

SUMMARY
Clubroot, caused by Plasmodiophora brassicae, is one of the most serious diseases of cultivated cruciferous crops in the world. However, the basis for pathogenicity in P. brassicae is not well understood. In this study, a serine protease gene (PRO1) was cloned from P. brassicae and its molecular characteristics were investigated. Southern analysis and specific polymerase chain reaction (PCR) amplification indicated that PRO1 is a single‐copy gene present in a broad range of P. brassicae pathotypes. Northern analysis revealed that the expression of PRO1 was induced during plant infection, and that the quantity of transcript fluctuated according to the stage of pathogenesis. Amino acid sequence analysis suggested that the encoded protein (Pro1) belongs to the S28 family of proteases, with a predicted signal peptide and a theoretical molecular mass of 49.4 kDa. The open reading frame (ORF) of PRO1 was transferred into Pichia pastoris and Pro1 was heterologously produced. Pro1 showed proteolytic activity on skimmed milk and N‐succinyl‐Ala‐Ala‐Phe‐7‐amido‐4‐methylcoumarin, and the activity could be inhibited by serine protease inhibitors and the chelating agent ethylenediaminetetraacetic acid. The optimal temperature of Pro1 was 25 °C, and it exhibited high activity at pH 6.0–6.4. These values coincide with the temperature and pH conditions favourable for P. brassicae resting spore germination in the field. When Pro1 was used to treat canola root exudates, it enhanced the stimulating effect of the root exudates on P. brassicae resting spore germination, indicating that Pro1 may play a role during clubroot pathogenesis by stimulating resting spore germination through its proteolytic activity.
INTRODUCTION
Clubroot, caused by Plasmodiophora brassicae Woronin, is one of the most serious diseases of cruciferous crops worldwide. In recent years, this disease has become established on canola (Brassica napus L.) in central Alberta, Canada (Strelkov et al., 2006), where it causes severe yield losses in heavily infected fields. A clubroot‐resistant canola cultivar was recently registered in Canada, but sources of resistance with different modes of action are urgently required. Enhancing our understanding of the biology of the pathogen and the mechanisms of pathogenesis could contribute to the development of novel sources of resistance and other control measures.
The obligate biotrophic nature of P. brassicae hampers the application of the majority of techniques for the study of the molecular mechanisms of pathogenesis. To date, there have been only a few reports on the molecular characterization of P. brassicae genes expressed during the growth of the pathogen in host tissues (Siemens et al., 2009a). 2006, 2007) constructed a suppression subtractive hybridization library between RNA from P. brassicae‐infected and uninfected Arabidopsis tissue. A putative serine protease (GenBank accession number AM411657) was identified among the P. brassicae genes that were expressed during infection. This protease carried a predicted signal peptide (SP) sequence and lacked homologues in other plant pathogens, which may indicate that this gene plays a unique role during clubroot pathogenesis. Our interest in P. brassicae pathogenicity factors led us to choose this gene as the target of the present study.
Proteases are enzymes that cleave proteins by the catalysis of peptide bond hydrolysis. They can be classified into five main classes on the basis of the reactive nucleophile in the active site of the enzyme: cysteine proteases, aspartate proteases, metalloproteases, threonine proteases and serine proteases. Enzymes in each class are further classified into evolutionarily distinct clans, and each clan is subdivided into families on the basis of sequence homology and the order of the catalytic triad (Rawlings and Barrett, 1993). Serine proteases are one of the most important and widely distributed families of proteases, which utilize a uniquely activated serine residue in the substrate‐binding pocket to catalytically hydrolyse peptide bonds. They are found in all organisms and carry out a diverse array of physiological functions, such as digestion, defence and the production of active peptides (Yousef et al., 2003). The pathogenicity function of serine protease has been well studied in protozoans that are pathogenic to humans, such as Plasmodium falciparum (Roggwiller et al., 1996), Trypanosoma cruzi (Burleigh et al., 1997) and Entamoeba species (Barrios‐Ceballos et al., 2005; Makioka et al., 2009).
Serine proteases have also been reported to be important for pathogenicity in plant pathogens. A serine protease and a metalloprotease from Xanthomonas campestris pv. campestris play important roles for pathogenesis in black rot on turnip leaves (Dow et al., 1990). In addition, a serine protease (pat‐1) has been shown to serve as a pathogenicity factor in Clavibacter michiganensis ssp. michiganensis, which causes bacterial wilt of tomato (Dreier et al., 1997). In the potato ring rot pathogen C. michiganensis ssp. sepedonicus, a secreted serine protease Chp‐7 has been identified and proven to be required for full virulence and induction of a nonhost hypersensitive response (Nissinen et al., 2009). Redman and Rodriguez (2002) demonstrated that an extracellular serine protease is required for the pathogenicity of Colletotrichum coccodes, and that the elimination of protease activity transforms a virulent pathogen to an avirulent endophyte. Among isolates of Phytophthora infestans, there was a positive correlation between serine protease‐specific activity and aggressiveness (Hamill et al., 2006). Despite these reports, generally neither the exact mode of action nor the substrate or target of the proteases is known.
In the present study, we cloned the protease sequence from P. brassicae based on sequence information from Bulman et al. (2007), and molecularly characterized the gene and the encoded protein. The objectives of the study were to evaluate the importance of this enzyme and its possible function(s) during pathogenesis in the canola–P. brassicae interaction.
RESULTS
PRO1 encodes a putative S28 family serine protease
Based on the nucleotide sequence of GenBank accession number AM411657 (Bulman et al., 2007), polymerase chain reaction (PCR) primers (Fig 1a,b) were designed for cloning and sequencing purposes. After sequencing, the DNA fragment amplified by primer pair F1/R1 contained an open reading frame (ORF) of 1847 bp in length. We named this ORF PRO1 and its deduced amino acid sequence Pro1. The sequence was submitted to GenBank under the accession number GU082362. Analysis of the PRO1 nucleotide sequence identified eight introns, ranging from 50 to 60 nucleotides in length. All of these intron sequences started with GT and ended with AG, consistent with those described for canonical spliceosomal introns (Rodriguez‐Trelles et al., 2006). A total of 16 nucleotides was found to differ in AM411657 and the exons of PRO1, with nine of these resulting in changes in the encoded amino acids. The translated amino acid sequence of Pro1 consisted of 467 amino acids, with the first 18 amino acids corresponding to a predicted SP and a potential N‐glycosylation site at N113. The sequence without the SP had a molecular mass of 49.4 kDa and a theoretical pI of 5.25. Search for homology by blast indicated that Pro1 was most similar to serine proteases belonging to the S28 family. The best hit of known proteins (e‐value = 8e‐83) was an S28 family protease (GenBank accession number 829777) from Arabidopsis thaliana, which shared 39% identity with Pro1. Alignment of Pro1 with three representatives of the S28 family of serine proteases [a Pro‐X carboxypeptidase (P42785), a dipeptidyl‐peptidase (Q9EPB1) and a thymus‐specific protease (Q9QXE5)] indicated that Pro1 contained the conserved catalytic triad (Ser152–Asp387–His418) of this family (data not shown).
Figure 1.
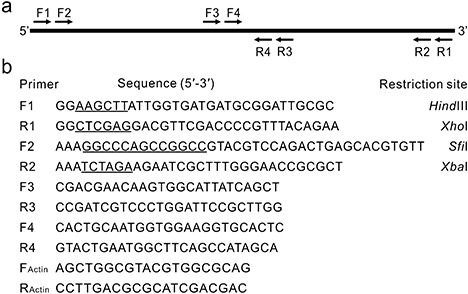
Primers used in this study. (a) Diagrammatic sketch illustrating the genomic position of the primers used to clone and sequence the protease gene PRO1 from Plasmodiophora brassicae. (b) Nucleotide sequences of the primers (with restriction enzyme cutting sites underlined).
PRO1 is a single‐copy gene that is widely present in genomes of P. brassicae
To investigate the genomic organization of PRO1 in the P. brassicae genome, we conducted a Southern analysis using PRO1 to hybridize with P. brassicae genomic DNA digested with selected restriction enzymes. Analysis of the genomic sequence of PRO1 indicated that there were no cutting sites for BamHI, EcoRV and XhoI, and one cutting site for SpeI located at position 1044. Hybridization produced a single band for each of the noncutting enzymes and two bands for SpeI, indicating that PRO1 is present as a single copy in the P. brassicae genome (Fig. 2a). The ubiquity of PRO1 among P. brassicae pathotypes was confirmed by PCR amplification using the primer pair F2/R4 (Fig. 1). A single band of approximately 900 bp was amplified from genomic DNA extracted from each of the four pathotypes tested, as well as from clubroot‐infested soil collected in the field (Fig. 2b).
Figure 2.
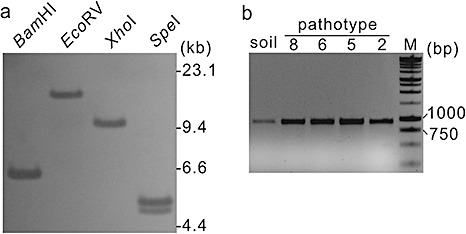
Copy number and occurrence of the protease gene PRO1 in the Plasmodiophora brassicae genome. (a) Southern analysis. Genomic DNA was digested with restriction enzymes as indicated at the top of each lane and hybridized with digoxigenin‐labelled PRO1. (b) Polymerase chain reaction (PCR) amplification of a PRO1 internal fragment from DNA of P. brassicae from soil and from single‐spore isolates of pathotypes P2–P8. The primers used were F2 and R4.
Expression of PRO1 is up‐regulated during pathogenesis
Northern analyses were performed to investigate the expression profile of PRO1 during the infection process. Total RNA was extracted from healthy canola roots and from infected roots at 4–42 days after inoculation (dai). The blot was hybridized successively with PRO1 and the P. brassicae actin gene, PbActin2, which served as a constitutively expressed control to monitor pathogen biomass. No transcript was detected from RNA of healthy canola roots for either PbActin2 or PRO1 (Fig. 3a), indicating the specificity of both probes. Transcript of PRO1, but not PbActin2, was detected at 4 dai (Fig. 3a). It is at this time that primary plasmodia of P. brassicae develop within the root hairs (Fig. 3b), following germination of the resting spores and infection by the primary zoospores (Aist and Williams, 1971). Expression of PRO1, but not of PbActin2, at 4 dai suggested the up‐regulation of the former during the primary infection stage.
Figure 3.
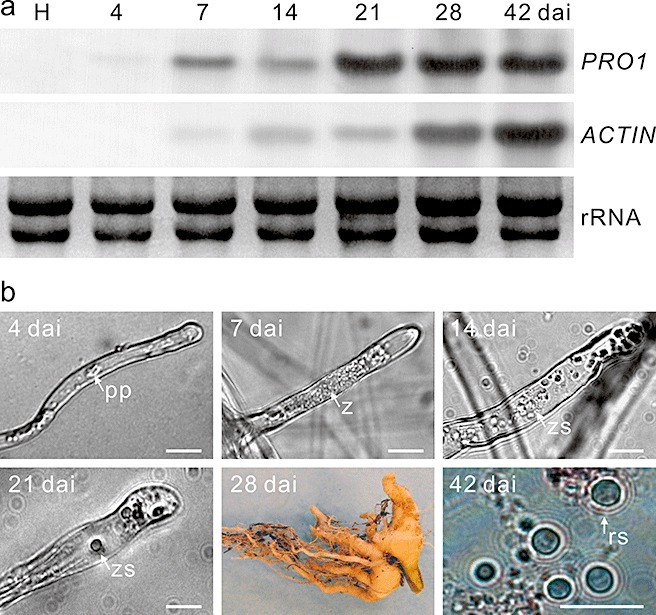
Expression analysis of the Plasmodiophora brassicae protease gene PRO1. (a) Total RNA was extracted from healthy canola roots (H) and P. brassicae‐infected roots collected at 4–42 days after inoculation (dai). The blot was probed successively with PRO1 and the PbActin2 gene (ACTIN). (b) The prevailing stages of P. brassicae in the root samples used for RNA extraction in Northern analysis: an infected root hair (4–21 dai), a clubroot (28 dai) and resting spores extracted from the clubroot and stained with orcein (42 dai). pp, primary plasmodium; rs, resting spore; z, zoosporangium; zs, zoospore. Bars, 10 µm.
Transcript of PbActin2 was detected at low but steady levels from 7 to 21 dai, indicating minimal changes in pathogen biomass during this period and consistent with the stages from secondary zoospore formation to the initiation of cortical infection (Asano et al., 1999; Fig. 3b). In contrast, the amount of PRO1 transcript increased at this time; two conspicuous peaks in expression were evident at 7 and 21 dai (Fig. 3a), which corresponded to the formation of zoosporangia and the initiation of cortical infection, respectively. The initiation of cortical infection at 21 dai was accompanied by the release of secondary zoospores from the zoosporangia (Fig. 3b), and the occasional occurrence of tiny root galls on the taproot (data not shown).
Pathogen biomass increased between 28 and 42 dai, as indicated by the increasing quantities of PbActin2 transcript (Fig. 3a). Transcript levels of PRO1 remained high during this period, but declined at 42 dai both with respect to PRO1 at 28 dai and to PbActin2 at 42 dai. The decline in PRO1 transcript levels coincided with resting spore formation by P. brassicae, and so it appears that expression of this gene declined in resting spores. Collectively, the data indicate that the expression of PRO1 is up‐regulated during pathogenesis, particularly during primary infection, zoosporangium formation and cortical infection, and subsequently declines.
Pro1 is a serine protease and possesses proteolytic activity on skimmed milk
Pro1 was heterologously expressed by transforming Pichia pastoris X‐33 cells with the plasmid pPICZαA‐Pro1, which contained the PRO1 ORF. As a control, transformation was also performed with the empty vector pPICZαA. Crude proteins were extracted from the transformants containing either pPICZαA‐Pro1 or pPICZαA and analysed by sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). A band of approximately 50 kDa was obtained from the pPICZαA‐Pro1 transformant, but not from the pPICZaA transformant, indicating that the former could heterologously produce Pro1 (Fig. 4a). To examine the proteolytic activity of the transformants, a plate assay was conducted with skimmed milk as the substrate. After 48 h of incubation, the pPICZαA‐Pro1 transformant was surrounded by a clear zone, indicating conversion of the casein contained in the skimmed milk into soluble nitrogenous compounds (Fig. 4b). No clear zone was observed in the vicinity of the pPICZαA transformant, confirming that PRO1 is the origin of the proteolytic activity.
Figure 4.
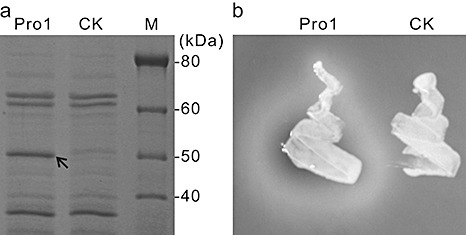
Heterologous expression of the Plasmodiophora brassicae protease Pro1. Pichia pastoris cells were transformed with pPICZαA (CK) or pPICZαA‐Pro1 (Pro1). (a) Crude protein extracted from transformants was fractionated on a sodium dodecylsulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) gel. The arrow indicates the extra band produced by the pPICZαA‐Pro1 transformant. (b) Transformants were cultured on solid media supplemented with 1% (w/v) skimmed milk for 48 h.
Pro1 showed high activity under conditions favourable for clubroot development
The proteolytic activity of the Pichia pastoris transformants was tested quantitatively under various pH and temperature regimes. Within the range of tested conditions, the highest activity was observed at 25 °C and pH 6.0–6.4 (Fig. 5a,b), which is consistent with the pH and temperature conditions that are favourable for clubroot development in the field (Dixon, 2009). Under optimum temperature (25 °C), proteolytic activity was highest at pH 6.0–6.4 and lowest at pH 7.6–8.0 (Fig. 5a). When assessed at pH 6.2, proteolytic activity increased sharply from 22 to 25 °C, declined sharply at 28 °C and then remained low from 31 to 37 °C (Fig. 5b).
Figure 5.
Quantitative assays of activity of the Plasmodiophora brassicae protease Pro1. Activity at a range of pH conditions at 25 °C (a) and at a range of temperatures at pH 6.2 (b), measured by the increased absorbance at 380 nm after 3 h of incubation. (c) Inhibition of protease activity by 1 mm diisopropylfluorophosphate (DFP), 1 mm phenylmethylsulphonylfluoride (PMSF) or 2 mm ethylenediaminetetraacetic acid (EDTA). The Ca2+ concentration in all assay media was 1 mm. (d) Activity in assay media containing different concentrations of Ca2+ and EDTA.
The proteolytic activity of Pro1 could be inhibited by the presence of the serine protease inhibitors diisopropylfluorophosphate (DFP) and phenylmethylsulphonylfluoride (PMSF), as well as the chelating agent ethylenediaminetetraacetic acid (EDTA) (Fig. 5c). PMSF appeared to be the strongest of the three inhibitors. The presence of calcium ions in the assay medium was important for enzyme activity, and this effect was dose dependent (Fig. 5d). The importance of calcium was further substantiated by the observation that the inhibition by EDTA could be restored through the addition of calcium (Fig. 5d).
Pro1 plays a role in resting spore germination
In a preliminary experiment, the germination of resting spores was microscopically investigated in order to develop a repeatable methodology for spore germination assays. We confirmed that germinated spores could be identified on the basis of the absence of refractile globules (Fig. 6a), as described by Naiki et al. (1987). Staining with orcein made nongerminated and germinating (i.e. spores in the germination process) spores easier to distinguish from germinated spores. Differentiation between nongerminated and germinating spores, however, was difficult. Thus, in our experiments, we focused on the spores after germination, which were empty.
Figure 6.
Effect of the protease Pro1 on the germination of resting spores of Plasmodiophora brassicae. (a) Resting spores viewed under a microscope after incubation in water for 2 days. Arrows and arrowheads indicate empty and nonempty spores, respectively. (b) Percentage empty spores in root exudates treated or not treated with Pro1. Means in the columns followed by the same letter do not differ on the basis of Duncan's multiple range test at P ≤ 0.05 (n = 10). MOPS, 3(N‐morpholino)propanesulphonic acid.
We examined the germination of resting spores incubated with either canola root exudates or canola root exudates treated with heterologously produced Pro1. At the beginning of the experiment (day 0), about 2% of the spores were empty in all treatments, and there were no differences among treatments. At day 2, the proportion of empty spores was higher in spores treated with root exudates plus Pro1 than in the other treatments or the control. At days 4 and 6, the presence of canola root exudates had stimulated resting spore germination, regardless of Pro1 treatment (Fig. 6b). Nonetheless, the proportion of empty spores was higher in those incubated with the root exudates plus Pro1 than in all the other treatments. Moreover, in the absence of root exudates, Pro1 itself did not have an effect on resting spore germination (Fig. 6b). These results confirm that resting spore germination can be stimulated by host root exudates, but, more importantly, Pro1 enhanced the stimulating effect of host root exudates by its proteolytic activity.
DISCUSSION
Molecular character of PRO1
The nucleotide sequence of the cloned PRO1 contains a large number of introns (eight within 1847 bp). Interestingly, the two previously reported P. brassicae genes, PbTPS1 (Brodmann et al., 2002) and PbSTKL1 (Ando et al., 2006), also contain one and 14 short introns, respectively. Bulman et al. (2007) concluded that intron‐rich genes are common in the P. brassicae genome, and suggested that P. brassicae might be an example of a eukaryote that has retained ancient intron numbers.
PRO1 is a single‐copy gene broadly present in P. brassicae pathotypes (Fig. 2). If (as we believe) PRO1 is important for pathogenicity, it could serve as a potential target for disease control, because the inhibition of the expression of this single gene could eliminate the corresponding function without interference from other gene copies.
Transcript of PRO1 was detected in host roots collected at 4 dai, but no PbActin2 transcript was detected at that time. There were also higher levels of PRO1 versus PbActin2 transcript in root samples collected at 7 and 21 dai (Fig. 3a). Up‐regulation of PRO1 expression suggests that Pro1 may be important in pathogenesis. To date, the expression profiles of P. brassicae genes correlated with particular stages of pathogenesis have been described in only a few studies (Ando et al., 2006; Brodmann et al., 2002; Ito et al., 1999; Siemens et al., 2009b), and the molecular function of most of these genes remains unknown.
Function of the Pro1 protein
Within the range of tested temperatures and pH conditions, Pro1 exhibited the highest enzyme activity at 25 °C and pH 6.0–6.4 (Fig. 5a). This coincides well with the optimal temperature and pH conditions for resting spore germination, which have been reported to be 24 °C and pH 6.0–6.7 (Dixon, 2009). The activity of Pro1 was shown to be dependent on the presence of calcium ions, probably because of the requirement for calcium as a cofactor for this enzyme. Interestingly, calcium has been reported to suppress clubroot in the soil (Webster and Dixon, 1991). Rather than a direct inhibition of resting spore germination, however, the suppression of P. brassicae by the application of calcium or calcium‐rich organic materials might result from enhanced host resistance (Webster and Dixon, 1991) and/or an increase in soil pH (Niwa et al., 2008). However, the stimulation of resting spore germination by the presence of calcium ions has been reported, and the stimulating effect is believed to be strongest for the germination of immature resting spores (Kageyama and Asano, 2009). Thus, the optimal conditions for Pro1 activity, observed in our study, are consistent with those reported for resting spore germination in the field. It is reasonable to hypothesize that naturally produced Pro1 plays a role in resting spore germination under field conditions. This hypothesis is supported by the observation from Northern analysis that the expression of PRO1 is high during pathogenesis.
Molecular mechanism of resting spore germination stimulation by Pro1
Plant roots can synthesize, accumulate and secrete a diverse array of compounds, such as amino acids, organic acids, polysaccharides and proteins, and these compounds are broadly referred to as root exudates (Flores et al., 1999). Compounds in the root exudates serve important roles as chemical attractants and repellants in the rhizosphere (Walker et al., 2003). Under natural conditions, dormant resting spores require external stimulants for germination (Dixon, 2009). Root exudates, from host as well as nonhost plants, have been reported to trigger the germination of P. brassicae resting spores (Friberg et al., 2005; Niwa et al., 2008; Suzuki et al., 1992), and the term ‘germination stimulating factors’ (GSFs) has been assigned to these triggering factors (Suzuki et al., 1992). In the present study, we confirmed that spore germination was enhanced by host root exudates, and showed that treatment with Pro1 increased germination only in combination with root exudates (Fig. 6). Thus, it appears that the proteolytic products resulting from Pro1 treatment are the true stimulating factors, and not the enzyme itself. It is possible that GSFs consist of two components: (i) compounds innate to the root exudates; and (ii) hydrolytes produced by the enzyme activities of Pro1 and/or other enzymes. Compounds in the first component may occur universally in the root exudates of hosts and nonhosts. It is probable that caffeic acid, coumalic acid and corilagin, identified by Ohi et al. (2003), are among these compounds. For the second GSF component, the involvement of enzymatic activity not only generates stimulatory chemicals, but also provides a potential avenue for the pathogen to recognize its host. Whether Pro1 activity is specific to hosts of P. brassicae requires further investigation.
Pro1 as a pathogenicity factor
As a pathogenicity factor, serine protease has been reported from various pathosystems (Dow et al., 1990; Dreier et al., 1997; Nissinen et al., 2009; Redman and Rodriguez, 2002). Dow et al. (1990) suggested that the role of bacterial proteases may be nutritional or, alternatively, that they could aid in the infection process through the proteolysis of structural proteins in plant cell walls. Although our data for Pro1 cannot distinguish between these two possibilities, they do indicate that the products of proteolysis serve as stimulators to enhance the germination of resting spores. It is also possible that Pro1 is utilized by P. brassicae as a tool to distinguish host from nonhost plants. Recognition and response to host surface environments are important for plant pathogens to establish a successful infection, especially during the early stages of pathogenesis. The fact that infection by P. brassicae is initiated by the germination of resting spores in the soil, and that secondary development promotes plant tissue disorganization (Devos et al., 2005), implies that P. brassicae, like many other phytopathogens, secretes proteins that effect host recognition and the establishment of infection.
The biotrophic nature of P. brassicae precludes gene disruption strategies to obtain a PRO1‐null mutant, thus hindering further investigation into its functions in clubroot development. Nevertheless, three lines of evidence suggest that Pro1 plays a role in the pathogenicity of P. brassicae: (i) the increased abundance of PRO1 transcript during pathogenesis; (ii) the correlation between conditions that are favourable for P. brassicae resting spore germination and for Pro1 activity; and (iii) the stimulation of resting spore germination by the proteolytic activity of Pro1. To our knowledge, PRO1 is the first gene to be functionally demonstrated to have a possible role in the pathogenicity of P. brassicae.
EXPERIMENTAL PROCEDURES
Chemicals and standard techniques
All chemicals were purchased from Fisher Scientific Canada (Ottawa, ON, Canada) unless otherwise specified. Restriction enzymes and PCR kits, including Taq polymerase, reaction buffer and deoxynucleoside triphosphates (dNTPs), were purchased from New England Biolabs (Ipswich, MA, USA). PCR primers (Fig. 1) were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Molecular techniques, if not specified, were performed according to the protocols described by Sambrook and Russell (2001). The accessibility of all websites listed in this paper was verified on the day of submission.
Plant material and P. brassicae isolates
The canola (B. napus) cultivar ‘SP621RR’ was used exclusively as the host in this study. Clubroots that had developed on plants of SP621RR after infection by P. brassicae were collected from experimental field plots near Leduc, AB, Canada (53.22°, −113.70°). Single‐spore isolates of P. brassicae were generated following the method of Xue et al. (2008). One single‐spore isolate, Led09, was used for gene cloning. Another five isolates that had been classified as pathotypes 2, 5, 6 and 8 (Xue et al., 2008) were used in PCR to confirm the ubiquity of PRO1 in the P. brassicae genome.
Nucleic acid extraction
Isolation of P. brassicae resting spores from diseased canola roots was conducted as described by Cao et al. (2007). For PCR, genomic DNA was extracted from the resting spores by a protocol described by Liu et al. (2000), with an additional step in which the resting spores were ground with a mortar and pestle in lysis buffer. For Southern hybridization, high‐molecular‐mass DNA was extracted from the resting spores using the proteinase K and phenol protocol. Purification of DNA from agarose gels was performed with a QIAquick Gel Extraction Kit (Qiagen Canada, Mississauga, ON, Canada).
To prepare the root samples for RNA extraction, seeds of SP621RR were surface sterilized in 1% NaOCl for 5 min, washed with distilled water and germinated on moistened filter paper for 6 days in a growth chamber maintained at 24 °C/18 °C (day/night) with a 16‐h photoperiod and 80% relative humidity. Inoculum of P. brassicae resting spores was prepared by homogenizing diseased roots in a blender and passing the resultant slurry through eight layers of cheesecloth. The prepared resting spores were used to inoculate Sunshine mix #4 soil (Sun Gro Horticulture, Vancouver, BC, Canada) at a concentration of 1 × 107 spores/mL soil. To ensure infection, roots of the seedlings were immersed in a resting spore suspension (5 × 107 spores/mL) for 5 min before transplanting into 15‐cm‐diameter pots filled with the inoculated soil. The pots were kept in trays in a growth chamber as described above, and watered from the bottom every second day with tap water at pH 6.4 (adjusted with HCl).
Healthy roots were collected from the plants grown in the noninoculated soil, 7 days after transplanting, and diseased roots were collected from the inoculated plants at 4, 7, 14, 21, 28 and 42 dai. Infection and development of P. brassicae in the roots were examined at each time point with a Zeiss AX10 microscope (Carl Zeiss, Thornwood, NY, USA). Total RNA was extracted from 0.5 g of healthy or diseased roots using a Promega PureYield RNA Midiprep Kit (Promega, Madison, WI, USA) according to the manufacturer's instructions.
Cloning
The primer pairs F1/R1 and F2/R2 (Fig. 1) were used to amplify two genomic sequences of the PRO1 gene: the ORF with the predicted SP and the ORF without the SP. To increase the fidelity of the amplification, the standard PCR was supplemented with Pfu polymerase (Stratagene, La Jolla, CA, USA) at 5 units/mL. The PCR products were inserted into the pGEM‐T easy vector (Promega) by TA cloning. The resulting vectors were named pPro1ORF and pPro1ORFSP–, respectively.
Sequencing
Sequencing PCR was conducted using plasmid DNA of pPro1ORF as the template with primers F1, F3, F4, R1, R3 or R4 (Fig. 1) in BigDye terminator reagent [Applied Biosystems (ABI), Foster City, CA, USA], following the manufacturer's instructions. PCR products were sequenced with an ABI PRISM 3100 automated DNA sequencer at the University of Alberta, Edmonton, AB, Canada. The complete PRO1 sequence was assembled manually based on the sequences obtained with each of the six primers.
Database search and computational analysis
The genomic DNA sequence of PRO1 was aligned to the cDNA sequence of GenBank accession number AM411657 using Spidey (http://www.ncbi.nlm.nih.gov/IEB/Research/Ostell/Spidey/). Translation of the DNA sequence to its corresponding amino acid sequence was performed with Transeq (http://www.ebi.ac.uk/Tools/emboss/transeq). Similarity searches for Pro1 were conducted with blastp against the National Center for Biotechnology Information (NCBI) database (http://blast.ncbi.nlm.nih.gov). The sequences of other proteases were retrieved from the Peptidase Database MEROPS (http://merops.sanger.ac.uk/). The SP was predicted with SignalP 3.0 software (http://www.cbs.dtu.dk). All other sequence analyses of Pro1 were performed using the appropriate software as accessed from http://ca.expasy.org.
Southern and Northern analyses
In the Southern and Northern analyses, the amount of nucleic acids loaded in each gel lane was 10 µg genomic DNA digested with BamHI, EcoRV, XhoI or SpeI, or 30 µg total RNA. Hybridization, stripping and detection were conducted using the DIG DNA Labelling and Detection Kit (Roche Applied Science Canada, Laval, QC, Canada) according to the manufacturer's instructions. In both analyses, the Dig‐labelled PRO1 ORF cut from pPro1ORF was used as a probe. A partial sequence of the PbActin2 gene (AY452179), amplified with the primer pair FActin/RActin (Siemens et al., 2009b; Fig. 1), was used as the biomass standard in the Northern analyses. The blot was hybridized successively with PRO1 ORF and PbActin2. Posthybridization washes were performed twice for 5 min at 22 °C with 2 × SSC (1 × SSC is 150 mm NaCl, 15 mm sodium citrate), 0.1% SDS and twice for 15 min at 65 °C with 0.5 × SSC, 0.1% SDS. Hybridization was conducted at 50 °C for 16 h and the time of colour development was 60 min.
Heterologous expression of Pro1 protein
The procedures and media recipes used for the work with Pichia pastoris were obtained from the manual accompanying the EasySelect™Pichia Expression Kit (Invitrogen, Carlsbad, CA, USA). For culturing of Pichia pastoris transformants, all media contained 100 µg/mL zeocin and the culture was incubated (dishes) or shaken (250 rpm, liquid cultures) at 30 °C in an incubator shaker.
To construct the vectors for expressing Pro1 in Pichia pastoris, plasmid DNA from pPro1ORFSP‐ was double digested with SfiI/XbaI, and the resulting fragment containing the Pro1 sequence was inserted into the same restriction site of pPICZαA. The constructed vector was termed pPICZαA‐Pro1. This vector, as well as pPICZαA, was digested with PmeI and transferred into Pichia pastoris X‐33 cells by EasyComp™ transformation. The transformants were selected on yeast peptone dextrose (YPD) medium containing 50 µg/mL zeocin and confirmed by PCR using the primer pair F2/R2.
SDS‐PAGE
Single colonies of Pichia pastoris transformants were shaken for 48 h in BMMY medium. Cells were removed by centrifugation at 4000 g for 5 min, and the supernatant was passed through a 0.22‐µm filter and then extracted with 0.5 vol of chloroform–methanol (2 : 1). Extracted proteins were concentrated by precipitation with 10% (w/v) trichloroacetic acid and washed with ice‐cold acetone. All samples were resuspended in the same volume of sample buffer (4% SDS, 0.2 m Tris‐HCl pH 6.8, 20% glycerol, 0.2% bromophenol blue, 1% 2‐mercaptoethanol). Samples were heated in boiling water for 10 min, and the proteins were separated by SDS‐PAGE in a 7% resolving gel using a Mini‐Protean Tetra Cell System (Bio‐Rad Canada, Mississauga, ON, Canada). Gels were stained with Coomassie blue to visualize the protein bands.
Assay for protease activity
Single colonies of the Pichia pastoris transformants on YPD were transferred to Petri dishes (diameter, 10 cm) of BMM medium containing 1% skimmed milk. The medium in each dish was supplemented with 50 µL methanol every 12 h. Degradation of skimmed milk was observed and photographed after 48 h.
Preparation of culture supernatant containing secreted Pro1
Single colonies of Pichia pastoris transformants were cultured in BMG medium for biomass generation for approximately 24 h until the optical density at 600 nm (OD600) reached 4–5. Cells were harvested by centrifugation at 3000 g for 5 min and resuspended in BMM medium to an OD600 of 1.0. The cultures were shaken and supplemented with 50 µL methanol every 12 h. After 48 h, the culture supernatants were collected by centrifugation at 5000 g for 5 min and stored at –20 °C for up to 1 week.
Quantitative protease activity assay
Proteolytic activity in Pichia pastoris culture supernatants was quantitatively evaluated using N‐succinyl‐Ala‐Ala‐Phe‐7‐amido‐4‐methylcoumarin (Suc‐AAF‐AMC; Sigma‐Aldrich Canada, Oakville, ON, Canada) as the substrate at a range of pH values (6.0–8.0) and temperatures (22–37 °C). Twenty microlitres of supernatant were added to 130 µL of 3(N‐morpholino)propanesulphonic acid (MOPS) buffer (0.1 m MOPS, 10 mm CaCl2, 5 mm MgCl2, pH 6.0–8.0) with 0.05 mm substrate and incubated for 3 h. Fluorescence was measured at 380 nm at 0 and 3 h using a NanoDrop 1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). Changes in fluorescence over time reflected the proteolytic activity in the supernatant. The inhibition assays were conducted under optimal temperature (25 °C) and pH (6.2). The inhibitors (DFP, PMSF or EDTA) were added to the assay buffer before mixing with the culture supernatants. All assays were conducted twice with similar results.
Preparation of root exudates from canola
Canola seeds were surface sterilized in 1% NaOCl for 5 min, rinsed three times in sterile distilled water and sown in sterile Pro‐Mix potting mixture (Premier Horticulture, Rivière‐du‐Loup, QC, Canada). The plants were maintained in a growth chamber (16 h light : 8 h dark; photoperiod at 23 °C : 19 °C light : dark; relative humidity 50%) and watered with tap water every other day. After 2 weeks, the plants were harvested and the roots were rinsed with tap water. To collect root exudates, the root systems were placed in a beaker filled with MOPS buffer (pH 6.2) for 24 h in the growth chamber, under the same conditions as described above. The plants were removed and the buffer containing the root exudates was passed through a 0.22‐µm filter and used immediately in the spore germination assays.
Resting spore germination assay
Resting spores isolated from diseased canola roots were surface disinfected following the method of Asano et al. (1999), with a 16‐h antibiotic treatment at 25 °C. The culture supernatants of Pichia pastoris transformants, containing pPICZαA or pPICZαA‐Pro1, were incubated with the same volumes of freshly prepared root exudates at 25 °C for 6 h. These mixtures, as well as MOPS buffer (pH 6.2), 0.5 × pPICZαA‐Pro1 supernatant and 0.5 × root exudates (both diluted with MOPS buffer), were used to treat the prepared resting spores in 1.5‐mL microcentrifuge tubes with 100 µL of root exudate mixture and 100 µL of resting spores. The tubes were incubated at 25 °C in darkness. The germination of resting spores in each treatment was quantified at 0, 2, 4 and 6 days. For each measurement, 10 spore samples were stained with orcein (Sigma‐Aldrich Canada) and 100 spores per sample were counted under a Zeiss AX10 microscope. The data were subjected to analysis of variance using PROC GLM of the SAS statistical package (version 9.1.3, SAS Institute, Cary, NC. USA), and differences among treatments at each time point were assessed using Duncan's multiple range test (P ≤ 0.05). The experiment was repeated, and similar results were obtained in each repetition.
ACKNOWLEDGEMENTS
We thank the funding contributions from the Canola Agronomic Research Program (Alberta Canola Producers Commission, Manitoba Canola Growers Association, SaskCanola and the Canola Council of Canada) and the Advancing Canadian Agriculture and Agri‐Food (ACAAF) Program. We are also grateful to Mr Q. Xiao for technical assistance.
REFERENCES
- Aist, J.R. and Williams, P.H. (1971) The cytology and kinetics of cabbage root hair penetration by Plasmodiophora brassicae . Can. J. Bot. 49, 2023–2034. [Google Scholar]
- Ando, S. , Yamada, T. , Asano, T. , Kamachi, S. , Tsushima, S. , Hagio, T. and Tabei, Y. (2006) Molecular cloning of PbSTKL1 gene from Plasmodiophora brassicae expressed during club root development. J. Phytopathol. 154, 185–189. [Google Scholar]
- Asano, T. , Kageyama, K. and Hyakumachi, M. (1999) Surface disinfestation of resting spores of Plasmodiophora brassicae used to infect hairy roots of Brassica spp. Phytopathology, 89, 314–319. [DOI] [PubMed] [Google Scholar]
- Barrios‐Ceballos, M.P. , Martínez‐Gallardo, N.A. , Anaya‐Velázquez, F. , Mirelman, D. and Padilla‐Vaca, F. (2005) A novel protease from Entamoeba histolytica homologous to members of the family S28 of serine proteases. Exp. Parasitol. 110, 270–275. [DOI] [PubMed] [Google Scholar]
- Brodmann, A. , Schuller, A. , Ludwig‐Muller, J. , Aeschbacher, R.A. , Wiemken, A. , Boller, T. and Wingler, A. (2002) Induction of trehalase in Arabidopsis plants infected with the trehalose producing pathogen Plasmodiophora brassicae . Mol. Plant–Microbe Interact. 15, 693–700. [DOI] [PubMed] [Google Scholar]
- Bulman, S. , Siemens, J. , Ridgway, H.J. , Eady, C. and Conner, A.J. (2006) Identification of genes from the obligate intracellular plant pathogen, Plasmodiophora brassicae . FEMS Microbiol. Lett. 264, 198–204. [DOI] [PubMed] [Google Scholar]
- Bulman, S. , Ridgway, H.J. , Eady, C. and Conner, A.J. (2007) Intron‐rich gene structure in the intracellular plant parasite Plasmodiophora brassicae . Protist, 158, 423–433. [DOI] [PubMed] [Google Scholar]
- Burleigh, B.A. , Caler, E.V. , Webster, P. and Andrews, N.W. (1997) A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+ signalling in mammalian cells. J. Cell Biol. 136, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, T. , Tewari, J. and Strelkov, S.E. (2007) Molecular detection of Plasmodiophora brassicae, causal agent of clubroot of crucifers, in plant and soil. Plant Dis. 91, 80–87. [DOI] [PubMed] [Google Scholar]
- Devos, S. , Vissenberg, K. , Verbelen, J.P. and Prinsen, E. (2005) Infection of Chinese cabbage by Plasmodiophora brassicae leads to a stimulation of plant growth: impacts on cell wall metabolism and hormone balance. New Phytol. 166, 241–250. [DOI] [PubMed] [Google Scholar]
- Dixon, G.R. (2009) Plasmodiophora brassicae in its environment. J. Plant Growth Regul. 28, 212–218. [Google Scholar]
- Dow, J.M. , Clarke, B.R. , Milligan, D.E. , Tang, J.L. and Daniels, M.J. (1990) Extracellular proteases from Xanthomonas campestris pv. campestris, the black rot pathogen. Appl. Environ. Microbiol. 56, 2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier, J. , Meletzus, D. and Eichenlaub, R. (1997) Characterization of the plasmid‐encoded virulence region pat‐1 of phytopathogenic Clavibacter michiganensis subsp. michiganensis . Mol. Plant–Microbe Interact. 10, 195–206. [DOI] [PubMed] [Google Scholar]
- Flores, H.E. , Vivanco, J.M. and Loyola‐Vargas, V.M. (1999) ‘Radicle’ biochemistry: the biology of root‐specific metabolism. Trends Plant Sci. 4, 220–226. [DOI] [PubMed] [Google Scholar]
- Friberg, H. , Lagerlöf, J. and Rämert, B. (2005) Germination of Plasmodiophora brassicae resting spores stimulated by a non‐host plant. Eur. J. Plant Pathol. 113, 275–281. [Google Scholar]
- Hamill, J.A. , Selby, C. and Cooke, L.R. (2006) Aggressiveness of Phytophthora infestans isolates correlates with their proteolytic activity. Acta Hortic. 725, 673–678. [Google Scholar]
- Ito, S. , Ichinose, H. , Yanagi, C. , Tanaka, S. , Kameya‐Iwaki, M. and Kishi, F. (1999) Identification of an in planta‐induced mRNA of Plasmodiophora brassicae . J. Phytopathol. 147, 79–82. [Google Scholar]
- Kageyama, K. and Asano, T. (2009) Life cycle of Plasmodiophora brassicae . J. Plant Growth Regul. 28, 203–211. [Google Scholar]
- Liu, D. , Coloe, S. , Baird, R. and Pedersen, J. (2000) Rapid mini‐preparation of fungal DNA for PCR. J. Clin. Microbiol. 38, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makioka, A. , Kumagai, M. , Kobayashi, S. and Takeuchi, T. (2009) Involvement of serine proteases in the excystation and metacystic development of Entamoeba invadens . Parasitol. Res. 105, 977–987. [DOI] [PubMed] [Google Scholar]
- Naiki, T. , Dixon, G.R. and Ikegami, H. (1987) Quantitative estimation of spore germination of Plasmodiophora brassicae . Trans. Br. Mycol. Soc. 89, 569–609. [Google Scholar]
- Nissinen, R. , Xia, Y. , Mattinen, L. , Ishimaru, C.A. , Knudson, D.L. , Knudson, S.E. , Metzler, M. and Pirhonen, M. (2009) The putative secreted serine protease Chp‐7 is required for full virulence and induction of a nonhost hypersensitive response by Clavibacter michiganensis subsp. sepedonicus . Mol. Plant–Microbe Interact. 22, 809–819. [DOI] [PubMed] [Google Scholar]
- Niwa, R. , Nomura, Y. , Osaki, M. and Ezawa, T. (2008) Suppression of clubroot disease under neutral pH caused by inhibition of spore germination of Plasmodiophora brassicae in the rhizosphere. Plant Pathol. 57, 445–452. [Google Scholar]
- Ohi, M. , Kitamura, T. and Hata, S. (2003) Stimulation by caffeic acid, coumalic acid and corilagin of germination of resting spores of the clubroot pathogen Plasmodiophora brassicae . Biosci. Biotechnol. Biochem. 67, 170–173. [DOI] [PubMed] [Google Scholar]
- Rawlings, N.D. and Barrett, A.J. (1993) Evolutionary families of peptidases. Biochem. J. 290, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman, R.S. and Rodriguez, R.J. (2002) Characterization and isolation of an extracellular serine protease from the tomato pathogen Colletotrichum coccodes, and its role in pathogenicity. Mycol. Res. 106, 1427–1434. [Google Scholar]
- Rodriguez‐Trelles, F. , Tarrio, R. and Ayala, F.J. (2006) Origins and evolution of spliceosomal introns. Annu. Rev. Genet. 40, 47–76. [DOI] [PubMed] [Google Scholar]
- Roggwiller, E. , Bétoulle, M.E.M. , Blisnick, T. and Braun‐Breton, C. (1996) A role for erythrocyte band 3 degradation by the parasite gpgp76 serine protease in the formation of the parasitophorous vacuole during invasion of erythrocytes by Plasmodium falciparum . Mol. Biochem. Parasitol. 82, 13–24. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. and Russell, D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd edn, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Siemens, J. , Bulman, S. , Rehn, F. and Sundelin, T. (2009a) Molecular biology of Plasmodiophora brassicae . J. Plant Growth Regul. 28, 245–251. [Google Scholar]
- Siemens, J. , Graf, H. , Bulman, S. , In, O. and Ludwig‐Müller, J. (2009b) Monitoring expression of selected Plasmodiophora brassicae genes during clubroot development in Arabidopsis thaliana . Plant Pathol. 58, 130–136. [Google Scholar]
- Strelkov, S.E. , Tewari, J.P. and Smith‐Degenhardt, E. (2006) Characterization of Plasmodiophora brassicae populations from Alberta, Canada. Can. J. Plant Pathol. 28, 467–474. [Google Scholar]
- Suzuki, K. , Matumiya, E. , Ueno, Y. and Mizutani, J. (1992) Some properties of germination‐stimulating factor from plants for resting spores of Plasmodiophora brassicae . Ann. Phytopathol. Soc. Jpn. 58, 699–705. [Google Scholar]
- Walker, T.S. , Bais, H.P. , Grotewold, E. and Vivanco, J.M. (2003) Root exudation and rhizosphere biology. Plant Physiol. 132, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, M.A. and Dixon, G.R. (1991) Calcium, pH and inoculum concentration influencing colonization by Plasmodiophora brassicae . Mycol. Res. 95, 64–73. [Google Scholar]
- Xue, S. , Cao, T. , Howard, R.J. , Hwang, S.F. and Strelkov, S.E. (2008) Isolation and variation in virulence of single‐spore isolates of Plasmodiophora brassicae from Canada. Plant Dis. 92, 456–462. [DOI] [PubMed] [Google Scholar]
- Yousef, G.M. , Kopolovic, A.D. , Elliott, M.B. and Diamandis, E.P. (2003) Genomic overview of serine proteases. Biochem. Biophys. Res. Commun. 305, 28–36. [DOI] [PubMed] [Google Scholar]