

Abstract
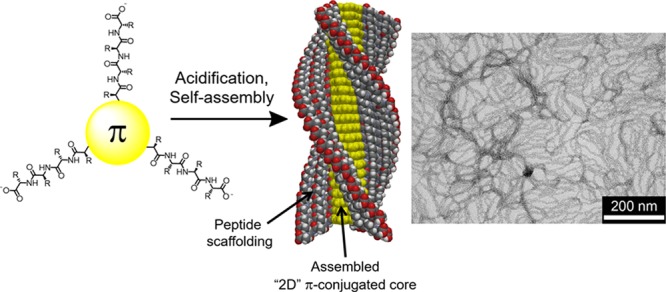
We present a completely solid-phase synthetic strategy to create three- and four-fold peptide-appended π-electron molecules, where the multivalent oligopeptide presentation is dictated by the symmetries of reactive handles placed on discotic π-conjugated cores. Carboxylic acid and anhydride groups were viable amidation and imidation partners, respectively, and oligomeric π-electron discotic cores were prepared through Pd-catalyzed cross-couplings. Due to intermolecular hydrogen bonding between the three or four peptide axes, these π-peptide hybrids self-assemble into robust one-dimensional nanostructures with high aspect ratios in aqueous solution. The preparation of these systems via solid-phase methods will be detailed along with their self-assembly properties, as revealed by steady-state spectroscopy and transmission electron microscopy and electrical characterization using field-effect transistor measurements.
Introduction
Disk-like two-dimensional (2-D) π-electron structures continue to be a subject of research focus, be it due to their established intermolecular interactions that are required for the formation of discotic liquid crystal phases with high carrier mobilities or their inherent relation to the cutting-edge graphene family of organic electronic materials. A classic example of the former would be the general family of hexa-alkoxy triphenylenes (HATs, Figure 1 left),1 where the enthalpic contributions of π-electron quadrupolar and dispersive interactions coupled with well-established van der Waals interactions among alkyl tails facilitate well-established discotic hexagonal mesophases. Such ordered assemblies are attractive targets for materials design in light of the exciting electronic conduction anticipated through these de facto supramolecular polymers.
Figure 1.

General structure of HAT, trialkoxy 1,3,5-benzene tricarboxamide (RO-BTA), and carbonyl-centered and N-centered BTA (C-BTA and N-BTA, respectively).
It therefore stands to reason that new motifs with even greater enthalpic affinities might lead to enhanced materials. For example, compounds with a threefold axis of rotation that are capable of intermolecular hydrogen bonding have been widely explored for their self-assembling abilities.2,3 A seminal example of multivalent discotic architectures capable of amide-driven self-assembly is the 1,3,5-benzene tricarboxamide (BTA, Figure 1), which have been explored by several groups.2,4−11 Due to their liquid-crystalline properties and the availability of three axes to participate in hydrogen bonding, BTAs and related molecules are able to form highly stable supramolecular assemblies. The substituents attached to the amide nitrogens can vary from simple alkyl or glycol chains12−15 to π-conjugated chromophores16−19 to amino acids and oligopeptides.20−26 Typically, these types of systems are known to form cylindrical shaped fibers arising from columnar stacking of the molecules, giving the structures one-dimensional (1-D) directionality and the potential for anisotropic properties which are attractive for organic electronic applications,16−18 especially when enhanced degrees of stacking can be enforced by crowded arenes (RO-BTA, Figure 1).27 Several studies have illustrated the advantages of three axes for the formation of chiral supramolecular assemblies, the enhancement of the thermodynamic strength of the self-assembled structures, and the lowering of the concentrations necessary to form organo- or hydrogels.4,11,28 Utilizing amino acid or oligopeptide side chains as self-assembly scaffolds is also beneficial due to their ability to create directional intermolecular hydrogen bonds, which work to strengthen supramolecular assemblies. They also provide handles to fine-tune self-assembly through altering the oligopeptide sequences.25
Our group previously utilized the self-assembly properties of peptide fragments as a means to assemble linearly π-conjugated semiconducting oligomers. To create these molecules, we developed synthetic procedures based on solid-phase double-couplings or “dimerizations” between π-conjugated chromophores and the N-termini of peptide fragments immobilized on a solid support, as illustrated in Figure 2a.29−31 These procedures begin with the solid-phase synthesis of the peptide fragment, followed by the incorporation of the π-conjugated unit onto the solid phase through either a double amidation (with π-conjugated dicarboxylic acids), double imidation (with dianhydrides), or a piecewise synthesis via N-terminus “capping” with an aryl halide, followed by palladium-catalyzed cross-coupling. All of these methods have proven versatile in the production of novel peptides embedded with a variety of π-conjugated cores. However, these methods thus far have solely been utilized to create twofold or linear peptide−π hybrids as opposed to multivalent 2-D systems, for example, discotic cores with three- or four-fold peptide attachment. These architectures can be accessed by expanding the previously developed solid-phase “dimerizations” into solid-phase multicoupling procedures, as depicted in Figure 2b,c. We preferred a solid-phase route to homogeneous solution reactions to minimize solubility issues or mutual reactivity of particular amino acid side chains when trying to couple a hydrophobic π-electron core with deprotected (and likely water soluble) oligopeptides under suitable carboxylic acid activation conditions. We envisioned that multiple peptide sequences might further strengthen the intermolecular π-electron interactions necessary for energy transport; however, at the same time, they might frustrate the fibrillization that is known to be associated with more linear peptide−π electron conjugates. Due to the potential benefits of creating more densely functionalized discotic cores in terms of possibly stronger electronic coupling and minimal fibrillization, we sought to expand our previously developed synthetic procedures to create these multivalent peptides and study their electronic and self-assembly properties.
Figure 2.
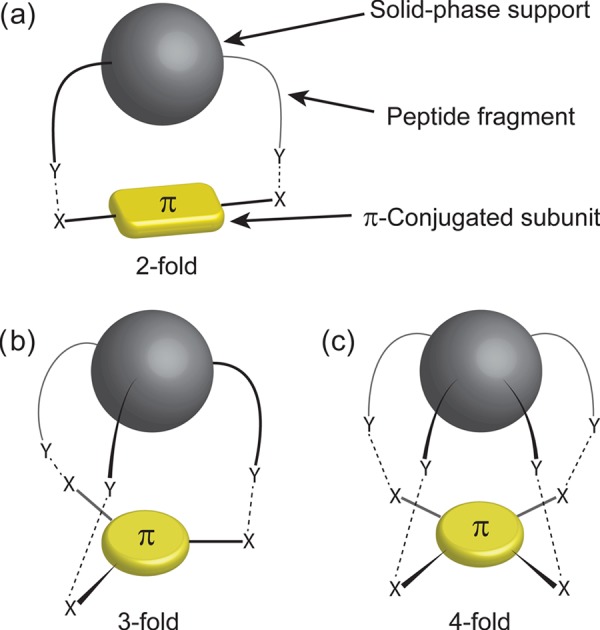
Illustrations of solid-phase (a) twofold, (b) threefold, and (c) fourfold couplings between solid-supported peptides and π-conjugated chromophores of various symmetries.
Results and Discussion
We explored the synthesis and self-assembly capabilities of four multivalent peptide oligomers embedded with three distinct discotic π-conjugated subunits: benzotrithiophene (BTT), decacyclene triimide, and meso-substituted porphyrin. The peptides were synthesized via standard solid-phase peptide synthesis, and π-conjugated subunits were introduced by employing three different solid-phase couplings (Stille cross-coupling, imidation, and amidation) to form the final multivalent architectures. Although analytically pure materials were available in fairly low yields, the quantities available are sufficient for future biomaterial investigations, and the electronic functions available to these hybrid bioelectronic materials could subsidize the material cost. The molecules thus formed would be expected to follow the BTA assembly paradigm, leading to columnar nanostructures through intermolecular hydrogen bonding between peptide scaffolds resulting in extensive intermolecular π-electron overlap (e.g. the generic depiction in Figure 3). In the case of appended peptides with ionizable amino acid residues, changes in pH can lead to variations in protonation that affect the overall net charge of the molecules and thus, their propensities to self-associate. For example, basic pH fosters a molecular state for a peptide with acidic residues (aspartic or glutamic acids) due to carboxylic acid deprotonation, whereas acidic pH would favor acid protonation and thus minimize the electrostatic repulsion and allow the peptide-directed hydrogen bonding to occur. At the same time, self-assembly is expected to bring the π-electron units into closer electronic communication. The self-assembly processes were followed using standard electronic spectroscopies and the nanostructural outcomes were visualized with electron microscopy. The 2-D chromophores appear to withstand both the acidic peptide cleavage conditions (TFA) and the acidic envrionments necessary to trigger assembly (HCl).
Figure 3.

General structure and idealized illustration of self-assembly of multivalent π–peptide hybrids.
BTT-Containing Peptide Trimers
BTT analogues have previously been investigated as donor molecules in molecular heterojunction solar cells.32 From a synthetic perspective, BTTs were attractive substrates because well-established thiophene functionalization chemistry should provide discotic cores compatible with solid-phase peptide ligation strategies. We synthesized a BTT core bearing three stannyl substituents under the expectation that it could be used to link peptides via on-resin Pd-catalyzed chemistry.30,31 To append three peptides to the BTT core, solid-supported peptides consisting of the amino acid sequence VEVAG-NH2 were prepared on a Wang resin and N-acylated with aryl bromides (either thiophene or bithiophene), as previously described.30,31 This peptide sequence was chosen based on our past experience that showed this to be a generally applicable “consensus sequence” able to promote the self-assembly of several different types of peptide−π-electron conjugates.29,33 Then, as shown in Scheme 1, stannylated BTT S4 was introduced onto the resin under Stille cross-coupling conditions, facilitating a triple coupling between S4 and three peptide fragments presented on a resin bead. Following cleavage from the resin, the desired peptide trimers 1 (T-BTT) and 2 (BT-BTT) were obtained.
Scheme 1. Synthesis of BTT-Containing Peptide Trimers 1 and 2 via Solid-Phase Stille Cross-Coupling.

The absorbance and photoluminescence changes observed for 1 and 2 upon assembly are somewhat dissimilar to those previously seen for peptides embedded with linear oligothiophene chromophores.29−31 Typically, the linear peptides exhibit a blue shift of the UV–vis absorbance λmax (usually accompanied by a weak, red-shifted shoulder) and significant quenching in the photoluminescence upon assembly, which is consistent with an H-like aggregation arrangement of the linear chromophores.34 However, due to the threefold nature of the BTT core, and hence the different symmetries and intermolecular interactions of the transition dipoles, the same perturbations in the optical spectra of the multivalent peptides were not expected. The absorption and photoluminescence spectra of 2 were red-shifted compared to those of 1 (absorption at 410 vs 363 nm and photoluminescence at 565 vs 520 nm) when unassembled at basic pH (Figure 4a,c, dotted lines), due to the increased conjugation afforded by the bithiophene linkers. Upon assembly, the absorption spectrum of 1 showed a slight (9 nm) blue shift accompanied by an enhancement of emission intensity with respect to the molar absorptivity of the assembled peptide (Figure 4a, solid lines). In contrast, 2 showed minimal absorption perturbations upon assembly, whereas the emission was much more significantly quenched (Figure 4c, solid lines). The behavior of 2 is roughly comparable to the properties reported for bithiophene-substituted BTT that indicated an evolution of vibronic features in the thin-film state that were slightly (15–20 nm, with an absorption onset of ca. 500 nm) to the red of the solution absorption.32 At acidic pH, the circular dichroism (CD) spectra of 1 and 2 displayed bisignate Cotton effects in the vicinity of the chromophore π–π* transitions (Figure 4b,d), suggesting that the transition dipoles of the self-assembled π-conjugated moieties interact within the chiral environment created by the assembly of the peptide substituents. Interestingly, even at a basic pH, where significant charge repulsion due to the deprotonated carboxylic acid endgroups and side chains would be expected to inhibit intermolecular assembly, Cotton effects were still evident, suggesting that some assembly occurs even at high pH. Many other pH-driven peptide assemblies, especially those with challenging hydrophobic π-electron components, are known to exhibit finite degrees of association under solution conditions that would be expected to favor molecular dissolution.35,36 Despite several attempts to achieve true molecularly dissolved solutions (addition of hydrogen-bond-disrupting hexafluoroisopropanol, measurement of serial dilutions, and even heating of the spectroscopic samples), signatures consistent with preassociating molecules were apparent. Dynamic light scattering also revealed the presence of assembled nanoparticulate structures at basic pH. These observations attest to the strong propensities these very hydrophobic 2-D π-electron platforms have for self-association in aqueous environments (see Figures S18 and S19). Furthermore, despite the similar structure of the two peptide−π hybrid molecules, the CD spectra depicted Cotton effects of opposite handedness and different intensities. Assembly 1 induced a fairly strong positive Cotton effect, suggesting an overall right-handed or clockwise helicity in the supramolecular assemblies, whereas 2 induced a weaker negative Cotton effect, proposing some bias toward left-handed or counterclockwise helicity.37−39 Inversions in global chirality are not unusual for peptide assemblies and unfortunately are not readily predictable.40,41
Figure 4.

UV–vis (left), photoluminescence (right) (a, c), and CD (b, d) spectra of unassembled (dotted lines, pH 8) and assembled (solid lines, pH 6) 1 (a, b) and 2 (c, d). UV–vis and photoluminescence spectra were each acquired at a concentration of 1.5 μM. CD spectra were acquired at a concentration of 22 μM. Photoluminescence spectra of 1 were obtained following excitation at 375 nm (pH 8) and 360 nm (pH 6). Photoluminescence spectra of 2 were obtained following excitation at 410 nm.
The assemblies of 1 and 2 were also visualized using transmission electron microscopy (TEM). Samples were prepared by assembling 1 mg/mL aqueous peptide solutions through diffusion of HCl vapor within an acid chamber (i.e., the sample was placed within a closed container containing a separate vial of concentrated HCl). Interestingly, the two related compounds seem to organize into quite different nanostructures. 1 assembles into long 1-D nanostructures, which appear to be on the order of a few microns in length (Figure 5a). The thinnest structures display widths of 5.0 ± 0.7 nm compared to the 3.7 nm width that would be the expected for a single columnar stack of the molecules in their most extended conformations. Most structures appear thicker, which is likely due to bundling interactions between two or more filaments. Alternatively, 2 (Figure 5b) appears to organize into relatively ill-defined aggregates, as opposed to well-defined 1-D nanostructures, under the same conditions. These differences could be due to variances in solubility due to the more extended and more hydrophobic core of 2.
Figure 5.

Transmission electron micrographs of assembled samples of (a) 1 and (b) 2.
Decacyclene Triimide-Containing Peptide Trimers
To complement the amide-bond-forming strategy above, we also examined anhydrides with a threefold axis of rotation. In particular, we were intrigued by Wudl’s recent work to synthesize decacyclene triimide (DTI) from the corresponding tris-anhydride (3).42 The DTI products are of interest for their self-assembling propensities and their potential to serve as n-channel semiconductors. Using the method previously developed in our lab for solid-phase coupling of peptides to anhydrides,29 a Wang resin prefunctionalized with a D(AD)4GG-NH2 sequence was treated with 3, thus leading to the peptide trimer 4 after resin cleavage according to Scheme 2. The multiple aspartic acid residues were required to enable solubilization and high-pressure liquid chromatography (HPLC) purification of the otherwise very insoluble peptide–DTI conjugates.
Scheme 2. Synthesis of Decacyclene Triimide-Containing Peptide Trimer 4 via Solid-Phase Triple Imidation.

The absorbance and photoluminescence changes observed for 4 upon assembly are again dissimilar to those seen for the previously studied linear peptide “dimers” based on diimide linkers.29 The λmax of peptide 4 displayed a decrease in the molar absorptivity upon acidification; however, this decrease did not coincide with a spectral shift (either bathochromic or hypsochromic), as shown in Figure 6a. The previous report on the DTI photophysics indicated that the UV–vis spectral properties become much more broadened in the solid state versus solution but the primary λmax remains the same.42 Furthermore, acidification of the sample caused a noticeable enhancement of the emission intensity, as opposed to the significant quenching that was previously seen for related linear constructs. The CD spectra of 4 did not show any meaningful absorptions in the chromophore π–π* region upon acidification (Figure 6b), although changes in the higher energy amide region were apparent. This suggests that the resulting supramolecular assemblies do not show any bias for right-handed or left-handed helicity.
Figure 6.

UV–vis (left), photoluminescence (right) (a), and CD (b) spectra of unassembled (dotted lines, pH 8) and assembled (solid lines, pH 4) 4. UV–vis and photoluminescence spectra were acquired at a concentration of 0.6 μM. CD spectra were acquired at a concentration of 26 μM. Photoluminescence spectra were obtained following excitation at 430 nm (pH 8) and 422 nm (pH 4).
The solution measurements did not show the obvious signs of the intermolecular electronic interactions that would be expected from the assembly of these types of multivalent molecules. Nevertheless, we studied the material that forms from a different assembly pathway, as a result of a combined pH-driven and evaporative assembly as the aqueous media under the necessary high-vacuum conditions is volatilized. TEM was employed to visualize the supramolecular assemblies of 4 and are shown in Figure 7. The 1-D nanostructures are very well defined and appear to be several microns in length, while maintaining a thickness of 5.1 ± 0.2 nm. The most extended conformation of 4 is expected to be approximately 4.2 nm in length, thus it is postulated that the nanostructures are composed of single columnar stacks of the molecules in this extended conformation, although more complex assembly motifs have been observed after DTI self-assembly in organic solvents.42
Figure 7.

TEM images of assembled 4 from 1 mg/mL solutions.
Porphyrin-Containing Peptide Tetramers
Porphyrins play natural roles as ligands, catalysts, light harvesters, photoinduced electron transfer donors, and photosensitizers that collectively find use in photosynthesis (e.g., chlorophyll) and a variety of enzymatic reactions. Synthetic porphyrins have likewise been utilized for a multitude of applications from biomedical therapy to electronic materials.43−51 Peptides have also been conjugated to porphyrins as a means to increase their cellular uptake for photodynamic therapy applications or for organization into arrays for photovoltaic devices.47−51
To create porphyrin–peptide conjugates that test the limitations of our previously developed solid-phase chromophore–peptide couplings, we constructed a free-base porphyrin core with fourfold peptide ligation. This was completed through a solid-phase PyBOP-mediated amidation procedure modified from our prior work,29 utilizing commercially available meso-tetra(4-carboxyphenyl)porphine (5) and Wang resin modified with DADGG-NH2 oligopeptides (Scheme 3) providing peptide tetramer 6.
Scheme 3. Synthesis of Porphyrin-Containing Peptide Tetramer 6 via Solid-Phase Quadruple Amidation.

As shown in Figure 8a, the UV–vis spectrum of 6 at pH 8 shows the Soret band at 415 nm as well as four Q-bands at 514, 553, 580, and 631 nm (dashed line). Upon acidification, the Soret band redshifts to 437 nm, and only two Q-bands are apparent, one at 595 nm and one at 649 nm (solid line). This loss of two Q-bands is indicative of a doubly protonated “diacid” version of the porphyrin core, due to increased symmetry upon protonation.52 Similar behavior is seen in the emission spectra of 6 at high and low pH (Figure 8b). The basic and acidic samples were excited at λmax of the Soret bands, resulting in emission peaks at 640 and 665 nm, respectively. To further investigate the assembly of 6, CD was employed. As shown in Figure 8c, no meaningful signal was seen in the spectra of samples prepared at pH 8, whereas that at pH 4 showed a weak signal near 450 nm, suggesting that chiral assembly occurs upon acidification, despite the protonation of the porphyrin core.
Figure 8.

UV–vis (a), photoluminescence (b), and CD (c) spectra of 6 in water at pH 8 (dashed lines) and pH 4 (solid lines). UV–vis and photoluminescence spectra were acquired at a concentration of 3.4 μM. CD spectra were acquired at a concentration of 33 μM. Photoluminescence spectra were obtained following excitation at 415 nm (pH 8) and 437 nm (pH 4).
To visualize the supramolecular assemblies created from acidification of solutions of 6, TEM was employed. The resulting images are shown in Figure 9. Using acidification as the assembly trigger resulted in 1-D nanostructures several microns in length, which undergo significant interstructure interactions to form larger bundled assemblies. Single structures range in thickness from 6.5 to 9 nm, whereas the expected width of the most extended conformation of 6 is approximately 5.0 nm.
Figure 9.

TEM images of 6 from 1 mg/mL solutions.
Electrical Properties
To evaluate the electrical properties of the semiconducting subunits embedded within the peptide backbones, the nanostructures obtained from these materials were incorporated as the active layers of field-effect transistors (FETs). A solution of the peptide was dropcast onto a Si/SiO2 substrate, and self-assembly was triggered by exposure to HCl vapor. We previously showed that the 1-D supramolecular assemblies form readily on SiOx dielectric surfaces. After drying, gold electrodes were evaporated onto the film through a TEM grid used as a mask. Figure 10 depicts the typical output characteristics obtained from the transistors fabricated using peptide 6, measured under ambient conditions. The hole mobility in the nanostructures was found to be 7.2 × 10–5 cm2 V–1 s–1, obtained by fitting the transfer curve (Figure 10b) data to the linear regime equation of the transistor current. This establishes that holes can be transported through the self-assembled nanostructure networks obtained from the peptide–porphyrin conjugate molecule. Previous studies on thin-film transistors fabricated using spray-coated films of 5,10,15,20-tetra-phenyl porphyrin, measured under vacuum, have demonstrated that the field-effect hole mobility in the core unit may be as high as 1.2 × 10–3 cm2 V–1 s–1.53 Single crystal based FETs fabricated using alkylated version of this metal-free porphyrin have been shown to exhibit hole mobilities of 1.8 × 10–3 cm2 V–1 s–1.54 The hole mobility obtained here, under ambient conditions, in the peptide–porphyrin conjugate seems reasonable, as the nonconducting peptide domain is expected to hinder the charge hopping mechanism within porphyrin units among neighboring nanostructures. Nevertheless, the measured mobilities within these nanomaterials suggest that they may be able to serve as active layers for carrier transport in future bioelectronics applications. The field-effect hole mobilities in peptides 1 and 2 were found to be comparable at 4.6 × 10–5 and 6.6 × 10–5 cm2 V–1 s–1, respectively (Figure S16).
Figure 10.

(a) Output characteristics and (b) transfer characteristics of the FET device fabricated from peptide 6.
In the case of peptide 4, however, no significant electron transport (or hole transport) could be measured under ambient conditions or vacuum. This may be due to a combination of molecular and macroscale impacts such as (i) the inherent electron deficiency imparted by the three planar diimide units in direct conjugation with the π-electron core inhibiting the hole-transport ability, (ii) the relatively larger volume fraction of the nonconducting peptide component in 4, as compared to the semiconducting unit, required to obtain water solubility, which may hinder charge hopping amongst adjacent nanostructures to a significantly greater degree, as compared to that observed in the other peptides, and/or (iii) the polycrystallinity of the dropcast film, as visualized from scanning electron microscopy (SEM) images (Figure S17), which inhibits the formation of a continuous network of the semiconducting units across the channel in the transistor. Although we could not record the hole mobility for peptide 4, it indicates that there is some macro- or meso-scale structural requirement for hole transport and it is not simply inherent to assembled peptide nanomaterials.
Conclusions
Multivalent self-assembling peptide−π hybrid materials containing a variety of tri- and tetravalent discotic cores (BTT, decacyclene triimide, and meso-substituted porphyrin) were investigated. Following the preparation of the π-conjugated subunits (if necessary), four multivalent peptide−π hybrid systems were synthesized by modifying solid-phase amidation, imidation, and Stille cross-coupling procedures previously developed in our lab, thus expanding these methodologies for the production of complex multivalent architectures. Although the synthesis yields are relatively low, we stress that our investigations to date have been enacted exclusively on commercial synthesis supports that are designed to minimize the occurrence of site–site peptide reactivity. It is conceivable that the yields reported here could be increased with resins presenting denser peptide loadings. These types of π-electron modifications should allow the resulting nanomaterials to serve as active materials for electrical device characterization in line with our past observations. Indeed, the hole transport measurements obtained from peptides 1, 2, and 6 suggest the presence of interactions among adjacent π-units within the 1-D nanostructures, which allow for migration of charge through the 1-D stacks.
Each multivalent peptide was capable of self-assembling into 1-D nanostructures; however, absorption, photoluminescence, and CD data suggested that the assemblies differed on the local supramolecular level. For instance, whereas the assembly of BTT-containing peptide 2 corresponded with a quenching of the BTT subunit’s photoluminescence, that of related peptide 1 and DTI-containing peptide 4 triggered enhancements in photoluminescence. Furthermore, the CD spectra of 1 and 2 showed Cotton effects upon aggregation of opposite sign, suggesting helical chirality of the assemblies with opposite handedness, whereas that of 4 showed no signal in the vicinity of the chromophore, signifying either a lack of helicity or directional bias of the helicity. The assembly characterization of porphyrin-containing peptide tetramer 6 was complicated by the protonation of the porphyrin core upon acidification, and could perhaps be tailored via the addition of metal ions within the porphyrin macrocycle. Our future work will seek to obtain a better understanding of the internal structure adopted within these assemblies from both an experimental (e.g., through diffraction and scattering measurements) and a computational (molecular dynamics simulations and electronic structure calculation based on these simulations) perspective.
Visualization with TEM also showed differences in the 1-D nanostructures formed by each multivalent peptide−π hybrid. 1, 4, and 6 each formed high aspect ratio 1-D nanostructures, each with widths corresponding to the length of the peptides in their most extended conformation, suggesting a 1-D stacking of the molecules in the supramolecular structures, whereas 2 appeared to form more undefined aggregates under the same conditions. Furthermore, the nanostructures differed in the amount of interstructure interactions, with 6 experiencing extreme bundling, 1 displaying minor structure intertwining, and 2 showing a relative lack of interstructure bundling. These differences may be due to the different multivalent architectures (trimeric vs tetrameric), as well as the variances in the conjugated cores’ structural rigidities.
Experimental Section
General Considerations
Dimethylformamide (DMF), pyridine, chloroform, and nitromethane were purchased from Sigma-Aldrich and dried over 4 Å molecular sieves. Toluene, tetrahydrofuran (THF), and dichloromethane (DCM) were acquired from an Innovative Technology Pure Solv solvent purification system. Solvents were degassed by sparging with nitrogen for 30–90 min before use. Reactions were performed under an inert N2 atmosphere and glassware was flame-dried prior to use. Tetrakis(triphenylphosphine)palladium was obtained from Strem Chemicals. FeCl3 was purchased from Acros. N-Methylpyrrolidone (NMP), Wang resin substituted with the first amino acid, and Fmoc-protected amino acids were obtained from Advanced ChemTech. N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate (HBTU), 2,3-dibromothiophene, and (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) were purchased from Oakwood Products Inc. All other reagents and starting materials were obtained from Sigma-Aldrich and were used as received. 3-(Tributylstannyl)thiophene (S4) was prepared using literature procedures.55
NMR Spectroscopy
1H NMR spectra were obtained using a Bruker Avance 400 MHz FT-NMR spectrometer and processed with Bruker Topspin 1.3. Chemical shifts were referenced to the residual protio-solvent peak (7.26 ppm for CDCl3, 4.79 ppm for D2O). Peptide 1H NMR spectra were acquired using a 1 s presaturation pulse to suppress water.
Electrospray Ionization Mass Spectrometry (ESI-MS)
ESI samples were collected using a Thermo Finnigan LCQ Deca Ion Trap Mass Spectrometer in negative ion mode. Samples were prepared in a 1:1 methanol/water solution with 0.1% ammonium hydroxide.
UV–Vis and Photoluminescence
UV–vis spectra were obtained using a Varian Cary 50 Bio UV–vis spectrophotometer. Photoluminescence spectra were obtained using a PTi Photon Technology International fluorometer with an Ushio xenon short arc lamp. Spectroscopic samples were prepared by diluting in Millipore water until achieving an absorbance near 0.1. The pH was then adjusted by adding 10 μL of either 1 M KOH (basic) or 1 M HCl (acidic).
CD
CD spectra were obtained using an AVIV 420 spectropolarimeter. Spectroscopic samples were prepared by diluting to the appropriate concentration in Millipore water. The pH was then adjusted by adding 10 μL of either 1 M KOH (basic) or 1 M HCl (acidic).
Due to uncertainties in determining the concentrations of “molecular” samples, the quantification of the spectral intensities (UV–vis absorption extinction coefficient, photoluminescence intensity, and CD molar absorptivity) should be viewed as approximate.
Reverse-Phase HPLC
HPLC purification was performed on a Varian PrepStar SD-1 (preparative) instrument using Luna 5 μm particle diameter C8 with TMS endcapping columns with silica solid support. An ammonium formate aqueous buffer (pH 8) and acetonitrile were used as the mobile phase.
TEM
Imaging was performed on a Philips EM 420 transmission electron microscope, equipped with an SIS Megaview III CCD digital camera. Aqueous peptide solutions (1 mg/mL) were rendered acidic after the diffusion of HCl vapor within an acid chamber (i.e., the sample was placed within a closed container containing a separate vial of concentrated HCl). The microscopy samples were prepared by pipetting a drop of these acidic solutions onto 200 mesh Formvar coated copper grids and incubating for 5 min at 25 °C. Excess solution was wicked off by touching the side of the grid to filter paper. The sample was then stained with a 2% uranyl acetate solution and excess moisture was wicked off. The grid was allowed to dry in air before imaging.
Device Fabrication
A neutral 0.5 wt % solution of the peptides was prepared by dissolving the peptides in MilliQ water. Of this solution, 5 μL were dropcast onto a piranha cleaned Si/SiO2 (300 nm) substrate. This was exposed to HCl vapor in a closed chamber for 5 min. After drying in air, 50 nm thick gold electrodes were thermally evaporated, at a rate of 0.4 Å/s, using a 200 mesh TEM grid as a shadow mask. The electrical measurements were made under ambient conditions using an Agilent 4155c semiconductor parameter analyzer. The mobility was calculated by fitting the transfer curve to the linear region of the FET equation
where Id is the drain current, μ is the hole mobility, Ci is the gate insulator capacitance per unit area, W and L are the width and length of the channel, respectively, Vd is the drain voltage, Vg is the gate voltage, and Vth is the threshold voltage. The capacitance value for the 300 nm SiO2 layer was 11.5 nF/cm2, and the electrodes had a width of 120 μm and a length of 12 μm.
SEM
SEM images of the films were obtained using a FEI Quanta 200 Environmental SEM. The samples were prepared so as to mimic the sample preparation for FET measurements and were sputtered with platinum before imaging.
VEVAG-T-BTT Peptide Trimer (1)
Wang resin-bound VEVAG-NH2 peptide capped with thiophene bromide (0.3 mmol) (synthesis detailed in ref (30)) was placed in a Schlenk flask and dried under vacuum. Pd(PPh3)4 (0.017 g, 0.015 mmol, 5 mol %) was added. S4 (0.113 g, 0.102 mmol) was dissolved in 7 mL DMF, and the solution was added to the reaction vessel via a syringe. The mixture was heated to 80 °C with constant nitrogen bubbling through the solution to agitate the resin beads for 20 h. The mixture was allowed to cool and the resin was transferred to a peptide chamber. The resin was subjected to a wash cycle (3× NMP, 3× DMF, 2× i-PrOH, 2× H2O, 2× (THF, i-PrOH), 2× acetonitrile, 2× ether, 2× hexanes). The resin was then treated with a mixture of 9.5 mL trifluoroacetic acid, 250 μL tri(isopropyl)silane, 250 μL H2O, and 5 mL DCM for 3 h. The resulting solution was separated from the resin and was concentrated to approximately 5 mL under reduced pressure; 90 mL of ether was added, and the orange solid was isolated via centrifugation. The solid was dissolved in approximately 5 mL of water and 10 μL NH4OH and lyophilized. The product was further purified by HPLC to give 1 (0.0071 mmol, 0.014 g, 7.1%) as a yellow powder. MS (ESI) m/z 1989.1 (M – H)− (calcd 1988.6), m/z 1016.2 (M – 4H + 2Na+)2– (calcd 1015.8), m/z 1005.3 (M – 3H + Na+)2– (calcd 1004.8), m/z 994.3 (M – 2H)2– (calcd 993.8), m/z 662.5 (M – 3H)3– (calcd 662.2).
VEVAG-BT-BTT Peptide Trimer (2)
Wang resin-bound VEVAG-NH2 peptide capped with bithiophene bromide (0.3 mmol) (synthesis detailed in ref (30)) was placed in a Schlenk flask and dried under vacuum. Pd(PPh3)4 (0.017 g, 0.015 mmol, 5 mol %) was added. S4 (0.113 g, 0.102 mmol) was dissolved in 7 mL DMF, and the solution was added to the reaction vessel via a syringe. The mixture was heated to 80 °C with constant nitrogen bubbling through the solution to agitate the resin beads for 20 h. The mixture was allowed to cool, and the resin was transferred to a peptide chamber. The resin was subjected to a wash cycle (3× NMP, 3× DMF, 2× i-PrOH, 2× H2O, 2× (THF, i-PrOH), 2× acetonitrile, 2× ether, 2× hexanes). The resin was then treated with a mixture of 9.5 mL trifluoroacetic acid, 250 μL tri(isopropyl)silane, 250 μL H2O, and 5 mL DCM for 3 h. The resulting solution was separated from the resin and was concentrated to approximately 5 mL under reduced pressure; 90 mL of ether was added, and the orange solid was isolated via centrifugation. The solid was dissolved in approximately 5 mL of water and 10 μL NH4OH and lyophilized. The product was further purified by HPLC to give 2 (0.0032 mmol, 0.0071 g, 3.2%) as an orange powder. MS (ESI) m/z 1117.2 (M – 2H)2– (calcd 1117.3), m/z 744.2 (M – 3H)3– (calcd 744.5).
DADADADADGG-DTI Peptide Trimer (4)
Using an imide bond-forming on-resin dimerization protocol,29 Wang resin-bound DADADADADGG-NH2 peptide (0.5 mmol) (synthesized by standard SPPS) was placed in a Schlenk flask and dried under vacuum. Decacyclene trianhydride (3)42 (0.110 g, 0.167 mmol) and pyridine (8 mL) were added. The mixture was heated to 65 °C. DIPEA (1.3 mL, 7.5 mmol) was added. The mixture was heated to 135 °C for 18 h with N2 bubbling constantly to agitate the resin. The reaction was allowed to cool and the resin was transferred to a peptide chamber. The resin was subjected to a wash cycle (3× NMP, 3× DMF, 2× i-PrOH, 2× H2O, 2× (THF, i-PrOH), 2× acetonitrile, 2× ether, 2× hexanes). The resin was then treated with a mixture of 9.5 mL trifluoroacetic acid, 250 μL tri(isopropyl)silane, 250 μL H2O, and 5 mL DCM for 3 h. The resulting solution was separated from the resin and was concentrated to approximately 5 mL under reduced pressure; 90 mL of ether was added, and the orange solid was isolated via centrifugation. The solid was dissolved in approximately 5 mL of water and 10 μL NH4OH and lyophilized to give the crude product that was further purified by HPLC to give 4 (0.033 g, 0.009 mmol, 5.5%) as an orange powder. MS (ESI) m/z 1193.5 (M – 3H)3– (calcd 1192.7), m/z 894.5 (M – 4H)4– (calcd 894.3), m/z 715.4 (M – 5H)5– (calcd 715.2), m/z 595.8 (M – 6H)6– (calcd 595.8), m/z 446.8 (M – 8H)8– (calcd 446.6).
DADGG-Porphyrin–Peptide Tetramer (6)
Using an imide bond-forming on-resin dimerization protocol,29 a resin-bound DADGG-NH2 peptide (0.5 mmol) within a peptide chamber was suspended in 5 mL of DCM. In a separate vial, meso-tetra(4-carboxyphenyl)porphine (5) (0.060 g, 0.075 mmol) and PyBOP (0.017 g, 0.33 mmol) was dissolved in 10 mL NMP. DIPEA (0.78 mL, 4.5 mmol) was added to the vial and the solution was agitated for 1 min. The solution was then added to the peptide chamber and mixed for 15 h. The resin was subjected to a washing cycle (3× NMP, 3× DMF, 2× i-PrOH, 2× water, 2× (2× THF, 2× i-PrOH), 2× acetonitrile, 2× ether, 2× hexanes). The resin was again swelled in 5 mL of DCM and subjected to a second round of coupling with meso-tetra(4-carboxyphenyl)porphine (0.040 g, 0.05 mmol) and PyBOP (0.12 g, 0.22 mmol) dissolved in 10 mL NMP, which was mixed with DIPEA (0.52 mL, 3.0 mmol), agitated for 1 min and added to the peptide chamber. The chamber was agitated for 24 h. The resin was again subjected to the same wash cycle. The resin was treated with 9.5 mL of trifluoroacetic acid, 250 μL tri(isopropyl)silane, and 250 μL water for 3 h. The peptide solution was filtered from the resin beads, which were then washed 3× with DCM, and the resulting solution was concentrated by evaporation under reduced pressure. The crude peptide was then precipitated from solution with 90 mL of diethyl ether and isolated through centrifugation. The resulting pellet was triturated with diethyl ether to yield the crude product, which was dissolved in approximately 2 mL of water and 30 μL ammonium hydroxide and lyophilized. The crude product was purified via HPLC to give 6 (0.028 g, 0.011 mmol, 9.2%) as a purplish-brown solid. MS (ESI) m/z 1224.9 (M – 2H)2– (calcd 1224.9), m/z 1236.6 (M – 3H + Na+)2– (calcd 1235.9), m/z 816.4 (M – 3H)3– (calcd 816.2), m/z 612.1 (M – 4H)4– (calcd 611.9).
Acknowledgments
We thank Johns Hopkins University and the Department of Energy Office of Basic Energy Sciences (DE-SC0004857, J.D.T. peptide nanomaterials synthesis) for financial support. A.M.S. was supported by the Harry and Cleio Greer Fellowship (JHU). We thank Dr. Toan Pho and Prof. Fred Wudl (University of California, Santa Barbara) for kindly supplying a sample of the decacyclene tris-anhydride (3).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00414.
NMR, ESI, HPLC, and additional TEM images of multivalent peptides (PDF)
The authors declare no competing financial interest.
Author Status
∥ Taken in part from the thesis of A.M.S. (Ph.D., Johns Hopkins, 2015). Current position: Postdoctoral Fellow (Cancer Research Training Award), National Cancer Institute, Frederick, Maryland 21702, United States (A.M.S.).
Supplementary Material
References
- Tabushi I.; Yamamura K.; Okada Y. Liquid Crystals II. The Tail Design via β-Oxygen Atom Modification for the Remarkably Enhanced Stability of Hexakis (alkanoyloxy) Triphenylene Mesophase. Tetrahedron Lett. 1987, 28, 2269–2272. 10.1016/S0040-4039(00)96098-2. [DOI] [Google Scholar]
- Cantekin S.; de Greef T. F. A.; Palmans A. R. A. Benzene-1,3,5-Tricarboxamide: A Versatile Ordering Moiety for Supramolecular Chemistry. Chem. Soc. Rev. 2012, 41, 6125. 10.1039/c2cs35156k. [DOI] [PubMed] [Google Scholar]
- Yasuda Y.; Takebe Y.; Fukumoto M.; Inada H.; Shirota Y. 4,4′,4″-Tris(stearoylamino)triphenylamine as a Novel Material for Functional Molecular Gels. Adv. Mater. 1996, 8, 740–741. 10.1002/adma.19960080907. [DOI] [Google Scholar]
- Bernet A.; Albuquerque R. Q.; Behr M.; Hoffmann S. T.; Schmidt H.-W. Formation of a Supramolecular Chromophore: A Spectroscopic and Theoretical Study. Soft Matter 2012, 8, 66–69. 10.1039/C1SM06789C. [DOI] [Google Scholar]
- Besenius P.; van den Hout K. P.; Albers H. M. H. G.; de Greef T. F. A.; Olijve L. L. C.; Hermans T. M.; de Waal B. F. M.; Bomans P. H. H.; Sommerdijk N. A. J. M.; Portale G.; Palmans A. R. A.; van Genderen M. H. P.; Vekemans J. A. J. M.; Meijer E. W. Controlled Supramolecular Oligomerization of C3-Symmetrical Molecules in Water: The Impact of Hydrophobic Shielding. Chem. – Eur. J. 2011, 17, 5193–5203. 10.1002/chem.201002976. [DOI] [PubMed] [Google Scholar]
- Brunsveld L.; Zhang H.; Glasbeek M.; Vekemans J. A. J. M.; Meijer E. W. Hierarchical Growth of Chiral Self-Assembled Structures in Protic Media. J. Am. Chem. Soc. 2000, 122, 6175–6182. 10.1021/ja0005237. [DOI] [Google Scholar]
- Chang J. Y.; Baik J. H.; Lee C. B.; Han M. J.; Hong S.-K. Liquid Crystals Obtained from Disclike Mesogenic Diacetylenes and Their Polymerization. J. Am. Chem. Soc. 1997, 119, 3197–3198. 10.1021/ja961193m. [DOI] [Google Scholar]
- Palmans A. R. A.; Vekemans J. A. J. M.; Havinga E. E.; Meijer E. W. Sergeants-and-Soldiers Principle in Chiral Columnar Stacks of Disc-Shaped Molecules with C3 Symmetry. Angew. Chem., Int. Ed. 1997, 36, 2648–2651. 10.1002/anie.199726481. [DOI] [Google Scholar]
- Palmans A. R. A.; Vekemans J. A. J. M.; Meijer E. W.; Palmans A. R. A.; Kooijman H.; Spek A. L. Hydrogen-Bonded Porous Solid Derived from Trimesic Amide. Chem. Commun. 1997, 2247–2248. 10.1039/a705339h. [DOI] [Google Scholar]
- Palmans A. R. A.; Vekemans J. A. J. M.; Fischer H.; Hikmet R. A.; Meijer E. W. Extended-Core Discotic Liquid Crystals Based on the Intramolecular H-Bonding in N -Acylated 2,2′-Bipyridine-3,3′-Diamine Moieties. Chem. – Eur. J. 1997, 3, 300–307. 10.1002/chem.19970030220. [DOI] [PubMed] [Google Scholar]
- van Gorp J. J.; Vekemans J. A. J. M.; Meijer E. W. C 3 -Symmetrical Supramolecular Architectures: Fibers and Organic Gels from Discotic Trisamides and Trisureas. J. Am. Chem. Soc. 2002, 124, 14759–14769. 10.1021/ja020984n. [DOI] [PubMed] [Google Scholar]
- Smulders M. M.; Schenning A. P. H. J.; Meijer E. W. Insight into the Mechanisms of Cooperative Self-Assembly: The “Sergeants-and-Soldiers” Principle of Chiral and Achiral C 3 -Symmetrical Discotic Triamides. J. Am. Chem. Soc. 2008, 130, 606–611. 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- Stals P. J. M.; Everts J. C.; de Bruijn R.; Filot I. A. W.; Smulders M. M. J.; Martín-Rapún R.; Pidko E. A.; de Greef T. F. A.; Palmans A. R. A.; Meijer E. W. Dynamic Supramolecular Polymers Based on Benzene-1,3,5-Tricarboxamides: The Influence of Amide Connectivity on Aggregate Stability and Amplification of Chirality. Chem. – Eur. J. 2010, 16, 810–821. 10.1002/chem.200902635. [DOI] [PubMed] [Google Scholar]
- Stals P. J. M.; Haveman J. F.; Martín-Rapún R.; Fitié C. F. C.; Palmans A. R. A.; Meijer E. W. The Influence of Oligo(ethylene Glycol) Side Chains on the Self-Assembly of Benzene-1,3,5-Tricarboxamides in the Solid State and in Solution. J. Mater. Chem. 2009, 19, 124–130. 10.1039/B816418E. [DOI] [Google Scholar]
- Stals P. J. M.; Smulders M. M. J.; Martín-Rapún R.; Palmans A. R. A.; Meijer E. W. Asymmetrically Substituted Benzene-1,3,5-Tricarboxamides: Self-Assembly and Odd-Even Effects in the Solid State and in Dilute Solution. Chem. – Eur. J. 2009, 15, 2071–2080. 10.1002/chem.200802196. [DOI] [PubMed] [Google Scholar]
- Narayan B.; Kulkarni C.; George S. J. Synthesis and Self-Assembly of a C 3 -Symmetric Benzene-1,3,5-Tricarboxamide (BTA) Anchored Naphthalene Diimide Disc. J. Mater. Chem. C 2013, 1, 626–629. 10.1039/C2TC00742H. [DOI] [Google Scholar]
- Paraschiv I.; Giesbers M.; van Lagen B.; Grozema F. C.; Abellon R. D.; Siebbeles L. D. A.; Marcelis A. T. M.; Zuilhof H.; Sudhölter E. J. R. H-Bond-Stabilized Triphenylene-Based Columnar Discotic Liquid Crystals. Chem. Mater. 2006, 18, 968–974. 10.1021/cm052221f. [DOI] [Google Scholar]
- van Herrikhuyzen J.; Jonkheijm P.; Schenning A. P. H. J.; Meijer E. W. The Influence of Hydrogen Bonding and π–π Stacking Interactions on the Self-Assembly Properties of C3-Symmetrical Oligo(p-Phenylenevinylene) Discs. Org. Biomol. Chem. 2006, 4, 1539. 10.1039/b517993a. [DOI] [PubMed] [Google Scholar]
- van Hameren R.; Schon P.; van Buul A. M.; Hoogboom J.; Lazarenko S. V.; Gerritsen J. W.; Engelkamp H.; Christianen P. C. M.; Heus H. A.; Maan J. C.; Rasing T.; Speller S.; Rowan A. E.; Elemans J. A. A. W.; Nolte R. J. M. Macroscopic Hierarchical Surface Patterning of Porphyrin Trimers via Self-Assembly and Dewetting. Science 2006, 314, 1433–1436. 10.1126/science.1133004. [DOI] [PubMed] [Google Scholar]
- Besenius P.; Portale G.; Bomans P. H. H.; Janssen H. M.; Palmans A. R. A.; Meijer E. W. Controlling the Growth and Shape of Chiral Supramolecular Polymers in Water. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17888–17893. 10.1073/pnas.1009592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P. P.; Drew M. G. B.; Das A. K.; Banerjee A. Formation of Triple Helical Nanofibers Using Self-Assembling Chiral Benzene-1,3,5-Tricarboxamides and Reversal of the Nanostructure’s Handedness Using Mirror Image Building Blocks. Chem. Commun. 2006, 3196. 10.1039/b606371c. [DOI] [PubMed] [Google Scholar]
- de Loos M.; van Esch J. H.; Kellogg R. M.; Feringa B. L. C3-Symmetric, Amino Acid Based Organogelators and Thickeners: A Systematic Study of Structure–property Relations. Tetrahedron 2007, 63, 7285–7301. 10.1016/j.tet.2007.02.066. [DOI] [Google Scholar]
- Frisch H.; Unsleber J. P.; Lüdeker D.; Peterlechner M.; Brunklaus G.; Waller M.; Besenius P. pH-Switchable Ampholytic Supramolecular Copolymers. Angew. Chem., Int. Ed. 2013, 52, 10097–10101. 10.1002/anie.201303810. [DOI] [PubMed] [Google Scholar]
- Matsuura K.; Murasato K.; Kimizuka N. Artificial Peptide-Nanospheres Self-Assembled from Three-Way Junctions of β-Sheet-Forming Peptides. J. Am. Chem. Soc. 2005, 127, 10148–10149. 10.1021/ja052644i. [DOI] [PubMed] [Google Scholar]
- van den Hout K. P.; Martín-Rapún R.; Vekemans J. A. J. M.; Meijer E. W. Tuning the Stacking Properties ofC3-Symmetrical Molecules by Modifying a Dipeptide Motif. Chem. – Eur. J. 2007, 13, 8111–8123. 10.1002/chem.200700630. [DOI] [PubMed] [Google Scholar]
- Veld M. A. J.; Haveman D.; Palmans A. R. A.; Meijer E. W. Sterically Demanding Benzene-1,3,5-Tricarboxamides: Tuning the Mechanisms of Supramolecular Polymerization and Chiral Amplification. Soft Matter 2011, 7, 524–531. 10.1039/C0SM00516A. [DOI] [Google Scholar]
- Bushey M. L.; Hwang A.; Stephens P. W.; Nuckolls C. Enforced Stacking in Crowded Arenes. J. Am. Chem. Soc. 2001, 123, 8157–8158. 10.1021/ja0104148. [DOI] [PubMed] [Google Scholar]
- Shikata T.; Ogata D.; Hanabusa K. Viscoelastic Behavior of Supramolecular Polymeric Systems Consisting of N, N′, N″-Tris(3,7-Dimethyloctyl)benzene-1,3,5-Tricarboxamide and N -Alkanes. J. Phys. Chem. B 2004, 108, 508–514. 10.1021/jp030510q. [DOI] [Google Scholar]
- Vadehra G. S.; Wall B. D.; Diegelmann S. R.; Tovar J. D. On-Resin Dimerization Incorporates a Diverse Array of π-Conjugated Functionality within Aqueous Self-Assembling Peptide Backbones. Chem. Commun. 2010, 46, 3947. 10.1039/c0cc00301h. [DOI] [PubMed] [Google Scholar]
- Sanders A. M.; Dawidczyk T. J.; Katz H. E.; Tovar J. D. Peptide-Based Supramolecular Semiconductor Nanomaterials via Pd-Catalyzed Solid-Phase “Dimerizations.”. ACS Macro Lett. 2012, 1, 1326–1329. 10.1021/mz3004665. [DOI] [PubMed] [Google Scholar]
- Sanders A. M.; Tovar J. D. Solid-Phase Pd-Catalysed Cross-Coupling Methods for the Construction of π-Conjugated Peptide Nanomaterials. Supramol. Chem. 2014, 26, 259–266. 10.1080/10610278.2013.852675. [DOI] [Google Scholar]
- de Bettignies R.; Nicolas Y.; Blanchard P.; Levillain E.; Nunzi J.-M.; Roncali J. Planarized Star-Shaped Oligothiophenes as a New Class of Organic Semiconductors for Heterojunction Solar Cells. Adv. Mater. 2003, 15, 1939–1943. 10.1002/adma.200305168. [DOI] [Google Scholar]
- Wall B. D.; Diegelmann S. R.; Zhang S.; Dawidczyk T. J.; Wilson W. L.; Katz H. E.; Mao H.-Q.; Tovar J. D. Aligned Macroscopic Domains of Optoelectronic Nanostructures Prepared via Shear-Flow Assembly of Peptide Hydrogels. Adv. Mater. 2011, 23, 5009–5014. 10.1002/adma.201102963. [DOI] [PubMed] [Google Scholar]
- Kasha M.; Rawls H. R.; El-Bayoumi M. A. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. 10.1351/pac196511030371. [DOI] [Google Scholar]
- Ardoña H. A. M.; Tovar J. D. Energy Transfer within Responsive Pi-Conjugated Coassembled Peptide-Based Nanostructures in Aqueous Environments. Chem. Sci. 2015, 6, 1474–1484. 10.1039/C4SC03122A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar J. D.; Claussen R. C.; Stupp S. I. Probing the Interior of Peptide Amphiphile Supramolecular Aggregates. J. Am. Chem. Soc. 2005, 127, 7337–7345. 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]
- Marty R.; Nigon R.; Leite D.; Frauenrath H. Two-Fold Odd–Even Effect in Self-Assembled Nanowires from Oligopeptide-Polymer-Substituted Perylene Bisimides. J. Am. Chem. Soc. 2014, 136, 3919–3927. 10.1021/ja412384p. [DOI] [PubMed] [Google Scholar]
- Thalacker C.; Würthner F. Chiral Perylene Bisimide–Melamine Assemblies: Hydrogen Bond-Directed Growth of Helically Stacked Dyes with Chiroptical Properties. Adv. Funct. Mater. 2002, 12, 209.. [DOI] [Google Scholar]
- Dehm V.; Chen Z.; Baumeister U.; Prins P.; Siebbeles L. D. A.; Würthner F. Helical Growth of Semiconducting Columnar Dye Assemblies Based on Chiral Perylene Bisimides. Org. Lett. 2007, 9, 1085–1088. 10.1021/ol0700963. [DOI] [PubMed] [Google Scholar]
- Ryan D. M.; Doran T. M.; Nilsson B. L. Complementary π–π Interactions Induce Multicomponent Coassembly into Functional Fibrils. Langmuir 2011, 27, 11145–11156. 10.1021/la202070d. [DOI] [PubMed] [Google Scholar]
- Wall B. D.; Zacca A. E.; Sanders A. M.; Wilson W. L.; Ferguson A. L.; Tovar J. D. Supramolecular Polymorphism: Tunable Electronic Interactions within π-Conjugated Peptide Nanostructures Dictated by Primary Amino Acid Sequence. Langmuir 2014, 30, 5946–5956. 10.1021/la500222y. [DOI] [PubMed] [Google Scholar]
- Pho T. V.; Toma F. M.; Chabinyc M. L.; Wudl F. Self-Assembling Decacyclene Triimides Prepared through a Regioselective Hextuple Friedel-Crafts Carbamylation. Angew. Chem., Int. Ed. 2013, 52, 1446–1451. 10.1002/anie.201207608. [DOI] [PubMed] [Google Scholar]
- Carvalho C. M. B.; Brocksom T. J.; de Oliveira K. T. Tetrabenzoporphyrins: Synthetic Developments and Applications. Chem. Soc. Rev. 2013, 42, 3302–3317. 10.1039/c3cs35500d. [DOI] [PubMed] [Google Scholar]
- Li W.-S.; Aida T. Dendrimer Porphyrins and Phthalocyanines. Chem. Rev. 2009, 109, 6047–6076. 10.1021/cr900186c. [DOI] [PubMed] [Google Scholar]
- Imahori H.; Umeyama T.; Kurotobi K.; Takano Y. Self-Assembling Porphyrins and Phthalocyanines for Photoinduced Charge Separation and Charge Transport. Chem. Commun. 2012, 48, 4032–4045. 10.1039/c2cc30621b. [DOI] [PubMed] [Google Scholar]
- Urbani M.; Grätzel M.; Nazeeruddin M. K.; Torres T. Meso-Substituted Porphyrins for Dye-Sensitized Solar Cells. Chem. Rev. 2014, 114, 12330–12396. 10.1021/cr5001964. [DOI] [PubMed] [Google Scholar]
- Kovaric B. C.; Kokona B.; Schwab A. D.; Twomey M. A.; de Paula J. C.; Fairman R. Self-Assembly of Peptide Porphyrin Complexes: Toward the Development of Smart Biomaterials. J. Am. Chem. Soc. 2006, 128, 4166–4167. 10.1021/ja056357q. [DOI] [PubMed] [Google Scholar]
- Venkatesh B.; Jayakumar R.; Pandian R. P.; Manoharan P. T. Surface Active Peptide-Mediated Porphyrin Aggregation. Biochem. Biophys. Res. Commun. 1996, 223, 390–396. 10.1006/bbrc.1996.0904. [DOI] [PubMed] [Google Scholar]
- Liu J. Y.; Schmidt J. A.; Bolton J. R. Intramolecular Photochemical Electron Transfer. 6. Bridge and Solvent Dependence of Electron Transfer in Covalently Linked Porphyrin-Peptide-Quinone Compounds. J. Phys. Chem. 1991, 95, 6924–6927. 10.1021/j100171a035. [DOI] [Google Scholar]
- Sibrian-Vazquez M.; Jensen T. J.; Vicente M. G. H. Synthesis, Characterization, and Metabolic Stability of Porphyrin–Peptide Conjugates Bearing Bifunctional Signaling Sequences. J. Med. Chem. 2008, 51, 2915–2923. 10.1021/jm701050j. [DOI] [PubMed] [Google Scholar]
- Hasobe T.; Kamat P. V.; Troiani V.; Solladié N.; Ahn T. K.; Kim S. K.; Kim D.; Kongkanand A.; Kuwabata S.; Fukuzumi S. Enhancement of Light-Energy Conversion Efficiency by Multi-Porphyrin Arrays of Porphyrin-Peptide Oligomers with Fullerene Clusters. J. Phys. Chem. B 2005, 109, 19–23. 10.1021/jp045246v. [DOI] [PubMed] [Google Scholar]
- Choi M. Y.; Pollard J. A.; Webb M. A.; McHale J. L. Counterion-Dependent Excitonic Spectra of Tetra (p-Carboxyphenyl) Porphyrin Aggregates in Acidic Aqueous Solution. J. Am. Chem. Soc. 2003, 125, 810–820. 10.1021/ja0274397. [DOI] [PubMed] [Google Scholar]
- Checcoli P.; Conte G.; Salvatori S.; Paolesse R.; Bolognesi A.; Berliocchi M.; Brunetti F.; D’Amico A.; Di Carlo A.; Lugli P. Tetra-Phenyl Porphyrin Based Thin Film Transistors. Synth. Met. 2003, 138, 261–266. 10.1016/S0379-6779(02)01308-5. [DOI] [Google Scholar]
- Ma P.; Chen Y.; Cai X.; Wang H.; Zhang Y.; Gao Y.; Jiang J. Organic Field Effect Transistors Based on 5,10,15,20-tetrakis(4-Pentyloxyphenyl)porphyrin Single Crystal. Synth. Met. 2010, 160, 510–515. 10.1016/j.synthmet.2009.11.040. [DOI] [Google Scholar]
- Leliège A.; Régent C.-H. Le; Allain M.; Blanchard P.; Roncali J. Structural Modulation of Internal Charge Transfer in Small Molecular Donors for Organic Solar Cells. Chem. Commun. 2012, 48, 8907. 10.1039/c2cc33921h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.