

Abstract
Vancomycin analogues bearing an A-ring trimethylammonium salt modification were synthesized and their antimicrobial activity against vancomycin-resistant Enterococci (VRE) was evaluated. The modification increased antimicrobial potency and provided the capability to induce bacteria cell membrane permeabilization, but both properties were weaker than that found with our earlier reported similar C-terminus modification. The results provide further insights on the additive effect and generalizability of the structural and site-specific nature of a peripheral quaternary trimethylammonium salt modification of vancomycin.
Keywords: glycopeptide antibiotics, antibiotic mechanism of action, membrane permeabilization, vancomycin peripheral modification, trimethylammonium salt
Graphical Abstract
1. Introduction
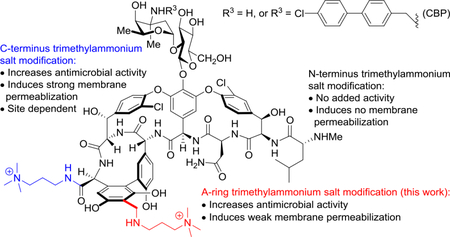
Vancomycin (1) and its related glycopeptide analogues are an invaluable class of antibiotics widely used in the clinic over the past 60 years [1–4]. They are effective against Gram-positive pathogens, especially methicillin-resistant Staphylococcus aureus (MRSA) where they are considered the antibiotics of the last resort [5,6]. The outstanding antimicrobial activity of vancomycin arises from its binding with the d-Ala-d-Ala residues on the C-terminus of peptidoglycan precursors, preventing the late-stage maturation and crosslinking step of bacteria cell wall biosynthesis [6–8]. Since the mechanism of action of vancomycin targets and sequesters a highly conserved enzyme substrate required for cell wall synthesis, antimicrobial resistance was slow to emerge and was only observed after 30 years of clinical use: first in Enterococci (VRE) [9–12] followed by the development of the resistance in Staphylococcus aureus (VRSA) [13–15]. The wide-spread antimicrobial resistance in VRE and the emergence of VRSA presents an urgent need for new glycopeptide antibiotics that overcome this resistance [16,17]. Peripheral modifications of vancomycin and related glycopeptide antibiotics have been found to increase the antimicrobial potency against both sensitive and resistant organisms by enhancing the interaction with bacterial membrane [18,19], or through added mechanisms of action independent of d-Ala-d-Ala/d-Ala-d-Lac binding [20–21]. Representative of this behavior, chlorobiphenylmethyl (CBP-)vancomycin (2) bears a lipophilic modification on vancosamine, which was found to enhance antimicrobial potency as much as 100-fold through an added mechanism of direct, competitive inhibition of transglycosylase [21–23] independent of d-Ala-d-Ala/d-Ala-d-Lac binding. The development and optimization of such semi-synthetic peripheral modifications of the glycopeptide antibiotics have provided three approved drugs that are now used in the clinic, oritavancin [24], dalbavancin [25], and telavancin [26]. Beyond simply improving the potency or spectrum of activity, such peripherally modified glycopeptide antibiotics that act by multiple mechanisms of action also display a greater durability [27]. Building on the total synthesis of the naturally occurring glycopeptide antibiotics[28–35] and the subsequent total synthesis of pocket modified vancomycin analogues designed to exhibit dual d-Ala-d-Ala/d-Ala-d-Lac binding[36–42], we have also examined their peripherally modified analogues. These not only regain activity against vancomycin-resistant organisms (VRE and VRSA) by virtue of the pocket modifications, but they also display added mechanisms of action not expressed by the underlying glycopeptide antibiotic due to the peripheral changes. Such analogues display a remarkable spectrum of activities, superb potencies, and stunning durabilities. This symposium-in-print is in honor of the 2019 Tetrahedron Young Investigator Awardee Ryan Shenvi who introduced the term “supernatural” products[27] to describe and distinguish such natural product analogues. Like the semisynthetic glycopeptide antibiotic drugs, such pocket and peripherally modified analogues are worthy of classification as “supernatural” products. Most notable of their characteristics is the antibiotic durability that may be attributed to the expression of multiple synergistic mechanisms of action, two of which are not expressed by the natural products themselves.
A trimethylammonium salt modification has been used as a method of introducing a permanent positive charge into a molecule while minimally altering its structure. This modification is often used to improve water solubility, and in some cases has been explored to improve antibacterial or antifungal activity[43–46]. Recently, we described an effective and useful quaternary trimethylammonium salt (C1) modification on the C-terminus of glycopeptide antibiotics [47]. The resulting vancomycin analogues (e.g., 3 and 4) were found to bear a third mechanism of action independent of d-Ala-d-Ala/d-Ala-d-Lac binding and transglycosylase inhibition (Fig. 1). This modification led to permeabilization of bacterial cell membrane, further enhancing both the antimicrobial potency and the durability of the antibiotics, and its impact was unique to the trimethylammonium salt (vs other trialkylammonium salts). We have also shown that a similar trimethylammonium salt modification on the N-terminus of vancomycin did not increase the potency of the parent antibiotics against either sensitive or resistant bacterial strains, implicating the site-specific nature of the modification [48]. In order to further probe this site-specific feature of the added substitution, as well as define new modification patterns that further enhance the antimicrobial potency of glycopeptide antibiotics, we examined and herein report a similar trimethylammonium salt modification installed on the resorcinol A-ring on vancomycin and its derivatives. Only a few examples of A-ring quaternary ammonium salt modifications on glycopeptide antibiotics have been reported[49–52], and the potency of such analogues was found to be comparable to, but not exceed that of, the parent antibiotics (chloroorienticin B and CBP-vancomycin) [49, 50]. Although the solubility of lipophilic glycopeptide antibiotics improved substantially with such modifications, the impact of this specific modification on the mechanism of action remains unclear. Herein, we report the synthesis and antimicrobial activity of vancomycin derivatives bearing an A-ring trimethylammonium salt or two such quaternary salt modifications. The investigation on mechanism of action of the newly synthesized derivatives provided additional insights on the site-specific nature of this peripheral modification of vancomycin.
Fig. 1.

Structure of vancomycin (1) and its CBP and C1 derivatives 2-4.
2. Results and discussion
Vancomycin bearing a trimethylammonium salt modification on the resorcinol A-ring was synthesized by a Mannich reaction between vancomycin, formaldehyde and a precursor to the C1 salt (H2NCH2CH2CH2NMe3+·Cl−) to provide 5 (C1Ar-vancomycin) by following a reported A-ring modification procedure [53]. A CBP group was then attached to the vancosamine site by reductive amination to give 6 (C1Ar-CBP-vancomycin). Vancomycin analogues that bear C1 modifications on both the A-ring and C-terminus were synthesized by coupling the A-ring modified vancomycins (5 and 6) with the C1 salt, providing C1-C1Ar-vancomycin (7) and C1- C1Ar-CBP-vancomycin (8), respectively (Scheme 1).
Scheme 1.

Synthesis of A-ring trimethylammonium salt modified vancomycins 5-8.
We examined the antimicrobial activity of 5-8 against vancomycin-resistant bacteria (VanA vancomycin-resistant enterococci, VRE) using a standard microdilution assay [54]. The results are summarized in Table 1 alongside vancomycin (1), CBP-vancomycin (2) and our earlier C-terminus trimethylammonium salt analogues (3 and 4). In such organisms, vancomycin analogues fail to express activity derived from d-Ala-d-Lac binding and any significant improvements are likely derived from alternative or added mechanisms of action endowed by the modification to the glycopeptide antibiotic. The A-ring trimethylammonium salt modification on vancomycin with 5 provided a 2 to 8-fold increase in antimicrobial potency against VRE strains, which approached but did not improve on the enhancement introduced by our C-terminus C1 modification (3). The doubly modified analogue 7, where the C-terminus and A-ring modifications were combined, was found to be 4 to 8-fold more potent than either 3 and 5, and 16 to 64-fold more potent than vancomycin. In contrast, the impact of the A-ring quaternary salt modification on the CBP-vancomycin derivatives (6 and 8) was found to be much subtler, where 6 displayed no (E. faecalis) or only a moderate increase in activity (E. faecium). By contrast, the analogous C-terminus modification in 4 provided a more substantial 5 to 10-fold increase in activity. The added C1 modification on C-terminus of 8 (compared with 6), was found to provide a subtle increase (2-fold) in antimicrobial potency against both strains.
Table 1.
Antimicrobial activity of 1–8.
| Compound | MIC (μg/mL) | |
|---|---|---|
| VanAa VRE | VanA VRE | |
| E. faecalisb | E. faeciumc | |
| vancomycin (1) | 250 | 250 |
| C1-vancomycin (3) | 63 | 31 |
| C1Ar-vancomycin (5) | 125 | 31 |
| C1-C1Ar-vancomycin (7) | 16 | 4 |
| CBP-vancomycin (2) | 2.5 | 2.5 |
| C1-CBP-vancomycin (4) | 0.25 | 0.5 |
| C1Ar-CBP-vancomycin (6) | 5 | 0.6 |
| C1-C1Ar-CBP-vancomycin (8) | 2.5 | 0.3 |
VanA: bacteria strains that are resistant to both vancomycin and teicoplanin.
BM 4166.
ATCC BAA-2317.
We have previously shown that C-terminus trimethylammonium salt modified vancomycin derivatives were capable of inducing bacteria cell membrane permeabilization independent of d-Ala-d-Ala/d-Ala-d-Lac binding [47]. The subsequent examination of the N-terminus trimethylammonium salt vancomycin derivatives, which had no impact on permeabilization, revealed that this effect was dependent on the site of the quaternary salt modification [48]. In order to further establish the site-specific nature of this modification, as well as to investigate the mechanistic implications of the A-ring quaternary salt modification, we examined the compounds for their ability to induce cell membrane permeabilization [55] (propidium iodide (PI) influx, Fig. 2). All three trimethylammonium salt modified CBP-vancomycins (4, 6 and 8) displayed the ability to induce bacteria membrane permeabilization, whereas CBP-vancomycin (2) itself does not. While the C-terminus trimethylammonium salt modified CBP-vancomycin (4) effectively induced permeabilization as previously reported, the same modification introduced on the A-ring resulted in a weaker permeabilization (6). The combination of these two modifications in compound 8 was found to display an additive effect, where the initial rate of membrane permeabilization was faster than that with either 4 or 6 (4 > 6).
Fig. 2.

Examination of cell membrane permeability induced by compounds 1, 2, and 4-8. (10 μM added at 5 min) in VanA VRE E. faecalis ATCC BAA-2317.
The trend in the ability of the trimethylammonium modified vancomycins to induce bacterial membrane permeabilization (8 > 4 > 6 > 2) paralleled their impact on antimicrobial potency (8 > 4 ≈ 6 > 2) in the same VanA VRE strain (ATCC BAA-2317). The combination of this trend with our previous observations on the effect of N-terminus modified vancomycins[48] generalizes the site-specific nature of the trimethylammonium salt modification on vancomycin at three possible modification sites with the antimicrobial potency enhancement and the extent of bacterial cell membrane permeabilization following a trend of C-terminus (strong permeabilization) > A-ring (weak permeabilization) > N-terminus (no permeabilization).
3. Conclusion
A series of vancomycin derivatives bearing an A-ring quaternary trimethylammonium salt modification were prepared and their antimicrobial potency was evaluated. This specific modification, either alone or combined with a C-terminus trimethylammonium salt modification, was found to enhance the antimicrobial activity of vancomycin against VRE, following a trend of double quaternary salt modification > C-terminus modification ≥ A-ring modification > N-terminus modification. The examination of mechanism of action of the compounds revealed the additive and site-specific nature of the peripheral modification, with the extent of bacterial cell membrane permeabilization following the same trend. Complementing our observations, it has also been shown that the presence of a permanent positive charge on the glucose residue (C6 position), including a trimethylammonium salt, resulted in reduced activity against vancomycin-resistant bacteria[56], further highlighting that the productive impact of a trimethylammonium salt in vancomycin analogues is dependent upon its location .
We view the trimethylammonium salt as a permanently charged surrogate for a protonated dimethylamino group. Its incorporation does not promote bacterial or mammalian cell wall lysis, does not correlate with bacterial cell wall depolarization,[47] and its effects are distinct and easily distinguishable from those of lipophilic quaternary ammonium salts. The exact mechanism or target by which it promotes the cell wall permeability independent of d-Ala-d-Ala/d-Ala-d-Lac binding and why its actions are synergistic with others that impact bacterial cell wall integrity are under further investigation.
4. Experimental section
4.1. General procedures
Reagents and solvents were purchased from commercial sources and used as received unless otherwise noted. Nuclear magnetic resonance spectra (1H NMR and 13C NMR) were recorded on a Bruker DRX-600 NMR spectrophotometer at 298 K. Residual solvent peaks were used as an internal reference. Coupling constants (J) (H, H) are given in Hz. Coupling patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet (m), or broad signal (br). High resolution mass spectra were measured with a TOF mass spectrometer. Optical rotation was measured on a Rudolph polarimeter. Analytical and preparative reverse-phase HPLC was performed using a Waters HPLC.
4.2. Synthesis of C1Ar-vancomycin (5)
A stirred solution of vancomycin (1, 100.0 mg, 67 μmol) in 0.33 M NaOH (750 μL) was treated with C1 (Me3N+(CH2)3NH2·Cl−, 76.3 mg, 500 μmol) and formaldehyde (37% solution in water, 5.0 μL, 67 μmol). The reaction mixture was stirred for 8 h at 5 °C in the dark before being quenched by water (2 mL). AcOH (60 μL) was then added to the mixture to adjust to pH 4. Semi-preparative reverse-phase HPLC (Nacalai Tesque, Inc., ARII-C18, 5 μm, 10 × 150 mm, 1–40% MeCN/H2O-0.07% TFA gradient over 40 min, 3 mL/min, tR = 18.2 min) afforded 5 (37.0 mg, 35%) as a white solid: 1H NMR (CD3OD, 600 MHz, 298 K) δ 9.34 (br, 1H), 8.91 (br, 2H), 7.88–7.80 (m, 1H), 7.77–7.72 (m, 2H), 7.60 (d, 2H, J = 8.3 Hz), 7.44 (s, 1H), 7.23 (s, 1H), 7.15 (d, 1H, J = 7.7 Hz), 6.75 (br, 1H), 6.62 (s, 1H), 6.14–6.03 (m, 2H), 5.59 (br, 1H), 5.50 (s, 1H), 5.42 (s, 1H), 5.38 (s, 1H), 5.27 (s, 1H), 4.75 (s, 1H), 4.72–4.68 (m, 1H), 4.44 (d, 1H, J = 13.4 Hz), 4.37 (d, 1H, J = 13.4 Hz), 4.26–4.21 (m, 2H), 4.06 (t, 1H, J = 6.7 Hz), 4.02–3.96 (m, 1H), 3.90–3.83 (m, 2H), 3.65 (d, 2H, J = 6.8 Hz), 3.43–3.39 (m, 2H), 3.37–3.34 (m, 2H), 3.14 (s, 9H), 3.12–3.09 (m, 1H), 3.03–2.98 (m, 1H), 2.90–2.87 (m, 1H), 2.77 (s, 3H), 2.30–2.23 (m, 1H), 2.22–2.14 (m, 1H), 1.94 (d, 2H, J = 11.3 Hz), 1.91 (br, 1H), 1.68–1.61 (m, 2H), 1.57 (s, 3H), 1.16 (s, 3H), 0.94 (d, 3H, J = 5.6 Hz), 0.90 (d, 3H, J = 5.6 Hz). 13C NMR (CD3OD, 150 MHz, 298 K) δ 175.7, 174.3, 172.8, 171.6, 169.9, 169.7, 169.4, 168.9, 162.7 (q, 2J = 35.0 Hz, CF3COO−), 158.3, 158.2, 157.1, 156.2, 156.0, 154.4, 150.8, 143.2, 141.5, 139.0, 138.0, 134.4, 130.0, 129.9, 129.4, 129.2, 129.1, 128.6, 128.4, 127.8, 126.3, 125.3, 124.9, 120.1, 119.3, 118.9, 118.0 (q, 1J = 291.5 Hz, CF3COO−), 108.2, 106.0, 102.2, 98.2, 79.1, 78.0, 74.1, 73.2, 72.5, 70.8, 64.9, 64.4, 64.1, 62.2, 61.9, 58.5, 56.4, 55.9, 55.4, 54.8, 53.9, 52.5, 49.6, 44.7, 43.5, 40.9, 40.3, 38.1, 37.1, 34.4, 32.9, 25.5, 23.6, 22.9, 20.3, 17.4. [α]D20 +20 (c 0.02, CH3OH). ESI-TOF HRMS m/z 1576.5676 ([M + H]+, [C73H91Cl2N11O24 + H]+ requires 1576.5688).
4.3. Synthesis of C1Ar-CBP-vancomycin (6)
Compound 5 (10.0 mg, 5.2 μmol), 4-(4’-chlorophenyl)benzaldehyde (2.2 mg, 10 μmol), and i-Pr2NEt (9.6 mg, 7.4 μmol) were dissolved in DMF (800 μL) and the mixture was stirred at 70 °C for 2 h. NaCNBH3 in THF (1.0 M, 154 μL, 154 μmol) was added to the solution and the mixture was stirred at 70 °C for another 5 h. The mixture was then cooled to 25 °C and diluted by addition of 50% MeOH in H2O (0.5 mL). Semi-preparative reverse-phase HPLC (Nacalai Tesque, Inc., ARII-C18, 5 μm, 10 × 150 mm, 20–80% MeCN/H2O-0.07% TFA gradient over 40 min, 3 mL/min, tR = 12.2 min) afforded 6 (2.0 mg, 18%) as a white solid: 1H NMR (CD3OD, 600 MHz, 298 K) δ 8.99 (s, 1H), 8.92 (s, 1H), 7.87 (s, 2H), 7.79–7.74 (m, 2H), 7.72 (d, 3H, J = 8.2 Hz), 7.65 (d, 3H, J = 8.5 Hz), 7.58 (d, 2H, J = 7.9 Hz), 7.48 (d, 3H, J = 8.5 Hz), 7.30 (s, 1H), 7.25–7.20 (m, 1H), 6.81 (s, 1H), 6.64 (s, 1H), 5.64 (br, 1H), 5.51 (s, 1H), 5.46 (s, 1H), 5.42 (s, 1H), 5.30 (s, 1H), 4.77 (br, 1H), 4.74 (d, 1H, J = 5.5 Hz), 4.47 (d, 1H, J = 13.4 Hz), 4.39 (d, 1H, J = 13.4 Hz), 4.30 (s, 1H), 4.24 (br, 1H), 4.21–4.16 (m, 2H), 4.09 (t, 1H, J = 5.0 Hz), 4.02 (br, 1H) 3.85 (t, 2H, J = 7.7 Hz), 3.69–3.64 (m, 2H), 3.63 (s, 1H), 3.48 (dd, 1H, J = 12.5 Hz, 4.3 Hz), 3.46–3.42 (m, 1H), 3.39 (dd, 1H, J = 12.2 Hz, 5.0 Hz), 3.17 (s, 9H), 3.14–3.11 (m, 3H), 3.04–3.02 (m, 1H), 2.88 (s, 1H), 2.79 (s, 3H), 2.32–2.26 (m, 1H), 2.23–2.17 (m, 2H), 2.07 (d, 1H, J = 13.2 Hz), 1.92 (br, 1H), 1.76 (s, 3H), 1.71–1.64 (m, 2H), 1.26 (s, 3H), 0.97 (d, 3H, J = 4.8 Hz) 0.94 (d, 3H, J = 4.8 Hz). ESI-TOF HRMS m/z 1776.6065 ([M + H]+, [C86H100Cl3N11O24 + H]+ requires 1776.6081).
4.4. Synthesis of C1-C1Ar-vancomycin (7)
A solution of 5 (1.4 mg, 0.73 μmol) in DMF/DMSO (1/1, 100 μL) was treated with C1 (Me3N+(CH2)3NH2·Cl−, 1 M in DMF/DMSO = 1/1, 4.3 μL, 4.3 μmol), N-methylmorpholine (distilled, 1 M in DMF/DMSO = 1/1, 25.5 μL, 25.5 μmol), and HBTU (1 M in DMF/DMSO = 1/1, 17.0 μL, 17.0 μmol) at 25 °C. The reaction mixture was stirred at 25 °C for 5 min and quenched with the addition of 50% MeOH in H2O (0.5 mL) at 25 °C. The mixture was purified by semi-preparative reverse-phase HPLC (Nacalai Tesque, Inc., ARII-C18, 5 μm, 10 × 150 mm, 1–40% MeCN/H2O-0.07% TFA gradient over 40 min, 3 mL/min, tR = 17.9 min) to afford 7 (1.8 mg, 29%) as a white solid: 1H NMR (CD3OD, 600 MHz, 298 K) δ 8.50 (s, 1H), 7.85–7.77 (m, 1H), 7.79 (d, 1H, J = 8.1 Hz), 7.37 (s, 1H), 7.26 (d, 1H, J = 7.8 Hz), 7.08 (s, 1H), 6.91 (d, 1H, J = 8.5 Hz), 6.57 (s, 1H), 5.83 (br, 1H), 5.55 (s, 1H), 5.48 (s, 1H), 5.42 (d, 1H, J = 3.5 Hz), 5.35 (s, 1H), 4.68 (br, 1H), 4.55 (d, 1H, J = 4.3 Hz), 4.47 (d, 1H, J = 13.3 Hz), 4.44–4.36 (m, 2H), 4.18 (br, 1H), 3.90–3.81 (m, 2H), 3.74 (br, 2H), 3.61–3.52 (m, 2H), 3.50–3.43 (m, 2H), 3.42–3.38 (m, 2H), 3.18 (s, 18H), 3.17–3.13 (m, 2H), 3.02 (br, 2H), 2.79 (s, 3H), 2.35–2.17 (m, 3H), 2.13–2.05 (m, 3H), 2.03–1.98 (m, 1H), 1.88–1.84 (m, 1H), 1.80–1.74 (m, 1H), 1,70–1.66 (m, 1H), 1.49 (s, 3H), 1.22 (d, 3H, J = 5.7 Hz), 1.01 (d, 3H, J = 6.0 Hz), 0.97 (d, 3H, J = 6.2 Hz). ESI-TOF HRMS m/z 837.8495 ([M + 2H]2+, [C79H105Cl2N13O23 + 2H]2+ requires 837.8485).
4.5. Synthesis of C1-C1Ar-CBP-vancomycin (8)
A solution of 6 (1.4 mg, 0.66 μmol) in DMF/DMSO (1/1, 100 μL) was treated with C1 (Me3N+(CH2)3NH2·Cl−, 1 M in DMF/DMSO = 1/1, 4.3 μL, 4.3 μmol), N-methylmorpholine (distilled, 1 M in DMF/DMSO = 1/1, 25.5 μL, 25.5 μmol), and HBTU (1 M in DMF/DMSO = 1/1, 17.0 μL, 17.0 μmol) at 25 °C. The reaction mixture was stirred at 25 °C for 5 min and quenched with the addition of 50% MeOH in H2O (0.5 mL) at 25 °C. The mixture was purified by semi-preparative reverse-phase HPLC (Nacalai Tesque, Inc., ARII-C18, 5 μm, 10 × 150 mm, 20–80% MeCN/H2O-0.07% TFA gradient over 40 min, 3 mL/min, tR = 12.0 min) to afford 8 (2.8 mg, 49%) as a white solid: 1H NMR (CD3OD, 600 MHz, 298 K) δ 7.88 (s, 1H), 7.70 (d, 2H, J = 8.2 Hz), 7.66 (d, 1H, J = 8.4 Hz), 7.62 (d, 2H, J = 8.5 Hz), 7.55 (d, 2H, J = 8.2 Hz), 7.46 (d, 2H, J = 8.5 Hz), 7.28 (d, 1H, J = 8.5 Hz), 7.20 (s, 1H), 7.06 (br, 1H), 6.89 (d, 1H, J = 8.5 Hz), 6.56 (s, 1H), 5.87 (br, 1H), 5.53 (s, 1H), 5.51 (s, 1H), 5.46 (d, 1H, J = 4.2 Hz), 5.38 (s, 1H), 5.32 (s, 1H), 4.68 (s, 1H), 4.57 (s, 1H), 4.45 (d, 1H, J = 13.4 Hz), 4.37 (d, 1H, J =13.4 Hz), 4.27 (s, 1H), 4.18 (d, 1H, J = 12.7 Hz), 4.12–4.06 (m, 2H), 3.85 (br, 1H), 3.78–3.71 (m, 1H), 3.63 (s, 1H), 3.53 (t, 1H, J = 6.4 Hz), 3.44–3.40 (m, 1H), 3.38–3.34 (m, 4H), 3.14 (s, 9H), 3.12 (s, 9H), 3.03–2.98 (m, 1H), 2.92–2.86 (m, 1H), 2.77 (s, 3H), 2.29 (br, 1H), 2.18 (dd, 2H, J = 13.5 Hz, 4.3 Hz), 2.11–2.00 (m, 3H), 1.88–1.82 (m, 1H), 1.79–1.73 (m, 1H), 1.70–1.63 (m, 1H), 1.68 (s, 3H), 1.26 (d, 3H, J = 6.4 Hz), 1.02 (d, 3H, J = 6.3 Hz), 0.98 (d, 3H, J = 6.3 Hz). ESI-TOF HRMS m/z 937.8679 ([M + 2H]2+, [C92H114Cl3N13O23 + 2H]2+ requires 937.8680).
4.6. In vitro antimicrobial assays[54]
One day before experiments were run, fresh cultures of vancomycin-resistant Enterococcus faecalis (VanA VRE, BM4166) and Enterococcus faecium (VanA VRE, ATCC BAA-2317) were inoculated and grown in an orbital shaker at 37 °C in 100% brain-heart infusion broth. After 24 h, the bacterial stock solutions were serial diluted with the culture medium (10% brain-heart infusion) to achieve a turbidity equivalent to a 1:100 dilution of a 0.5 M McFarland solution. This diluted bacterial stock solution was then inoculated in a 96-well glass coated flat-bottom non-treated microtiter plate (Corning 3370), supplemented with serial diluted aliquots of the antibiotic solution in DMSO (4 μL), to achieve a total assay volume of 100 μL. The plate was then incubated at 37 °C for 16 h, after which minimum inhibitory concentrations (MICs) were determined by monitoring the cell growth (observed as a pellet) in the wells. The lowest concentration of antibiotic (in μg/mL) capable of eliminating cell growth in the wells is the reported MIC value. The reported MIC values for the vancomycin analogues were determined against vancomycin as a standard in the first well. For VanA E. faecalis (VanA VRE, BM 4166): resistant to erythromycin, gentamicin, chloramphenicol, and ciprofloxacin as well as vancomycin and teicoplanin; sensitive to daptomycin. For VanA E. faecium (VanA VRE, ATCC BAA-2317): resistant to ampicillin, benzylpenicillin, ciprofloxacin, erythromycin, levofloxacin, nitrofurantoin, and tetracycline as well as vancomycin and teicoplanin, insensitive to linezolid; sensitive to tigecycline and dalfopristine.
4.7. Cell membrane permeability assays[55]
One day before experiments were run, cultures of vancomycin-resistant Enterococcus faecalis (VanA VRE, BM4166) and Enterococcus faecium (VanA VRE, ATCC BAA-2317) were inoculated and grown in an orbital shaker at 37 °C in 100% brain-heart infusion broth for 12 h. The above bacterial solution was subjected to a subculture to obtain fresh mid log phase bacterial cells (incubation time = 6 h). The bacterial suspension was diluted to a total volume of 7 mL with OD600 = 0.6). After the cultured bacteria was harvested (3000 rpm, 4 °C, 20 min), the white bacterial precipitate was washed and resuspended in 5 mM glucose and 5 mM HEPES buffer (1:1, 5.00 mL, pH = 7.2). This bacterial suspension (130 μL) was charged in a 96-well black plate with a clear bottom (Corning 3651). The propidium iodide dye (10 μL, 150 μM DMSO solution) was added to the above suspension and the fluorescence was monitored at 25 °C for 5 min at 30 second intervals using a microplate reader (Molecular Devices®, Max Gemini EX) at an excitation wavelength of 535 nm and an emission wavelength of 617 nm. The test compound (10 μLm 150 μM buffer solution) was added to the cell suspension and the fluorescence was monitored at 25 °C for an additional 15 min.
Acknowledgements
We gratefully acknowledge the financial support of the National Institute of Health (CA041101).
References
- 1.McCormick MH, Antibiot. Annu 3 (1956) 606. [PubMed] [Google Scholar]
- 2.Harris CM, Kopecka H, Harris TM, J. Am. Chem. Soc 105 (1983) 6915 DOI 10.1021/ja00361a029. [DOI] [Google Scholar]
- 3.Kahne D, Leimkuhler C, Lu W, Walsh C, Chem. Rev 105 (2005) 425 DOI 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 4.Nagarajan R, Glycopeptide Antibiotics. Marcel Dekker: New York, 1994. [Google Scholar]
- 5.Hubbard BK, Walsh CT, Angew. Chem. Int. Ed 42 (2003) 730 DOI 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 6.Barna JCJ, Williams DH, Annu. Rev. Microbiol 38 (1984) 339 DOI 10.1146/annurev.mi.38.100184.002011. [DOI] [PubMed] [Google Scholar]
- 7.Williams DH, Bardsley B, Angew. Chem. Int. Ed 38 (1999) 1172. DOI [DOI] [PubMed] [Google Scholar]
- 8.Perkins HR, Pharmacol. Ther 16 (1982) 181 DOI 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 9.Courvalin P, Clin. Infect. Dis 42 (2006) S25 DOI: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 10.Leclercq R, Derlot E, Duval J, Courvalin P, New Engl. J. Med 319 (1988) 157 DOI 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 11.Van Bambeke F, Van Laethem Y, Courvalin P, Tulkens PM, Drugs 64 (2004) 913 DOI 10.2165/00003495-200464090-00001. [DOI] [PubMed] [Google Scholar]
- 12.Pootoolal J, Neu J, Wright GD, Annu. Rev. Pharmacol. Toxicol 42 (2002) 381 DOI 10.1146/annurev.pharmtox.42.091601.142813. [DOI] [PubMed] [Google Scholar]
- 13.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC, Science 302 (2003) 1569 DOI 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- 14.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML, Clin. Microbiol. Rev 23 (2010) 99 DOI 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh TR, Howe RA, Annu. Rev. Microbiol 56 (2002) 657 DOI 10.1146/annurev.micro.56.012302.160806. [DOI] [PubMed] [Google Scholar]
- 16.Willyard C, Nature 543 (2017) 15 DOI 10.1038/nature.2017.21550. [DOI] [PubMed] [Google Scholar]
- 17.van Harten RM, Willems RJL, Martin NI, Hendrickx APA, Trends Microbiol 25 (2017) 467 DOI 10.1016/j.tim.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Yarlagadda V, Sarkar P, Samaddar S, Haldar J, Angew. Chem. Int. Ed. 55 (2016) 7836 DOI 10.1002/anie.201601621. [DOI] [PubMed] [Google Scholar]
- 19.Yarlagadda V, Akkapeddi P, Manjunath GB, Haldar J, J. Med. Chem 57 (2014) 4568 DOI 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- 20.Yarlagadda V, Samaddar S, Paramanandham K, Shome BR, Haldar J, Angew. Chem. Int. Ed 54 (2015) 13644 DOI 10.1002/anie.201507567. [DOI] [PubMed] [Google Scholar]
- 21.Allen NE, Hobbs JN, Nicas TI, Antimicrob. Agents Chemother 40 (1996) 2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D, Science 284 (1999) 507 DOI 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D, Proc. Natl. Acad. Sci. U.S.A 100 (2003) 5658 DOI 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markham A, Drugs A 74 (2014) 1823 DOI 10.1007/s40265-014-0295-4. [DOI] [PubMed] [Google Scholar]
- 25.Anderson VR, Keating GM, Drugs 68 (2008) 639 DOI 10.2165/00003495-200868050-00006. [DOI] [PubMed] [Google Scholar]
- 26.Corey GR, Stryjewski ME, Weyenberg W, Yasothan U, Kirkpatrick P, Nat. Rev. Drug Discov 8 (2009) 929 DOI 10.1038/nrd3051. [DOI] [PubMed] [Google Scholar]
- 27.Wan KK, Shenvi RA, Synlett 27 (2016) 1145 DOI 10.1055/s-0035-1561329. [DOI] [Google Scholar]
- 28.Okano A, Isley NA, Boger DL, Chem. Rev 117 (2017) 11952 DOI 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boger DL, Med. Res. Rev 21 (2001) 356 DOI 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]
- 30.L Boger D, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q, J. Am. Chem. Soc 121 (1999) 10004 DOI 10.1021/ja992577q. [DOI] [Google Scholar]
- 31.Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL, J. Am. Chem. Soc 121 (1999) 3226 DOI 10.1021/ja990189i. [DOI] [Google Scholar]
- 32.Nakayama A, Okano A, Feng Y, Collins JC, Collins KC, Walsh CT, Boger DL, Org. Lett 16 (2014) 3572 DOI 10.1021/ol501568t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng JH, Mori Y, Rogel O, Castle SL, McAtee JJ, J. Am. Chem. Soc 122 (2000) 7416 DOI 10.1021/ja001663j. [DOI] [Google Scholar]
- 34.Boger DL, Kim SH, Mori Y, Weng JH, Rogel O, Castle SL, McAtee JJ, J. Am. Chem. Soc 123 (2001) 1862 DOI 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 35.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL, J. Am. Chem. Soc 126 (2004) 4310 DOI 10.1021/ja039795a. [DOI] [PubMed] [Google Scholar]
- 36.James RC, Pierce JG, Okano A, Xie J, Boger DL, ACS Chem. Biol 7 (2012) 797 DOI 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crowley BM, Boger DL, J. Am. Chem. Soc 128 (2006) 2885 DOI 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie J, Pierce JG, James RC, Okano A, Boger DL, Am DLJ. Chem. Soc 133 (2011) 13946 DOI 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL, J. Am. Chem. Soc 134 (2012) 1284 DOI 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okano A, Nakayama A, Schammel AW, Boger DL, J. Am. Chem. Soc 136 (2014) 13522 DOI 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okano A, Nakayama A, Wu K, Lindsey EA, Schammel AW, Feng Y, Collins KC, Boger DL, J. Am. Chem. Soc 137 (2015) 3693 DOI 10.1021/jacs.5b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McComas CC, Crowley BM, Boger DL, J. Am. Chem. Soc 125 (2003) 9314 DOI 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 43.Kumar MR, Muzzarelli R, Muzzarelli C, Sashiwa H, Domb AJ, Chem. Rev 104 (2004) 6017 DOI 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 44.Xu T, Xin M, Li M, Huang H, Zhou S, Liu J, Carbohydr. Res 346 (2011) 2445 DOI 10.1016/j.carres.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Giurleo D, Parhi A, Kaul M, Pilch DS, LaVoie EJ, Bioorg. Med. Chem. Lett 23 (2013) 2001 DOI 10.1016/j.bmcl.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Middleton MP, Armstrong SA, Bicker KL, Bioorg KL. Med. Chem. Lett 28 (2018) 3514 DOI: 10.1016/j.bmcl.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okano A, Isley NA, Boger DL, Proc. Natl. Acad. Sci. U.S.A 114 (2017) E5052 DOI 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Z-C, Isley NA, Boger DL, ACS Infect. Dis 4 (2018) 1468 DOI 10.1021/acsinfecdis.8b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida O, Yasukata T, Sumino Y, Munekage T, Narukawa Y, Nishitani Y, Bioorg. Med. Chem. Lett 12 (2002) 3027 DOI 10.1016/S0960-894X(02)00664-9. [DOI] [PubMed] [Google Scholar]
- 50.Radhakrishnan B, Sherrill RG, James KD Jr., US Patent 2014171357A1, 2014.
- 51.Osamu Y, Tsutomu I, JP Patent 2009029730A, 2009.
- 52.Balzarini J, Preobrazhenskaya M, Clercq ED, U.S. Patent 2005250677A, 2005.
- 53.Leadbetter MR, Adams SM, Bazzini B, Fatheree PP, Karr DE, Krause KM, Lam BMT, Linsell MS, Nodwell MB, Pace JL, Quast K, Shaw J-P, Soriano E, Trapp SG, Villena JD, Wu TX, Christensen BG, Judice JK, Antibiot J. 57 (2004) 326 DOI 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- 54.Wikler MA, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard, 7th ed, CLSI document M07-A8, Clinical and Laboratory Standards Institute: Wayne, PA, 2009. [Google Scholar]
- 55.Uppu DS, Akkapeddi P, Manjunath GB, Yarlagadda V, Hoque J, Haldar J, Chem. Commun 49 (2013) 9389 DOI 10.1039/C3CC43751E. [DOI] [PubMed] [Google Scholar]
- 56.Blizzard TA, Kim RM, Morgan JD II, Chang J, Kohler J, Kilburn R, Chapman K, Hammond ML, Bioorg. Med. Chem. Lett 12 (2002) 849 DOI 10.1016/S0960-894X(02)00049-5. [DOI] [PubMed] [Google Scholar]