

Abstract

Glycopolypeptide-based self-assembled nano-/microstructures with surface-tethered carbohydrates are excellent mimics of glycoproteins on the cell surface. To expand the broad repertoire of glycopolypeptide-based supramolecular soft structures such as polymersomes formed via self-assembly of amphiphilic polymers, we have developed a new class of polyionic complex vesicles (PICsomes) with glycopolypeptides grafted on the external surface. Oppositely charged hydrophilic block copolymers of glycopolypeptide20-b-poly-l-lysine100 and PEG2k-b-poly-l-glutamate100 [PEG = poly(ethylene glycol)] were synthesized using a combination of ring-opening polymerization of N-carboxyanhydrides and “click” chemistry. Under physiological conditions, the catiomer and aniomer self-assemble to form glycopolypeptide-conjugated PICsomes (GP-PICsomes) of micrometer dimensions. Electron and atomic force microscopy suggests a hollow morphology of the PICsomes, with inner aqueous pool (core) and peripheral PIC (shell) regions. Owing to their relatively large (∼micrometers) size, the hollowness of the supramolecular structure could be established via fluorescence microscopy of single GP-PICsomes, both in solution and under dry conditions, using spatially distributed fluorescent probes. Furthermore, the dynamics of single PICsomes in solution could be imaged in real time, which also allowed us to test for multivalent interactions between PICsomes mediated by a carbohydrate (mannose)-binding protein (lectin, Con-A). The immediate association of several GP-PICsomes in the presence of Con-A and their eventual aggregation to form large insoluble aggregate clusters reveal that upon self-assembly carbohydrate moieties protrude on the outer surface which retains their biochemical activity. Challenge experiments with excess mannose reveal fast deaggregation of GP-PICsomes as opposed to that in the presence of excess galactose, which further establishes the specificity of lectin-mediated polyvalent interactions of the GP-PICsomes.
1. Introduction
The interaction between carbohydrates present as glycoconjugates on cell surfaces and extracellular proteins (lectins) plays several complex roles in vivo, particularly in biomolecular recognition events such as cell–cell recognition, adhesion, cancer cell metastasis, pathogen invasion, and toxin mediation.1−4 Multivalent interaction of carbohydrates and their corresponding receptors are key to the high specificity of these important biomolecular interactions.5,6 In this context, a vast number of multivalent synthetic glycoconjugates have been prepared over the past decades, including dendritic scaffolds, linear polymers, micelles, and nanoparticles.7−10 Among these, glycopolypeptides, where sugar units are attached to a polypeptide backbone with well-defined secondary structures, mimic the molecular composition of proteoglycans and thus represent suitable candidates for biological applications.11−16 The construction of supramolecular nano-/microstructures (such as micelles or vesicles) from glycopolypeptides is advantageous as they are better mimics of densely populated carbohydrates on the cell surface than the individual glycopolypeptides. Such self-assembled structures can then be used to understand carbohydrate–protein interaction, which would be more realistic mimics of living systems.
We and others have reported on synthetic amphiphilic glycopolypeptide-based block copolymers that undergo self-assembly to generate morphologically different micro-/nanostructures.17−21 By varying factors such as molecular weight, chemical composition, functionality, and architecture of the amphiphilic block copolymers, the self-assembly could be controlled to afford size selection and a broad variety of morphologies. In vitro studies have shown that glycopolypeptide-based polymeric vesicles (≤100 nm) are endocytosed into MCF-7/MDA-MB-231 breast cancer cells using an overexpressed MRC2 receptor (mannose receptor C-type 2), which opens up the possibility of using these nanocarriers for receptor-mediated drug delivery to cancer cells.22
Typically, the fabrication of polymersomes using conventional self-assembly of amphiphilic block copolymers is tedious and involves procedures such as multiple dialysis, sonication, and heating. This, combined with the use of organic solvents, can cause damage to the loaded cargo (sensitive biomolecules such as proteins, enzymes, or drugs) and may also induce toxicity in biological systems.23,24 In addition, for the delivery of hydrophilic macromolecules, their poor solubility in nonaqueous medium and low penetration through the middle hydrophobic membrane in amphiphilic polymersomes limit their functionality as semipermeable containers.25 To overcome these limitations, the utilization of polyion complexes (PICs) can provide better solution because PIC formation can be carried out in aqueous media without the use of organic solvents, and the preparation process is remarkably straightforward.26,27 Oppositely charged hydrophilic block copolymers undergo self-assembly process via electrostatic interactions leading to the formation of PIC assemblies or vesicles, the morphology of which depends on the length and composition of ionic blocks as well as the length of aliphatic spacer side chains.28−30 Although PIC vesicles (PICsomes) possess a hollow spherical structure similar to polymersomes self-assembled from amphiphilic polymers, the PIC membrane sandwiched between outer and inner hydrophilic shell layers is semipermeable to hydrophilic solutes as opposed to the hydrophobic shell layer of amphiphilic polymersomes. In the PIC family, Kataoka and co-workers first demonstrated the formation of PIC micellar nanostructures and subsequently, unilamellar polymersomes in aqueous media by the electrostatic self-assembly of oppositely charged double hydrophilic block copolymers (or charged homopolymers) composed of biocompatible poly(ethylene glycol) (PEG) and poly(amino acid)s.31−36
In the quest to expand the broad repertoire of glycopolypeptide-based nano-/microstructures formed via self-assembly of homo and block amphiphilic glycopolypeptides, we envisioned the development of a new class of glycopolypeptide-based PICsomes in which glycopolypeptides are decorated on the surface, so that they can potentially interact with specific carbohydrate-binding proteins (lectins). Herein, we report the efficient synthesis of glycopolypeptide-based positively charged polyamino acids and their self assembly with PEG-based negatively charged polyamino acids in an aqueous medium to generate bioactive glycopolypeptide PICsomes (GP-PICsomes) (Figure 1). The micrometer dimensions of these GP-PICsomes, with a high surface density of high-molecular-weight glycopolypeptides, are close mimics of the protein-bound carbohydrates found on cell surfaces. In this context, it is important to understand their surface bioactivity, which has typically been studied for smaller supramolecular soft structures using techniques such as surface plasmon resonance, calorimetric assays, and dynamic light scattering (DLS).37−41 The formation of large PICsomes allows for the visualization (using optical microscopy) of their interaction in the presence of specific lectins and aggregation/deaggregation dynamics in solution. Such measurements can demonstrate whether carbohydrate-decorated PICsomes retain their biological activity (and specificity) apart from probing the role of lectins in mediating inter-PICsome interactions.
Figure 1.
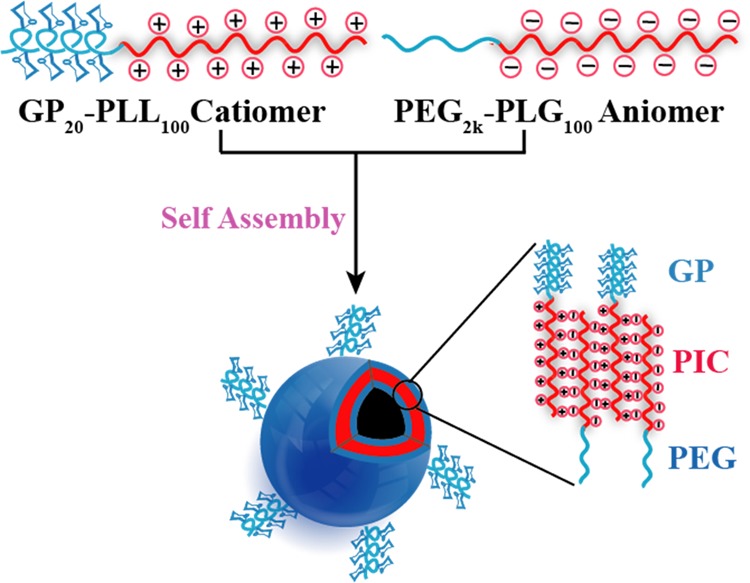
Simplified schematic representation depicting the structures of hydrophilic-charged block copolymers and their self-assembly into glycopolypeptide-based PICsomes (GP-PICsomes) under aqueous environments. Possibly, some of the PEG2k chains and glycopolypeptide moieties may be present in the outer layers and the inner layers, respectively. Although a high density of glycopolypeptides is present on the surface, only a few of them are blown up in the cartoon depiction for clarity.
Recently, the lectin-induced aggregation kinetics of dextran-coated colloidal silica (hard) particles have been investigated on a PEGylated glass surface using confocal microscopy.42 Although this provides insights into the aggregation mechanisms of mixed synthetic biomaterials induced by specific biomolecular interactions, the polyvalent binding of lectins with glycoprotein mimics such as glycopolypeptides grafted on soft polymersomes and their resulting aggregation behaviors have not been studied yet. Using fluorescent GP-PICsomes, we report real-time imaging of dynamic events induced by the binding of GP-PICsomes with a receptor (lectin), leading to the formation of various higher-order aggregates, which disassemble only in the presence of excess (monomeric) mannose, demonstrating biochemical specificity of pendant groups on the surface.
2. Materials and Methods
Poly(ethylene glycol) monomethyl ether (PEG-OH, Mn = 2000 g/mol), rhodamine B isothiocyanate (RITC), and fluorescein isothiocyanate (FITC)-dextran (Mn = 5000) were purchased from Sigma-Aldrich. Glyco-N-carboxyanhydride was prepared using our previously published methodology.43 Ligand tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) was synthesized by following the method of Finn and co-workers.44 All other chemicals used were obtained from Merck, India. UV–vis spectra were recorded on a Cary 300 UV–vis spectrometer using a 1 cm quartz cuvette at 25 °C. Fourier transform infrared (FT-IR) spectra were recorded on PerkinElmer FT-IR spectrum GX instrument using KBr pellets that were prepared by mixing 3 mg sample with 97 mg KBr. 1H NMR and 13C NMR spectra were recorded using a Bruker spectrometer (200.13 MHz, 400.13 MHz). Size-exclusion chromatography of the protected polymer was performed on a Viscotek TDA 305-040 triple detector array GPC/SEC module. Separations were achieved by using three columns (T6000M, General Mixed Org 300 × 7.8 mm2) and one guard column (TGAURD, Org Guard Col 10 × 4.6 mm2) at 0.025 M LiBr with dimethylformamide (DMF) as an eluant at 60 °C. Gel permeation chromatography (GPC)/light scattering samples were prepared at concentrations of 5 mg/mL. A constant flow rate of 1 mL/min was maintained, and the instrument was calibrated using poly(methylmethacrylate) (PMMA) standards. Polydispersity index (PDI) values were calculated using OmniSEC software. GPC measurements of ionic block copolymers were performed using a Waters Alliance 2690 separation module (RI Detector 2410). Separations were achieved by using three columns (G2000PW, G3000PW and G4000PW) using 10 mM phosphate buffer with 100 mM NaCl (pH 7.4) at a flow rate of 0.5 mL/min at room temperature (RT). GPC samples were prepared at concentrations of 2.5 mg/mL. Transmission electron microscopy (TEM) measurements were performed at 100 kV on an FEI Tecnai T20 instrument. Atomic force microscopy (AFM) measurements and analysis were performed on a Nanoscope IV (Veeco). Scanning electron microscopy (SEM) measurements were performed using a Quanta 200 3D SEM. The details of sample-preparation procedures for TEM, AFM, and SEM are provided in the Supporting Information. Fluorescence measurements were performed using a Horiba Jobin Yvon Fluorolog-3 spectrophotometer.
2.1. General Procedure for the Synthesis of Cationic and Anionic Block Copolymers
2.1.1. Synthesis of Glycopolypeptide-b-poly-l-lysine (GP20-PLL100)
(GP20-PLL100) was synthesized by conjugating hydrophilic glycopolypeptide block and poly-l-lysine cationic block using “click chemistry”. The blocks were first synthesized separately and then were conjugated using click chemistry.
2.1.1.1. Synthesis of Alkyne Functionalized Mannose Glycopolypeptide (Pr-GP20) as a Hydrophilic Block (1d)
First, Pr-AcGP20 was synthesized by the ring-opening polymerization (ROP) of mannose N-carboxyanhydrides monomer by propargylamine as an initiator using our previously published methodology.43 Furthermore, Pr-AcGP20 was deprotected using hydrazine hydrate in tetrahydrofuran (THF) to yield Pr-GP20 (Scheme S1).
Pr-GP20Polymer (1d): 1H NMR (400 MHz, D2O): δ [ppm]: 1.35–2.12 (br m, 6H), 2.28 (s, 1H), 3.00–3.2 (br m, 2H), 3.60–3.90 (br m, 5H), 3.95–4.45 (br m, 2H), 5.80–5.95 (br m, 1H). FT-IR (cm–1): 3328, 2938, 1706, 1652, 1548, 1275.
2.1.1.2. Synthesis of Azido-Terminated Poly-l-lysine (N3-PLL100) as Cationic Block (2d)
To a solution of Nε-trifluoroacetyl-l-lysine NCA (2b) in dry DMF (100 mg/mL) was added azido–PEG–amine (DMF solution 0.5 M) as the initiator (M/I = 100) inside of the glove box (Scheme S2). The progress of the polymerization was monitored using FT-IR spectroscopy by comparing with the intensity of the initial NCA anhydride stretching at 1785 and 1858 cm–1 (Figure S6b). The reaction was generally complete within 36 h. The resulting polymer was reprecipitated in deionized (DI) water for couple of times. The precipitated polymer was collected by centrifugation and washed with DI water. A white solid was obtained and dried under high vacuum at 50 °C to yield N3-PLL(TFA)100 (2c) in almost 90% yield.
N3-PLL(TFA)100Polymer (2c): 1H NMR (400 MHz, DMSO-d6): δ [ppm]: 1.25–2.12 (br m, 6H), 3.00–3.25 (br m, 2H), 3.48–3.68 (m, for CH2CH2O unit in initiator 24H), 3.7–4.4 (br m, 1H). FT-IR (cm–1): 2116, 1342, 1649, 1552, 1264, 1059, 1107.
Deprotection of N3-PLL(TFA)100Polymer (2d): The removal of labile triflouroacetyl (TFA)-protecting groups of lysine was achieved by dissolving the polymer (100 mg) in THF (4 mL) followed by dropwise addition of KOH solution (1.5 equiv) and stirring for an additional 24 h at RT. After complete deprotection, the solvent was removed using rotary evaporator to afford a solid residue. The residue was redissolved in DI water and transferred to a dialysis tubing (3.5 kDa MWCO). The samples were dialyzed against 0.01 N HCl for 1 day and then with water for another day with several water changes. Dialyzed polymers were lyophilized to yield N3-PLL100 (2d) as white solids in the form of hydrochloride salt.
N3-PLL100Polymer (2d): 1H NMR (400 MHz, D2O): 1.35–1.9 (m, 6H), 2.95–3.2 (m, 2H), 3.48–3.68 (m, for CH2CH2O unit in initiator 24H), 4.3–4.7 (br m, 1H). FT-IR (cm–1): 3328, 2938, 2116, 1726, 1649, 1552, 1324, 1264.
2.1.1.3. Synthesis of Glycopolypeptide-b-poly-l-lysine (GP20-PLL100) Cationic Polymer (4)
Azide-functionalized deprotected poly-l-lysine (2d) (0.8 equiv) was added to a solution of deprotected alkyne-terminated glycopolypeptide (1d) in phosphate buffer (pH = 7.4, 50 mM), and the resultant reaction mixture was degassed by three freeze–pump–thaw cycles. A premixed solution of CuSO4 (0.25 equiv) and ligand THPTA (1.25 equiv) was added followed by the addition of sodium ascorbate (2.5 equiv), and the reaction was allowed to proceed for 24 h at RT (Scheme 1). The progress of the reaction was monitored by the disappearance of the azide stretch at 2115 cm–1 using FT-IR (Figure S1). After completion of the reaction (disappearance of azide stretch ≥95%), the reaction mixture was directly transferred into a dialysis tube (12.5 kDa MWCO) and dialyzed against EDTA solution for 1 day with solution (water) change every 4 h and followed by dialysis against DI water solution for another day. Finally, the reaction mixture was dialyzed against 0.01 N HCl for an additional 1 day. Dialyzed polymers were lyophilized to yield glycopolypeptide-b-poly-l-lysine (GP20-PLL100) cationic polymers as white fluffy solids in the form of hydrochloride salt (Figure S2).
Scheme 1. Synthesis of Glycopolypeptide-b-poly-l-lysine (GP20-PLL100) Cationic Polymers.

GP20-PLL100Polymer (4): 1H NMR (400 MHz, D2O): 1.29–1.96 (br m, 12H), 2.78–2.95 (br m, 2H), 3.0–3.3 (br m, 2H), 3.4–3.53 (m, 24H), 3.56–3.72 (br m, 5H), 3.75–3.95 (br m, 2H), 4.2–4.45 (br m, 1H), 5.72–5.79 (br m, 1H). FT-IR (cm–1): 3328, 2950, 1752, 1650, 1552, 1250.
2.1.2. Synthesis of PEG2k-b-poly-l-glutamate (PEG2k-PLG100) as Anionic Block Copolymers
PEG2k-PLG100 was synthesized by ring-opening polymerization of benzyl-l-glutamate NCA by amine-terminated polyethylene glycol (PEG2k-NH2). PEG2k-NH2 monomethyl ether (2000) was synthesized by following the method of Loos and co-workers.45
2.1.2.1. Synthesis of PEG2k-PLG100 Anionic Polymers by ROP (3c)
To a solution of benzyl-l-glutamate NCA in dry DMF (3a) (100 mg/mL) was added PEG2k-NH2 (DMF solution, 0.5 M) as the initiator (M/I = 100) inside of the glove box (Scheme 2). The progress of the polymerization was monitored using FT-IR spectroscopy by quantifying the decrease in the intensity of the initial NCA anhydride stretching at 1785 and 1858 cm–1 (Figure S6c). The reaction was generally complete within 24 h. The resulting polymer was reprecipitated in DI water for a couple of times, collected by centrifugation, and washed with DI water. The white solid obtained was dried under high vacuum at 50 °C to afford PEG2k-PBLG100 (3b) in almost 90% yield.
Scheme 2. Synthesis of PEG2k-PLG100 Anionic Polymers by the ROP of Bn-Glu-NCA Using PEG2k-NH2 as the Macroinitiator.

PEG2k-PBLG100Polymer (3b): 1H NMR (400 MHz, CDCl3): δ [ppm]: 1.5–2.6 br (m, 4H), 3.39 (br m, 3H), 3.4–3.7 (m, 180H for CH2CH2O unit in initiator), 3.93–4.2 (br m, 1H), 4.9–5.3 (br m, 2H), 7.42 (s, 5H). FT-IR (cm–1): 3328, 1652, 1548, 1342, 1167, 1107, 749, 697.
Deprotection of PEG2k-PBLG100Polymer (3c): A solution of PEG2k-PBLG100 polymer (100 mg) in THF (4 mL) was placed in 50 mL round-bottom flask and cooled to 4 °C. To the above-mentioned solution, NaOH solution (0.5 mL, 2 M; ∼1.5 equiv per carboxylic group) was added dropwise at 4 °C, which resulted in a turbid solution after few minutes. The solution was left under vigorous stirring for 16 h at RT. After complete deprotection, the solvent was removed using rotary evaporator to obtain the crude product. The crude product was redissolved in deionized water and transferred into a dialysis tubing (3.5 kDa MWCO), which was then dialyzed against DI water for 2 days with six water changes. Dialyzed polymers were lyophilized to yield PEG2k-PLG100 (white solid) as a sodium salt (3c) (Figure S3).
PEG2k-PLG100Polymer (3c): 1H NMR (400 MHz, D2O): δ [ppm]: 1.80–2.40 (br m, 4H), 2.14–2.38 (m, 2H), 3.39 (br m, 3H), 3.4–3.7 (m, 180H for CH2CH2O unit in initiator), 4.26–4.31 (br m, 1H). FT-IR (cm–1): 3440, 3228, 1649, 1544, 1407, 1342.
2.1.2.2. Synthesis of Rhodamine-Labeled PEG2k-PLG100 Polymer (3d)
Et3N (16.9 μmol, 22 μL) and RITC (6.7 mg, 3 equiv) were added to a solution of PEG2k-PBLG100 polymer (100 mg, 4.16 μmol) in dry DMF (5 mL), and stirred for 48 h. The reaction mixture was then reprecipitated in methanol (at least five times) to remove excess RITC. Finally, the rhodamine-labeled polymer was deprotected and purified by dialysis as stated above. Dialyzed polymers were lyophilized to yield a rhodamine-labeled anionic polymer (RhB-PEG2k-PLG100) as pink solids.
RhB-PEG2k-PLG100Polymer (3d): 1H NMR (400 MHz, D2O): 1.5–2.6 (br m, 4H), 3.39 (s, 3H), 3.4–3.7 (m, 180H for CH2CH2O unit in initiator), 3.93–4.2 (br m, 1H), 4.9–5.3 (br m, 2H), 7.42, 7.47 (m, H for rhodamine phenyl protons). FT-IR (cm–1): 3440, 3228, 1648, 1530, 1407, 1342, 794.
2.1.2.3. Homoanionic Copolymer (Hx-PLG100)
Homoanionic copolymer (Hx-PLG100) was synthesized by polymerizing benzyl-l-glutamate NCA with hexylamine as an initiator in dry DMF. The deprotection of the benzyl group to yield Hx-PLG100 was carried out using the same protocol as described for the deprotection of PEG2k-PBLG100 polymer.
Hx-PBLG100Polymer (5a): 1H NMR (400 MHz, CDCl3): δ [ppm]: 0.83 (t, 3H for initiator), 1.5–2.6 (br m, 4H), 3.93–4.2 (br m, 1H), 4.9–5.3 (br m, 2H), 7.42 (s, 5H). FT-IR (cm–1): 3440, 3228, 1648, 1542, 1407, 749, 697.
Hx-PLG100Polymer (5b): 1H NMR (400 MHz, D2O): δ [ppm]: 0.83 (t, 3H for initiator), 1.80–2.40 (m, 4H), 2.14–2.38 (m, 2H), 4.26–4.3 (br, 1H). FT-IR (cm–1): 3440, 3228, 1648, 1542, 1407.
2.2. Self-Assembly of Charged Block Polypeptides
Cationic block copolymers (GP20-PLL100) at a concentration of 1.0 mg/mL and anionic block copolymers (PEG2k-PLG100) at a concentration of 0.73 mg/mL were individually solubilized in 50 mM phosphate buffer (pH 7.4). The solutions were filtered through a 0.22 μm membrane filter to remove dust particles and any other insoluble portions. GP20-PLL100 solutions were added in one portion to a solution of PEG2k-PLG100 at RT in an equal charge ratio of −COO– and −NH3+ and were mixed thoroughly using a vortex mixer for 2 min. The resulting polymer solution was incubated for 24 h at RT to generate the GP-PICsomes. RhB-labeled PICs were formed from rhodamine-labeled block aniomer (RhB-PEG2k-PLG100) with block catiomer (GP20-PLL100), following a similar methodology.
2.3. Dye Encapsulation
2.3.1. Preparation of FITC-Encapsulated GP-PICsomes
The encapsulation of hydrophilic molecule FITC-dextran into the GP-PICsomes was performed using the coassembly method. First, filtered solutions of PEG2k-PLG100 (1.46 mg/mL) and GP20-PLL100 (1 mg/mL) were prepared separately in 50 mM phosphate buffer. 500 μL of GP20-PLL100 solution (1 mg/mL) was added to a 500 μL solution containing 0.75 mg FITC-dextran (Mn = 5000) and 0.73 mg PEG2k-PLG100 in phosphate buffer (50 mM, pH 7.4) and was mixed thoroughly using a vortex mixer. The solution was incubated for 24 h at RT to encapsulate FITC-dextran into GP-PICsomes. The mixture was then placed into a dialysis membrane (50 kDa MWCO) and dialyzed against phosphate buffer (10 mM) at 4 °C for 24 h while changing the buffer at a 4 h interval. Within 24 h, all unencapsulated FITC-dextran was excluded from the dialysis membrane. After the removal of free FITC-dextran, the mixture was dialyzed against DI water for 24 h.
2.4. Dye Release Studies
Immediately after the removal of excess FITC-dextran, the dye-loaded GP-PICsome solution was transferred into a fresh dialysis tube and then immersed into 30 mL of phosphate buffer solution (10 mM) and stirred at RT for 2 days. At predefined time points, aliquots (1 mL) were removed from the buffer solution outside of the dialysis membrane, which was replaced with an equal volume of fresh buffer. The concentration of FITC-dextran in the aliquots was estimated using fluorescence spectroscopy. Finally, the release of FITC-dextran from the GP-PICsomes was reported as a percentage of the total amount released from the dialysis tubes.
2.5. Fluorescence Imaging of GP-PICsomes
An epifluorescence/total internal reflection fluorescence (TIRF) microscopy setup built on an inverted microscope (Nikon Eclipse TE2000-U) was used for imaging GP-PICsomes and studying the process of aggregation and deaggregation. Details on the home-built setup are provided elsewhere.18 In brief, a 488 nm argon ion laser (Melles Griot, model: 35-MAP-321-240) and a 532 nm DPSS laser (Melles Griot, model: LD-WL206) were used to illuminate the same area of the sample (field of view ∼1000 μm2) via an objective lens (Nikon, 1.49 NA, 60× TIRF). The emission from the sample was collected by the same objective and passed through appropriate dichroic/longpass filters and detected by a CCD camera (DVC 1412AM). For colocalization studies of individual GP-PICsomes labeled with two fluorophores, FITC and RhB were selectively excited at 488 and 532 nm, and their emission was collected using 445–530 and 545–630 nm bandpass emission filters, respectively. The excitation power (50–200 μW) was varied for the two lasers using neutral density filters such that the comparable intensity of emission (within a factor of 3) is observed in the two energetically separated detection channels (colored green for FITC and red for RhB). Typically, movies were collected with an exposure time of 100 ms and averaged over 100 frames before further image processing. To monitor the aggregation dynamics of GP-PICsomes (labeled with RhB) in the presence of Con-A and their deaggregation in excess mannose, movies were collected at 5 Hz. ImageJ was used to obtain montage images from the acquired movies, which show single-frame sequential snapshots at regular time interval. To visualize the trajectories of single GP-PICsomes and their aggregates in solution, maximum projection (MP) images,46 rather than time average images, were generated over a certain time interval using ImageJ. All measurements were performed at 295 K.
2.6. Sample Preparation for Fluorescence Imaging of GP-PICsomes
2.6.1. For Hollowness and Dye Encapsulation in GP-PICsomes
Fluorescence imaging in solution was performed by placing 20 μL of FITC-dextran-encapsulated RhB-GP-PICsomes in between two glass coverslips and incubating for 10 min. For dry-state measurements, the sample was spin-coated (20 μL, 1000 rpm, 1 min; Ducom PRS-6K) on a clean coverslip and imaged after 30 min.
2.6.2. Sample Preparation for Fluorescence Imaging of GP-PICsomes Aggregation and Deaggregation
A solution (1.0 mg/mL) of Con-A was prepared in 100 mM phosphate buffer (pH 7.4) containing 0.1 mM MnCl2, 0.1 mM CaCl2, and 0.1 M NaCl. The aggregation experiment was performed by preincubating RhB-GP-PICsomes with Con-A and by sandwiching this solution in between two glass coverslips. A reservoir of buffer solution was maintained on both sides of the solution to prevent solvent evaporation. Alternatively, Con-A was introduced between the coverslips and incubated for 10 min for adsorption on the glass surface, and excess Con-A solution was washed out by capillary action using a filter paper at one end while maintaining enough buffer in the reservoir on the other end. Finally, RhB-GP-PICsome solution was introduced from reservoir end of the coverslips while imaging. Movies were collected at 3 Hz to visualize the aggregates. For deaggregation studies, preformed aggregates were prepared by mixing Con-A solution (50 μL) and RhB-GP-PICsome solution (100 μL) for different times (10–30 min). This mixture was then introduced into a homemade flow chamber through an injection pump, where flow rates can be controlled. After visualizing the aggregates of RhB-GP-PICsomes, enough buffer was injected to wash off excess aggregates and unbound Con-A, followed by the introduction of buffer containing 300 mM mannose. Flow of the solution was controlled using the syringe pump. The control experiment for deaggregation process with 300 mM galactose solution was performed not only under identical conditions but also for much longer time (10 min) incubation with excess galactose.
3. Results and Discussion
3.1. Synthesis of Cationic and Anionic Block Copolymers
GP-PICsomes were synthesized by the self-assembly of oppositely charged double hydrophilic block copolymer via the electrostatic interaction between GP-containing catiomer and PEG-containing aniomer. Kataoka et al. had previously demonstrated that uniform PICsomes were formed by mixing cationic PEG2k-b-poly(aspartamide)100 and PEG2k-b-poly(aspartate)100.29 They subsequently demonstrated that the usage of a large volume of PEGs, or a higher PEG weight fraction (fPEG) in the block copolymers, prevented the growth of PIC and drove micelle formation because of the immiscibility of a PEG part and PIC part as well as large steric hindrance of the PEG strands.30,31 We decided to attempt the synthesis of GP-PICsomes based on a double hydrophilic catiomer GP20-b-poly(l-lysine)100 and aniomer PEG2k-b-poly(l-glutamate)100, such that upon mixing, 100-mer poly-l-lysine and poly-l-glutamate would generate the stable PIC membrane, where most of the GP20 and PEG2k moieties would form the outer and inner layers, respectively. Because the carbohydrate side chains are very bulky (MW 7 kDa), we avoided using GP in both catiomer and aniomer to avoid the formation of micelles. A short PEG2k was used in the aniomer instead to help the PIC transform into PICsomes.
The catiomer was obtained by independently synthesizing alkyne-labeled GP20 and azide-labeled PLL100 and then by conjugating them using click chemistry (Scheme 1). GP20-containing pendant carbohydrate moieties with an alkyne at the chain end were synthesized by following our previously developed methodology that involves propargylamine-initiated ROP of the mannosyl-N-carboxyanhydride (NCA) monomer.43 The removal of acetyl protection followed by purification lead to fully water-soluble alkyne-terminated GP (Scheme S1). The cationic PLL polymer containing azide at the chain end was synthesized from TFA-protected l-lysine (Scheme S2). All of the protected copolymer building blocks, which included alkyne-AcGP20 and azide-PLL(TFA)100, had low PDI (Figure S4) and the chain length of each polymer (from Mn) was determined from 1H NMR spectra using end-group analysis (Table 1 and Supporting Information figure: NMR section). The Mn values determined matched closely to that expected from M/I values. After the deprotection of copolymers, both alkyne-GP20 and azide-PLL100 were conjugated using copper-catalyzed azide–alkyne click reaction to synthesize cationic block-co-glycopolypeptides (GP20-PLL100). The progress of the reaction was monitored using FT-IR spectra by following the decrease in the characteristic azide stretch at 2117 cm–1 (Figure S1). After the reaction was complete, dialysis against EDTA solution was performed to remove all residual copper followed by dialysis (using 12 kDa MWCO dialysis membrane) against DI water to remove the excess GP (the molecular weight of deprotected GP is 7 kDa) used in the click reaction. We have performed an SEC analysis of this purified GP20-PLL100 block copolypeptides to ascertain the efficiency of click reaction. The aqueous GPC of the purified “clicked” product (Figure S5) displayed a new peak at higher molecular weight, which was distinctly shifted from the parent GP and polylysine, suggesting the formation of the clicked product. Moreover, the complete absence of characteristic organo azide stretch (2115 cm–1) in IR analysis (Figure S1) suggests a near-quantitative click reaction. The exact composition of the block copolypeptide was determined from 1H NMR by comparing the resonance characteristics present in the GP and PLL segment that was present in the expected molar ratio of 1:1 (Figure S2).
Table 1. Synthesis of Alkyne-AcGP, Azide-PLL (TFA), and PEG2k-PBLG and Their Corresponding Deprotected Polymers.
| entry no. | initiator (I) | protected polymers | M/Ia | Mnb (103 g/mol) | DPc | Mw/Mnd | DPe | deprotected polymers | DPf |
|---|---|---|---|---|---|---|---|---|---|
| 1 | propargyl amine | AcGP20 | 20 | 11.0 | 22 | 1.15 | 24 | GP20 | 22 |
| 2 | N3PEGNH2 | PLL(TFA)100 | 100 | 22.9 | 102 | 1.12 | 106 | PLL100 | 102 |
| 3 | PEG(2k)-NH2 | PBLG100 | 100 | 24.9 | 105 | 1.10 | 110 | PLG100 | 105 |
| 4 | hexylamine | PBLG100 | 100 | 21.8 | 97 | 1.11 | 111 | PLG100 | 97 |
| 5 | GP20-PLL100 | 22 + 102 |
Monomer to initiator ratio.
Number average molecular weight calculated from GPC.
DP was calculated from 1H NMR.
Mw/Mn were calculated from GPC.
DP was calculated from GPC.
DP was calculated from 1H NMR (Supporting Information).
For anionic block copolypeptides, ROP of γ-benzyl-l-glutamate N-carboxyanhydride was performed using PEG2k-NH2 as the macroinitiator (M/I = 100) in dry DMF (Scheme 1 and Table 1). The completion of the reaction was confirmed by the complete disappearance of the anhydride stretch of initial NCA at 1785 and 1858 cm–1 in the FT-IR spectra (Figure S6c). The number average molecular weight (Mn) was calculated using 1H NMR from the relative intensity of the peak at 3.68 ppm because of the characteristic proton present in the initiator PEG-amine (−OCH2CH2) with the proton peaks of the benzyl group (−C6H5) present in the benzyl ester moiety of the polymer (7.30 ppm). The molecular weight distribution observed from GPC was monomodal and the PDI calculated was reasonably narrow (Figure S4c and Table 1). PEG2k-PLG100 was prepared by the benzyl-ester deprotection of PEG2k-PBLG100 using base hydrolysis. 1H NMR of the resulting polymer showed complete disappearance of the signals corresponding to the benzyl group and rest of the signals were assigned to PEG2k-PLG100 (Figure S3).
3.2. Self-Assembly of Charged Polymers
GP-PICsomes were prepared by mixing 0.5 mg/mL solution of GP20-PLL100 and 0.36 mg/mL solution of PEG2k-PLG100 in 50 mM pH 7.4 phosphate buffer such that equimolar amounts of l-lysine and glutamic acid residues were present in solution (electrostatically neutralized condition). The mixture was first vortexed for 15 min and then incubated overnight at RT. To confirm the types of macroscopic structures formed upon self-assembly, SEM was performed on GP-PICsomes cast on a surface under solvent-evaporated conditions. SEM measurements showed the presence of a spherical morphology (Figure 2a) having an average diameter of 1.67 μm with sizes between 1.2 and 2.2 μm (Figure 2b). GP-PICsomes were also characterized using TEM after adding a drop of GP-PICsome solution onto carbon-coated copper grid and negatively staining with uranyl acetate (0.2 wt %) solution. TEM analysis also indicated the presence of well-dispersed PICsomes with a spherical structure having a characteristic dark rim on the outer surface, indicating their hollow morphology (Figure 2c). Further evidence of hollow morphology was obtained from AFM measurements on a silicon wafer (Figure 2d), which revealed a wall thickness of ∼30 nm and a height/diameter ratio of ∼0.08, which is consistent with a spherical hollow vesicular morphology.47 Because all of the above microscopies were performed in the dried state, we analyzed the size of the PICsomes formed after 24 h incubation of the GP20-PLL100 and PEG2k-PLG100 using DLS from which an average diameter of 1.46 μm was determined. This was again consistent with SEM and TEM measurements (Figure S8). The size and distribution of GP-PICsomes were also determined using optical micrograph, which also showed an average diameter of 1.43 μm (Figure S9). The vesicle wall thickness of 25–30 nm obtained from both TEM and AFM measurements approximately equals the size of a fully extended chain length of the constituent polyelectrolyte strands (∼25 nm), roughly estimated by summation of the average backbone length of each amino acid unit in the polypeptide chain (C–C–N, 0.24 nm).33 This suggests that the vesicle wall is composed of a monolayer of PIC sandwiched by PEG and GPs palisades or the mixture of both PEG and GP part in the inner and outer surface.
Figure 2.

GP-PICsomes formed via self-assembly upon the addition of equimolar amounts of GP20-PLL100 and PEG2k-PLG100. (a) SEM image of GP-PICsomes, (b) size distribution of GP-PICsomes from SEM images, (c) TEM image of GP-PICsomes, and (d) AFM image of the GP-PICsomes drop-cast from aqueous solution on the silicon wafer and their corresponding height profile.
We believe that in our system, the growth of GP-PICsomes in aqueous medium is initiated by uPIC formation, which is a result of the bilayer formed owing to the interaction between cationic amino groups in (GP20-PLL100) and anionic glutamate groups in PEG2k-PLG100, resulting in charge neutralization.34 This uPIC units grow over time resulting in an initial formation of a three-layered membrane structure consisting of a middle PIC phase sandwiched by external and internal hydrophilic shell layers (PEG and GP). When the membrane-like aggregates become so large that the interfacial energy is predominant, the membrane-like bilayer closes to form vesicles. The bending of the three-layered lamellar structure would reduce the interfacial free energy and enclose it into a vesicular morphology.48−51 Because the uPIC unit is composed of 100-mer PLL and 100-mer PLG, it would behave as a stiff polymer unit with low internal degrees of freedom and low formational entropy. Following the general rule that increasing order inherently leads to an increased bending modulus κ of the bilayers, a vesicle with relatively low interfacial curvature leading to large sizes (micron diameter) is expected. We note that the molecular weight of the shell layer GP20 (MW 7 kDa) is significantly higher than that of PEG2k and such high weight fractions have been shown earlier to prevent the formation of PICsomes. However, their self-assembly into PICsomes is likely attributed to the efficient packing of the helical GP20 unit (see circular dichroism data in Figure S7) at the surface of GP-PICsomes, which induces morphology bearing a lower curvature as observed earlier with amphiphilic glycopolypeptides.17,18,20,22 In GP-PICsomes, three possibilities exist during the formation of the three-layered lamellar structure with respect to the position of GP20 and PEG2k in the inner layer (I) or outer layer (O) of the PICsomes: [(I)PEG-PIC-(O)GP], [(I)PEG, GP-PIC-(O)PEG, GP], [(I)GP-PIC-(O)PEG]. Because GP20 is much more bulky than PEG2k, the assemblies will prefer a morphology with less curvature and hence GP20 chains are expected to be present on the outer surface of the PICsomes (the first two of the three possible structures mentioned above). The presence of glycopolypeptide part on PICsomes outer surface will be later confirmed by investigating their interaction with carbohydrate-binding protein (lectins). It is possible that some of the PEG2k chains are also present in the outer layer, however, we were unable to determine the extent of heterogeneity. The presence of PEG2k in the aniomer was critical to the formation of PICsomes. Upon mixing equimolar solutions of GP20-PLL100 with the homoaniomer Hx-PLG100, formation of well-defined structures was not observed (Figure S10).
3.3. Stability and Dye Encapsulation Studies of GP-PICsomes
In an aqueous solution, the hollow nature of the spherical particles was tested by encapsulating hydrophilic macromolecule FITC-dextran. UV–vis spectra of FITC-dextran-encapsulated GP-PICsomes show a characteristic peak (λmax) at 490 nm (Figure S11). The encapsulation efficiency (EE) was determined as EE = (W1/W2) × 100, where W1 is the amount of FITC-dextran encapsulated and W2 is the amount of FITC-dextran added initially for encapsulation. The value of W1 was calculated by measuring the optical density at 493 nm (molar absorption coefficient of FITC-dextran in water is taken as 75 000 L mol–1). The percentage EE of FITC-dextran was determined to be ∼2.06%.
To test the release of FITC-dextran from GP-PICsomes, the aqueous solution of FITC-dextran-encapsulated GP-PICsomes was dialyzed against phosphate buffer solution for 2 days. Emission spectra of the phosphate buffer outside of the dialysis tube indicated the leakage of FITC-dextran with the passage of time (Figure S12a). After 48 h, the leakage of FITC-dextran was estimated by comparing the absorption spectra of FITC-dextran-encapsulated GP-PICsomes inside of the dialysis tubing before and after the release experiment. The percentage of FITC-dextran released in this period of 48 h was determined to be approximately 63% (Figure S12b), which indicates its slow release from the GP-PICsomes.
3.4. Visualization of Individual GP-PICsomes
Although both AFM and electron microscopy measurements provide crucial information on the size distributions and shapes of the GP-PICsomes under dry conditions, it is difficult to conclusively comment whether the solution structures are indeed preserved upon solvent evaporation (for drop-cast samples). Furthermore, it is also challenging to conclude directly from AFM or TEM measurements whether the morphology of GP-PICsomes is indeed hollow, as indicated by dye encapsulation studies. Since the dimensions of these GP-PICsomes are much larger than the diffraction limit, the most direct way to test their morphology (and dimensions) in solution is to image individual PICsomes with two fluorescent dyes located in different spatial locations, specifically the central aqueous pool and the PIC shell regions.
To study colocalization behaviors, RhB-labeled GP-PICsomes were synthesized by the self-assembly of block aniomer (RhB-PEG2k-PLG100) and block catiomer (GP20-PLL100), following which hydrophilic FITC-dextran dye was encapsulated within the RhB-GP-PICsomes. Careful optimization on the relative proportion of encapsulated FITC-dextran and sequential excitation of the two dyes using 488 and 532 nm lasers allowed us to visualize GP-PICsomes in solution with comparable emission intensity (within a factor of 3) in energetically separated detection channels. Figure 3a–c shows two such individual FITC-encapsulated RhB-GP-PICsomes in buffer solution. It is important to note that the vast majority of the GP-PICsomes undergo diffusion in solution; however, it was possible to image few spatially segregated PICsomes, which adhere to the glass surface and do not undergo any translational motion. The images obtained from both emission channels show that in solution, GP-PICsomes are quasispherical and have dimensions of 1–2 μm, consistent with AFM and electron microscopy measurements (Figure 2). More importantly, the spatial intensity profiles in the two detection channels display contrasting (filled and ringlike) emission patterns (Figure 3a,b), which can be readily visualized in the pseudocolor overlay images (Figure 3c) and in the intensity line profiles, as expected for a hollow morphology.
Figure 3.

Fluorescence images of individual GP-PICsomes labeled with both FITC and RhB in solution (a–c) and spin-cast on glass coverslips (d–f), collected through bandpass filters for FITC (a,d) and RhB (b,e) emission (colored green and red, respectively), along with pseudocolor images (c,f) obtained via quantitative superposition of intensity images acquired for the same area through two detection channels. The line profiles (color-coded) of single GP-PICsomes (identified in c and f) on the top (a–c) and bottom (d–f) depict the emission intensity in each detection channel and the normalized emission for the overlay images (c,f) in solution and under dry conditions.
In contrast, the emission behaviors of GP-PICsomes in the absence of solvents are quite different, as observed from dual-color imaging of FITC-encapsulated RhB-GP-PICsomes spin-cast on glass coverslips from the solution (Figure 3d–f). First, solvent evaporation leads to higher density of individual PICsomes that can be imaged in the same field of view. Interestingly, the relative background emission in the FITC channel (Figure 3d) is significantly higher as compared to that in solution (Figure 3a), which suggests partial leaching of encapsulated FITC upon solvent removal. This inference is further supported by the images of single GP-PICsomes (and corresponding line profiles) observed in the RhB channel (Figure 3e), which have much higher intensity and spatial uniformity in emission (edges and central regions) as compared to that in solution, presumably owing to the shrinkage (flattenning) of GP-PICsomes during dehydration (leaching). Furthermore, it is also evident from the overlay image (Figure 3f) and intensity line profiles that the emission originating from FITC within GP-PICsomes is far more localized (300–500 nm) as compared with those in buffer solution. This indicates the existence of water pools of relatively smaller dimensions within flattened PICsomes, where remaining hydrophilic dextran-FITC accumulates and increases the effective local concentration, thereby producing a relatively intense localized emission from constricted GP-PICsomes. Nonetheless, both of these measurements performed in a buffer solution and under dry conditions provide direct evidence on the existence of hollow morphology.
3.5. Dynamics of GP-PICsomes and Their Interaction with Con-A
To address whether glycopolypeptides were displayed on the outer surface of the mannose-containing PICsomes and verify that the surface remained bioactive upon self-assembly, we studied the aggregation behaviors of GP-PICsomes upon the addition of the mannose-specific lectin, Con-A, and investigated deaggregation of clustered PICsomes in excess of free mannose. Simple turbidimetic assays were not performed, as free GP20-PLL100, if still present in solution, could also cross-link with Con-A to form turbid solutions. The relatively large (∼micrometers) size of the GP-PICsomes, which are expected to diffuse sluggishly in solution, allowed us to probe Con-A-induced aggregation dynamics of individual assemblies in buffer solution using epifluorescence video microscopy. As schematically depicted in Figure 4, if the mannose moieties are present on the outer surface of the GP-PICsomes, their polyvalent interaction with the tetrameric Con-A would lead to the formation of large PICsome aggregates. In contrast, if the GP moieties are present in the inner membrane post self-assembly, the addition of Con-A would not lead to the association of two or more GP-PICsomes and therefore would not induce aggregation.
Figure 4.

Schematic depiction of the multivalent association of GP-PICsomes mediated by mannose-binding lectin (Con-A), which leads to aggregation, and disassembly of aggregates only in the presence of excess free mannose (not galactose) in solution.
The dynamics of two single RhB-GP-PICsomes in buffer solution in the absence of any external hydrodynamic force is shown in Figure 5a, where each PICsome undergoes 3D-translational motion as evidenced from the spatial intensity distributions of their trajectories. The diffusion dynamics of GP-PICsomes can be easily captured at 0.5–1 Hz because of their relatively large size; however, we refrain from extracting diffusion constants because it is unclear whether they interact with the glass surface intermittently. Upon incubation (for 15 min) with 0.5 mg/mL lectin receptor (Con-A), which has four mannose-binding sites, rarely do we detect individual PICsomes. Rather, the diffusion of small aggregates constituting of few (typically 3–6) GP-PICsomes can be readily visualized (Figure S13). The tumbling dynamics (without any hydrodynamic drag) of one such small aggregate assembly, constituting of five or six GP-PICsomes attached to each other, is shown as single-frame sequential snapshots in Figure 5b (0–20 s) along with their trajectory (see Movie M3). We noticed that this small aggregate eventually gets attached to the surface (after ∼20 s), presumably because of binding to surface-adsorbed Con-A. To test this, Con-A solution (1 mg/mL) was introduced on a glass coverslip and incubated for 10 min, so that some of it adsorbs on the substrate. After the removal of excess Con-A with buffer solution, GP-PICsomes were introduced in the flow chamber. Here, surface attachment of aggregates and their eventual immobilization in the presence of slight hydrodynamic flow can be observed frequently, as exemplified using another aggregate shown in Figure 5c. We noticed that in the presence of excess (1 mg/mL) Con-A in solution, higher order aggregation is induced (Figure 5d), where all GP-PICsomes within the aggregate tumble (and move) synchronously, and the entire aggregate assembly diffuse sluggishly even in the presence of weak hydrodynamic flow.
Figure 5.

(a) Dynamics of individual RhB-labeled GP-PICsomes in the absence of any external hydrodynamic force. Top and bottom rows represent ten single-frame sequential snapshots for two representative GP-PICsomes in solution. Left and right panels represent corresponding maximum projection (MP) images46 of the same area (at 2× magnification) for trajectory duration of 40.5 and 13.5 s. Movies M1 and M2 are provided in Supporting Information. (b–d) Solution dynamics and eventual surface association of GP-PICsome aggregates of various sizes formed upon the addition of Con-A, in the absence (b) and presence (c,d) of weak hydrodynamic flow. Rightmost panels in (b–d) depict MP images over entire trajectories from which representative single-frame sequential snapshots are shown for each aggregate. Movies M3, M4, and M5 are provided in Supporting Information. (e) Variety of multiple scattered GP-PICsome aggregates attached on to glass surface with blowups (5 × 5 μm2), depicting the presence of many GP-PICsomes within each aggregate. (f) Formation of very large extended aggregate clusters of GP-PICsomes upon incubation with Con-A for 30 min. Movie M6 provided in Supporting Information depicts the clustering of aggregates in three dimensions.
Typically, over several tens of minutes, we found that more number of smaller aggregates coalesce with each other and eventually adhere directly to the surface or attach onto existing surface-bound aggregates (Figure 5e). Furthermore, we noticed that some of these aggregates (similar to those zoomed-in at the bottom as shown in Figure 5e) tend to undergo anchored or restricted motion in solution even when attached to the surface (Figure S13). Such a restricted anchored movement may also be responsible for the Con-A-induced bridging (or connection) of two or more nearby aggregates, eventually leading to the formation of higher-order aggregates, as shown in Figure 5e (blow up on top). It is worth mentioning that estimation of the number of GP-PICsomes within each surface-bound aggregate using static fluorescence images (such as Figure 5e) can be very challenging if not impossible; for instance, single snapshot image at 25 s in Figure 5b seems to portray the existence of only 2–3 GP-PICsomes within the aggregate, whereas observation of the entire movie reveals a more realistic picture. In fact, all large aggregates are 3D assemblies, which constitute much more number of PICsomes as compared with those observed from the static fluorescence images. This is further evident for very large extended clusters of aggregates (Figure 5f) that are generated on the surface over several hours in the presence of Con-A, and such 3D assembly of PICsomes in these large aggregate clusters can be readily visualized upon changing the (z-) focus of the objective during fluorescence imaging (see Movie M6).
To ensure that the aggregation of RhB-GP-PICsomes observed was indeed induced by specific interaction of the mannose-binding domain in Con-A interacting with the surface-grafted mannose moieties of GP-PICsomes (Figure 4), we have performed challenge experiments with excess (300 mM) of free mannose and monitored surface-bound large aggregates and aggregate-clusters using fluorescence video imaging. The results of such a competition experiment are depicted in Figure 6a, where a large aggregate cluster of RhB-GP-PICsomes is imaged under hydrodynamic flow in the absence and presence of excess mannose. We find that within a few seconds in the presence of the mannose-containing buffer solution (at ∼6 s), the aggregate cluster disassembles rapidly (∼seconds) and only a small fraction of GP-PICsomes (or their smaller aggregates) still remain bound to the surface. A possible reason for the presence of these firmly adhered smaller aggregates is that mannose was unable to access all Con-A-binding sites (buried within aggregates) with equal ease. As a control, the same experiments were performed with galactose, a carbohydrate which has nominal binding affinity toward Con-A (Figure 4). We find that excess (300 mM) of galactose does not affect the aggregates or large aggregate clusters in any way. For instance, as shown in Figure 6b, neither deaggregation occurred nor any GP-PICsome disassembled from Aggregate-1 under continuous flow of buffer-containing excess galactose. We were able to identify a few large GP-PICsome aggregate clusters, parts of which were not firmly anchored to the glass surface, such as Aggregate-2 in Figure 6b. Relatively long time (5 min) incubation with excess galactose followed by intermittent buffer flow (momentarily turned on and turned off) could only induce a part of this aggregate cluster to swivel rather than disassemble, which recoiled in the absence of the hydrodynamic drag without altering the remaining structure. From these experiments, we conclude that the self-assembled PICsomes displayed part or all of the GP on their outer surface, and the mannose residues of GP specifically interacted with it’s lectin rendering the surface of PICsomes biochemically active.
Figure 6.

(a) Dissociation/deaggregation of an immobile RhB-GP-PICsome aggregate cluster upon the addition of excess monomeric mannose solution. The first panel (0–4.5 s) depicts the time-average image of the aggregate-cluster in the absence of flow, whereas other panels are sequential single-frame snapshots under flow in the absence (5–6 s) and presence (>6 s) of mannose (300 mM) containing buffer. Movie M7 is provided in Supporting Information. (b) Sequential single-frame snapshots of an immobile GP-PICsome aggregate cluster (Aggregate-1) attached to glass surface under continuous flow of 300 mM galactose-containing buffer. Single-frame snapshots of a larger aggregate cluster (Aggregate-2, right) incubated with 300 mM galactose for 5 min, under momentary buffer flow (suddenly turned on and off, as represented by arrowheads). Movie M8 is provided in Supporting Information.
4. Conclusions
To develop mimics of glycoproteins-bearing cell surfaces, we have synthesized oppositely charged hydrophilic block copolymers (catiomer and aniomer) based on glycopolypeptide-b-poly-l-lysine and PEG2k-b-poly-l-glutamate, which self-assemble in an aqueous medium to form glycopolypeptide-based polyionic complex vesicles (GP-PICsomes). Imaging of these microscopic assemblies in solution and in the dry state reveals a hollow structural morphology of few micrometer dimensions, where several carbohydrate functional groups protrude out in solution, reminiscent of a biological cell. More importantly, the surface-grafted mannose moieties retain biochemically activity, as evident from multivalent interaction dynamics of the GP-PICsomes to form aggregates, mediated by a carbohydrate-binding protein (lectin). Our results demonstrate that the bioactivity of surface-grafted carbohydrate groups of PICsomes is highly specific to the lectin Con-A that has multiple binding sites and thereby acts as an effective bridge between GP-PICsomes to induce strong interparticle interactions. Using fluorescently labeled GP-PICsomes, we have been able to visualize the aggregation dynamics in the presence of Con-A, which show the formation of extended, three-dimensional higher-order aggregate clusters from the association of individual PICsomes over time, at times mediated by Con-A bound to the glass surface. Finally, we show that the PICsome-aggregate clusters disassemble (deaggregate) spontaneously only in the presence of excess mannose in solution, reiterating the specificity of biomolecular interactions of surface pendant groups. We speculate that these large surface bioactive GP-PICsomes (or further modifications thereof) have the potential to be used as suitable models to study various interactions on cellular surfaces and intercellular recognition.
Acknowledgments
B.P. thanks CSIR New Delhi, J.M. thanks IRCC IIT Bombay, and K.B.C. thanks DST New Delhi for fellowships. S.S.G. thanks CSIR 12th FYP project CSC0302 for funding. A.C. acknowledges IRCC IIT Bombay for initial funding. We gratefully acknowledge Dr. P. Rajamohanan for NMR support, Mrs. D. Dhoble for GPC measurements, and Mr. Amit. K. Yadav for CD measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00142.
Synthetic procedures for all monomers and polymers with characterization using 1H NMR, 13C NMR, FT-IR, GPC, fluorescence spectra, CD spectra, and movies for interaction dynamic studies (PDF)
Wide field movie of a single RhB-GP-PICsome diffusing randomly in buffer solution (AVI)
Random motion of another single Rh labeled GP-PICsome in buffer solution (AVI)
RhB-GP-PICsome in presence of Con-A, showing a small cluster undergoing tumbling motion in buffer solution (AVI)
Small RhB-GP-PICsome aggregate in presence of unidirectional buffer flow (AVI)
Dynamics of a larger RhB-GP-PICsome aggregate in the presence of a buffer flow (AVI)
RhB-GP-PICsome aggregate-clusters while changing the Z focus during movie collection (AVI)
Deaggregation of a RhB GP-PICsome aggregate upon addition of excess mannose (AVI)
Dynamics of a RhB-GP-PICsome aggregate-clusters incubated with excess galactose depicting its swivel motion by intermittent buffer flow (AVI)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bertozzi C. R.; Kiessling L. L. Chemical Glycobiology. Science 2001, 291, 2357–2364. 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- Mammen M.; Choi S.-K.; Whitesides G. M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. . [DOI] [PubMed] [Google Scholar]
- Boltje T. J.; Buskas T.; Boons G.-J. Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat. Chem. 2009, 1, 611–622. 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L.; Vosseller K.; Hart G. W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378. 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- Ambrosi M.; Cameron N. R.; Davis B. G. Lectins: Tools for the molecular understanding of the glycocode. Org. Biomol. Chem. 2005, 3, 1593–1608. 10.1039/b414350g. [DOI] [PubMed] [Google Scholar]
- Ting S. R. S.; Chen G.; Stenzel M. H. Synthesis of glycopolymers and their multivalent recognitions with lectins. Polym. Chem. 2010, 1, 1392–1412. 10.1039/c0py00141d. [DOI] [Google Scholar]
- Schatz C.; Louguet S.; Meins J.-F. L.; Lecommandoux S. Polysaccharide-Block-Polypeptide Copolymer Vesicles: Towards Synthetic Viral Capsids. Angew. Chem., Int. Ed. 2009, 48, 2572–2575. 10.1002/anie.200805895. [DOI] [PubMed] [Google Scholar]
- Wu D.-Q.; Lu B.; Chang C.; Chen C.-S.; Wang T.; Zhang Y.-Y.; Cheng S.-X.; Jiang X.-J.; Zhang X.-Z.; Zhuo R.-X. Galactosylated Fluorescent Labelled Micelles as a Liver Targeting Drug Carrier. Biomaterials 2009, 30, 1363–1371. 10.1016/j.biomaterials.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Cho C. S.; Seo S. J.; Park I. K.; Kim S. H.; Kim T. H.; Hoshiba T.; Harada I.; Akaike T. Galactose-Carrying Polymers as Extracellular Matrices for Liver Tissue Engineering. Biomaterials 2006, 27, 576–585. 10.1016/j.biomaterials.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Chen G.; Amajjahe S.; Stenzel M. H. Synthesis of Thiol-Linked Neoglycopolymers and Thermo-Responsive Glycomicelles as Potential Drug Carrier. Chem. Commun. 2009, 10, 1198–1200. 10.1039/b900215d. [DOI] [PubMed] [Google Scholar]
- Bonduelle C.; Lecommandoux S. Synthetic Glycopolypeptides as Biomimetic Analogues of Natural Glycoproteins. Biomacromolecules 2013, 14, 2973–2983. 10.1021/bm4008088. [DOI] [PubMed] [Google Scholar]
- Herzner H.; Reipen T.; Schultz M.; Kunz H. Synthesis of glycopeptides containing carbohydrate and peptide recognition motifs. Chem. Rev. 2000, 100, 4495–4538. 10.1021/cr990308c. [DOI] [PubMed] [Google Scholar]
- Pratt M. A.; Bertozzi C. R. Synthetic glycopeptides and glycoproteins as tools for biology. Chem. Soc. Rev. 2005, 34, 58–68. 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Dordick J. S.; Linhardt R. J. Synthesis and application of carbohydrate-containing polymers. Chem. Mater. 2002, 14, 3232–3244. 10.1021/cm0200137. [DOI] [Google Scholar]
- Kramer J. R.; Deming T. J. Recent advances in glycopolypeptide synthesis. Polym. Chem. 2014, 5, 671–682. 10.1039/C3PY01081C. [DOI] [Google Scholar]
- Krannig K.-S.; Schlaad H. Emerging bioinspired polymers: Glycopolypeptides. Soft Matter 2014, 10, 4228–4235. 10.1039/c4sm00352g. [DOI] [PubMed] [Google Scholar]
- Pati D.; Kalva N.; Das S.; Kumaraswamy G.; Gupta S. S.; Ambade A. V. Multiple Topologies from Glycopolypeptide–Dendron Conjugate Self-Assembly: Nanorods, Micelles, and Organogels. J. Am. Chem. Soc. 2012, 134, 7796–7802. 10.1021/ja300065f. [DOI] [PubMed] [Google Scholar]
- Das S.; Sharma D. K.; Chakrabarty S.; Chowdhury A.; Gupta S. S. Bioactive polymersomes self-assembled from amphiphilic PPO-glyco polypeptides: Synthesis, characterization, and dual-dye encapsulation. Langmuir 2015, 31, 3402–3412. 10.1021/la503993e. [DOI] [PubMed] [Google Scholar]
- Dhaware V.; Shaikh A. Y.; Kar M.; Hotha S.; Gupta S. S. Synthesis and self-assembly of amphiphilic homoglycopolypeptide. Langmuir 2013, 29, 5659–5667. 10.1021/la400144t. [DOI] [PubMed] [Google Scholar]
- Kramer J. R.; Rodriguez A. R.; Choe U.-J.; Kamei D. T.; Deming T. J. Glycopolypeptide Conformations in Bioactive Block Copolymer Assemblies Influence Their Nanoscale Morphology. Soft Matter 2013, 9, 3389–3395. 10.1039/c3sm27559k. [DOI] [Google Scholar]
- Bonduelle C.; Huang J.; Ibarboure E.; Heise A.; Lecommandoux S. Synthesis and Self-Assembly of “Tree-Like”Amphiphilic Glycopolypeptides. Chem. Commun. 2012, 48, 8353–8355. 10.1039/c2cc32970k. [DOI] [PubMed] [Google Scholar]
- Pati D.; Das S.; Patil N. G.; Parekh N.; Anjum D. H.; Dhaware V.; Ambade A. V.; Gupta S. S. Tunable nanocarriers morphologies from glycopolypeptide-based amphiphilic biocompatible star copolymer and their carbohydrate specific intracellular delivery. Biomacromolecules 2016, 17, 466–475. 10.1021/acs.biomac.5b01354. [DOI] [PubMed] [Google Scholar]
- Onaca O.; Nallani M.; Ihle S.; Schenk A.; Schwaneberg U. Functionalized nanocompartments (synthosomes): Limitation and prospective applications in industrial biotechnology. Biotechnol. J. 2006, 1, 795–805. 10.1002/biot.200600050. [DOI] [PubMed] [Google Scholar]
- Yow H. N.; Routh A. F. Formation of liquid core–polymer shell microcapsules. Soft Matter 2006, 2, 940–949. 10.1039/B606965G. [DOI] [PubMed] [Google Scholar]
- Discher B. M.; Won Y.-Y.; Ege D. S.; Lee J. C.-M.; Bates F. S.; Discher D. E.; Hammer D. A. Polymersomes: Tough vesicles made from diblock copolymers. Science 1999, 284, 1143–1146. 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Kataoka K. Biosignal-sensitive polyion complex micelles for the delivery of biopharmaceuticals. Soft Matter 2009, 5, 3810–3817. 10.1039/b909934d. [DOI] [Google Scholar]
- Kataoka K.; Harada A. Supramolecular assemblies of block copolymers in aqueous media as nanocontainers relevant to biological applications. Prog. Polym. Sci. 2006, 31, 949–982. 10.1016/j.progpolymsci.2006.09.004. [DOI] [Google Scholar]
- Hartig S. M.; Greene R. R.; Dikov M. M.; Prokop A.; Davidson J. M. Multifunctional nanoparticulate polyelectrolyte complexes. Pharm. Res. 2007, 24, 2353–2369. 10.1007/s11095-007-9459-1. [DOI] [PubMed] [Google Scholar]
- Harada K.; Kataoka K. Formation of Polyion Complex Micelles in an Aqueous Milieu from a Pair of Oppositely-Charged Block Copolymers with Poly(ethy1ene glycol) Segments. Macromolecules 1995, 28, 5294–5299. 10.1021/ma00119a019. [DOI] [Google Scholar]
- Chuanoi S.; Kishimura A.; Dong W.-F.; Anraku1 Y.; Yamasaki Y.; Kataoka K. Structural factors directing nanosized polyion complex vesicles (Nano-PICsomes) to form a pair of block aniomer/homo catiomers: Studies on the aniomer segment length and the catiomer side-chain structure. Polym. J. 2014, 46, 130–135. 10.1038/pj.2013.82. [DOI] [Google Scholar]
- Koide Y.; Kishimura A.; Osada K.; Jang W.-D.; Yamasaki Y.; Kataoka K. Semipermeable Polymer Vesicle (PICsome) Self-Assembled in Aqueous Medium from a Pair of Oppositely Charged Block Copolymers: Physiologically Stable Micro-/Nanocontainers of Water-Soluble Macromolecules. J. Am. Chem. Soc. 2006, 128, 5988–5989. 10.1021/ja057993r. [DOI] [PubMed] [Google Scholar]
- Dong W.-F.; Kishimura A.; Anraku Y.; Chuanoi S.; Kataoka K. Monodispersed Polymeric Nanocapsules: Spontaneous Evolution and Morphology Transition from Reducible Hetero-PEG PICmicelles by Controlled Degradation. J. Am. Chem. Soc. 2009, 131, 3804–3805. 10.1021/ja808419b. [DOI] [PubMed] [Google Scholar]
- Anraku Y.; Kishimura A.; Oba M.; Yamasaki Y.; Kataoka K. Spontaneous Formation of Nanosized Unilamellar Polyion Complex Vesicles with Tunable Size and Properties. J. Am. Chem. Soc. 2010, 132, 1631–1636. 10.1021/ja908350e. [DOI] [PubMed] [Google Scholar]
- Anraku Y.; Kishimura A.; Yamasaki Y.; Kataoka K. Living Unimodal Growth of Polyion Complex Vesicles via Two-Dimensional Supramolecular Polymerization. J. Am. Chem. Soc. 2013, 135, 1423–1429. 10.1021/ja3096587. [DOI] [PubMed] [Google Scholar]
- Kishimura A.; Koide A.; Osada K.; Yamasaki Y.; Kataoka K. Encapsulation of Myoglobin in PEGylated Polyion Complex Vesicles made from a Pair of Oppositely Charged Block Ionomers: A Physiologically Available Oxygen Carrier. Angew. Chem., Int. Ed. 2007, 46, 6085–6088. 10.1002/anie.200701776. [DOI] [PubMed] [Google Scholar]
- Oana H.; Kishimura A.; Yonehara K.; Yamasaki Y.; Washizu M.; Kataoka K. Spontaneous Formation of Giant Unilamellar Vesicles from Microdroplets of a Polyion Complex by Thermally Induced Phase Separation. Angew. Chem., Int. Ed. 2009, 48, 4613–4616. 10.1002/anie.200900721. [DOI] [PubMed] [Google Scholar]
- Munoz E. M.; Correa J.; Riguera R.; Fernandez-Megia E. Real-Time Evaluation of Binding Mechanisms in Multivalent Interactions: A Surface Plasmon Resonance Kinetic Approach. J. Am. Chem. Soc. 2013, 135, 5966–5969. 10.1021/ja400951g. [DOI] [PubMed] [Google Scholar]
- Su L.; Zhao Y.; Chen G.; Jiang M. Polymeric vesicles mimicking glycocalyx (PV-Gx) for studying carbohydrate–protein interactions in solution. Polym. Chem. 2012, 3, 1560–1566. 10.1039/c2py20110k. [DOI] [Google Scholar]
- Yilmaz G.; Becer C. R. Glyconanoparticles and their interactions with lectins. Polym. Chem. 2015, 6, 5503–5514. 10.1039/C5PY00089K. [DOI] [Google Scholar]
- Nagasaki Y.; Yasugi K.; Yamamoto Y.; Harada A.; Kataoka K. Sugar-Installed Block Copolymer Micelles: Their Preparation and Specific Interaction with Lectin Molecules. Biomacromolecules 2001, 2, 1067–1070. 10.1021/bm015574q. [DOI] [PubMed] [Google Scholar]
- Pati D.; Shaikh A. Y.; Das S.; Nareddy P. K.; Swamy M. J.; Hotha S.; Gupta S. S. Controlled Synthesis of O-Glycopolypeptide Polymers and Their Molecular Recognition by Lectins. Biomacromolecules 2012, 13, 1287–1295. 10.1021/bm201813s. [DOI] [PubMed] [Google Scholar]
- Duncan G. A.; Bevan M. A. Tunable Aggregation by Competing Biomolecular Interactions. Langmuir 2014, 30, 15253–15260. 10.1021/la503772g. [DOI] [PubMed] [Google Scholar]
- Das S.; Pati D.; Tiwari N.; Nisal A.; Gupta S. S. Synthesis of Silk Fibroin–Glycopolypeptide Conjugates and Their Recognition with Lectin. Biomacromolecules 2012, 13, 3695–3702. 10.1021/bm301170u. [DOI] [PubMed] [Google Scholar]
- Hong V.; Presolski S. I.; Ma C.; Finn M. G. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew. Chem., Int. Ed. 2009, 48, 9879–9883. 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlist J. V. D.; Faber M.; Loen L.; Dijkman T. J.; Asri L. A. T. W.; Loos K. Synthesis of Hyperbranched Glycoconjugates by the Combined Action of Potato Phosphorylase and Glycogen Branching Enzyme from Deinococcus geothermalis. Polymers 2012, 4, 674–690. 10.3390/polym4010674. [DOI] [Google Scholar]
- Bhattacharya S.; Sharma D. K.; Saurabh S.; De S.; Sain A.; Nandi A.; Chowdhury A. Plasticization of poly(vinylpyrrolidone) thin films under ambient humidity: Insight from single-molecule tracer diffusion dynamics. J. Phys. Chem. B 2013, 117, 7771–7782. 10.1021/jp401704e. [DOI] [PubMed] [Google Scholar]
- Kazakov S.; Kaholek M.; Kudasheva D.; Teraoka I.; Cowman M. K.; Levon K. Poly(N-isopropylacrylamide-co-1-Vinylimidazole) Hydrogel Nanoparticles Prepared and Hydrophobically Modified in Liposome Reactors: Atomic Force Microscopy and Dynamic Light Scattering Study. Langmuir 2003, 19, 8086–8093. 10.1021/la030066i. [DOI] [Google Scholar]
- Mai Y.; Eisenberg A. Self-Assembly of Block Copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Armes S. P.; Ryan A. J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and Their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. 10.1002/marc.200800713. [DOI] [PubMed] [Google Scholar]
- Antonietti M.; Förster S. Vesicles and Liposomes: A Self-Assembly Principle Beyond Lipids. Adv. Mater. 2003, 15, 1323–1333. 10.1002/adma.200300010. [DOI] [Google Scholar]
- Discher D. E.; Eisenberg A. Polymer Vesicles. Science 2002, 297, 967–973. 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.