

Abstract
Organic dyes can be excellent catalysts for photoredox chemistry, offering low price, low toxicity, and an exceptional range of available materials. Their use has been limited because in comparison to their transition-metal catalysts the spectroscopic, kinetic, and electrochemical information available is far more limited. To remediate this situation, we have determined the necessary data for 14 readily available dyes with excellent potential as photoredox catalysts. We have also demonstrated the utility of these dyes through visible-light-mediated reductive dehalogenation and Aza-Henry reactions. We envision that this collection of data will lead to an increase in the use of cationic dyes in photoredox processes because users will find the necessary information readily available.
Introduction
In recent years, the field of light-mediated redox catalysis has experienced remarkable growth because light can provide spatial and temporal control in organic synthesis1−3 under generally mild conditions. Transition-metal complexes, such as Ru(II) and Ir(III) bypiridyl complexes, have been extensively employed in these transformations. The use of organic dyes as photocatalysts for these reactions has also been examined in a few cases,4,5 including examples from the König group with Eosin Y,6 the Nicewicz group with acridinium salts,7−9 and our own group with methylene blue.10,11
Given the low price, low toxicity, and demonstrated ability to perform as redox photoctalysts, the limited use of organic dyes is rather surprising. We hypothesize that one of the primary reasons underlying the popularity of Ru(II) and Ir(III) complexes is that both their photophysical and electrochemical properties have been readily available for over 30 years.12−14 A few organic dyes are just as well understood, for example, methylene blue, but this is not the case for many of the dye options available. In fact, whereas in some cases organic dyes can be even more reactive than their transition-metal counterparts,10,15,16 the same exhaustive collection of photophysical data does not exist in the literature for these photosensitizers. In screening organic dyes for our own work, we realized that this information may be extremely valuable for those in the field and that some of the measurements that we performed with ease in our laboratory are not widely available.
In this article, we provide essential photochemical and electrochemical data for a collection four distinct classes of cationic dyes (Figure 1) that may aid in increasing their use in photoredox catalysis.17−19 We provide a detailed information chart for each of the 14 organic dyes along with the 2 most common Ru and Ir photosensitizers. In addition to these 16 charts, the Supporting Information includes all of the original data for those requiring details in addition to those summarized in the Appendix.
Figure 1.
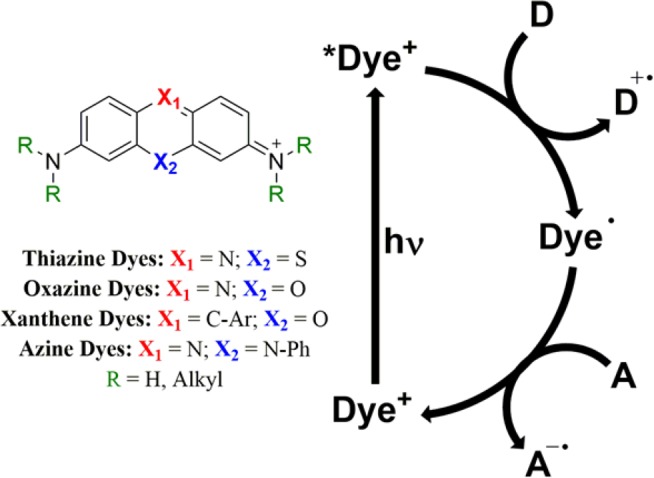
Basic structural representation of the organic dyes investigated in this work and a typical photoredox cycle.
Cationic dyes are excellent candidates for photocatalysis because they provide the advantage of being economically viable while also displaying improved photophysical properties such as increased light absorption across the visible spectrum and longer triplet excited-state lifetimes in comparison to their transition-metal counterparts. In this work, we compare the efficiencies of our cationic dyes with two ubiquitous photoredox catalysts, Ru(bpy)3Cl2 and fac-Ir(ppy)3, along with the 9-mesityl-10-methylacridinium cation originally synthesized by Fukuzumi and co-workers.20 The photosensitizers were compared in both the reduction of meso-1,2-dibromo-1,2-diphenylethane originally studied by Willner in 199021 and the light-mediated Aza-Henry reaction originally studied by Stephenson in 2010.22 Importantly, we also demonstrate that favorable kinetics of electron transfer for mechanistically key steps can correlate to increased reaction efficiency, highlighting the importance of investigating not only the thermodynamic feasibility but also the kinetic feasibility of the catalytic system. Finally, we provide a comprehensive summary of both the photochemical and electrochemical properties of all of the cationic dyes studied in this work, which are summarized in the Appendix.
Experimental Section
General Information
All dyes were purchased from chemical suppliers (Sigma-Aldrich, Alfa Aesar, TCI America, Fisher Scientific) and used without further purification. meso-1,2-Dibromo-1,2-diphenylethane and N,N,N′,N′-tetramethyl ethylenediamine (TMEDA) were purchased from Sigma-Aldrich and Acros Organics, respectively, and used as received. All reactions were irradiated with two 90 W warm-white LEDs purchased from LedEngin unless otherwise noted. (See the SI for the power spectrum.) Flash column chromatography was performed using 230–400 mesh silica gel. All 1H and 13C NMR spectra were recorded on a Bruker AVANCE 400 spectrometer. Chemical shifts (δ) are reported in ppm from the solvent resonance as the internal standard (CDCl3: δ 7.26 ppm).
General Procedure for the Dehalogenation of meso-1,2-Dibromo-1,2-diphenylethane
An oven-dried 10 mL Schlenk tube was charged with meso-1,2-dibromo-1,2-diphenylethane (0.3 mmol, 102 mg) and a photosensitizer (0.003 mmol). The contents were dissolved in 5 mL of dry DMF, followed by the addition of TMEDA (0.6 mmol, 90 μL). (meso-1,2-Dibromo-1,2-diphenylethane is sparingly soluble in DMF; however, as the reaction proceeds, it becomes fully homogeneous.) The reaction was purged with argon for 15 min, followed by 5 min of irradiation with two warm-white LEDs. The reaction mixture was extracted with ether (three times) and washed with brine (five times). The organic phase was dried with MgSO4 and concentrated via rotary evaporation. Percent conversions were determined by 1H NMR analysis.
General Procedure for Aza-Henry Reactions
An oven-dried 10 mL Schlenk flask was charged with 2-phenyl-1,2,3,4-tetrahydrosisoquinoline (0.3 mmol, 63 mg) and a photosensitizer (0.003 mmol). The contents were dissolved in 5 mL of a 4:1 mixture of MeCN/MeNO2. The reaction was then irradiated for 1 h with two warm-white LEDs and diluted with ether (20 mL) and H2O (20 mL). The aqueous phase was extracted with ether (two times), and the combined organic phases were washed with brine, dried with MgSO4, and concentrated by rotary evaporation. All yields are reported as isolated yields.
General Procedure for Laser Flash Photolysis Experiments
The triplet quenching experiments were performed using either a Nd:YAG laser (532) or a Surelite plus OPO (450–700 nm) in a LFP-111 laser flash photolysis sytem (Luzchem Research Inc., Ottawa, Canada) and 1 × 1 cm2 quartz cuvettes. Samples were prepared with a total volume of 3 mL and an absorbance of 0.1 at the excitation wavelength.
General Procedure for Steady-State Quenching Experiments
The fluorescence emission measurements required for the singlet quenching experiments were carried out in a Photon Technology International (PTI) spectrofluorimeter at room temperature using 1 × 1 cm2 quartz cuvettes. The fluorescence lifetime was measured in an Easy-Life (PTI) system and calculated using integrated Easy-Life software. Samples were prepared with a final absorbance of 0.1 at the excitation wavelength. The substrates used in the quenching studies were also prepared in this solution to ensure that the observed quenching is not due to dilution of the fluorophore.
Results and Discussion
Dehalogenation of meso-1,2-Dibromo-1,2-diphenylethane
The dehalogenation of vicinal dibromo compounds employing photoredox techniques has been extensively studied in the literature, including examples from the Willner and Rieser groups employing Ru(bpy)3Cl2 as the photosensitizer21,23 and more recently our own group employing α-sexithiophene.24 Because of the analytical simplicity of this reaction, we decided that it would be an ideal system to test the efficiency of our cationic organic dyes, and it also allowed for kinetic measurements to be performed with ease.
We began by examining the dehalogenation of meso-1,2-dibromo-1,2-diphenylethane under visible-light irradiation by employing methylene blue as the photosensitizer. After only 1 h of irradiation, we could observe nearly quantitative conversion to the dehalogenated product, trans-stilbene (SI). To compare the photocatalytic efficiency of methylene blue with other cationic dyes, a time point was required in order to compare the initial reaction efficiencies of the dyes. We found that decreasing the irradiation time to only 5 min was enough to decrease the conversion of the dibromo compound to 66%. It is important to note here that in these cases only the trans isomer of stilbene was observed under short periods of irradiation time.
The reaction mechanism for this transformation is shown in Scheme 1. Upon excitation with visible light, the photosensitizer’s excited state is generated (either singlet or triplet depending on the photosensitizer studied), which can be quenched by an amine, in our case TMEDA, generating the reduced form of the dye and an amine radical cation. The amine radical cation will be in equilibrium with its deprotonated form, the α-amino radical, which along with the reduced form of the dye can reduce the dibromo compound.24,25 Consistent with this mechanism, control experiments in which the photosensitizer or visible light is omitted do not produce any conversion. Importantly, experiments performed under UV irradiation in the prescence of TMEDA but in the absence of a photosensitizer efficiently dehalogenate the dibromo compound. This is consistent with the proposed chain mechanism in Scheme 1 in which a liberated Br· produced in the reduction of meso-1,2-dibromo-1,2-diphenylethane can abstract a hydrogen from the amine to yield an α-amino radical, which can in turn propagate the chain (SI).24,26,27
Scheme 1. Proposed Catalytic Cycle and Chain Reaction for the Reductive Dehalogenation of meso-1,2-Dibromo-1,2-diphenylethane Using Cationic Organic Photosensitizers.
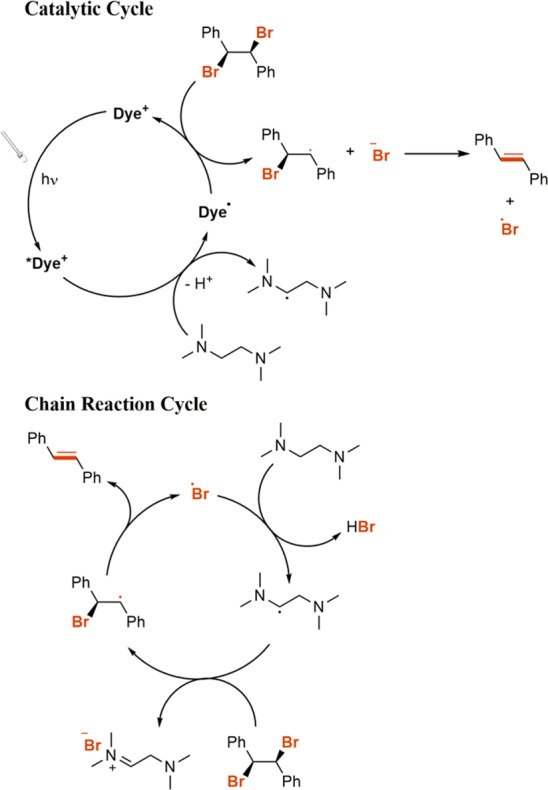
We then proceeded to test a variety of cationic organic photosensitizers (Table 1) whose detailed photophysical and electrochemical properties can be found in the Appendix. Importantly, we also obtained the bimolecular quenching constant (kq) for each cationic organic dye with TMEDA, the sacrificial electron donor. As demonstrated in Table 1, the overall efficiency of the reaction correlates well with the magnitude of the bimolecular quenching constant, indicating the importance that the kinetics of this electron-transfer step plays in the overall mechanism. One exception is the reactivity of 9-mesityl-10-methyl acridinium perchlorate, which should be among the most reactive photosensitizers based on the kinetic data listed in Table 1. It is possible, however, that this is a thermodynamic issue because the reduction potential of 9-mesityl-10-methyl acridinium perchlorate (−0.49 V vs SCE) is not sufficiently negative to reduce meso-1,2-dibromo-1,2-diphenylethane (−1.10 V vs SCE).20,28 The xanthene and oxazine dyes are also less active than their initial bimolecular quenching constants would suggest because of their short singlet-state lifetimes (ns time scales), which greatly decreases the probability of electron -transfer events compared to those for thiazine and azine dyes with longer triplet-state lifetimes (μs time scales). This demonstrates the importance of using triplet photosensitizers to increase the probability of the excited state interacting with a quencher before relaxing back to the ground state, and this will be expanded on further vide infra. It is also important to note here that the kq value observed for rhodamine B is greater than the diffusion control limit of DMF, which can be attributed to static quenching due to ground-state complexation with TMEDA. Once again, it is important to note that the trans isomer was the only isomer observed for all examples.
Table 1. Reductive Dehalogenation of meso-1,2-Dibromo-1,2-diphenylethane Using Cationic Organic Dyes.

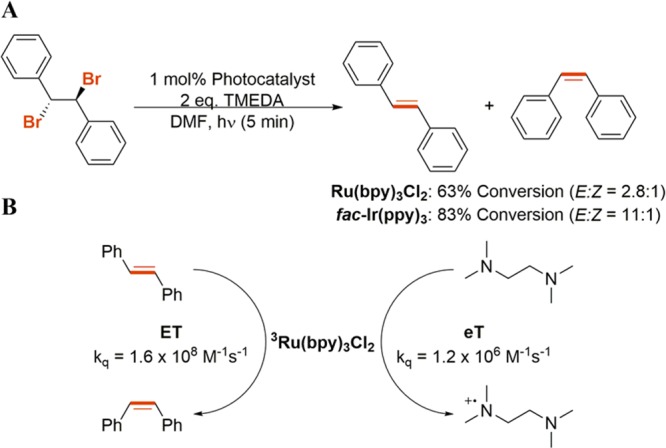
We also examined the reductive dehalogenation of our model vicinal dibromo compound employing Ru(bpy)3Cl2 and fac-Ir(ppy)3, two ubiquitous complexes employed in the field of photoredox catalysis. Although both complexes performed well under the standard employed conditions, we were surprised to observe both the cis and trans isomers of stilbene (Scheme 2A). However, by examining the kinetics of the system for the Ru(bpy)3Cl2 example, we have determined that the loss in selectivity stems from the unfavorable kinetics of the initial electron-transfer step. As shown in Scheme 2B, the bimolecular quenching constant for trans-stilbene and Ru(bpy)3Cl2 is 2 orders of magnitude higher than the corresponding bimolecular quenching constant with TMEDA, the sacrificial electron donor. Therefore, any trans-stilbene formed is able to outcompete TMEDA in the quenching of 3Ru(bpy)3Cl2, resulting in energy transfer and isomerization around the double bond. In good agreement with this, irradiating trans-stilbene in the presence of either Ru(bpy)3Cl2 or fac-Ir(ppy)3 results in the same E/Z ratio observed in the reductive dehalogenation experiments (SI).
Scheme 2. (A) Reductive Dehalogenation Experiments Employing Ru(bpy)3Cl2 and fac-Ir(ppy)3 as the Photosensitizer and (B) Bimolecular Quenching Constants for the Ru(bpy)3Cl2-Catalyzed Reaction.
Aza-Henry Reaction
The visible-light-mediated Aza-Henry reaction of 2-phenyl-1,2,3,4-tetrahydroisoquinoline (PhTHIQ) with nitromethane (MeNO2) as the nucleophile was first studied by Stephenson and co-workers in 2010 and since has become one of the most studied reactions in the field of photoredox catalysis (Scheme 3).22,29 In fact, this reaction has become one of the gold standards when testing the activity of newly developed photocatalysts.30−36 Because of the popularity of the reaction and the availability of in-depth mechanistic studies,37 we decided it would be another excellent test reaction for the cationic dyes employed in this study. Moreover, the simplicity of the reaction greatly facilitates the kinetic analysis of our catalytic systems, allowing us to examine any possible correlations between the xcited-state quenching efficiency and the overall efficiency of the reaction. Because the only quenchers in our system are PhTHIQ and molecular oxygen, by measuring the bimolecular quenching constants of both, we can calculate the probability at which the excited state of the photocatalyst will be quenched by PhTHIQ. The corresponding bimolecular quenching constants can be found in the Appendix.
Scheme 3. Proposed Mechanism for the Visible-Light-Mediated Aza-Henry Reaction.

Note that reactions of 1O2 are viable only for examples in which triplet-state dyes are employed.
The results for the Aza-Henry photocatalyzed reaction are summarized in Table 2. Here, the difference is more pronounced between the triplet- and singlet-state dyes, highlighting the importance of selecting a photocatalyst with a long excited-state lifetime. Once again, 9-mesityl-10-acridinium perchlorate was shown to be much less efficient than the high bimolecular quenching constant would suggest, which we attribute to the lack of reactivity of the intermediate acridinium radical toward molecular oxygen (E1/2 = −0.8 V vs SCE),38 the required step for catalytic turnover. Control experiments were also performed with methylene blue as the photocatalyst, and it was demonstrated that the reaction did not proceed in the absence of O2, consistent with the previously reported mechanisms (SI). More specifically, it has been proposed that either singlet oxygen (1O2) or superoxide (O2•–) plays a prominent role in the overall mechanism (Scheme 3),22,37 and upon addition of 1,3-diphenylisobenzofuran (DPBF), an efficient 1O2 and O2•– quencher,39,40 the reactivity is substantially diminished, consistent with the proposed mechanism of previous reports (SI). Laser flash photolysis studies confirmed that the diminished reactivity does indeed arise from the quenching of the reactive oxygen species by DPBF and not from the quenching of 3MB by DPBF.
Table 2. Light-Mediated Aza-Henry Reaction of 2-Phenyl-1,2,3,4-tetrahydroisoquinoline with MeNO2 Using a Cationic Organic Dye.

With all of the kinetic data in hand, we can employ eq 1 to determine the probability that the excited state of the dye (*PC) reacts with PhTHIQ under our initial reaction conditions.
| 1 |
This calculation was performed with all 13 cationic dyes that were examined in this study, and the results were plotted against the yield of the corresponding Aza-Henry reaction (Figure 2). Although the correlation is not strong, this plot still demonstrates that a majority of the more efficient reactions are for the larger probabilities of excited-state quenching. It is also important to note that the highest probabilities for *PC quenching by PhTHIQ occur when triplet-state dyes (blue • and green ■) are employed, which one would predict considering the effect that the excited-state lifetime plays in eq 1. Furthermore, the rate-limiting step of this reaction is the addition of MeNO2 and not the initial excited-state electron transfer to the amine, which could affect the correlation observed in Figure 2.22,37 However, it is still clear that by optimizing the mechanistically key steps of the system, even if it is not the rate-limiting step, one can increase the overall efficiency of the reaction.
Figure 2.
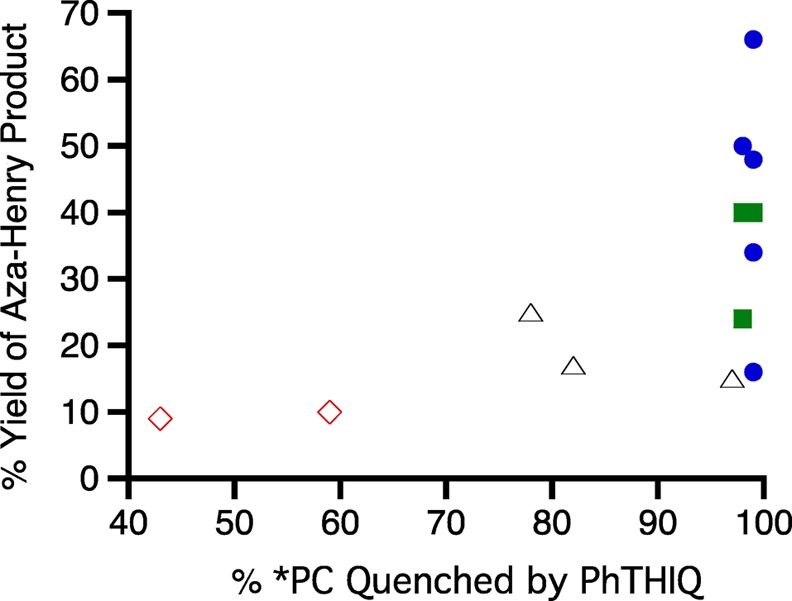
Plot of percent yield of the Aza-Henry product versus the percent of *dye quenched by PhTHIQ for all cationic dyes examined in this study. Dye legend: thiazine (blue •), oxazine (red ◊), xanthene (Δ), and azine (green ■).
We can also perform a similar analysis on the thermodynamic feasibility of these reactions. Because the oxidation potential of PhTHIQ is known (0.90 V vs SCE),41 we can use this value along with the ground-state reduction potentials and the excited-state energies of each dye to calculate the Gibbs free energy of photoinduced electron transfer (ΔGeT) for each reaction using the following equation:
| 2 |
This calculation was performed for all 13 cationic dyes examined in this study, and the results were plotted against the yield of the Aza-Henry product (Figure 3). We would typically expect that a more negative ΔGeT would lead to a more favorable and therefore more efficient reaction. However, we essentially see the opposite trend in Figure 3, as the more favorable reactions (more negative ΔGeT) give the lowest yields after 2 h of irradiation. Upon further examination, we can see that the more negative ΔGeT corresponds to the singlet excited-state dyes (red ◊ and Δ), which can be correlated to their higher excited-state energies compared to those of the triplet-state dyes. However, because of their short singlet-state lifetimes, the probability of electron transfer is greatly decreased, as seen in Figure 2, even though the electron-transfer event is more favorable. This highlights the importance of performing kinetics studies because even though a reaction can have favorable thermodynamics it is ultimately kinetics that determines to what extent the reaction proceeds.
Figure 3.
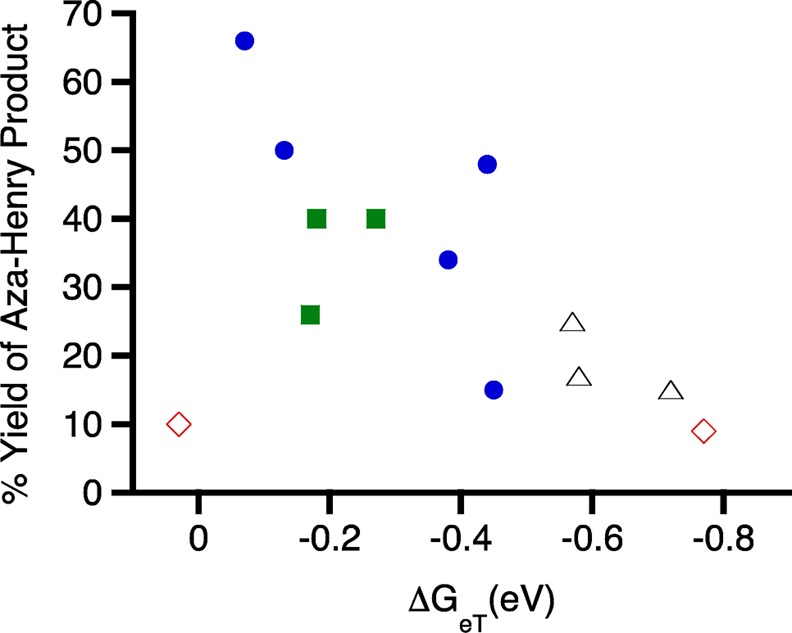
Plot of percent yield of the Aza-Henry product versus the Gibbs’ free energy for photoinduced electron transfer (ΔGeT) for all cationic dyes examined in this study. Dye legend: thiazine (blue •), oxazine (red ◊), xanthene (Δ), and azine (green ■).
Similar to the reductive dehalogenation reactions, we also performed the light-mediated Aza-Henry reaction employing both Ru(bpy)3Cl2 and fac-Ir(ppy)3, which gave 27 and 26% yields of the final Aza-Henry product, respectively. One would expect similar results for both photocatalysts because their rate constants for bimolecular quenching with PhTHIQ are of the same order of magnitude (2.9 × 107 and 3.1 × 107 M–1s–1 for Ru(bpy)3Cl2 and fac-Ir(ppy)3, respectively).
Conclusions
In this work, we have demonstrated that cationic dyes can act as viable metal-free alternatives to transition-metal complexes for visible-light-mediated photoredox transformations. We have demonstrated the utility of these dyes in both the reductive dehalogenation of a vicinal dibromo compound and the visible-light-mediated Aza-Henry reaction of PhTHIQ. In the majority of these examples, improved kinetics of electron transfer between the excited state of the photocatalyst and the amine resulted in an overall increase in the reaction efficiency. This highlights the importance of optimizing the kinetics of each mechanistically key step, even if it is not the rate-limiting step of the reaction.
Importantly, we have also provided a summary of all of the photophysical and electrochemical properties of these cationic dyes, which are readily available in the Appendix. With this information in hand, we envision that this will result in an increase in popularity of these cationic dyes being employed in photoredox processes because this information will now be readily available to laboratories who may lack the necessary equipment and/or expertise to perform these studies.
Acknowledgments
We acknowledge the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), and the Canada Research Chairs program as the primary supporters of this work. S.P.P. and C.D.M. thank the NSERC for providing Canadian Graduate Studies-Doctoral (CGS-D) scholarships.
Appendix




Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00058.
Warm-while LED lamp spectrum, details of experimental conditions for test reactions, quenching plots, NMR spectra, determination of triplet energies, cyclic voltammograms, and quenching conditions for 50 and 90% quenching efficiencies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D. M.; Yoon T. P. Solar synthesis: prospects in visible light photocatalysis. Science 2014, 343, 1239176. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Nicewicz D. A.; Nguyen T. M. Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014, 4, 355–360. 10.1021/cs400956a. [DOI] [Google Scholar]
- Ravelli D.; Fagnoni M.; Albini A. Photoorganocatalysis. What for?. Chem. Soc. Rev. 2013, 42, 97–113. 10.1039/C2CS35250H. [DOI] [PubMed] [Google Scholar]
- Hari D. P.; Konig B. Synthetic applications of eosin Y in photoredox catalysis. Chem. Commun. 2014, 50, 6688–6699. 10.1039/C4CC00751D. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Margrey K. A.; Tay N. E.; Nicewicz D. A. Site-selective arene C-H amination via photoredox catalysis. Science 2015, 349, 1326–1330. 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Mechanistic Insight into the Photoredox catalysis of Anti-Markovnikov alkene hydrofunctionalization reactions. J. Am. Chem. Soc. 2014, 136, 17024–17035. 10.1021/ja506228u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilger D. J.; GrandjeanJean-Marc M.; Lammert T. R.; Nicewicz D. A. The direct anti-Markovnikov addition of mineral acids to styrenes. Nat. Chem. 2014, 6, 720–726. 10.1038/nchem.2000. [DOI] [PubMed] [Google Scholar]
- Pitre S. P.; McTiernan C. D.; Ismaili H.; Scaiano J. C. Mechanistic insights and kinetic analysis for the oxidative hydroxylation of arylboronic acids by visible light photoredox catalysis: A metal-free alternative. J. Am. Chem. Soc. 2013, 135, 13286–13289. 10.1021/ja406311g. [DOI] [PubMed] [Google Scholar]
- Pitre S. P.; McTiernan C. D.; Ismaili H.; Scaiano J. C. Metal-free photocatalytic radical trifluoromethylation utilizing Methylene Blue and visible light irradiation. ACS Catal. 2014, 4, 2530–2535. 10.1021/cs5005823. [DOI] [Google Scholar]
- Juris A.; Balzani V.; Barigelletti F.; Campagna S.; Belser P.; von Zelewsky A. Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- Flamigni L.; Barbieri A.; Sabatini C.; Ventura B.; Barigelletti F.. Photochemistry and photophysics of coordination compounds: Iridium. In Photochemistry and Photophysics of Coordination Compounds II; Balzani V., Campagna S., Eds.; Springer: Berlin, 2007; pp 143–203. [Google Scholar]
- Ballardini R.; Varani G.; Indelli M. T.; Scandola F.; Balzani V. Free energy correlation of rate constants for electron transfer quenching of excited transition metal complexes. J. Am. Chem. Soc. 1978, 100, 7219–7223. 10.1021/ja00491a017. [DOI] [Google Scholar]
- Guo S.; Zhang H.; Huang L.; Guo Z.; Xiong G.; Zhao J. Porous material-immobilized iodo-Bodipy as an efficient photocatalyst for photoredox catalytic organic reaction to prepare pyrrolo[2,1-a]isoquinoline. Chem. Commun. 2013, 49, 8689–8691. 10.1039/c3cc44486d. [DOI] [PubMed] [Google Scholar]
- Huang L.; Zhao J.; Guo S.; Zhang C.; Ma J. Bodipy derivatives as organic triplet photosensitizers for aerobic photoorganocatalytic oxidative coupling of amines and photooxidation of dihydroxylnaphthalenes. J. Org. Chem. 2013, 78, 5627–5637. 10.1021/jo400769u. [DOI] [PubMed] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 2016, 27, 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- Cosa G.; Focsaneanu K. S.; McLean J. R. N.; McNamee J. P.; Scaiano J. C. Photophysical properties of fluorescent DNA-dyes bound to single- and double-stranded DNA in aqueous buffered solution. Photochem. Photobiol. 2001, 73, 585–599. . [DOI] [PubMed] [Google Scholar]
- Shaath N. A. Encyclopedia of UV absorbers for sunscreen products. Cosmetics & Toiletries 1987, 102, 21–37. [Google Scholar]
- Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. Electron-transfer state of 9-Mesityl-10-methylacridinium ion with a much longer lifetime and higher energy than that of the natural photosynthetic reaction center. J. Am. Chem. Soc. 2004, 126, 1600–1601. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- Willner I.; Tsfania T.; Eichen Y. Photocatalyzed and electrocatalyzed reduction of vicinal dibromides and activated ketones using ruthenium(I) tris(bipyridine) as electron-transfer mediator. J. Org. Chem. 1990, 55, 2656–2662. 10.1021/jo00296a023. [DOI] [Google Scholar]
- Condie A. G.; González-Gómez J. C.; Stephenson C. R. J. Visible-light photoredox catalysis: Aza-Henry reactions via C–H functionalization. J. Am. Chem. Soc. 2010, 132, 1464–1465. 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]
- Maji T.; Karmakar A.; Reiser O. Visible-light photoredox catalysis: dehalogenation of vicinal dibromo-, α-halo-, and α,α-dibromocarbonyl compounds. J. Org. Chem. 2011, 76, 736–739. 10.1021/jo102239x. [DOI] [PubMed] [Google Scholar]
- McTiernan C. D.; Pitre S. P.; Scaiano J. C. Photocatalytic dehalogenation of vicinal dibromo compounds utilizing Sexithiophene and visible-light irradiation. ACS Catal. 2014, 4, 4034–4039. 10.1021/cs501030f. [DOI] [Google Scholar]
- Wayner D. D. M.; McPhee D. J.; Griller D. Oxidation and reduction potentials of transient free radicals. J. Am. Chem. Soc. 1988, 110, 132–137. 10.1021/ja00209a021. [DOI] [Google Scholar]
- Scaiano J. C.; Barra M.; Krzywinski M.; Sinta R.; Calabrese G. Laser flash photolysis determination of absolute rate constants for reactions of bromine atoms in solution. J. Am. Chem. Soc. 1993, 115, 8340–8344. 10.1021/ja00071a048. [DOI] [Google Scholar]
- Scaiano J. C.; Barra M.; Sinta R. Chain amplified photoacid generation from vicinal dibromides. A general strategy for the efficient generation of hydrogen bromide across the ultraviolet and visible spectrum. Chem. Mater. 1996, 8, 161–166. 10.1021/cm950337d. [DOI] [Google Scholar]
- Bosser G.; Paris J. Dehalogenation of vicinal dibromides by electrogenerated polysulfide ions in dimethylacetamide. J. Chem. Soc., Perkin Trans. 2 1992, 2057–2063. 10.1039/p29920002057. [DOI] [Google Scholar]
- Hu J.; Wang J.; Nguyen T. H.; Zheng N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. 10.3762/bjoc.9.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.-X.; Li Y.; Wu X.; Xiao J.; Adams D. J.; Cooper A. I. Conjugated microporous polymers with Rose Bengal dye for highly efficient heterogeneous organo-photocatalysis. Macromolecules 2013, 46, 8779–8783. 10.1021/ma402104h. [DOI] [Google Scholar]
- Pan L.; Xu M.-Y.; Feng L.-J.; Chen Q.; He Y.-J.; Han B.-H. Conjugated microporous polycarbazole containing tris(2-phenylpyridine)iridium(iii) complexes: phosphorescence, porosity, and heterogeneous organic photocatalysis. Polym. Chem. 2016, 7, 2299–2307. 10.1039/C5PY01955A. [DOI] [Google Scholar]
- Wang B.; Shelar D. P.; Han X.-Z.; Li T.-T.; Guan X.; Lu W.; Liu K.; Chen Y.; Fu W.-F.; Che C.-M. Long-lived excited states of zwitterionic Copper(I) complexes for photoinduced cross-dehydrogenative coupling Reactions. Chem. - Eur. J. 2015, 21, 1184–1190. 10.1002/chem.201405356. [DOI] [PubMed] [Google Scholar]
- Wang J.-L.; Wang C.; deKrafft K. E.; Lin W. Cross-linked polymers with exceptionally high Ru(bipy)32+ loadings for efficient heterogeneous photocatalysis. ACS Catal. 2012, 2, 417–424. 10.1021/cs300027n. [DOI] [Google Scholar]
- Xie Z.; Wang C.; deKrafft K. E.; Lin W. Highly stable and porous cross-linked polymers for efficient photocatalysis. J. Am. Chem. Soc. 2011, 133, 2056–2059. 10.1021/ja109166b. [DOI] [PubMed] [Google Scholar]
- Zhang W.-Q.; Li Q.-Y.; Zhang Q.; Lu Y.; Lu H.; Wang W.; Zhao X.; Wang X.-J. Robust metal–organic framework containing benzoselenadiazole for highly efficient aerobic cross-dehydrogenative coupling reactions under visible light. Inorg. Chem. 2016, 55, 1005–1007. 10.1021/acs.inorgchem.5b02626. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zhang C.; Chin K. F.; Pytela O.; Wei G.; Liu H.; Bures F.; Jiang Z. Dicyanopyrazine-derived push-pull chromophores for highly efficient photoredox catalysis. RSC Adv. 2014, 4, 30062–30067. 10.1039/C4RA05525J. [DOI] [Google Scholar]
- Liu Q.; Li Y.-N.; Zhang H.-H.; Chen B.; Tung C.-H.; Wu L.-Z. Reactivity and mechanistic insight into visible-light-induced aerobic cross-dehydrogenative coupling reaction by organophotocatalysts. Chem. - Eur. J. 2012, 18, 620–627. 10.1002/chem.201102299. [DOI] [PubMed] [Google Scholar]
- Juillard J. Dimethylformamide: purification, tests for purity and physical properties. Pure Appl. Chem. 1977, 49, 885–892. [Google Scholar]
- Ohyashiki T.; Nunomura M.; Katoh T. Detection of superoxide anion radical in phospholipid liposomal membrane by fluorescence quenching method using 1,3-diphenylisobenzofuran. Biochim. Biophys. Acta, Biomembr. 1999, 1421, 131–139. 10.1016/S0005-2736(99)00119-4. [DOI] [PubMed] [Google Scholar]
- Farmilo A.; Wilkinson F. On the mechanism of quenching of singlet oxygen in solution. Photochem. Photobiol. 1973, 18, 447–450. 10.1111/j.1751-1097.1973.tb06448.x. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Kee C. W.; Chen L.; Tan C.-H. Dehydrogenative coupling reactions catalysed by Rose Bengal using visible light irradiation. Green Chem. 2011, 13, 2682–2685. 10.1039/c1gc15489c. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.