

Abstract
In this article, we argue that the primary role of isoprene is to remove the singlet delta oxygen (O21Δg) that forms inside plants by ultraviolet excitation rather than to provide heat protection or scavenge ozone, OH, or other reactive oxygen species (ROS) in the gas phase. By deploying a quantum chemical framework, we address for the first time the exact mode of isoprene reactions with O21Δg, the most prominent ROS that causes damage to leaves. Initial reactions of isoprene with O21Δg comprise its addition at the two terminal carbon atoms. The two primary open-shell adducts that appear in these reactions undergo 1,2-cycloaddition to generate methyl vinyl ketone and methacrolein, the sole products detected from in-house (i.e., inside of plants) oxidation of isoprene. Formation of other products, comprising the peroxy O–O bonds, is kinetically insignificant. Furthermore, these adducts are thermodynamically too unstable to diffuse outside of plants. Oxidation of isoprene with O21Δg does not produce new ROS (such as OH or HO2), supporting the well-documented role of isoprene as an effective ROS scavenger. Deploying a solvation model reduces the energy requirements for the primary pathways in the range of 10–56 kJ/mol. The present results indicate that plants attach significant value to the in-home protection against O21Δg by investing carbon and energy into the formation of isoprene, in spite of the appearance of the cytotoxic methyl vinyl ketone as one of the reaction products. (The same chemical species also form in unrelated gas-phase reactions involving isoprene and other ROS.) This finding explains the primary reason for the appearance of the dynamic biosphere–atmosphere exchange of methyl vinyl ketone.
1. Introduction
Emission of isoprene (2-methyl-1,3-butadiene) from many terrestrial plants accounts for nearly one-third of the annual global budget of volatile organic compounds (VOCs) from all biogenic and anthropogenic sources combined.1 Owing to its high chemical reactivity, once emitted to the atmosphere, isoprene plays a crucial role in many atmospheric phenomena.2 This includes modifying the mechanisms of the atmospheric removal of other VOCs, influencing the formation of secondary organic aerosol and affecting the production of ozone. Although isoprene exerts no greenhouse gas (GHG) effect on its own right, its fast reaction with OH radicals can change the atmospheric oxidation capacity and the atmospheric lifetime of methane (the third most important GHG). But this is not the reason why plants invest their carbon and energy resources to synthesize isoprene.
A significant debate has been sweeping the field on reasons for plants to produce isoprene in an operation that is both biochemically expensive and energy dependent.3 Mounting experimental evidence demonstrates that isoprene undergoes complex heat-tolerant mechanisms in plants. The direct production of isoprene from light-dependent photosynthesis indicates a plausible thermoslike protection of leaves from certain types of heat shocks.4 However, the most discussed role of isoprene is the mitigation of the effect of reactive oxygen species (ROS) in plants.5 By acting as an ROS scavenger, isoprene can prevent visible damage caused by exposure of leaves to ozone or OH radicals and reduce the loss in their photosynthetic capacity. Nonetheless, isoprene–ROS reactions remain to be elucidated, especially those occurring inside plants.
The lowest excited state of singlet delta oxygen (O21Δg) constitutes the most prominent ROS causing damage to leaves6 in such a way that this exotic species accounts for over 80% of lipid oxidation within leaf tissues.7 Photo-induced activation produces O21Δg in plants, mainly mediated by the excited state of the triplet chlorophyll (3Chl*).8 The literature has documented well the O21Δg scavenging properties of isoprene.9 Isoprene acts as an effective quenching agent for many ROS, including the O21Δg.3b Through the use of rose bengal as a photo-sensitizer, experiments9a,9b have demonstrated that isoprene protects plants against the damaging effect of O21Δg. In fact, ultraviolet (UV) excitation of an isoprene/ground state oxygen mixture results in the formation of O21Δg.10 Because the biochemical synthesis of isoprene displays light-dependency, the resistance to O21Δg correlates positively with the light intensity.3a However, the exact mode of isoprene attacks on O21Δg remains largely unclear. Jardine et al.11 have experimentally simulated a “within-plant” oxidation process of isoprene to report methyl vinyl ketone and methacrolein as the sole oxidation products. The authors linked the formation of these two compounds with the functionality of isoprene as an ROS scavenger but without pointing to O21Δg, in particular.
Here, we suggest that the main role of isoprene is to remove O21Δg that forms inside of plants by the photo-induced activation process. From this perspective, by deploying quantum chemical calculations, this article provides a detailed mechanistic account of the reactions of O21Δg with isoprene. Because of the highly transient nature of O21Δg (lifetime of a few microseconds),12 the reaction chemistry of O21Δg constitutes an excellent candidate for a purely theoretical scrutiny. The reaction mechanism and the kinetic parameters developed herein will assist in formulating an atomic-scale understanding of the protective functions of isoprene in plants against ROS, particularly O21Δg.
2. Methodology
Normally, treatment of the biradical character exhibited by the adduct of isoprene and O21Δg would require the application of multireference wave function methods such as CASSAF and CASSTP2.13 The pioneering work by the Yamaguchi14 group has demonstrated that the energies of the biradical systems computed by any single-determinant method can be significantly improved by the application of a simple approximate spin-projection (AP) scheme. In this scheme, an approximate spin-projected energy (EAP) can be derived from the energies of the broken-symmetry (EBS) and pure high-spin (EHS) states
where fAP denotes the spin-projection factor,
and ⟨S2⟩HS and ⟨S2⟩BS signify the expectation values of spin contamination pertinent to the pure high-spin and broken-symmetry states, respectively. Application of the AP methodology to various biradical systems yields B3LYP-derived energies to within 4.2–12.6 kJ/mol of the corresponding values obtained with more expensive multireference methods.15 In a series of contributions,16 Cremer and co-workers showed that unrestricted density functional theory (UDFT) methods perform surprisingly well in deriving energies and geometries of singlet biradicals.
Accordingly, we deploy the density functional theory (DFT) functional of B3LYP with the extended 6-311+G(d,p)17 basis set to investigate the biradical system of isoprene and O21Δg. Final energies are corrected using the AP approach as described above, whenever the reacting species (including reactants, products, and transition states) attain a biradical character. As an initial accuracy benchmark, we calculate the triplet–singlet delta enthalpic gap of the oxygen molecule to be 85.4 kJ/mol, in a relatively good agreement with the corresponding experimental value at 94.1 kJ/mol.12 Standard UB3LYP calculations result in a significantly larger enthalpy gap of 161.5 kJ/mol, demonstrating the necessity of applying the AP approach. Table S1 enlists the calculated thermal enthalpies and the expected spin contamination values, that is, values required in calculations based on the AP methodology. We carry out all structural optimizations and energy calculations using the Gaussian 09 suite of programs.18 We have carefully confirmed the identity of each transition structure by performing intrinsic reaction coordinate (IRC) calculations.
The ChemRate code facilitates the calculation of the reaction rate constants, in the temperature range of 300–600 K.19 We obtain the Fukui20 indices for electrophilic attack (f-1) and the electronic Hirshfeld21 charges with the aid of DMol3 code.22 The Fukui f–1 indices serve as a molecular descriptor, reflecting the tendency of a certain site in a chemical species to undergo an electrophilic addition.
3. Results and Discussion
The structure of isoprene features a conjugated π system, where the digits 1 and 2 denote two sites for the addition of O21Δg (Figure 1). Figure 2 depicts the mechanism of the reactions of isoprene with O21Δg, with the geometries of intermediates and transition structures portrayed separately in Figures S1 and S2. The electrophilic properties of O21Δg enable the introduction of oxygen atoms into olefins, dienes, and aromatic structures through three main categories of reactions: 1,2-cycloaddition to double bonds, 1,4-cycloaddition to aromatic rings, and the so-called concerted ene reaction. Generally,23 the nonconcerted mechanism underlying the reactions of typical dienes with the O21Δg passes through diradical intermediates. For instance, the diradical peroxy adduct has been previously observed in the case of cis-1,3-butadiene.13b Similarly, in this study, the simulations led to two diradical intermediates (M1 and M14).
Figure 1.

Isoprene chemical structure.
Figure 2.

Reaction routes in the isoprene + O21Δg system obtained by the calculation level of B3LYP/6-311+G(d,p). The values in bold and italics signify reaction and activation enthalpies computed at 298.15 K, respectively (in kJ/mol).
All attempts to locate the transition structures preceding 1,2-/1,4-cyloadditions or a direct abstraction of a methyl hydrogen atom (i.e., an ene reaction) converge to the peroxy adducts M1 and M14, in which the oxygen molecule binds to the terminal carbon atoms at sites 1 and 14, respectively. If it operates in our system, ene reaction should result in the formation of an allylic hydroperoxide upon a shift in the position of the most electron-rich double bond (i.e., species M3 in Figure 2). If the occurrence of an ene is a feasible opening channel, this would violate the role of isoprene as an ROS scavenger because it results in the generation of another potent ROS species, namely the OH radical, through dissociation of the weak O–OH bond.
Although peroxirane and zwitterionic intermediates were detected on the potential energy surface in the reaction between butadiene and O21Δg,24 despite our best efforts, we were unable to locate these intermediates. Nonetheless, conversion of the initially formed diradical peroxy-type adduct into peroxirane was shown to be a highly endothermic process by about 100 kJ/mol. Analogously, a related study25 predicted that a zwitterionic adduct resides nearly 40 kJ/mol above the initially formed diradical intermediates.
Rotation of the peroxy O–O bond in M1 and M14 adducts could potentially generate syn and anti conformers. However, performing a partial optimization along the corresponding dihedral angles leads to structures in which the outer oxygen atom either abstracts a hydrogen atom or adds to the carbon skeleton (one of the products in Figure 2); viz., stable syn and anti conformers do not arise in M1 and M14.
The M1 and M14 moieties, in addition to their transition structures TS1 and TS12, possess an open-shell character. On the basis of AP-corrected enthalpies (298.15 K), the reaction barrier of TS12 (99 kJ/mol) overshoots that of TS1 by 51 kJ/mol. To elucidate the different reactivities for the addition of O21Δg at sites 1 and 2, Figure 3 displays the calculated Fukui indices and electronic charges on isoprene. The higher electron densities and f–1 values at site 2 in comparison with the analogous estimates at site 1 concur with the lower activation barrier of TS1 with reference to TS12.
Figure 3.
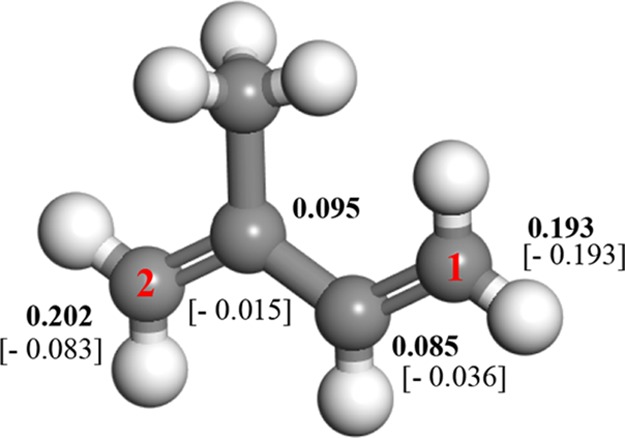
Fukui f–1 indices (in bold) and Hirshfeld charges (in brackets).
The two initial adducts, M1 and M14, reside at a similar level above the entrance channel. As Figure 2 depicts, the initial adduct M1 branches into seven exit routes, whereas M14 launches six channels. In the main channel, a 1,2-cycloaddition occurs via TS7 overcoming a modest enthalpic barrier of 46 kJ/mol and forms the M7 adduct. A very exothermic C–C bond fission (−376 kJ/mol) in M7 provides the experimental product for in-house oxidation of isoprene, the methyl vinyl ketone molecule. This process takes place through a barrier of 92 kJ/mol embedded in TS8.
The transition states TS2, TS4, and TS6 from the main channel, along with TS13 and TS14 signify H-migration reactions, in which the outer oxygen atom abstracts a hydrogen atom from the neighboring carbon atoms through sizable reaction barriers amounting to 151, 166, 80, 86, and 143 kJ/mol, respectively. Fission of the relatively weak O–OH bond in the structure of hydroperoxide intermediates (such as M5 and M16) requires modest bond dissociation enthalpies in the range of 141–147 kJ/mol. Nonetheless, significantly higher barriers for the formation of allylic hydroperoxide (80–166 kJ/mol) with reference to the barrier of the main route (46 kJ/mol) indicate that the formation of the propagating OH radical from the unimolecular rearrangement of M1 is highly unlikely. All our attempts to find transition states connecting M1 directly with M3 and M5 have led to TS2 and TS4, respectively. This interpretation has been guided by detailed IRC calculations and vibration analysis. The formation of hydroperoxide M6 from M1, as well as M15 from M14, occurs via H abstraction from the CH2 site by the terminal oxygen atom of the peroxy group, with modest reaction barriers of 80 kJ/mol (TS7) and 86 kJ/mol (TS14), respectively.
Transition structures TS9 (137 kJ/mol) and TS10 (67 kJ/mol) engender 1,3- and 1,4-cycloadditions commencing from M1. Formation of the six-membered ring, M11 structure, from M1 and M14 proceeds via similar reaction barriers through TS10 and TS18. The peroxy O–O bonds in the molecules are typically very weak, that is, 150 kJ/mol.26 This explains the absence of M10, M11, and M19 from the products of isoprene oxidations initiated by O21Δg.
Both M1 and M14 are capable of forming short-lived dioxirane species M13 and M21 via a two-step pathway, initiated by 1,2-hydrogen transfer characterized by the TS11 (115 kJ/mol) and TS19 (117 kJ/mol) transition states. The corresponding biradical intermediates of M12 and M20 then pass through a barrierless unimolecular cyclization to form the highly stable heterocyclic peroxides M13 and M21. Because the reaction barriers associated with TS11 and TS19 significantly overshoot the barriers of the primary channels (TS7 and TS15), synthesis of dioxiranes from the reaction of isoprene with O21Δg is largely negligible.
On the basis of the reaction and activation barriers displayed in Figure 2, the channel associated with the attack of O21Δg molecule on the double bond at site 2 produces methyl vinyl ketone predominantly. Likewise, addition of oxygen at site 1 preferentially forms the second experimentally detected product of methacrolein. The kinetic parameters listed in Table 1 reveal that the overall isoprene + O21Δg reaction proceeds mainly by the addition of O21Δg at site 2 (M1) and forms methyl vinyl ketone molecule in a three-step mechanism (M1 → M7 → M8 + M9). All other channels display negligible importance, including isomerization of M1 into the six-membered cyclic peroxide M11.
Table 1. Arrhenius Parameters for Reactions Encountered during Oxidation by O21Δg.
| reaction | A (s–1 or cm3 molecule–1 s–1) | Ea (kJ/mol) |
|---|---|---|
| isoprene + O2 1Δg → M1 | 2.60 × 10–12 | 55 |
| M1 → M2 | 2.36 × 1012 | 132 |
| M2 → M3 | 4.55 × 1013 | 22 |
| M1 → M4 | 1.22 × 1013 | 147 |
| M4 → M5 | 1.80 × 1013 | 26 |
| M1 → M6 | 1.17 × 1013 | 61 |
| M1 → M7 | 2.58 × 1012 | 27 |
| M7 → M8 + M9 | 3.51 × 1013 | 180 |
| M1 → M10 | 1.24 × 1012 | 118 |
| M1 → M11 | 8.17 × 1011 | 48 |
| M1 → M12 | 1.37 × 1013 | 96 |
| isoprene + O21Δg → M14 | 1.16 × 10–12 | 105 |
| M14 → M15 | 1.39 × 1013 | 66 |
| M14 → M16 | 1.69 × 1012 | 122 |
| M14 → M17 | 4.88 × 1012 | 29 |
| M17 → M18 + M9 | 2.55 × 1013 | 143 |
| M14 → M19 | 1.31 × 1012 | 108 |
| M14 → M20 | 2.47 × 1012 | 39 |
| M14 → M21 | 1.88 × 1013 | 97 |
To investigate the solvent effect on the reaction energetics for the two main pathways (i.e., formation of M8 and M18 from M1 and M14, respectively), we deploy a polarizable continuum model (PCM)27 at the B3LYP/6-311g+(d,p) level of theory. Figure 4 contrasts the energies obtained in the gaseous and aqueous media. The PCM approach simulates the solvent effect by generating multiple overlapping spheres inside of a dielectric continuum, around the atoms within the molecule. The PCM predicts a systematic energy reduction in the range of 10 kJ/mol (TS1) to 56 kJ/mol (TS12) for transition states, intermediates, and final products along the two primary channels. Overall, the effect of the solvent is limited to reducing the energy penalties whereas the product distribution (i.e., based on kinetic values in Table 1) remains unchanged with reference to the gas-phase system.
Figure 4.

Potential energy diagram of isoprene + O21Δg reaction with (red) and without (black) solvent (water) effects.
Methyl vinyl ketone also constitutes a major product from the OH-initiated atmospheric oxidation of isoprene.28 However, in accordance with the experimental finding of Jardine et al.,11 we have shown that methyl vinyl ketone also appears from a within-plant oxidation of isoprene with O21Δg. As Jardine et al.11 have indicated, this calls for a reconsideration of the biogenic life cycle of isoprene and its oxidation products. Clearly, plants devote a significant effort into their protection against O21Δg by producing isoprene in a diurnal cycle29 that reflects the formation of O21Δg, in spite of the appearance of the cytotoxic methyl vinyl ketone as the reaction product.
Acknowledgments
This study has been supported by grants of computing time from the National Computational Infrastructure (NCI) and from the Pawsey Supercomputing Centre, Australia, as well as funds from the Australian Research Council (ARC). N.Z. and J.A.-N. thank Murdoch University for the award of postgraduate scholarships.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00025.
Table S1 (thermal enthalpies and spin contamination values for all species), Figures S1 (geometries of stable products and adducts), and Figure S2 (geometries of transition structures) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Guenther A. Atmospheric chemistry: Are plant emissions green?. Nature 2008, 452, 701–702. 10.1038/452701a. [DOI] [PubMed] [Google Scholar]
- a Claeys M.; Graham B.; Vas G.; Wang W.; Vermeylen R.; Pashynska V.; Cafmeyer J.; Guyon P.; Andreae M. O.; Artaxo P.; Maenhaut W. Formation of Secondary Organic Aerosols Through Photooxidation of Isoprene. Science 2004, 303, 1173–1176. 10.1126/science.1092805. [DOI] [PubMed] [Google Scholar]; b Fuentes J. D.; Gu L.; Lerdau M.; Atkinson R.; Baldocchi D.; Bottenheim J. W.; Ciccioli P.; Lamb B.; Geron C.; Guenther A.; Sharkey T. D.; Stockwell W. Biogenic Hydrocarbons in the Atmospheric Boundary Layer: A Review. Bull. Am. Meteorol. Soc. 2000, 81, 1537–1575. . [DOI] [Google Scholar]
- a Sharkey T. D.; Wiberley A. E.; Donohue A. R. Isoprene Emission from Plants: Why and How. Ann. Bot. 2008, 101, 5–18. 10.1093/aob/mcm240. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vickers C. E.; Gershenzon J.; Lerdau M. T.; Loreto F. A Unified Mechanism of Action for Volatile Isoprenoids in Plant Abiotic Stress. Nat. Chem. Biol. 2009, 5, 283–291. 10.1038/nchembio.158. [DOI] [PubMed] [Google Scholar]
- Sharkey T. D.; Singsaas E. L. Why Plants Emit Isoprene. Nature 1995, 374, 769. 10.1038/374769a0. [DOI] [Google Scholar]
- Monson R. K.; Jones R. T.; Rosenstiel T. N.; Schnitzler J.-P. Why Only Some Plants Emit Isoprene. Plant, Cell Environ. 2013, 36, 503–516. 10.1111/pce.12015. [DOI] [PubMed] [Google Scholar]
- Triantaphylidès C.; Havaux M. Singlet Oxygen in Plants: Production, Detoxification and Signaling. Trends Plant Sci. 2009, 14, 219–228. 10.1016/j.tplants.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Triantaphylidès C.; Krischke M.; Hoeberichts F. A.; Ksas B.; Gresser G.; Havaux M.; Van Breusegem F.; Mueller M. J. Singlet Oxygen is the Major Reactive Oxygen Species Involved in Photooxidative Damage to Plants. Plant Physiol. 2008, 148, 960–968. 10.1104/pp.108.125690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingey D. T.; Manning M.; Grothaus L. C.; Burns W. F. The Influence of Light and Temperature on Isoprene Emission Rates from Live Oak. Physiol. Plant. 1979, 47, 112–118. 10.1111/j.1399-3054.1979.tb03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Affek H. P.; Yakir D. Protection by Isoprene Against Singlet Oxygen in Leaves. Plant Physiol. 2002, 129, 269–277. 10.1104/pp.010909. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Velikova V.; Edreva A.; Loreto F. Endogenous Isoprene Protects Phragmites Australis Leaves Against Singlet Oxygen. Physiol. Plant. 2004, 122, 219–225. 10.1111/j.0031-9317.2004.00392.x. [DOI] [Google Scholar]; c Loreto F.; Velikova V. Isoprene Produced by Leaves Protects the Photosynthetic Apparatus against Ozone Damage, Quenches Ozone Products, and Reduces Lipid Peroxidation of Cellular Membranes. Plant Physiol. 2001, 127, 1781–1787. 10.1104/pp.010497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyryaeva A. P.; Goldort V. G.; Kochubei S. A.; Baklanov A. V. Singlet Oxygen O2(1Δg) Formation via UV-Excitation of Isoprene-Oxygen C5H8–O2 Encounter Complexes in Gas Phase. Chem. Phys. Lett. 2014, 610–611, 8–13. 10.1016/j.cplett.2014.07.021. [DOI] [Google Scholar]
- Jardine K. J.; Monson R. K.; Abrell L.; Saleska S. R.; Arneth A.; Jardine A.; Ishida F. Y.; Serrano A. M. Y.; Artaxo P.; Karl T.; Fares S.; Goldstein A.; Loreto F.; Huxman T. Within-Plant Isoprene Oxidation Confirmed by Direct Emissions of Oxidation Products Methyl Vinyl Ketone and Methacrolein. Global Change Biol. 2012, 18, 973–984. 10.1111/j.1365-2486.2011.02610.x. [DOI] [Google Scholar]
- Clennan E. L.; Pace A. Advances in Singlet Oxygen Chemistry. Tetrahedron 2005, 61, 6665–6691. 10.1016/j.tet.2005.04.017. [DOI] [Google Scholar]
- a Abe M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. 10.1021/cr400056a. [DOI] [PubMed] [Google Scholar]; b Bobrowski M.; Liwo A.; Ołdziej S.; Jeziorek D.; Ossowski T. CAS MCSCF/CAS MCQDPT2 Study of the Mechanism of Singlet Oxygen Addition to 1,3-Butadiene and Benzene. J. Am. Chem. Soc. 2000, 122, 8112–8119. 10.1021/ja001185c. [DOI] [Google Scholar]
- Yamanaka S.; Kawakami T.; Nagao H.; Yamaguchi K. Effective Exchange Integrals for Open-Shell Species by Density Functional Methods. Chem. Phys. Lett. 1994, 231, 25–33. 10.1016/0009-2614(94)01221-0. [DOI] [Google Scholar]
- a Datta S. N.; Trindle C. O.; Illas F.. Theoretical and Computational Aspects of Magnetic Organic Molecules; Imperial College Press: Singapore, 2014; pp 1–335. [Google Scholar]; b Saito T.; Nishihara S.; Kataoka Y.; Nakanishi Y.; Kitagawa Y.; Kawakami T.; Yamanaka S.; Okumura M.; Yamaguchi K. Reinvestigation of the Reaction of Ethylene and Singlet Oxygen by the Approximate Spin Projection Method. Comparison with Multireference Coupled-Cluster Calculations. J. Phys. Chem. A 2010, 114, 7967–7974. 10.1021/jp102635s. [DOI] [PubMed] [Google Scholar]
- a Cramer C. J.; Thompson J. Quantum Chemical Characterization of Singlet and Triplet Didehydroindenes. J. Phys. Chem. A 2001, 105, 2091–2098. 10.1021/jp004379n. [DOI] [Google Scholar]; b Gräfenstein J.; Cremer D. The combination of density functional theory with multi-configuration methods—CAS-DFT. Chem. Phys. Lett. 2000, 316, 569–577. 10.1016/S0009-2614(99)01326-3. [DOI] [Google Scholar]
- Montgomery J. A.; Ochterski J. W.; Petersson G. A. A Complete Basis Set Model Chemistry. IV. An Improved Atomic Pair Natural Orbital Method. J. Chem. Phys. 1994, 101, 5900–5909. 10.1063/1.467306. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, A.1; Gaussian, Inc: Wallingford, CT, 2009.
- Mokrushin V.; Bedanov V.; Tsang W.; Zachariah M.; Knyazev V.. ChemRate, V.1.19; NIST: Gaithersburg, MD, 2002.
- Fukui K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. 10.1126/science.218.4574.747. [DOI] [PubMed] [Google Scholar]
- Hirshfeld F. L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Acta 1977, 44, 129–138. 10.1007/BF00549096. [DOI] [Google Scholar]
- Delley B. From Molecules to Solids with the DMol3 Approach. J. Chem. Phys. 2000, 113, 7756–7764. 10.1063/1.1316015. [DOI] [Google Scholar]
- Leach A. G.; Houk K. N. Diels–Alder and Ene Reactions of Singlet Oxygen, Nitroso Compounds and Triazolinediones: Transition States and Mechanisms from Contemporary Theory. Chem. Commun. 2002, 1243–1255. 10.1039/b111251c. [DOI] [PubMed] [Google Scholar]
- Maranzana A.; Ghigo G.; Tonachini G. Diradical and Peroxirane Pathways in the [π2+ π2] Cycloaddition Reactions of 1Δg Dioxygen with Ethene, Methyl Vinyl Ether, and Butadiene: A Density Functional and Multireference Perturbation Theory Study. J. Am. Chem. Soc. 2000, 122, 1414–1423. 10.1021/ja990805a. [DOI] [Google Scholar]
- Bobrowski M.; Liwo A.; Ołdziej S.; Jeziorek D.; Ossowski T. CAS MCSCF/CAS MCQDPT2 Study of the Mechanism of Singlet Oxygen Addition to 1,3-Butadiene and Benzene. J. Am. Chem. Soc. 2000, 122, 8112–8119. 10.1021/ja001185c. [DOI] [Google Scholar]
- Luo Y.-R.Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press, 2002. [Google Scholar]
- Barone V.; Cossi M.; Tomasi J. A New Definition of Cavities for the Computation of Solvation Free Energies by the Polarizable Continuum Model. J. Chem. Phys. 1997, 107, 3210–3221. 10.1063/1.474671. [DOI] [Google Scholar]
- Biesenthal T. A.; Shepson P. B. Observations of Anthropogenic Inputs of the Isoprene Oxidation Products Methyl Vinyl Ketone and Methacrolein to the Atmosphere. Geophys. Res. Lett. 1997, 24, 1375–1378. 10.1029/97GL01337. [DOI] [Google Scholar]
- Warneke C.; Holzinger R.; Hansel A.; Jordan A.; Lindinger W.; Pöschl U.; Williams J.; Hoor P.; Fischer H.; Crutzen P. J.; Scheeren H. A.; Lelieveld J. Isoprene and Its Oxidation Products Methyl Vinyl Ketone, Methacrolein, and Isoprene Related Peroxides Measured Online over the Tropical Rain Forest of Surinam in March 1998. J. Atmos. Chem. 2001, 38, 167–185. 10.1023/a:1006326802432. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.