

Abstract
Six ionic liquid (IL)-forming ions (choline, tetrabutylphosphonium, tetrabutylammonium, and trimethylhexadecylammonium cations, and chloride and docusate anions) were paired with acyclovir as the counterion to form four low melting solid salts and two waxes; five of these compounds could be classified as ILs. All of the newly synthesized acyclovir ILs exhibited increased aqueous solubilities by at least 2 orders of magnitude when compared to that of neutral acyclovir. For three of the prepared compounds, the solubilities in simulated body fluids (phosphate-buffered saline, simulated gastric, and simulated intestinal fluids) were also greatly enhanced when compared to that of neutral acyclovir. Acyclovir in its anionic form was more water- or buffer-soluble than acyclovir in its cationic form, though this might be the effect of the particular ions, indicating that the solubilities can be finely tuned by proper choice of the cationic or anionic form of acyclovir and the counterion paired with it.
Introduction
Acyclovir (9-[(2-hydroxyethoxy)methyl]guanine), a nucleoside analogue of the guanosine family, is the most commonly used antiviral drug.1 It is primarily used in tablets, topical creams, intravenous injections, and ophthalmic ointments as an anti-Herpes drug.2 Although widely used, neutral acyclovir suffers from limited solubility in water (1.2–1.6 mg/mL at 22–25 °C3,4), and its absorption into the gastrointestinal tract is only 10–30% of an oral dose, with the low systemic bioavailability being a result of this limited aqueous solubility and low permeability.4−7 Because of such low bioavailability, it has to be administered in very high doses (from 200 to 1000 mg three to four times daily) or intravenously.8 In addition, acyclovir exists in multiple polymorphic and solvate forms, four polymorphs and two hydrates, each reported to have different dissolution rates.3,9,10
Several active strategies are currently being explored to improve acyclovir bioavailability. The most common strategy is the formation of acyclovir prodrugs (e.g., esters11), such as valacyclovir, the valine ester of acyclovir, which has an oral bioavailability that is 3 times higher than that of acyclovir itself (ca. 55%).12 Although efficient, the prodrug approach involves multistep synthesis with chemical modification of the active pharmaceutical ingredient (API) and requires either metabolic activation or hydrolysis in the body for the prodrug to be converted to the final active drug form.
Another common strategy used by the pharmaceutical industry to overcome acyclovir’s low solubility is to use microemulsions as delivery strategies (often stabilized by surfactants).13,14 ILs can also be used as a solvent delivery vehicle as they can be tailored to solvate a specific drug, increasing its concentration for delivery,15 or providing otherwise low shelf-life drugs with stability, and contributing to the delivery of many “hard-to-dissolve” drugs.16−18 For example, McCrary et al. described an IL delivery system for poorly water-soluble amphiphilic APIs and demonstrated its suitability for solubilization of amphotericin B and itraconazole.19 For acyclovir, Moniruzzaman showed the use of IL microemulsions for transdermal delivery using dimethylimidazolium dimethylphosphate ([C1mim][(MeO)2PO2]), in which acyclovir was dissolved 500 times more efficiently than in water, although diffusion of the drug into the skin was negligible.20 Further addition of an oil phase to the system created an IL-in-oil microemulsion, resulting in topical and transdermal drug delivery that was 6 times greater than that of current acyclovir creams.20 Although the approach of using ILs for solubilization can offer an improvement in API delivery, the obstacle in using ILs as a solvent for APIs remains the mostly unknown toxicity profiles of the majority of ILs under study.
The formation of acyclovir salts or co-crystals with higher solubility21 is also actively being investigated. As acyclovir is amphoteric, both acidic and basic approaches have been utilized for salt formation,22 and acyclovir salts derived from metal cations (e.g., sodium or lithium22) or some pharmaceutically acceptable anions (e.g., nitrate,23 arylsulfonates,24 or maleate21) have been well investigated. Nonetheless, although the aqueous solubility of acyclovir sodium is 70–80 times higher than that of acyclovir, formation of acyclovir salts has been focused on the crystalline salt forms, with all of the advantages and disadvantages of solids.
We and others have previously reported on the IL approach to transform solid pharmaceuticals into their liquid salts (ILs, API–ILs, liquids at or below the average body temperature of 37 °C)25−27 where the state of an API (liquid vs solid) has a direct effect on API solubility.28,29 However, perhaps because of the pharmaceutical industry’s reliance on crystalline forms of APIs,30 there appears to be no work on forming acyclovir API–ILs either as a cation or as an anion. Here, we explore the use of the IL approach to increase the water solubility of acyclovir and report the synthesis, characterization, and solubility (aqueous and in physiological fluids) of acyclovir ILs.
Results and Discussion
Synthesis
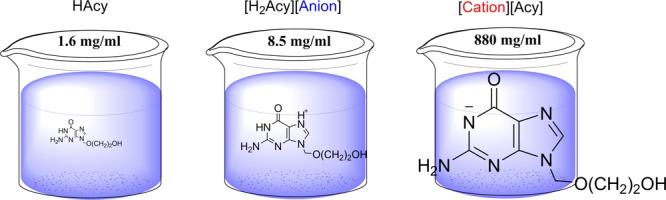
Based on its amphoteric character, acyclovir (Figure 1), which possesses two acidic protons (pKa1 = 2.27; pKa2 = 9.2531,32) and two nitrogen basic sites (pKb not experimentally determined but typical values for guanine and imidazole bases are reported to be 0.433 and −0.5,34 respectively), can be easily both deprotonated and protonated to form the corresponding anion or cation. In this study, acyclovir ions were paired with permanent IL-forming counterions to form acyclovir ILs. The choline cation ([Cho]+) was selected due to its hydrophilicity and safety as it belongs to the “generally regarded as safe” (GRAS) list,35 the Food Additive List,36 and the Everything Added to Food in the United States (EAFUS) list.37 It is also well known that the aqueous solubilities of choline-based salts of APIs are substantially higher than those of the neutral parent drugs; for example, in a recent study, we showed that choline sulfasalazine not only exhibited increased solubility but also bioavailability and improved oral exposure (compared to those of the nonmodified drug).29
Figure 1.

Synthetic scheme for 1–6.
Three other cations, tetrabutylphosphonium ([P4,4,4,4]+), tetrabutylammonium ([N4,4,4,4]+), and trimethylhexadecylammonium ([N1,1,1,16]+), were selected for their additional biological activity (antibacterial activity against both Gram-positive and Gram-negative bacteria, viruses, protozoans, and fungi)38 to complement the antiviral activity of acyclovir. In addition, these three cations possess surfactant/membrane-disruptive properties due to the presence of long alkyl chains.
Two anions, docusate and chloride, were chosen to pair with acyclovir in its cationic form. The docusate anion ([Doc]−) was chosen for its emollient activity, and to improve the absorption and membrane transfer of the drug if applied transdermally. The chloride anion ([Cl]−), one of the most common anions used in pharmaceutical salts,39 was chosen as an intermediate to allow ready metathesis to the docusate salt.
Compounds containing the acyclovir anion ([Cho][Acy] 1, [P4,4,4,4][Acy] 2, [N4,4,4,4][Acy] 3, and [N1,1,1,16][Acy] 4) were synthesized via acid–base reactions between acyclovir hydrate and the hydroxide salts of the desired cations, using water or methanol as the reaction medium. Clear solutions were obtained after stirring for 0.25–1 h at room temperature, concentrated, then dried under reduced pressure to afford either a wax ([Cho][Acy] 1) or low melting crystalline powders ([P4,4,4,4][Acy] 2, [N4,4,4,4][Acy] 3, and [N1,1,1,16][Acy] 4). Acyclovir hydrochloride ([H2Acy]Cl 5) was obtained as a crystalline solid by reacting equimolar amounts of neutral acyclovir and HCl, and used as a precursor for preparing acyclovir docusate ([H2Acy][Doc] 6). The docusate [H2Acy][Doc] 6 was obtained by a metathesis reaction between [H2Acy]Cl 5 and silver docusate (Ag[Doc]) via stirring the mixture of their equimolar amounts in the dark overnight followed by filtration through Celite and solvent evaporation, yielding a white wax. All of the compounds were obtained in high yields (>93%) and with high purities (>95%), as determined by NMR and Fourier transform infrared (FT-IR) spectroscopy. Purity was determined using 1H NMR integration and evaluation of satellites in 13C NMR (satellites from the peaks, if present, indicate that any peaks lost in noise have to be less than 0.5% of the intensity of the main peak). No remaining starting materials or remaining solvent were observed after isolation of the compounds in the corresponding 1H NMR spectra, and there were no signals in the NMR spectra without reasonable attribution. In addition, all but one reaction did not result in any inorganic impurities; however, the metathesis of [Acy]Cl to [Acy][Doc] resulted in solid, water-insoluble AgCl, filtered out from the reaction. Water content was also estimated using the 1H NMR spectra (through integration of the water peak in DMSO-d6 located at 3.33 ppm) and was found to be below 1%, except for the highly hydrophilic [Cho][Acy], which was observed to contain 3% water as isolated.
While the formation of all compounds and their 1:1 stoichiometry was proven using NMR (1H and 13C NMR) spectroscopy, ionization was assessed using FT-IR spectroscopy. FT-IR spectra were recorded for the neat compounds 1–6, and compared with the FT-IR spectra of free acyclovir hydrate (3Acy·2H2O) and fully ionized sodium acyclovirate (Figures S2 and S3 in the Supporting Information (SI)). The main region of difference among the compounds was from 1600 to 1200 cm–1. Characterization by FT-IR spectroscopy (Figures S2 and S3) revealed that upon deprotonation, the peak of the carbonyl in the six-membered ring of neutral acyclovir (C–NH–C(O)–C) shifted from ca. 1600 to 1560 cm–1 and greatly increased in intensity; both the value and intensity are similar to that of Na[Acy] (1575 cm–1). This is the most indicative peak related to ionization of HAcy, and the shift can be explained by the weakening of the C=O bond due to deprotonation of the adjacent NH group and the appearance of the corresponding resonance structure ((C–N––C(O)–C) <−> C–N=C(O–)–C).
Thermal Characterization
The prepared compounds were characterized through thermogravimetric analysis (TGA) to establish the decomposition temperatures, and differential scanning calorimetry (DSC) to determine their melting points (Tm) and/or phase transitions (glass, Tg, or solid–solid, Ts-s) (see SI). All of the synthesized compounds showed similar or lower thermal stabilities than that of the neutral, crystalline acyclovir hydrate (T5% of 240 °C, see Table 1). The highest thermal stability was obtained for [P4,4,4,4][Acy] 2, where the T5% of ca. 243 °C was at least 100 °C higher than those of the ILs [Cho][Acy] 1, [N4,4,4,4][Acy] 3, and [N1,1,1,16][Acy] 4. Both the [N4,4,4,4][Acy] 3 and [N1,1,1,16][Acy] 4 ILs had a similar thermal stability, although a slightly higher T5% was observed for the symmetric [N4,4,4,4]+ cation in 3 (ca. 130 °C) than that for the asymmetric, long-chain [N1,1,1,16]+ cation in 4 (ca. 120 °C); these values are in agreement with reported data: stability decreases with increasing length of the alkyl groups for ammonium or phosphonium ions.40 [Cho][Acy] 1 also showed low thermal stability (T5% = 131 °C). The chloride salt [H2Acy]Cl 5 underwent the same two-step decomposition observed for acyclovir, although at a much lower temperature (T5% 156 °C).
Table 1. Thermal Analyses of Acyclovir and 1–6.
| DSC |
TGA | ||||
|---|---|---|---|---|---|
| compound | app.a | Tg (°C)b | Tm (°C)c | Ttransition (°C)d | T5% (°C)e |
| acyclovir | cryst.f | NDg | 256.5 (dec.)h | ND | 240.1 |
| [Cho][Acy] 1 | wax | –50.2 | ND | ND | 131.0 |
| [P4,4,4,4][Acy] 2 | cryst. | –35.4i | 95.1i | 100.4j | 242.9 |
| [N4,4,4,4][Acy] 3 | cryst. | 45.1 | 65.4i | 59.5j, 70.4j, 76.6j | 132.1 |
| [N1,1,1,16][Acy] 4 | cryst. | –22.92i | 48.3j | 55.8j, 84.1j | 119.9 |
| [H2Acy]Cl 5 | cryst. | ND | >290k | ND | 156.1 |
| [H2Acy][Doc] 6 | wax | ND | ND | ND | 122.1 |
Appearance at 25 °C.
Tg, glass transition.
Tm, melting point.
Ts–s, solid–solid transition,
T5%, decomposition of 5% of the sample.
Crystalline.
ND, not detected in the range −60 to 120 °C.
In agreement with ref (41).
Observed during second and third DSC cycles.
Observed only during first DSC cycle.
Using Fisher–Johns melting point apparatus.
Although five of the six prepared salts could be classified as ILs by the common literature definition of melting below 100 °C, we only met our target for the API–ILs of being liquid below average body temperature42 for [Cho][Acy] 1 and [H2Acy][Doc] 6 (see Table 1). The four salts obtained with the acyclovir anion were amorphous ([Cho][Acy] 1) or had much lower melting points ([P4,4,4,4][Acy] 2, [N4,4,4,4][Acy] 3, and [N1,1,1,16][Acy] 4) compared to that of acyclovir. All but [Cho][Acy] 1 (which showed only a glass transition, Tg = −50.2 °C) and [H2Acy][Doc] 6 (which showed no transitions in the investigated temperature range) exhibited different transitions in the first and consecutive cycles of the DSC curve.
Thus, [P4,4,4,4][Acy] 2 melts through a transition with multiple peaks, a major one at 95.1 °C and a smaller one at 100.4 °C, both on the first heating. It was suggested that [Acy]− converts to HAcy with the change in ratio of [P4,4,4,4]+ and Acy from 1:1 to 1:2, indicating disproportionation of the [P4,4,4,4][Acy] IL to [P4,4,4,4][Acy]·HAcy and at least one other unidentified crystalline phase, and this is discussed in detail below, in the Single-Crystal X-ray Diffraction section. After the first melting occurred, the compound exhibited only a glass transition at Tg = −35.4 °C during the two consecutive DSC cycles.
Similarly, [N4,4,4,4][Acy] 3 showed three separate transitions on first heating, at 59.5, 70.4, and 76.6 °C, respectively, which can also be explained as the melting of multiple coinciding solid phases followed by crystallization of only a single solid form that shows only one melting event at 65.4 °C. Accordingly, [N1,1,1,16][Acy] 4 had a sharp melting peak at Tm = 48.3 °C and a few transitions (55.8, 84.1 °C) on first heating observed after the melting event, which could indicate multiple solid forms melting on the first cycle. On subsequent cooling the compound appeared to solidify into a different form (Tg = −22.92 °C), which undergoes overlapping exothermic and endothermic transitions on warming, immediately followed by melting.
Acyclovir hydrochloride, [H2Acy]Cl 5 (acyclovir cation Cl– intermediate), was obtained as a high melting crystalline material with a Tm of >290 °C (the limit of our Fisher–Johns melting point apparatus), and as mentioned above, there was no melting or other transition observed for the docusate IL, [H2Acy][Doc] 6.
Single-Crystal X-ray Diffraction
Further insight into the nature of the complicated thermal behavior of the salts of [Acy]− was obtained from [P4,4,4,4][Acy] 2 which, although initially isolated as a waxy solid, eventually gave large crystals on storing. Single-crystal X-ray diffraction on one of these crystals identified it as [P4,4,4,4][Acy]·HAcy, which crystallized in the monoclinic space group P21/c with one [P4,4,4,4]+ cation, one neutral HAcy molecule, and one deprotonated [Acy]− anion in the asymmetric unit (Z = 4, Figure 2, top). The acidic N–H atom of the neutral acyclovir molecule is positioned on the ring nitrogen atom α to the carbonyl group (N3), which is the same as in the tautomer of guanine found in nucleic acids,43 and it makes a hydrogen bond with the equivalent position on the [Acy]− anion (N3–H···N9). The [P4,4,4,4]+ cation has three transoid butyl chains and one chain that is all transoid except for the terminal methyl group (C8), which is in a gauche conformation. It can be seen from the crystal structure that, once deprotonated, [Acy]− actually has the right geometry to engage in complementary Watson–Crick-type base pairing with HAcy, similar to that observed in co-crystals of HAcy with 5-fluorouracil,44 and the resulting complex behaves in some ways as a single ion (Figure 2, bottom).
Figure 2.

Fifty percent probability ellipsoid plot of asymmetric unit of [P4,4,4,4][Acy]·HAcy (top) and the [H(Acy)2]− anion (bottom).
The CSD45 contains no examples of a free acyclovir anion. The bond distances of the two acyclovir moieties in [P4,4,4,4][Acy]·HAcy show that, upon going from neutral HAcy to the anion, the N–C single bonds become significantly shorter and the C=N and C=O double bonds become longer. This is consistent with the anion being a hybrid of three resonance forms where the negative charge resides at N8, N9, or O6. The C26–N10 single bond in the anion is significantly longer than that in the neutral molecule, likely because the presence of a partial negative charge on N9 forces the lone pair on N10 to be more localized, making it more like an amine and less like an amidine. In the neutral HAcy molecule, the C18–N3 and C18–N5 bonds are longer than those reported in the crystal structures of pure HAcy44 or HAcy·2/3H2O.46 This may be the result of some transfer of the negative charge on [Acy]− to HAcy through the hydrogen affecting these bonds, respectively, by decreasing the polarity of the C18–N3 bond and reducing the delocalization of the lone pair on N5. The differences in bond distances of [Acy]− and HAcy are comparable in magnitude to the bond distance differences between certain bonds in the three crystallographically unique HAcy molecules of 3Acy·2H2O (see SI).
Furthermore, [P4,4,4,4]+ forms short contacts with three anions and one neutral HAcy molecule (Figure 3). Two of these anions and the HAcy molecule sandwich the cation by stacking above and below it, whereas the third anion forms short contacts with one of the butyl groups through the C–H groups on its ether side chain. The anion makes short contacts with three cations, four anions, and two neutral molecules of acyclovir. The anion donates and receives hydrogen bonds between the −OH group of the side chain and the free nitrogen position on the imidazole ring, which are the shortest hydrogen bonds in the structure. The other two anions interact by either donating or receiving a hydrogen bond from the imidazole C–H group to the alcohol −OH group. Atoms O6, N9, and N10 are all involved in hydrogen bonding with a single complementary HAcy neighbor.
Figure 3.

Short (less than the sum of the van der Waals radii) contact environments around the cation (top left), anion (top right), and neutral HAcy molecule (bottom).
The other neighboring HAcy molecule interacts by donating and receiving hydrogen bonds between the amino group and nonprotonated ring nitrogen atoms, forming a symmetric eight-membered hydrogen bonded cycle. The HAcy molecule makes short contacts with one cation and two anions through the aforementioned interactions as well as with five other HAcy molecules. Two neighboring HAcy molecules interact to form a chain through hydrogen bonds between the −OH groups and carbonyl oxygen atoms. One neighbor engages in a dimeric interaction where the imidazole C–H group is donated in a weak hydrogen bond to the imidazole N atom. The remaining two HAcy molecules either donate or receive weak hydrogen bonds between C–H groups and the −OH group oxygen atom.
The complementary hydrogen bonding between HAcy and [Acy]− is similar to that seen in the cocrystal of HAcy and 5-fluoruracil,47 but because the amino groups involved in these hydrogen bonds are pyramidal, the resulting H-bonded complex is V-shaped rather than flat (Figure 4). These individual complexes are linked along c into infinite ribbons through anion–anion O–H···N hydrogen bonds and through the cyclic hydrogen bonding between the amino and pyrimidine ring nitrogen atoms of HAcy and [Acy]−. The adjacent H-bonded complexes in these ribbons are oriented with the vertex of the V-shape pointed in opposite directions.
Figure 4.

Packing diagrams. (Top) Hydrogen bonded ribbon with cations. (Bottom) 2 × 2 × 2 unit cell packing diagram viewed down. Green lines indicate short contacts.
Cations reside in the cleft of the V and appear to interact with both HAcy and [Acy]−, so although the structures of the molecules indicate localization of the charge to one ion, they act together as an oligomeric ion in the lattice. The weaker hydrogen bonds involving the imidazole rings and the alcohol side chains connect the ribbons along ±a to form anionic zigzag layers, and the cations reside above and below these layers along b and bridge them to complete the 3-D network. The alternating stacking of cations over anions and the layer network can be observed when viewed down the c axis.
Although small amounts of adventitious water could provide the protons to convert [Acy]− to HAcy without being easily detected by spectroscopy, the change in ratio of [P4,4,4,4]+ and Acy from 1:1 to 1:2 indicated that the IL itself must be undergoing disproportionation. This was further confirmed by optical polarizing microscope photographs (see SI), which showed the presence of both crystalline and liquid phases, and powder X-ray diffraction (PXRD), which confirmed the presence of [P4,4,4,4][Acy]·HAcy as well as at least one other unidentified crystalline phase (see SI). This disproportionation is likely stabilized by the complementarity of the interaction between [Acy]− and HAcy. On the one hand, such dimers are likely to form in the other [Acy]− salts and may play a role in preventing their crystallization, as has been seen in cases where pharmaceutical salts were deliberately combined with excess acid or base.48 On the other hand, this structure demonstrated that, like the challenge of “disappearing polymorphism” in solids,47 ILs are capable of crystallizing over time through unforeseen mechanisms that change their composition. The unpredictability of long-term stability is a major problem for solid APIs yet is often overlooked in the design of ILs for pharmaceutical applications, although we previously noted it in the irreversible hydration of the IL procaine acetate to give its crystalline hydrate.49
Solubilities
The determination of the aqueous solubilities of acyclovir, [P4,4,4,4][Acy] 2, [N4,4,4,4][Acy] 3, [N1,1,1,16][Acy] 4, and [H2Acy]Cl 5 in deionized (DI) H2O was conducted via the “shake-flask method”50 at room temperature and atmospheric pressure. An equilibrium solubility (the concentration of acyclovir compounds in a saturated aqueous solution in the presence of undissolved solid) was determined by placing an excess of solid into DI H2O followed by stirring for 24 h, to establish equilibrium before separating the saturated solution from the precipitate by filtration through a 0.2 μm Teflon syringe. The filtrate was then analyzed using either ultraviolet–visible (UV–vis) spectroscopy and previously prepared calibration curves ([P4,4,4,4][Acy] 2, [N1,1,1,16][Acy] 4, and [H2Acy]Cl 5) or quantitative NMR ([N4,4,4,4][Acy] 3), see Methods section.
The solubility of [Cho][Acy] (1, wax) in DI H2O was also determined; however, the procedure was modified for this sample because it absorbs small amounts of water to become an oil. After an excess of [Cho][Acy] 1 was placed into DI water, the mixture was centrifuged for 150 min, and allowed to equilibrate until phase separation between the oil of [Cho][Acy] 1 (lower layer) and the saturated aqueous solution (upper layer) was achieved (15 days). After phase separation, a small amount of aqueous upper layer was removed and analyzed by UV–vis spectroscopy.51 (Please note that care must be taken to prevent any of the bottom layer (the oil) from contaminating the top layer.)
For the study of the solubility of waxy [H2Acy][Doc] 6, DI H2O was added first (ca. 0.3 mL) and an emulsion was formed. More water was then added in small increments (2–4 drops) while the emulsion was sonicated for ∼30 min after each water addition. This dilution procedure was repeated multiple times until all of [H2Acy][Doc] 6 had dissolved and a clear solution was obtained. The solution was then subjected to UV–vis spectroscopy to determine the concentration of [H2Acy][Doc] 6 using the corresponding calibration curve.
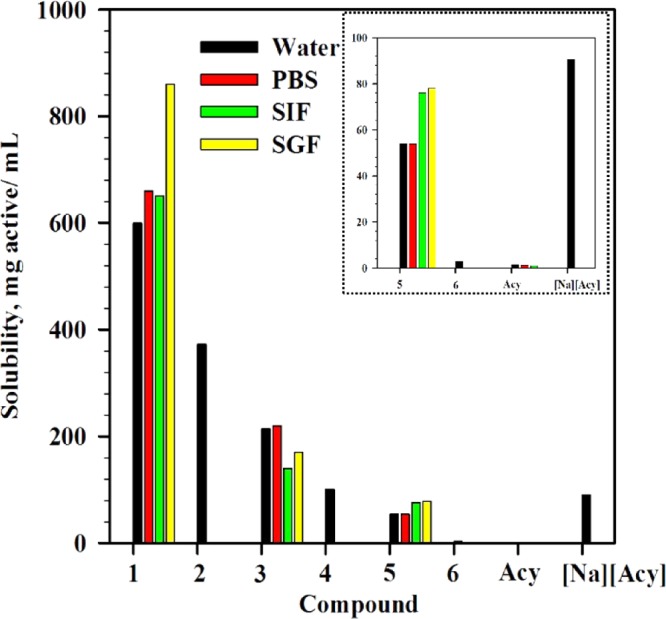
All six compounds showed orders of magnitude higher solubility in DI water (pH = 6.92) than that of neutral acyclovir (1.57 mg acyclovir active/mL, Table 2, Figure 5) and all [Cation][Acyclovir] 1–4 compounds showed higher solubilities than the published value for acyclovir sodium, the commercial form of acyclovir (90.68 mg acyclovir active/mL, Table 2, Figure 5). As expected, the highest water solubility was observed for [Cho][Acy] 1, with a maximum solubility of 600(10) mg acyclovir active/mL, which is almost 400 times higher than that of acyclovir and 6 times higher than that of the acyclovir sodium salt. Our results are in agreement with the data reported in the literature; there are numerous examples of choline salts of marketed drugs that have higher solubility compared to that of the neutral form (e.g., choline naproxen is 6700 times more water soluble and choline tolmetin is 8000 times more water soluble than their corresponding free bases52).
Table 2. Solubility of Acyclovir and 1–6 in Water and in Simulated Body Fluids.
| buffer or DI | mol/L | mg/mL | mg active/mL | |
|---|---|---|---|---|
| acyclovir (3Acy·2H2O) | water | 0.00699(1) | 1.574(2) | 1.495(2) |
| PBS | 0.00631(3) | 1.4(1) | 1.33(1) | |
| SIF | 0.00444(1) | 1.0(1) | 0.95(1) | |
| Na[Acy] | water | 0.404(5) | 100.0 | 90.68 |
| [Cho][Acy] (1) | water | 2.68(5) | 880(20) | 600(10) |
| PBS | 2.92(5) | 960(20) | 660(10) | |
| SIF | 2.83(9) | 930(30) | 650(20) | |
| SGF | 3.8(1) | 1300(500) | 860(30) | |
| [P4,4,4,4][Acy] (2) | water | 1.66(3) | 800(10) | 373(7) |
| [N4,4,4,4][Acy] (3) | water | 0.952(8) | 444.7 | 213.7 |
| PBS | 0.9867(2) | 490(30) | 220(20) | |
| SIF | 0.6394(2) | 300(20) | 140(10) | |
| SGF | 0.7416(8) | 350 (2) | 170(20) | |
| [N1,1,1,16][Acy] (4) | water | 0.4496(25) | 229(1) | 101.3(5) |
| [H2Acy]Cl (5) | water | 0.241(1) | 63.1(30) | 54.4(3) |
| PBS | 0.244(7) | 64(2) | 55(2) | |
| SIF | 0.339(7) | 89(2) | 76(2) | |
| SGF | 0.348(3) | 91.0(8) | 78.4(7) | |
| [H2Acy][Doc] (6) | water | 0.01305(1) | 8.461(5) | 2.948(4) |
Figure 5.
Solubilities of compounds 1–6 in water and buffers.
Improvements in acyclovir solubility were also observed even when more hydrophobic cations were used; for example the solubility of [P4,4,4,4][Acy] 2 (372.7 mg acyclovir active/mL) was ca. 200 times higher than that of acyclovir. The solubilities of the ammonium salts ([N4,4,4,4][Acy] 3, 213.7 mg acyclovir active/mL and [N1,1,1,16][Acy] 4, 101.3 mg acyclovir active/mL) were somewhat lower compared to those of either [Cho][Acy] 1 or [P4,4,4,4][Acy] 2, yet 60–100 times higher than that of acyclovir. The relative solubilities of [P4,4,4,4][Acy] 2, [N4,4,4,4][Acy] 3, and [N1,1,1,16][Acy] 4 appear to be directly related to the aqueous solubilities of the phosphonium and ammonium ions.
The salts of the acyclovir cation were also more soluble than neutral acyclovir, but to a much smaller degree (54.3 mg acyclovir active/mL for [H2Acy]Cl 5 and 2.9 mg acyclovir active/mL for [H2Acy][Doc] 6). When compared to the sodium salt, chloride [H2Acy]Cl 5 was comparable in solubility to Na[Acy], and [H2Acy][Doc] 6 was less soluble than the sodium salt. We would like to note here that although [H2Acy][Doc] 6 had the lowest water solubility observed for any of the salts prepared here, it could still deliver more than twice the concentration of acyclovir to solution than that from the neutral form of this API. We should also note that our original strategy of using the docusate anion was to reduce the water solubility to enhance transdermal transport, and generally, high hydrophobicity is observed for docusate salts.27
The solubilities of [Cho][Acy] 1, [N4,4,4,4][Acy] 3, and [H2Acy]Cl 5 were also investigated in different physiologically relevant aqueous environments: phosphate-buffered saline (PBS, pH = 7.4, commercially available), simulated intestinal fluid (SIF, pH = 6.8, prepared according to United States Pharmacopeia (USP) 2653,54), and simulated gastric fluid (SGF, pH = 1.2, prepared according to USP 30).54 As acyclovir reacts with stoichiometric amounts of HCl to form [H2Acy]Cl 5, the solubility of neutral acyclovir was studied only in PBS and SIF. For these tests, [Cho][Acy] 1 was chosen as its aqueous solubility was the highest of all of the acyclovir compounds. Compound [H2Acy]Cl 5 was chosen to test the cationic form of acyclovir given the difficulty in the study of [H2Acy][Doc] 6. Out of the three similarly behaving ammonium and phosphonium cationic acyclovir compounds, 3 was chosen to represent them.
Solubility studies in the buffers were conducted as noted above, however, the method used to determine the solubility of [Cho][Acy] 1 again had to be slightly modified. When it was mixed with each buffer, no clear phase separation was observed in any of the fluids (i.e., no evidence of precipitation was noted). After the solutions stood for 7 days at room temperature, phase separation was noted, where the top layer was a buffer solution of [Cho][Acy] 1 and the bottom layer was neat [Cho][Acy] (confirmed by 1H NMR). The top phase was removed, filtered, and analyzed using UV–vis spectroscopy.
All of the salts studied demonstrated much higher solubility compared to that of neutral acyclovir in all three physiological media, with similar trends ([Cho][Acy] 1 > [N4,4,4,4][Acy] 3 > [H2Acy]Cl 5) to the aqueous solubility data discussed above. The solubilities of [Cho][Acy] 1 were again the highest and similar in the fluids with similar pH (H2O, PBS, and SIF, respectively). In PBS, [Cho][Acy] 1 showed a solubility enhancement of ca. 450 times that of neutral acyclovir (657.0 mg acyclovir active/mL for [Cho][Acy] 1 vs 1.421 mg acyclovir active/mL for acyclovir, Table 2). In SIF, [Cho][Acy] 1 was ca. 650 times more soluble than neutral acyclovir (649.1 mg acyclovir active/mL, Table 2). An even higher solubility for [Cho][Acy] 1 was observed for the lower pH buffer (861.8 mg acyclovir active/mL, Table 2). Salt [N4,4,4,4][Acy] 3 delivered ca. 150 times more acyclovir active than that of acyclovir itself in all three media used. Acyclovir in its anionic form was more buffer-soluble than acyclovir in its cationic form independent of the media chosen; however, even compound [H2Acy]Cl 5 delivered ca. 40–80 times more acyclovir than the neutral API itself, depending on the medium.
Conclusions
Because poorly soluble drugs such as acyclovir are known to be absorbed more slowly than more soluble APIs, current research has focused on finding methods to increase the solubility of these drugs. Here, we have shown how one can take advantage of the amphoteric character of acyclovir and form the corresponding ILs or salts of the acyclovir cation or anion to achieve improved solubility over acyclovir or acyclovir sodium in water. The solubility of two of the ILs and the hydrochloride salt in simulated body fluids also showed great improvement when compared to that of the neutral acyclovir. It was found that acyclovir in its anionic form (i.e., [Cat][Acy]) was more water- or buffer-soluble than acyclovir in its cationic form ([H2Acy][Anion]) although this might be the effect of these particular ions. Overall, the data suggest not only that solubility enhancements can be achieved, but that the solubilities can be finely tuned over a wide range of solubilities by proper choice of the cationic or anionic form of acyclovir and the counterion paired with it.
Methods
Chemicals
Acyclovir was purchased from TCI America (Portland, OR), and was determined to be a hydrate according to its PXRD pattern, described below (3Acy·2H2O, 95% acyclovir active). Choline hydroxide (46 wt % solution in water) was purchased from Aldrich Chemical Company (Dorset, U.K. and Saint Louis, MO). Tetrabutylphosphonium hydroxide (40 wt % in water) and tetrabutylammonium hydroxide (40 wt % in water) were purchased from Alfa Aesar (Ward Hill, MA). The exact concentration of ammonium and phosphonium hydroxide solution was determined prior to use via titration. DI H2O used in the solubility experiments, in the preparation of buffers, and in determining the calibration curves was obtained with a specific resistivity of 17.38 MΩ cm from a commercial deionizer (Culligan Water Systems, Chattanooga, TN). Celite, anhydrous ethanol, and methanol were obtained from Aldrich Chemical Company (Saint Louis, MO). Monobasic potassium phosphate (KH2PO4), sodium chloride (NaCl), sodium hydroxide (NaOH), and hydrochloric acid (Hydrochloric Acid, Certified ACS Plus, Fisher Chemical) were obtained from Fisher Scientific (Hampton, NH). PBS tablets were obtained from Aldrich Chemical Company (Saint Louis, MO). All chemicals were used as received without any further purification unless otherwise noted.
Preparation of Buffers
PBS (pH = 7.4) was prepared from the commercially available tablets by dissolving 1 tablet in 200 mL of DI H2O. SIF (pH = 6.8, no pancreatic enzyme added) was prepared following the USP guidelines53 where, in a volumetric flask, 0.6805 g of KH2PO4 and 0.0896 g of NaOH were dissolved and diluted with DI H2O to 100 mL. SGF (pH = 1.2, no pepsin enzyme added) was prepared following USP guidelines,53 where, in a volumetric flask, 0.200 g of NaCl and 0.7 mL of concentrated HCl (to adjust to pH = 1.2) were dissolved and diluted with DI H2O to 100 mL.
Characterization
NMR Spectroscopy
NMR data were recorded in DMSO-d6 at 25 °C on a Bruker (Coventry, U.K.) 300 DRX spectrometer and the solvent peak was used as the reference, and on a Bruker (Madison, WI) spectrometer 500 MHz Bruker Avance Spectrometer Bruker/Magnex UltraShield 500 MHz magnet (operating at 500 MHz for 1H, 125 MHz for 13C, and 202.5 MHz for 31P spectra, respectively).
FT-IR Spectroscopy
FT-IR data were recorded using a Bruker α FT-IR instrument, Bruker Optics Inc. (Billerica, MA) featuring an attenuated total reflection sampler equipped with a diamond crystal. Spectra were obtained in the range of 600–4000 cm–1.
UV–Vis Spectra
UV–vis spectra were recorded on a Perkin Elmer Lambda XLS UV–Visible Spectrophotometer (Waltham, MA).
TGA
TGA was performed on a Mettler-Toledo TGA/DSC 1 (Columbus, OH) under a flow of nitrogen. The instrument’s internal temperature was calibrated by observing the melting point of Au, Zn, and In standards (Tm 1064, 419.5, and 155.6, respectively). Samples (5–20 mg) were analyzed in 70 μL platinum pans and were heated from 25 to 800 °C and measured in the dynamic heating regime, using a constant heating ramp of 5 °C/min with a 30 min isotherm at 75 °C. Decomposition temperatures are reported as the 5% onset (T5%onset).
PXRD
Data were collected on a Bruker (Madison, WI) D2 Phaser powder X-ray diffractometer and Bruker D8 Advance diffractometer with a Lynxeye linear position-sensitive detector (Bruker-AXS). A spatula-tip full of material was taken from the sample vial and ground by hand in an agate mortar and pestle. The resulting soft, tacky solid was smeared onto a silicon wafer cut to an off-Bragg plane. PXRD was recorded in Bragg–Brentano geometry using Ni-filtered Cu Kα radiation. The sample was rotated at 15 rpm during data collection. Data were collected using 1 s/0.02° steps and a 2θ range of 5–40 or 10–35°.
DSC Analyses
Thermal transitions for the compounds were measured on a TA Instrument-2000 DSC unit (New Castle, DE) under a stream of nitrogen. The sample (2–4 mg) was placed into T Zero pans (Part # 901683.901), covered with hermetic aluminum lids (Part # 900794.901), sealed using a press (T Zero Sample Press), and heated at a heating rate of 5 °C/min, followed by a 5 min isotherm, then cooled at a rate of 5 °C/min to −60 °C, again followed by a final 5 min isotherm at −60 °C.
(Acyclovir itself was taken from 25 to 300 °C, and docusate 6 from −60 to 150 °C, at the same rate with isotherms at the lowest and highest temperature.) The cycle was repeated twice to ensure consistency of the observed thermal transitions. The protocol used in the case of solid samples was as follows: (a) ramp up to the target temperature (Ttarget) at 5 °C/min; (b) isotherm for 5 min to ensure equilibration of the temperature in the cell; (c) ramp down at 5 °C/min to about −60 °C; (d) isotherm for 5 min to ensure equilibration of the temperature in the cell; (e) repeat steps (a)–(d) twice (i.e., the entire cycle was repeated three times).
Single-Crystal X-ray Diffraction Data (SCXRD)
SCXRD was collected on a Bruker D8 Venture diffractometer with a Photon 100 CMOS detector using Mo Kα radiation from an IμS microfocus source (Bruker-AXS, Madison, WI). A crystalline prism measuring 0.82 × 0.15 × 0.15 mm3 was mounted in a Kapton loop using heavy hydrocarbon oil (MiTeGen, Ithaca, NY) and cooled to 100 K under a cold stream of nitrogen using an Oxford N-Helix cryostat (Oxford Cryosystems, Oxford, U.K.). A hemisphere of unique data was collected through strategies of scans about the phi and omega axes. The Apex3 software suite was used for data collection, unit cell determination, data reduction, integration, and scaling.55
The crystal structure was solved by intrinsic phasing and refined by full-matrix least squares refinement against F2. Nonhydrogen atoms were refined anisotropically. Hydrogen atoms bonded to N or O were located from the difference map, their coordinates were refined freely, and their thermal parameters constrained to ride on the carrier atoms. Hydrogen atoms bonded to carbon were placed in calculated positions and constrained to ride on the carrier atoms. Methyl groups were refined using a riding–rotating model. Structure solution and refinement were done using the Bruker SHELXTL software suite56 and SHELXL-2014.57
Microscope Photographs
Microscope photographs were taken on an Axiolab POL microscope at 40× magnification (Carl-Zeiss AG, Oberken, Germany).
Syntheses
General Syntheses of Cation Acyclovir, [Cation][Acy] 1–4
Acyclovir powder was suspended in anhydrous ethanol and an equimolar (with respect to pure acyclovir, i.e., 0.95 mol/mol with respect to hydrate) amount of ammonium or phosphonium hydroxide (aqueous or methanolic, respectively) was added dropwise using a glass Hamilton syringe. The suspension was stirred using a Teflon-coated magnetic stirrer for 15 min at room temperature, until a clear solution was obtained. After that, the solvent was evaporated using a Buchi rotary evaporator and the remaining volatile materials were removed further under high vacuum (0.01 mbar, 50 °C) to yield the desired compound.
Choline Acyclovir, [Cho][Acy] 1
Yellow wax, 97% yield. 1H NMR (300 MHz, DMSO-d6) δ (ppm) = 7.4 (s, 1H), 5.2 (s, 2H), 4.9 (br s, 2H), 3.8 (s, 2H), 3.4 (m, 6H), 3.0 (s, 9H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) = 167.9, 161.8, 134.5, 118.9, 71.9, 70.4, 67.7, 60.3, 55.6, 53.5.
Tetrabutylphosphonium Acyclovir, [P4,4,4,4][Acy] 2
White glassy solid, 95% yield. 1H NMR (500 MHz, DMSO-d6) δ (ppm) = 7.4 (s, 1H), 5.2 (s, 2H), 4.9 (br s, 1H), 3.4 (s, 4H), 2.2 (m, 8H), 1.4 (m, 16H), 0.8 (m, 12H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) = 167.72, 161.81, 151.97, 133.88, 119.22, 71.83, 70.29, 60.44, 23.71, 23.05, 17.71, 13.64; 31P NMR (202.5 MHz, DMSO-d6) δ (ppm) = 34.07 (s).
Tetrabutylammonium Acyclovir, [N4,4,4,4][Acy] 3
White crystalline solid, 96% yield. 1H NMR (500 MHz, DMSO-d6) δ (ppm) = 7.39 (s, 1H), 5.27 (s, 2H), 4.88 (br, 2H), 3.46 (s, 4H), 3.17 (m, 8H), 1.57 (m, 8H), 1.30 (m, 8H), 0.93 (m, 12H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) = 134.08, 71.95, 71.19, 61.00, 58.00, 23.55, 19.70, 13.93.
Hexadecyltrimethylammonium Acyclovir, [N1,1,1,16][Acy] 4
White crystalline solid, 98% yield. 1H NMR (500 MHz, DMSO-d6) δ (ppm) = 7.49 (s, 1H), 5.8 (s, 1H), 5.29 (s, 2H), 3.46 (s, 4H), 3.27 (m, 3H), 3.05 (s, 9H), 1.64 (m, 2H), 1.24 (m, 26H), 0.85 (m, 3 H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) = 166.31, 160.79, 152.29, 134.99, 118.53, 72.03, 70.49, 65.74, 60.51, 52.58, 31.75, 29.51, 29.47, 29.41, 29.27, 29.15, 28.97, 26.22, 22.54, 22.51, 14.39.
Syntheses of Acyclovir Anion, [Acy][Anion] 5, 6
Synthesis of Acyclovir Hydrochloride, [H2Acy]Cl 5
Acyclovir (3.00 g, 13.3 mmol) was suspended in 30 mL of isopropanol, in a 50 mL round-bottom flask equipped with a magnetic stirring bar, an addition funnel, and an ice bath. Separately, an aqueous solution of concentrated HCl (13.3 mmol, 1.32 mL) in 15 mL of isopropanol was prepared and placed into the addition funnel. After that, HCl solution was added dropwise to the acyclovir–isopropanol suspension, with external ice-bath cooling. After the addition, the cooling bath was removed, and the reaction mixture was stirred for 30 min at room temperature. After the reaction was complete, the solvent was evaporated using a Buchi rotary evaporator and the obtained solid was further dried using high vacuum with no heating (0.01 mbar, 20 °C). White crystalline solid, 94% yield.
1H NMR (500 MHz, DMSO-d6) δ (ppm) = 11.68 (s, 1H), 8.99 (s, 1H), 7.31 (s, 2H), 5.51 (s, 2H), 3.58 (t, 2H), 3.47 (t, 2H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) =155.92, 154.56, 150.56, 138.02, 110.28, 74.13, 71.68, 60.30.
Synthesis of Acyclovir Docusate, [H2Acy][Doc] 6
Acyclovir hydrochloride 5 (1.5 g, 5.73 mmol) and silver docusate (3.03 g, 5.73 mmol) were suspended in 80 mL of methanol and the resulting mixture was stirred using a Teflon-coated magnetic stirrer at room temperature for 10 h in the dark. The obtained suspension was filtered through Celite and the resulting solution was evaporated using a Buchi Rotary evaporator at 40 °C. The obtained residue was further dried under high vacuum (0.01 mbar, 40 °C). Off-white wax, 80% yield.
1H NMR (500 MHz, DMSO-d6) δ (ppm) = 11.30 (s, 1H), 8.97 (s, 1H), 7.01 (s, 2H), 5.49 (s, 2H), 3.89 (m, 4H), 3.65 (dd, 1H), 3.58 (m, 2H), 3.48 (m, 2H), 2.91 (dd, 1H), 2.81 (dd, 1H), 1.49 (m, 2H), 1.27 (m, 18H), 0.84 (m, 12H); 13C NMR (125 MHz, DMSO-d6) δ (ppm) = 171.47, 168.78, 155.67, 154.68, 150.65, 138.13, 110.16, 74.22, 71.75, 66.68, 66.61, 66.57, 66.54, 61.92, 60.32, 38.65, 38.61, 38.57, 34.56, 30.21, 30.09, 30.03, 28.80, 23.67, 23.64, 23.49, 22.86, 22.83, 14.35, 14.32, 11.26, 11.23, 11.19.
Solubility
Construction of Calibration Curves
Calibration curves in DI H2O and buffers were obtained by analyzing five times each, five to eight different solutions of known concentrations (0–5 × 10–4 M) of the corresponding acyclovir compound. Aqueous (or corresponding buffer) stock solutions were first prepared by mass, and then several standard dilutions were made to obtain a series of known concentrations. The absorbance at 254 nm was selected for analysis and the plots of concentration against absorbance at 254 nm were fit to a straight line passing through the origin (correlation coefficients of R2 ≥ 0.99 were obtained). The resultant concentration was back calculated from the obtained data against the calibration curve to calculate the solubility values.
Determination of Solubility Based on Phase Separation for [Cho][Acy] 1
Solubility of [Cho][Acy] (1) in DI water was determined as follows. In a 20 mL screw top vial, 5.500 g of [Cho][Acy] 1 was mixed with 2 mL of DI H2O. The mixture was stirred for 24 h at room temperature resulting in the formation of a very viscous solution. The solution was centrifuged for 150 min at 2500 rpm to facilitate phase separation, but no phase separation was initially observed. The mixture was allowed to stand for 15 days when a thin layer of a more viscous mixture (excess of [Cho][Acy] 1 according to NMR analysis) was phase-separated from the solution, at the bottom of the vial (see Figure S1).
Then, 0.5 mL of the top phase (aqueous solution of solution [Cho][Acy] 1) was taken out from the top phase, filtered (Teflon syringe filter membranes, 0.2 μm pore size), and a 0.1 mL aliquot was withdrawn. The withdrawn aliquot was diluted to 100 mL with DI H2O using a volumetric flask and the absorbance at 254 nm was measured via UV–vis spectroscopy. The obtained absorbance value was too large and outside of the spectrometer range. Therefore, the solution was further diluted as follows: 0.25 mL of this solution was transferred into a 25 mL volumetric flask and DI water was added up to the mark. The absorbance at 254 nm of the obtained solution was measured and the concentration determined using a predetermined calibration curve of the compound in water. Solubility values were recalculated taking into the account the multiple dilutions described above. The experiment was done in duplicate.
Determination of Solubility for Acyclovir, [P4,4,4,4][Acy] 2, [N1,1,1,16][Acy] 4, and [Acy]Cl 5 via the Flask-Shake Method
The solubility of acyclovir in both DI water and PBS, solubility of [P4,4,4,4][Acy] 2 and [N1,1,1,16][Acy] 4 in DI water, and solubility of [Acy]Cl 5 in both DI water and all three physiological fluids (PBS, SGF, or SIF) were determined as follows. In a screw top vial, the crystalline powder (acyclovir, [P4,4,4,4][Acy] 2, [N1,1,1,16][Acy] 4, and [Acy]Cl 5) was suspended in DI H2O and stirred overnight at room temperature. Equilibrium between the solid–liquid phases was reached by the end of 12 h, leaving some remaining solid undissolved. The remaining solid was removed by filtration (Teflon syringe filter membranes, 0.2 μm pore size) and the filtrate was analyzed using UV–vis spectroscopy, against a predetermined calibration curve.
Determination of Solubility of [N4,4,4,4][Acy] 3 via Flask-Shake Method Followed by Quantitative 1H NMR
The solubilities of [N4,4,4,4][Acy] 3 in DI water and all three physiological fluids (PBS, SGF, or SIF) were determined as follows. 0.1 g of the compound was placed into 0.5 mL of DI water, and the solution was stirred at room temperature for 24 h. The resulting suspension was filtered through a 0.2 μm Teflon syringe filter to obtain a clear solution, and 0.1 mL of this solution was combined with 0.4 mL of DMSO-d6 0.05% (v/v) TMS (corresponding to 1.836 × 10–6 mol TMS). 1H NMR spectra were recorded and the final solubility was determined using integration, that is, the molar ratio between the peaks corresponding to the compound and TMS was determined, followed by calculation of the molar amount of the dissolved compound.
Determination of Solubility via the Flask-Shake Method for [H2Acy][Doc] 6
The solubility of [H2Acy][Doc] 6 in DI water was determined as follows. In a screw top vial, 0.2 g of [Acy][Doc] was suspended in 1.5 mL of DI H2O. The mixture was stirred for 24 h at room temperature resulting in the formation of a stable emulsion (stable for at least 48 h at room temperature). To about 0.3 mL of this emulsion, water was added dropwise, two to four drops at a time (with vigorous mixing each time after water was added), and the mixture was sonicated for about 30 min. This process (addition of water, mixing, sonication) was repeated until a clear solution was obtained. The solution was analyzed through UV–vis spectroscopy, against a predetermined calibration curve.
Acknowledgments
This work was initiated at the Queen’s University Ionic Liquids Laboratory (QUILL), the Queen’s University, Belfast, Northern Ireland. This research was undertaken, in part, thanks to funding from the Canada Excellence Research Chairs Program.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00554.
FT-IR spectroscopy data, structural description and analysis of [P4,4,4,4][Acy]·HAcy (product of disproportionation of [P4,4,4,4][Acy]), transmission optical microscopy images of [P4,4,4,4][Acy], PXRD data, TGA data, DSC data, calibration curves for acyclovir hydrate (3Acy·2H2O) and all synthesized compounds (PDF)
Author Present Address
∇ Hudson Data, LLC, 122 North Genesee Street, Suite 302 A, Geneva, New York 14456, United States. E-mail: SPWallace@crimson.ua.edu (S.P.W.).
Author Present Address
# Institute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: Katharina.Schroeder@tuwien.ac.at (K.B.).
Author Present Address
⊥ Tennessee Tech University, 1 William L Jones Dr, Cookeville, Tennessee 38505, United States. E-mail: OCojocaru@TNTech.edu (O.A.C.).
The authors declare the following competing financial interest(s): RDR and GG have partial ownership of 525 Solutions; JLS is an employee of 525 Solutions. RDR and GG are named inventors on related patent application. The University of Alabama and McGill University maintain approved Conflict of Interest Management Plans.
Supplementary Material
References
- Elion G. B.; Furman P. A.; Fyfe J. A.; De Miranda P.; Beauchamp L.; Schaeffer H. J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 5716–5720. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.nlm.nih.gov/medlineplus/druginfo/meds/a681045.html (last accessed April 19, 2017).
- Kristl A. Estimation of aqueous solubility for some guanine derivatives using partition coefficient and melting temperature. J. Pharm. Sci. 1999, 88, 109–110. 10.1021/js980180z. [DOI] [PubMed] [Google Scholar]
- Bergström C. A.; Norinder U.; Luthman K.; Artursson P. Experimental and computational screening models for prediction of aqueous drug solubility. Pharm. Res. 2002, 19, 182–188. 10.1023/A:1014224900524. [DOI] [PubMed] [Google Scholar]
- AHFS Drug Information; American Society of Hospital Pharmacists (AHFS): Bethesda, MD, 2004; p 765. [Google Scholar]
- Kristl A.; Srcic S.; Vrecer F.; Sustar B.; Vojnovic D. Polymorphism and pseudopolymorphism: Influencing the dissolution properties of the guanine derivative acyclovir. Int. J. Pharm. 1996, 139, 231–235. 10.1016/0378-5173(96)04601-7. [DOI] [Google Scholar]
- Nair A. B.; Attimarad M.; Al-Dhubiab B. E.; Wadhwa J.; Harsha S.; Ahmed M. Enhanced oral bioavailability of acyclovir by inclusion complex using hydroxypropyl-β-cyclodextrin. Drug Delivery 2014, 21, 540–547. 10.3109/10717544.2013.853213. [DOI] [PubMed] [Google Scholar]
- http://samwald.info/medical_microdata/dailymed_resource_drugs_652.html (last accessed on April 19, 2017).
- Sohn Y. T.; Kim S. H. Polymorphism and pseudopolymorphism of acyclovir. Arch. Pharmacal Res. 2008, 31, 231–234. 10.1007/s12272-001-1146-x. [DOI] [PubMed] [Google Scholar]
- Lutker K. M.; Quiñones R.; Xu J.; Ramamoorthy A.; Matzger A. J. Polymorphs and hydrates of acyclovir. J. Pharm. Sci. 2011, 100, 949–963. 10.1002/jps.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla L.; De Clercq E.; Busson R.; Vanderhaeghe H. Synthesis and antiviral activity of water-soluble esters of acyclovir [9-[(2-hydroxyethoxy)methyl]guanine). J. Med. Chem. 1983, 26, 602–604. 10.1021/jm00358a029. [DOI] [PubMed] [Google Scholar]
- MacDougall C.; Guglielmo B. J. Pharmacokinetics of valaciclovir. J. Antimicrob. Chemother. 2004, 53, 899–901. 10.1093/jac/dkh244. [DOI] [PubMed] [Google Scholar]
- Arnal J.; Gonzalez-Alvarez I.; Bermejo M.; Amidon G. L.; Junginger H. E.; Kopp S.; Midha K. K.; Shah V. P.; Stavchansky S.; Dressman J. B.; Barends D. M. Biowaiver monographs for immediate release solid oral dosage forms: acyclovir. J. Pharm. Sci. 2008, 97, 5061–5073. 10.1002/jps.21392. [DOI] [PubMed] [Google Scholar]
- Maitra A.; Feldmann G.; Bisht S.. Water-Dispersible Oral, Parenteral, and Topical Formulations for Poorly Water Soluble Drugs Using Smart Polymeric Nanoparticles. U.S. Patent Appl. US20080107749, 2008.
- Shamshina J. L.; Barber P. S.; Rogers R. D. Ionic liquids in drug delivery. Expert Opin. Drug Delivery 2013, 10, 1367–1381. 10.1517/17425247.2013.808185. [DOI] [PubMed] [Google Scholar]
- Forte A.; Melo C. I.; Bogel-Łukasik R.; Bogel-Łukasik E. A favourable solubility of isoniazid, an antitubercular antibiotic drug, in alternative solvents. Fluid Phase Equilib. 2012, 318, 89–95. 10.1016/j.fluid.2012.01.022. [DOI] [Google Scholar]
- Lourenço K.; Melo C. I.; Bogel-Łukasik R.; Bogel-Łukasik E. Solubility advantage of pyrazine-2-carboxamide: application of alternative solvents on the way to the future pharmaceutical development. J. Chem. Eng. Data 2012, 57, 1525–1533. 10.1021/je300044x. [DOI] [Google Scholar]
- Meloa C. I.; Bogel-Łukasik R.; da Ponte M. N.; Bogel-Łukasik E. Ammonium ionic liquids as green solvents for drugs. Fluid Phase Equilib. 2013, 338, 209–216. 10.1016/j.fluid.2012.11.029. [DOI] [Google Scholar]
- McCrary P. D.; Beasley P. A.; Gurau G.; Narita A.; Barber P. S.; Cojocaru O. A.; Rogers R. D. Drug specific tuning of an ionic liquid’s hydrophilic-lipophilic balance to improve water solubility of poorly soluble active pharmaceutical ingredients. New J. Chem. 2013, 37, 2196–2202. 10.1039/c3nj00454f. [DOI] [Google Scholar]
- Moniruzzaman M.; Tahara Y.; Tamura M.; Kamiya N.; Goto M. Ionic liquid-assisted transdermal delivery of sparingly soluble drugs. Chem. Commun. 2010, 46, 1452–1454. 10.1039/b907462g. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Chen J.-M.; Lu T.-B. Simultaneously enhancing the solubility and permeability of acyclovir by crystal engineering approach. CrystEngComm 2013, 15, 6457–6460. 10.1039/c3ce41017j. [DOI] [Google Scholar]
- Guazzi G.Acyclovir Dihydrate Sodium Salt and the Preparation Thereof. U.S. Patent US6040445, 2000.
- Del Soldato P.; Benedini F.; Antognazza P.. Nitrate Salts of Antimicrobial Agents. PCT Int. Appl., WO20010546912001.
- Powers J. P.Arylsulfonic Acid Salts of Pyrimidine-Based Antiviral Agents. PCT Int. Appl., WO2001051485, 2001.
- Hough W. L.; Rogers R. D. Ionic liquids then and now: from solvents to materials to Active Pharmaceutical Ingredients. Bull. Chem. Soc. Jpn. 2007, 80, 2262–2269. 10.1246/bcsj.80.2262. [DOI] [Google Scholar]
- Rogers R. D.; Daly D. T.; Swatloski R. P.; Hough W. L.; Davis J. H. Jr.; Smiglak M.; Pernak J.; Spear S. K.. Multi-Functional Ionic Liquid Compositions for Overcoming Polymorphism and Imparting Improved Properties for Active Pharmaceutical, Biological, Nutritional, and Energetic Ingredients. U.S. Patent US8232265 B2, 2012.
- Hough W. L.; Smiglak M.; Rodriguez H.; Swatloski R. P.; Spear S. K.; Daly D. T.; Pernak J.; Grisel J. E.; Carliss R. D.; Soutullo M. D.; Davis J. H. Jr.; Rogers R. D. The third evolution of ionic liquids: active pharmaceutical ingredients. New J. Chem. 2007, 31, 1429–1436. 10.1039/b706677p. [DOI] [Google Scholar]
- Sahbaz Y.; Williams H. D.; Nguyen T.-H.; Saunders J.; Ford L.; Charman S. A.; Scammells P. J.; Porter C. J. H. Transformation of poorly water-soluble drugs into lipophilic ionic liquids enhances oral drug exposure from lipid based formulations. Mol. Pharmaceutics 2015, 12, 1980–1991. 10.1021/mp500790t. [DOI] [PubMed] [Google Scholar]
- Shadid M.; Gurau G.; Shamshina J. L.; Chuang B.-C.; Hailu S.; Guan E.; Chowdhury S. K.; Wu J.-T.; Rizvi S. A. A.; Griffin R. J.; Rogers R. D. Sulfasalazine in ionic liquid form with improved solubility and exposure. Med. Chem. Commun. 2015, 6, 1837–1841. 10.1039/C5MD00290G. [DOI] [Google Scholar]
- Shamshina J. L.; Kelley S. P.; Gurau G.; Rogers R. D. Chemistry: Develop ionic liquid drugs. Nature 2015, 528, 188–189. 10.1038/528188a. [DOI] [PubMed] [Google Scholar]
- Raymond G. G.; Born J. L. An updated pKa listing of medicinal compounds. Drug Intell. Clin. Pharm. 1986, 20, 683–686. 10.1177/106002808602000910. [DOI] [PubMed] [Google Scholar]
- Balon K.; Riebesehl B. U.; Muller B. W. Drug liposome partitioning as a tool for the prediction of human passive intestinal absorption. Pharm. Res. 1999, 16, 882–888. 10.1023/A:1018882221008. [DOI] [PubMed] [Google Scholar]
- Griffiths M. Z.; Alkorta I.; Popelier P. L. A. Predicting pKa values in aqueous solution for the guanidine functional group from gas phase Ab Initio bond lengths. Mol. Inf. 2013, 32, 363–376. 10.1002/minf.201300008. [DOI] [PubMed] [Google Scholar]
- Superbases for Organic Synthesis: Guanidines, Amidines and Phosphazenes and Related Organocatalysts; Ishikawa T., Ed.; Wiley: West Sussex, U.K., 2009. [Google Scholar]
- http://www.ift.org/knowledge-center/focus-areas/product-development-and-ingredient-innovations/~/media/Food%20Technology/pdf/2009/06/0609feat_GRAS24text.pdf (last accessed April 19, 2017).
- http://www.fda.gov/Food/IngredientsPackagingLabeling/oodAdditivesIngredients/ucm091048.htm#ftnC (last accessed on April 19, 2017).
- http://www.usc.es/caa/EdulcWeb/EAFUS.pdf (last accessed on April 19, 2017).
- Ohta Y.; Kondo Y.; Kawada K.; Teranaka T.; Yoshino N. Synthesis and antibacterial activity of quaternary ammonium salt-type antibacterial agents with a phosphate group. J. Oleo Sci. 2008, 57, 445–452. 10.5650/jos.57.445. [DOI] [PubMed] [Google Scholar]
- Haynes D. A.; Jones W.; Samuel Motherwell W. D. Occurrence of pharmaceutically acceptable anions and cations in the Cambridge Structural Database. J. Pharm. Sci. 2005, 94, 2111–2120. 10.1002/jps.20441. [DOI] [PubMed] [Google Scholar]
- Prasad M. R. R.; Krishnan K.; Ninan K. N.; Krishnamurthy V. N. Thermal decomposition of tetraalkyl ammonium tetrafluoroborates. Thermochim. Acta 1997, 297, 207–210. 10.1016/S0040-6031(96)03070-5. [DOI] [Google Scholar]
- The Merck Index, 12th ed.; Budavari S., Ed.; Merck & Co.: Whitehouse Station, NJ, 1996; p 27. [Google Scholar]
- Ionic Liquids: Industrial Applications for Green Chemistry; Rogers R. D., Seddon K. R., Eds.; ACS Symposium Series 818; American Chemical Society: Washington, DC, 2002. [Google Scholar]
- Crick F. H. C.; Watson J. D. The complementary structure of deoxyribonucleic acid. Proc. R. Soc. A 1954, 223, 80–96. 10.1098/rspa.1954.0101. [DOI] [Google Scholar]
- Tutughamiarso M.; Guido W.; Egert E. Cocrystals of 5-fluorocytosine. I. Coformers with fixed hydrogen-bonding sites. Acta Crystallogr., Sect. B: Struct. Sci. 2012, 68, 431–443. 10.1107/S010876811202561X. [DOI] [PubMed] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci. 2016, 72, 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum G. I.; Cygler M.; Shugar D. Conformational features of acyclonucleosides: structure of acyclovir, an antiherpes agent. Can. J. Chem. 1984, 62, 2646–2652. 10.1139/v84-449. [DOI] [Google Scholar]
- Bučar D. K.; Lancaster L. W.; Bernstein J. Disappearing polymorphs revisited. Angew. Chem., Int. Ed. 2015, 54, 6972–6993. 10.1002/anie.201410356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bica K.; Rogers R. D. Confused ionic liquid ions—a “liquification” and dosage strategy for pharmaceutically active salts. Chem. Commun. 2010, 46, 1215–1217. 10.1039/b925147b. [DOI] [PubMed] [Google Scholar]
- Cojocaru O. A.; Kelley S. P.; Rogers R. D. Procainium acetate versus procainium acetate dihydrate: irreversible crystallization of a room-temperature Active Pharmaceutical-Ingredient ionic liquid upon hydration. Cryst. Growth Des. 2013, 13, 3290–3293. 10.1021/cg400686e. [DOI] [Google Scholar]
- Higuchi T.; Connors K. A. Phase solubility techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]
- Sintra T. E.; Luís A.; Rocha S. N.; Ferreira A. I. M. C. L.; Gonçalves F.; Santos L. M. N. B. F.; Neves B. M.; Freire M. G.; Ventura S. P. M.; Coutinho J. A. P. Enhancing the antioxidant characteristics of phenolic acids by their conversion into cholinium salts. ACS Sustainable Chem. Eng. 2015, 3, 2558–2565. 10.1021/acssuschemeng.5b00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Sadrzadeh N.; Constantinides P. P.. Water-Insoluble Drug Formulation; Liu R., Ed.; Interpharm Press: Denver, CO, 2000. [Google Scholar]
- United States Pharmacopeia and National Formulary, 26th ed.; United States Pharmacopeia Convention Inc.: Rockville, MD, 2003. [Google Scholar]
- United States Pharmacopeia and National Formulary, 30th ed.; United States Pharmacopeia Convention Inc.: Rockville, MD, 2007. [Google Scholar]
- APEX3, AXScale and SAINT, version 2015; Bruker AXS, Inc.: Madison, WI, 2015.
- Sheldrick G. M.SHELXTL (2001), Structure Determination Software Suite, v.6.10; Bruker AXS Inc: Madison, WI, 2001.
- Sheldrick G. M.. SHELXTL XLMP Crystal Structure Refinement Multi-CPU Version, v.2014/7; Bruker AXS Inc.: Madison, WI, 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.