser, A. , Keel, C. and Maurhofer, M. (2008) Detection of plant‐modulated alterations in antifungal gene expression in Pseudomonas fluorescens CHA0 on roots by flow cytometry. Appl. Environ. Microbiol. 74, 1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H. , Roberts, N. , Singleton, F.L. , Attwell, R.W. , Grimes, D.J. and Colwell, R.R. (1982) Survival and viability of nonculturable Escherichia coli and Vibrio cholerae in the estuarine and marine environment. Microb. Ecol. 8, 313–323. [DOI] [PubMed] [Google Scholar]
- Yeater, K.M. , Grau, C.R. and Rayburn, A.L. (2002) Flow cytometric analysis of Phialophora gregata isolated from soybean plants resistant and susceptible to brown stem rot. J. Phytopathol. 150, 258–262. [Google Scholar]
- Yu, W. , Dodds, W.K. , Banks, M.K. , Skalsky, J. and Strauss, E.A. (1995) Optimal staining and sample storage time for direct microscopic enumeration of total and active bacteria in soil with 2 fluorescent dyes. Appl. Environ. Microbiol. 61, 3367–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolan, M.E. (1995) Chromosome length polymorphism in fungi. Microbiol. Rev. 59, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]