

Abstract
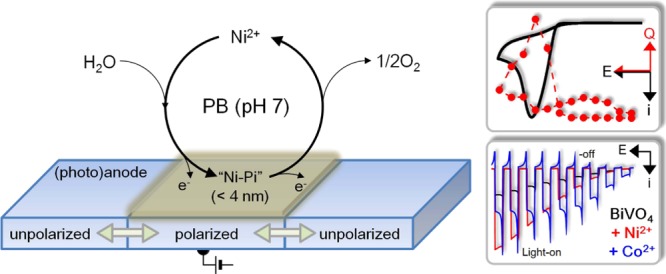
Water oxidation electrocatalyzed by Ni2+ under neutral conditions was investigated using various electrochemical analyses. The addition of Ni2+ in a phosphate-buffered solution catalyzed the oxidation of water, as confirmed by the detection of oxygen generation via scanning electrochemical microscopy. A combination of cyclic voltammetry, coulometric titration, and electrochemical quartz microbalance measurements identified the catalysis as heterogeneous and the catalyst as a Ni-based ultrathin (<4 nm) layer (“Ni-Pi”). Analysis of the potential- and pH-dependency of the titrated amount of charge revealed that the catalyst was deposited only under anodic polarization conditions and was removed under unpolarized conditions; the catalyst may be Ni(III) oxide, and its formation and oxidation appeared to be chemically irreversible. The diffusion-limited nature of water oxidation catalyzed by Ni2+ was closely related to the phosphate ions involved in the catalyst formation and the accompanying catalysis. Although the catalytic performance of Ni2+ alone was not remarkable, it exhibited a synergetic effect with BiVO4 for photoelectrochemical water oxidation, which can compete with Co-Pi-decorated BiVO4.
Introduction
A robust water-splitting catalyst is fundamental for the effective conversion of solar energy into chemical fuel (H2) via electrochemical and photoelectrochemical water splitting.1−3 In water splitting (2H2O → 2H2 + O2, ΔE° = 1.23 V), the oxygen evolution reaction (OER or water oxidation, 2H2O → O2 + 4H+ + 4e–) is known to be more complicated than the hydrogen evolution reaction (HER, 2H+ + 2e– → H2) and its slow kinetics limits the overall water-splitting rate.2,4 Therefore, using a water-oxidation catalyst based on earth-abundant elements is essential for cost-effective production of chemical fuel; numerous homogeneous and heterogeneous catalysts made of transition metals have been developed toward this end.5−9
Transition-metal-based catalysts are also frequently combined with photoelectrodes (p- and n-type semiconductors) to utilize solar energy for water splitting.10 Neutral pH conditions, rather than acidic or alkaline conditions, are favored to ensure the long-term stability of photoelectrodes and metallic electrodes.11−14 However, transition-metal-oxide-based heterogeneous catalysts are thermodynamically unstable and less active at neutral pH; accordingly, most water-oxidation studies have been performed under alkaline conditions.15−18 Recently, Nocera et al.2,19 reported a cobalt phosphate (Co-Pi) heterogeneous electrocatalyst that consists of amorphous cobalt oxide coordinated with a phosphate ion. This catalyst displayed vigorous activity toward water oxidation at pH 7 and triggered extensive research on conjugating it with various photoelectrodes such as silicon,11,20 Fe2O3,21,22 WO3,13 Ta3N5,23,24 and BiVO4.25,26
In this study, we evaluate the electrocatalytic properties of Ni2+ for water oxidation at neutral pH. This is inspired by the catalysis of water oxidation by Co2+ in a phosphate buffer.2 The electrocatalytic performance of Ni-oxide heterogeneous catalysts has not been investigated under neutral pH conditions, which is probably due to its thermodynamic instability; there are only a few reports on homogeneous water-oxidation catalysis (WOC) by Ni(II) complexes at neutral pH.27,28 Herein, we found an ultrathin (<4 nm) heterogeneous electrocatalyst (“Ni-Pi”), and its formation amount varied with the applied potential and pH. The formation of the ultrathin catalyst was verified by electrochemical analyses that were so sensitive in detecting the signals from the monolayer adsorption of electroactive species or from the tiny mass change (ng/cm2 scale) of electrode associated with the electrochemical reaction when it is combined with a quartz crystal microbalance. Electrochemical analysis was also used to investigate the nature of the catalyst and the catalysis mechanism, which included the irreversible reaction steps and the diffusion of phosphate ions. Studying the catalyst nature is important not only for understanding the WOC mechanism but also for successfully combining the catalyst with a photoelectrode; decoration of heterogeneous catalysts on photoelectrodes should be controlled with respect to the surface coverage29−31 and optical properties.32,33 In this study, we also tested the catalytic properties of ultrathin “Ni-Pi” in conjunction with a BiVO4 photoelectrode and investigated the synergetic effect between the two materials.
Results and Discussion
The WOC by Ni2+ was examined under various pH conditions (Figure 1a). At neutral and high pHs, the evolution of an oxidation current on a fluorine-doped tin oxide (FTO) electrode in an aqueous phosphate buffer solution containing Ni2+ was observed using cyclic voltammetry (CV) and compared to that in the absence of Ni2+. The evolution of oxidation current with the addition of Ni2+ was negligible under acidic conditions (pH < 5) but started to appear at pH 5.69 and increased in magnitude with increasing pH. When the pH exceeded 10, which is outside the phosphate buffering range (pH 6–8), the oxidation current increased continuously without peaking. The emerging prewaves (black arrows, Figure 1a) at pH 11 indicate the formation of a surface species; this must be the heterogeneous catalyst, Ni(III)Ox(s) or Ni(IV)Ox(s), formed via Ni2+ oxidation. The same CV features were reported in the studies on the in situ formation of Co-Pi2 and Ni-Bi.34 After repeated CV cycling, the surface peak became more apparent and an appreciable amount of a brown-colored surface deposit was observed concurrently with the apparent electrocatalytic water oxidation (Figure S1a,b). X-ray diffractometry, energy-dispersive spectroscopy, and X-ray photoelectron spectroscopy measurements revealed that the deposit was amorphous and composed of Ni, O, and P (Figure S1b–e). Fe was not detected in any of these analyses (Figure S1c,e), indicating that the effect of Fe incorporation is not significant in this study. An attenuated total reflectance infrared spectrum of the deposit (please see the Supporting Information for the details of measurement) displayed a distinct Ni(OH)2-group Ni–O stretch peak (680 cm–1)35 and a phosphate P=O stretch peak (1020 cm–1) (Figure S1d).36 Moreover, the P=O stretch peak was slightly shifted compared to that of the ionic phosphate (Na2HPO4 or NaH2PO4, 1070–1050 cm–1),36 which indicates that the phosphate in the deposit has a covalent character.37 These results show that the deposit may be NiOx coordinated with a phosphate ion; this is referred to as “Ni-Pi” by analogy with Co-Pi. “Ni-Pi” and its efficient WOC in alkaline solution have recently been reported elsewhere.38 A distinct color change in the surface deposit was observed during potential scanning, with the deposit being transparent at low oxidation potentials and becoming brownish and darker at higher potentials (Figure S1a). This color change corresponds to a phase transformation from a transparent Ni(II) species (NiO, α-Ni(OH)2, β-Ni(OH)2) to a brownish Ni(III) species (Ni(OH)3, β-NiOOH, oxidation state: 2.7–3.0, porous) and probably up to the more dense γ-NiOOH (3.7).39,40 The color change indicates a change in the oxidation state of the deposited oxide, which is also intimately related to the Ni2+ electrocatalytic water oxidation mechanism.
Figure 1.

(a) Cyclic voltammograms (scan rate: 20 mV/s) of FTO (surface area: 0.0314 cm2) in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution of varying pHs containing 2 mM Ni2+. The electrolyte pH was adjusted by adding 0.1 M H2SO4 or 0.1 M NaOH. The black solid lines show cyclic voltammograms measured in Ni-free solutions at each pH. (b) Cyclic voltammogram of a GC substrate in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution containing Ni2+ (scan rate: 20 mV/s) (bottom) and the respective change in the current on a Au tip electrode (top) held at −0.5 V during potential scanning of the GC substrate. The small cathodic peak at −0.5 V in the inset is attributed to the reduction of oxygen absorbed on the electrode surface.
The Ni(II) oxide is thermodynamically unstable below pH 7.45 ([Ni2+] = 2 mM) due to the following reaction41
| 1 |
After repeated CV cycling, or continuous electrolysis, with Ni2+ under these conditions, no visible surface deposit or precipitate was found on the FTO surface (Figure S2a). The instability of Ni oxide at neutral pH was also confirmed upon observation of the Ni-Pi film, which had been previously formed at pH 11, fading away at pH 6.8 (Figure S2b). However, the evolution of an oxidation current at neutral pH is still due to catalytic water oxidation. The peak current (ipeak) with 2 mM Ni2+ was 2.4 × 10–4 A, which is about 40 times bigger than the theoretical peak current derived from the Nernstian one-electron transfer of 2 mM Ni2+ (Ni3+ + e– ↔ Ni2+) at 20 mV/s (ipeak, 2 mM = 6.15 × 10–6 A, where DNi2+ = 6.61 × 10–6 cm2/s42 and electrode area = 0.0314 cm2). A large oxidation current was also observed on other substrates such as GC and Au (Figure S2c,d). The WOC by Ni2+ was verified by electrochemical detection of the evolved oxygen using scanning electrochemical microscopy (SECM) (Figure 1b); SECM has been previously used to characterize electrocatalytic4 and photoelectrochemical23,43,44 water oxidation. Oxygen evolved on the GC substrate diffuses to a Au disk electrode, located 5 μm above the GC substrate, and arrives in a few milliseconds (t = d2/2D = 0.0125 s, where d is the distance between the tip and the substrate, which is 5 μm, and D is the diffusion coefficient of oxygen in water, which is 2 × 10–5 cm2/s45). The oxygen-reduction reaction (ORR) current at the Au UME tip was monitored by holding the tip at a constant potential of −0.5 V (vs Ag/AgCl), whereas the substrate potential was cycled from 0.4 to 1.8 V. In the black SG-TC CV, a reduction current was observed on the Au tip only when an oxidation current developed with the positive polarization, indicating that the oxidation current was attributed to oxygen evolution. Under the same condition, the magnitude of water-oxidation current at the substrate and corresponding oxygen-reduction current at the tip electrode increased significantly with the addition of Ni2+, which confirmed that Ni2+ catalyzed the water-oxidation reaction. More vigorous evolution of oxygen with the addition of Ni2+ was also found in gas chromatography measurements (Figure S3, please see the Supporting Information for the experimental details), and it would be a quantitative evidence for the WOC by Ni2+.
The CV response for Ni2+ WOC at neutral pH was investigated at various Ni2+ concentrations, scan rates (v), and phosphate concentrations (Figure 2). The oxidation current increased gradually with increasing Ni2+ concentration but did not vary significantly above 2 mM Ni2+ (Figure 2a). A light-green precipitate, presumably Ni3(PO4)2, was found when >4 mM Ni2+ was added to the phosphate solution. The peak current was almost linearly proportional to v1/2 (Figure 2b), which is a general characteristic of diffusion-controlled electrochemical reactions.46 Moreover, the peak potential was shifted positively with increasing v, which indicates that the kinetics of the relevant electrochemical reaction is somewhat irreversible.46 Interestingly, no catalytic current was observed in the absence of phosphate ions, and the current was proportional to the phosphate ion concentration (Figure 2c). The negligible catalytic function in the absence of buffer is similar to that seen for water oxidation using Co-Pi47 and Ni-Bi34 heterogeneous catalysts. Shinagawa et al.48 studied the effects of the buffer on the reaction kinetics and mass transfer of water splitting. Meanwhile, a tiny reduction peak was observed on the reverse scan at 1.2–1.0 V (vs Ag/AgCl); this peak correlated linearly with v (Figure 2d), which indicates that it corresponds to a surface reaction. The total charge obtained via integration of the surface peak was about 230 μC/cm2, which is equivalent to a few atomic layers of electroactive surface species. Contrary to the CV data at pH >10, the surface-peak magnitude did not increase during repeated scanning (Figure S2a) or after continuous oxidation, which indicates that the surface deposit did not grow further. This may be due to removal of the deposit by a reduction reaction or due to the instability of Ni(II)Ox under neutral conditions.
Figure 2.

Cyclic voltammograms of FTO (surface area: 0.0314 cm2) in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution of varying (a) Ni2+ concentrations, (b) phosphate concentrations, and (c) scan rates (ν). (d) The surface-related reduction peak in the cyclic voltammogram.
The change in the electrode surface on a molecular scale (e.g., adsorption, phase change, and underpotential deposition) can be evaluated by electrochemical quartz crystal microbalance (EQCM) measurements. Herein, we used an EQCM to observe the formation of a heterogeneous catalyst on Au-coated quartz (Figures 3 and S4). Unfortunately, Au-oxide formation and oxygen-bubble evolution were expected to perturb the oscillation frequency of quartz, and two distinct changes were observed during a potential scan in a nickel-free phosphate buffer solution (black line); the slight increase in mass at potentials >0.8 V (vs Ag/AgCl) is attributed to the formation of Au oxide, as reported elsewhere.49 The mass started to decrease during the reverse scan at potentials >0.6 V, which corresponds to Au-oxide reduction, and eventually the mass was less than that before potential cycling, which is attributed to the loss of Au. Bubble formation was found to have a negligible impact on the quartz frequency. A catalytic oxidation current accompanied the mass increase when Ni2+ was present, which indicates the formation of a thin heterogeneous catalyst layer. Interestingly, a mass decrease (the removal of a catalyst layer) was also observed during the reverse scan.
Figure 3.
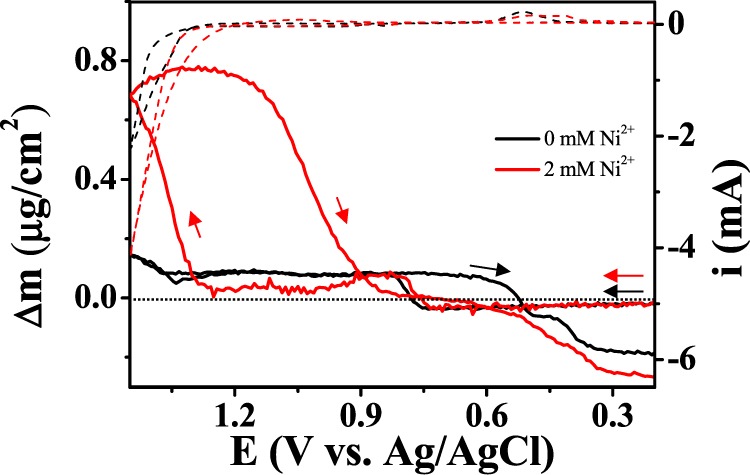
In situ change in the mass of a Au-coated quartz crystal during CV. The electrolyte was a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution containing 2 mM Ni2+.
In general, the first step for resolving heterogeneity of the catalysis would be to search for an electrode deposit or nanoparticle formation occurring during the water oxidation. However, this is difficult when the nanoparticles are very small (<10 nm) or the deposit is unstable under ambient conditions. Thus, distinguishing between homogeneous and heterogeneous catalytic reactions is sometimes difficult and has resulted in occasional scientific arguments.50−54 In addition, simply changing the pH caused a transition between homogeneous and heterogeneous catalyses by Co2+ in aqueous solution.55 Although we did not detect a deposit spectroscopically in this study, our electrochemical analyses clearly revealed the heterogeneity of Ni2+ WOC under neutral conditions.
The peak-shaped CV and removal of the surface deposit indicate that the Ni-Pi-based heterogeneous catalyst (e.g., Ni(OH)2 or NiOOH coordinated with the phosphate), responsible for the enhanced water oxidation, is unstable; this must be closely related to the solution pH. As mentioned above, Ni(II) oxides are thermodynamically unstable at neutral pH, and the higher-order oxides (Ni(III)Ox or Ni(IV)Ox) are stable only when anodically polarized, as evident from the Ni Pourbaix diagram (Figure S5). Accordingly, electrochemical syntheses of Ni-based heterogeneous catalysts have been reported in solutions with a pH ranging from 9.2 (Ni-Bi)34 to 14.15,17,56,57 The mass change in the electrode measured by the EQCM was about 1.0 × 10–6 g/cm2. Assuming that the species growing on the surface was NiOOH (the most common oxide form of Ni(III) oxide) deposited via the reaction41
| 2 |
and that it coordinates with a phosphate ion and water molecules (where its molecular weight ranges from 100 to 150 g/mol), the relevant charge would be 500–600 μC/cm2. This is in the same range as that of the charge obtained from the surface peak in the CV measurements (Figure 2d).
The correlation between the CV surface peak and the mass change measured using the EQCM indicates that the surface species can be titrated coulometrically. Variation of the amount of surface species during CV was monitored by coulometric titration (with a 0.9 V vs Ag/AgCl potential applied for 20 s immediately after stopping the potential scanning), and the total titration charge was then calculated. The amount of charge collected in the presence of Ni2+ was considerably more than that collected in the absence of Ni2+, as shown in Figure 4a. The titration charge is plotted against the potential at which the CV was stopped in Figure 4b. The charge collected in the absence of Ni2+ (black square points) was very small (<1 μC/cm2) even when there was a considerable oxidation current flow; the collected charge in this case is associated with an electrical double-layer charge and reducible species on, or near, the FTO surface (e.g., the •OH radical). Conversely, a noticeable amount of charge was collected in the presence of Ni2+, and the amount of surface species varied with the applied potential. At 1.3 V versus Ag/AgCl, the collected charge increased rapidly during the oxidative scan but started to decrease beyond 1.7 V. This trend resembles the CV features, which confirms that the surface species act as electrocatalysts for water oxidation and the change in the catalytic current originates from changes in the amount of surface species. Nevertheless, the decrease in the amount of heterogeneous catalyst during the forward scan is unusual as continuous growth was expected. Figure 4c shows the potential- and time-dependency of the amount of surface electrocatalyst. In this time span, the electrocatalyst grew slowly at low potentials (<1.5 V), whereas it grew rapidly at high potentials (>1.6 V), and eventually its amount reached a steady state. The steady-state values decreased with increasing applying potential, and at potentials over 1.5 V, those were less than 46.3 μC (1470 μC/cm2), which corresponds to a 3.41 nm thick layer of NiOOH (mass density ∼4 g/cm3).
Figure 4.

(a) Current–time response during coulometric titration (0.9 V vs Ag/AgCl for 20 s). (b) A plot of the amount of collected charge with respect to the potential at the end point of cyclic voltammetric scanning. (c) A plot of the amount of collected charge with respect to the time duration of the various potential steps prior to the titration step. (d) A plot of the amount of charge collected at saturation versus the applied potential prior to the titration step.
Interpreting the aforementioned unusual decrease, or saturation, in the amount of surface electrocatalyst requires understanding the Ni2+ WOC mechanism. This would also provide a better insight into the changes in the amount of surface species with respect to the formation conditions. Related studies have suggested that the catalysis of water oxidation by nickel oxides is based on the electrochemical oxidation of Ni2+ (NiO or Ni(OH)2) to higher oxidation states, Ni3+ or Ni4+ (β- or γ-NiOOH), that then oxidize water and form Ni2+ again.15,16,18,34,58 Electrocatalytic water oxidation by Ni2+ can be explained in a similar manner, though the Ni2+ involved in electrocatalytic water oxidation is assumed to be in aqueous solution rather than an oxide. Gerken et al.55 proposed the water oxidation catalyzed by aqueous Co2+ under low pH conditions. By analogy with their work, we propose the following reaction pathway for heterogeneous electrocatalytic water oxidation catalyzed by Ni2+ at neutral pH
| 3.a |
| 3.b |
| 3.c |
| 3.d |
| 3.e |
| 1a |
| 3.f |
| 3.g |
We used nickel oxides Ni(OH)3 and Ni(OH)4 in 3.b–3.e, respectively, from a thermodynamic point of view, though the actual nickel oxides (β- and γ-NiOOH) has a nonstoichiometry in elemental composition. We presume that Ni(OH)3 (s) is β-NiOOH, where the combination of eqs 3.a and 3.b is equivalent to eq 2. Equations 3.c and 3.d describe the transition from Ni(III) oxide to Ni(IV) oxide via a proton-coupled electron transfer (PCET) to balance the charge neutrality. γ-NiOOH is the most plausible form of Ni(IV) oxide, and it can be thermodynamically expressed as Ni(OH)4 or NiO2. It is known that the transformation from β-NiOOH to γ-NiOOH is associated with an irreversible structural change;56,59,60 thus, eqs 3.c and 3.d can be regarded as irreversible reactions. As the solution was buffered with phosphate, the proton generation in eqs 3.b and 3.d was coupled with eq 3.g.
Ni(IV) oxide is very active toward the water-oxidation reaction in comparison with Ni(III) oxide;62 thus, it would have a relatively short lifetime and we can assume that most of the charge collected during the coulometric titration is associated with Ni(OH)3 (that is, β-NiOOH). Subsequently, approaching a constant titration charge corresponds to reaching the steady-state activity (or coverage, θ) of Ni(OH)3 (dθNi(OH)3/dt = 0). Assuming that the reaction in eq 3.c is electrochemically irreversible, dθNi(OH)3/dt can be expressed as
| 4 |
where kc°, Ec°, and αc are the standard rate constant, standard reduction potential, and transfer coefficient, respectively, for the reaction in eq 3.c.
Therefore, at the steady state
| 5 |
As Ni3+ was not detected at any scan rate in our CV measurements (Figure S6) and it has not been reported elsewhere, we expect its concentration to be very low, such that the rate constant for the backward reaction of eq 3.b would be small compared to that of the forward reaction. Moreover, the H+ concentration would be very small at neural pH. Therefore, eq 5 becomes
| 6 |
Although the surface charge started collecting at potentials >1.3 V (vs Ag/AgCl), the steady-state conditions were reached at a relative large potential (1.5 V vs Ag/AgCl) in about 100 s (Figure 4c). Therefore, at steady state, the electrode is significantly polarized toward the anodic reaction of eq 3.a. As the Ni3+ concentration would remain low during water oxidation, the Ni3+ produced should be immediately consumed and, therefore, the requisite conditions would be
| 7 |
where ka° and αa are the standard rate constant and the transfer coefficient, respectively, for the reaction in eq 3.a. By substituting eq 7 into eq 6, we obtain
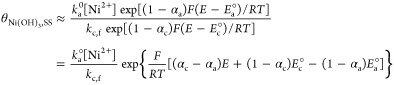 |
8 |
Equation 8 expresses the linear relationship between the logarithm of the collected charge at steady state and the applied potential, with a proportional factor of (αc – αa)F/RT. Figure 4d plots the steady-state amount of the catalyst (presumed to be θt=100) against the applied potential. The analyzed potential range was narrowed to exclude mass transfer effects. From eq 8, the negative slope of the plot of ln(θNi(OH)3,SS) versus applied potential over 1.5 V (vs Ag/AgCl) might indicate that αa is larger than αc, whereby the reaction in eq 3.c is more favorable as the oxidation reaction, though the estimated difference between αa and αc is as small as 0.1.
When the electrode is not anodically polarized (<1.4 V vs Ag/AgCl), the surface concentrations of Ni2+ and Ni3+ satisfy the Nernst relationship
| 9 |
where Ea° is the standard reduction potential of the reaction in eq 3.a. Combining eqs 6 and 9 gives
| 10 |
At low potentials (<1.4 V vs Ag/AgCl), the collected charge actually increased with increasing potential and varied more significantly compared to the charge collected at high potentials (Figure 4d). This is consistent with eq 10 where the proportional factor would be αcF/RT, though the collected charges were not steady-state values. This analysis indicates that the formation of Ni(III) oxide and its subsequent oxidation are electrochemically and chemically irreversible. Accordingly, the removal of Ni(III) oxide during the titration may not proceed via the back reactions in eqs 3.a and 3.b but via a different pathway consisting of electrochemical reduction to Ni(II) oxide (eq 3.e) followed by dissociation (eq 1).
Charge from the surface deposit was also collected in pH 5–7 electrolyte solutions at potentials where the oxidation current with the addition of Ni2+ evolved; thus, the surface deposit (catalyst layer) is related to the oxidation current (Figures 1a and 5). The collected charge and relevant catalytic current decreased (Figure 5a,b) with increasingly acidic electrolyte. Figure 5c maps the charge onto the potential-pH plane (Pourbaix diagram). The boundary slope of the charge-collection region is close to −177 mV/pH (3 × −59 mV/pH), which indicates that the surface-species formation involves the generation of three protons, such that the surface species would be regarded as Ni(OH)3 (or NiOOH) in eq 2. The boundary in Figure 5c is shifted positively from the theoretical boundary (E = 2.044–0.177 × pH V vs Ag/AgCl at 2 mM Ni2+); this might be due to the overpotential required to form a Ni(OH)3 film on FTO.
Figure 5.
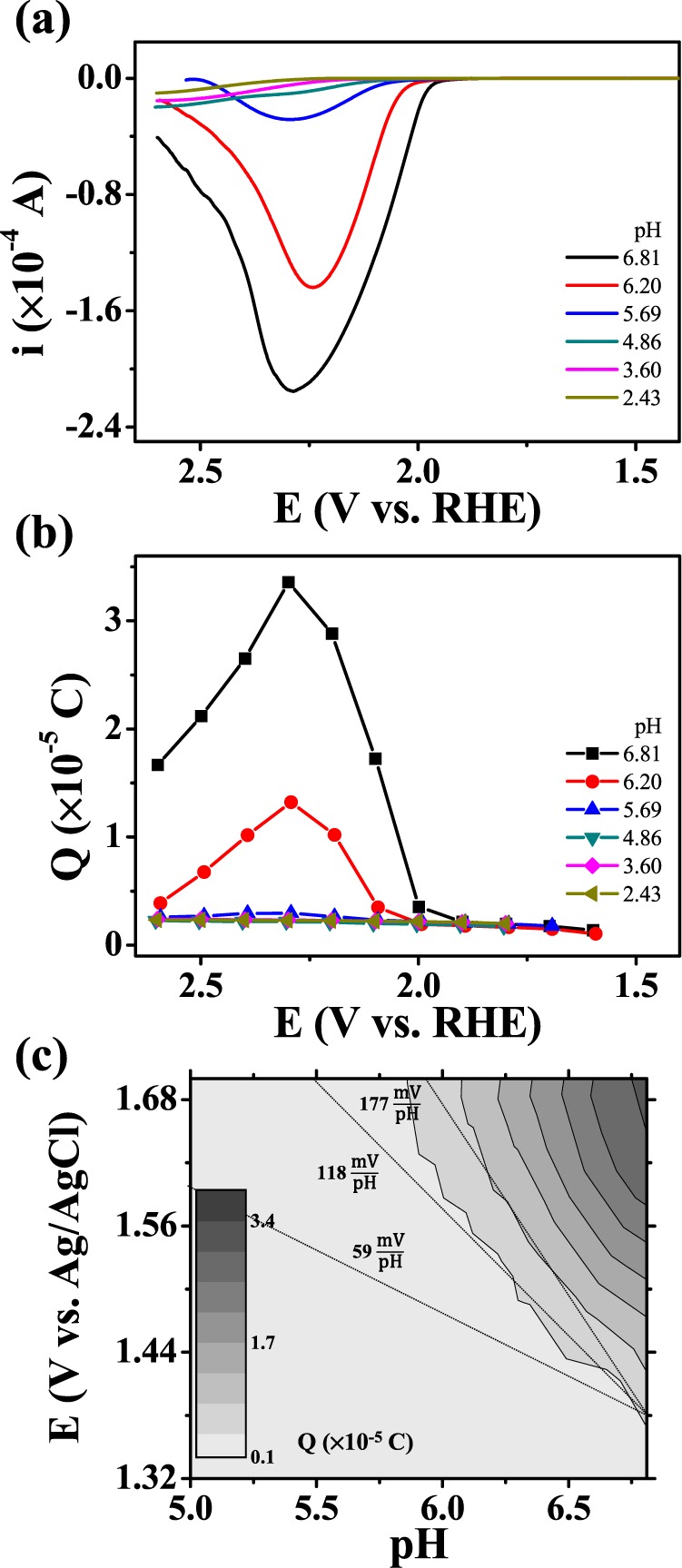
(a) Cyclic voltammograms (scan rate: 20 mV/s) of FTO (surface area: 0.0314 cm2) and (b) the relevant charge collected, in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution of varying pHs and containing 2 mM Ni2+. (c) The collected charge mapped on the potential-pH plane (Pourbaix diagram).
Proton generation in an unbuffered solution would decrease the local pH at the electrode, which creates an unfavorable environment for nickel oxide formation near the electrode. Indeed, the electrocatalytic effect of Ni2+ was not observed under unbuffered conditions (Figure 2b) and a buffering agent is essential for Co-Pi and Ni-Bi formation.2,34 The magnitude of the catalytic current as a function of the phosphate concentration also reflects the importance of the phosphate buffer in Ni2+-based WOC. Interestingly, Ni2+ did not display a noticeable catalytic effect in a pH 7 tris buffer solution (Figure S7). The coordination of phosphate ions with Ni-oxide films at high pH is similar to the coordination of cobalt oxides with phosphate and borate ions (Figure S1), which implies that the ability of the buffer ion to coordinate to the solid deposit is also important in determining the catalytic effect of the solid deposit; the phosphate ion is known to mediate PCET through the catalyst.61 As phosphate ions play an important role in the catalyst-layer formation, the decreasing amount of catalyst observed during potential cycling might be associated with a change in the phosphate ion concentration. According to the proposed mechanism, two dibasic phosphate ions (HPO42–, 0.1 M in the electrolyte) are consumed during oxidation of a single water molecule (eqs 3.a → 3.e, two-electron process), whereas Ni2+ is regenerated with a single turnover. It is noticeable that the catalytic current peak (2.4 × 10–4 A) observed during CV is of the same magnitude as that of the current associated with the one-electron reversible charge-transfer reaction of 0.1 M reactant (3.1 × 10–4 A, Figure S8a). In addition, the chronoamperogram of Ni2+-based WOC under anodic polarization is very similar to the response expected from 0.1 M of reactant based on the Cottrell equation (Figure S8b). These similarities indicate that the diffusion of dibasic phosphate ions affects Ni2+-based WOC. The decline in the catalytic current after reaching the CV peak may be due to the depletion of phosphate ions, which would induce a decrease in the local pH near the electrode, producing conditions that are unfavorable for catalyst formation. Considering the phosphate ion diffusion can also explain the catalytic-current dependency on the phosphate concentration (Figure 2b) and v1/2 (Figure 2c). Shinagawa et al.48 also emphasized the effect of buffering-agent diffusion on the water-splitting reaction. Digital simulations of electrocatalytic water oxidation catalyzed by Ni2+ would provide a more clear insight into the CV features, but surface-related heterogeneous electrochemical reactions are one of the most complicated problems to simulate. Nevertheless, as phosphate-ion diffusion is only partially involved in the electrocatalytic reaction and the surface deposit is an intermediate rather than the final product, it is plausible to consider the overall reaction as a diffusion-controlled ECEC route, where E and C represent electrochemical and chemical reactions, respectively. Simulations actually predicted emerging catalytic currents with various scan rates and the phosphate concentrations, and they are quite similar to the measured CVs (Figure S9).
We examined the interaction between Ni2+ catalysis and photoelectrodes using a BiVO4 n-type semiconductor, with the results presented in Figure 6. The photoelectrochemical OER increased upon addition of Ni2+ in a phosphate buffer solution (Figure 6a). A synergetic effect was also confirmed upon detection of enhanced oxygen evolution via SECM combined with a ring optical-fiber disk electrode,62 as shown in Figure 6b. The synergetic effect can be understood by analogy with the synergetic effect between n-type photoelectrodes and Co-Pi25,26 or Ni-Bi.63 We did not observe an increase in the photoresponse upon adding Ni2+ in the presence of a sulfite (SO32–) hole scavenger (Figure S10); this indicates that the synergetic effect originates from improving the water-oxidation reaction kinetics rather than modifying the physical properties of the semiconductor. Likewise, improvement of the incident photon conversion efficiency (IPCE) by Ni2+ was observed within the range of wavelengths over which BiVO4 was active (Figure S11).
Figure 6.
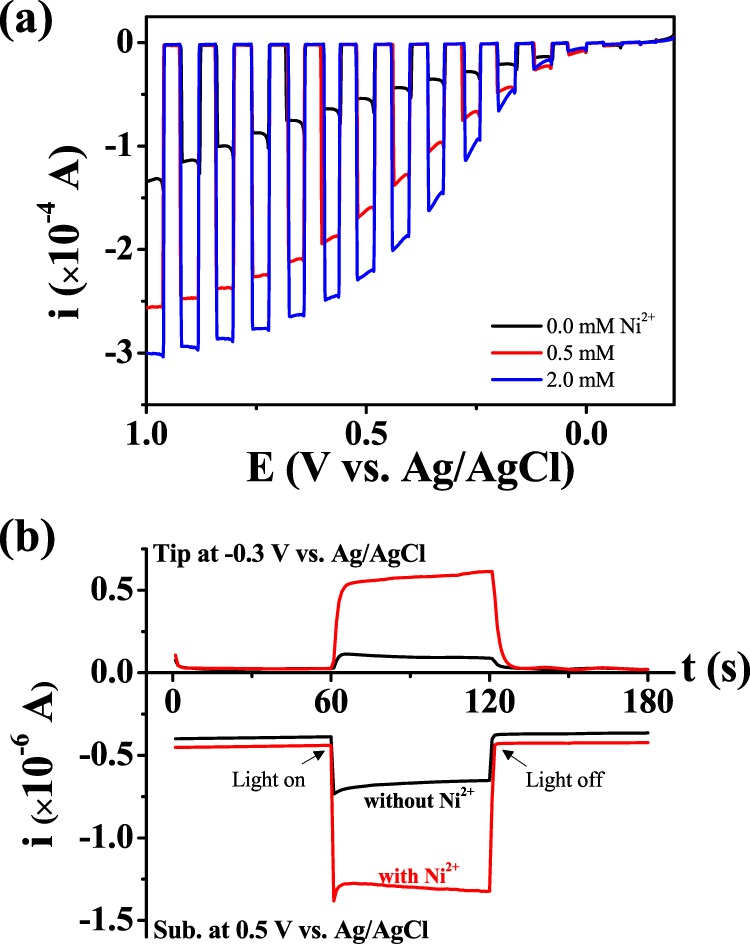
(a) Linear sweep voltammograms (scan rate: 20 mV/s) of BiVO4 (surface area: 0.27 cm2) in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution containing Ni2+, with chopped UV–visible light irradiation (xenon lamp, 100 mW/cm2). (b) SECM chronoamperograms, collected in the TC/SG mode, measuring photoelectrochemical water oxidation on a BiVO4 substrate in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution containing Ni2+ (bottom) and detection of the generated oxygen by a Au ring electrode (top). Measurements started in the dark, and UV–vis light irradiation started at 60 s and finished at 120 s.
In general, the performance of a heterogeneous catalyst is evaluated with respect to the onset potential (or overpotential), and the stability of the catalyst and the performance of Ni2+ in pH 7 phosphate buffer are not very comparable to those of Co2+ in this sense. When using a Ni2+ catalyst, the overpotential (η) of ∼0.8 V was required to achieve a current density of 1 mA/cm2 (Figure S12) compared to η = 0.4–0.5 V when using a Co2+ catalyst.2 Moreover, the turnover frequency for Ni2+ catalysis was roughly estimated to be <1 s–1 (at η > 0.9 V; please see the Supporting Information for detailed calculations), which is small compared to the recently reported turnover frequency of Co-Pi (>2 s–1 at η = 0.41 V).64,65 However, when the heterogeneous catalyst is coupled with a photoelectrode, the onset potential for the photoinduced reaction was mainly determined by the flat-band potential of the photoelectrode; the role of the catalyst was limited to minimization of the deteriorative potential shift originating from the surface recombination.25,46 To synergistically interact with a photoelectrode, the heterogeneous catalyst layer must be ultrathin and discontinuous, such that it does not affect the nature of the semiconductor–electrolyte interface.29,66,67 Recent studies have raised the issue of parasitic light absorption by the heterogeneous catalyst decorated on the photoelectrode and a subsequent loss in efficiency as the catalyst thickness increases.32,33Figure 7 presents a comparison between photocatalytic water oxidation by Ni2+ and Co2+ on BiVO4. The presence of Co2+ results in a higher photocurrent, compared to that in the presence of Ni2+, during the early stages of potential scanning. However, the Co2+-decorated BiVO4 displays a significant diminishing transient as potential scanning proceeds, with no increase in the water oxidation current (Figure 7a). Moreover, as potential scanning is repeated, the photocurrent becomes lower and is eventually lower than that of bare BiVO4 (Figure 7c). This deterioration in the photocatalytic activity of Co-Pi on BiVO4 has been reported previously44,68 and was attributed to thickening of the Co-Pi layer, causing an increase in the surface recombination of the photogenerated electron–hole pair.25 Abdi et al. reported that the synergetic effect was maximized when the layer of Co-Pi on BiVO4 was 30 nm thick,68 and many studies have stated that optimizing the thickness of the Co-Pi layer on the photoanode is necessary.25,26,44,68 Conversely, the photo-oxidation enhancement by Ni2+ was consistent and repeatable (Figure 7a,b). As the thickness of the Ni-based catalyst layer is naturally limited to <4 nm (calculated from the collected charge), it enhances the water oxidation without any negative impact on the photoelectrode. With the exception of a slight underperformance at low potentials, the catalysis by Ni2+ on BiVO4 was still comparable to that of Co-Pi on BiVO4 even after optimizing the Co-Pi layer (Figure 7d). This indicates that Ni2+ enhances the photocatalytic properties of semiconductor electrodes without any pretreatment or optimization and, moreover, the catalyst would exhibit a stable performance during water oxidation because of its continuous regeneration.
Figure 7.

(a) Linear sweep voltammograms (scan rate: 20 mV/s) of BiVO4 (surface area: 0.27 cm2) in a pH 6.8 phosphate buffer (0.1 M HPO42–/0.1 M H2PO4–) solution containing Ni2+ and Co2+, with chopped UV–visible light irradiation (xenon lamp, 100 mW/cm2). (b–c) Changes in the photocurrent responses with repeating a potential sweep. (d) Linear sweep voltammograms of BiVO4 with the highest photocurrent achieved by Ni2+ or Co2+. In the case of Ni2+, the photocurrent was measured in a phosphate buffer containing 2 mM Ni2+, and for Co2+, BiVO4 was immersed in the phosphate-buffered solution containing 0.05 mM Co2+ with the potential sweeping from 0.2 to 1.0 V (vs Ag/AgCl) (four times) under UV–vis light illumination, and then, the pretreated BiVO4 was tested in a Co2+-free phosphate solution.
Conclusions
The catalysis of water oxidation by Ni2+ under neutral conditions was identified as heterogeneous using various electrochemical analyses. SECM was used to probe the catalyzed oxygen evolution, and CV and EQCM measurements revealed that the ultrathin (<4 nm) catalyst layer was deposited only when the electrode was anodically polarized, with no deposition observed under the nonpolarized conditions. The amount of the surface catalyst present during water oxidation was monitored by coulometric titration, and analysis of its dependence on the potential and pH indicated the formation of Ni(III) oxide and the irreversible kinetics of the catalyst formation and the following water-oxidation reaction. The diffusion-limited characteristics of water oxidation catalyzed by Ni2+ are associated with the phosphate-ion mass transport. The electrocatalytic effect of Ni2+ on water oxidation was also observed on a BiVO4 photoanode, and a synergetic effect was observed.
Experimental Section
Materials
Ni(NO3)2·6H2O (>99%), Co(NO3)2·6H2O (>99%), Na2HPO4 (>98%, ACS reagent), and NaH2PO4 (>98%, ACS reagent) were purchased from Acros Organics (Fair Lawn, NJ). Na2SO4 (99.3%), Na2SO3 (99.6%), and ethylene glycol were purchased from Fisher Scientific (Pittsburgh, PA). Bi(NO3)3·5H2O (99.999%) and (NH4)10H2(W2O7)6·xH2O (99.99%) were obtained from Strem Chemicals (Newburyport, MA). VCl3 (99%, Alfa-Aesar, Ward Hill, MA) was used as received. FTO (<14 Ω, TEC 15; Pilkington, Toledo, OH) was used as the working electrode in all electrochemical measurements and as the substrate for photoelectrode preparation. The solvent in all electrochemical experiments was deionized Milli-Q water (18.2 MΩ cm), and its impurity level was double-checked via conductivity measurements (Orion Star A215). As Fe impurities have a profound effect on the catalytic activity of Ni oxides,69−71 its presence in the electrochemical cell was thoroughly controlled and the Fe concentration level was estimated to be <280 ppb.
Electrochemical Analysis
A CH Instruments Model 631E Electrochemical Analyzer was used for electrochemical analysis. For the electrochemical measurements, a FTO working electrode was mounted on Teflon cell with O-ring, which defined the surface area of 0.0314 cm2. A platinum wire (1 mm diameter) counter electrode and a Ag/AgCl reference electrode in a saturated KCl solution were used to complete the three-electrode configuration. A Au disk (2 mm diameter) and a glassy carbon disk (GC, 4 mm diameter) were also used as working electrodes. The electrolyte was composed of 0.2 M sodium phosphate (HPO42–/H2PO4– = 1:1, pH 6.8), 0.1 M sodium sulfate, and varying concentrations of nickel nitrate. Prior to the electrochemical measurement, the electrolyte was purged with N2 flow for at least 15 min to remove the oxygen dissolved in the electrolyte. An EQCM (Q-Sense Explorer) was used to measure the change in electrode mass. A Au-coated quartz crystal was used as the working electrode (exposed area = 1 cm2), and its mass change (Δm in g/cm2) was estimated from the change in frequency (Δf in Hz) using the following equation
| 11 |
where Cf is the sensitivity factor for the crystal, which is 56.6 Hz/μg cm2 for a 5 MHz AT-cut quartz crystal at room temperature.
Preparation and Characterization of a W-Doped BiVO4 Photoelectrode
BiVO4 films were prepared using a drop-casting method as reported elsewhere.43 Briefly, Bi(NO3)3 and VCl3 were dissolved in ethylene glycol to prepare the precursor solution and (NH4)10H2(W2O7)6·xH2O was added to increase the photoactivity of the synthesized BiVO4 photoelectrode. The concentrations of Bi, W, and V in the precursor solution were 4.5, 0.5, and 5.0 mM, respectively.43 The precursor solution (200 μL) was spread over a 15 mm × 20 mm FTO substrate followed by thermal annealing at 550 °C for 3 h after heating up in increments of 1 °C/min, which produced a ∼200 nm thick monoclinic W-doped BiVO4 film on FTO. The photoactivity of the deposited BiVO4 was measured in a 0.2 M sodium phosphate buffer solution (pH 6.8) containing Ni2+ under UV–vis light irradiation (Xe lamp, 100 mW/cm2). All measurements were carried out in a borosilicate glass cell, which has three compartments for the photoelectrode, Pt counter electrode, and Ag/AgCl reference electrode that are separated with a glass frit.
Detection of Oxygen Evolution via SECM
Oxygen evolved from water oxidation at the working electrode was detected by a tip electrode, positioned a few micrometers above the electrode, using SECM operated in the substrate-generation–tip-collection (SG-TC) mode.4,72 Two kinds of tip electrodes were used in this experiment. A Au ultramicroelectrode (UME, dia. 10 μm) was employed to characterize the electrocatalytic water oxidation. To prepare the Au UME, a 10 μm dia. gold wire (Goodfellow, 99.99% purity) was sealed in a borosilicate glass capillary (inner dia. 0.75 mm/outer dia. 1 mm) under vacuum. One end was then polished with 600-mesh sandpaper (Buehler, Lake Bluff, IL) until the metal disk was exposed, and it was then smoothed with 1200-mesh abrasive SiC paper and alumina suspensions in water down to 0.3 μm (Buehler). A Au-ring optical-fiber disk electrode was used to characterize the photoelectrochemical water oxidation. It was prepared from a commercial Au-coated optical fiber (Fiberguide Industries, Inc., Stirling, NJ) with a glass seal. The tip position in SECM measurements was controlled by an xyz-stepper motor (T-LA28A; Zaber Technologies Inc., Vancouver, Canada) on a SECM stage. The tip was gently touched to the substrate to make initial contact and then retracted by 5 μm (the distance between the tip and the substrate) using the stepper motor. Oxygen detection was performed in a 0.2 M sodium phosphate buffer solution with the tip held at −0.5 V (vs Ag/AgCl) to induce the ORR, whereas the substrate potential was scanned or held constant to induce the OER. The tip potential was sufficiently negative to ensure the collection of diffusive oxygen without any secondary reactions occurring, for example, HER. The photoelectrode was illuminated with UV–vis light via a tip-embedded optical fiber from a halogen lamp. The sizes of the Au-ring optical-fiber components were as follows: inner diameter of optical fiber, 200 μm; inner diameter of Au ring, 240 μm; outer diameter of Au ring, 275 μm; and the diameter of the whole optical fiber including the glass insulator, 600 μm.
Acknowledgments
The authors would like to thank Prof. Allen J. Bard (UT-Aution) for helpful discussions and experimental support. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2015R1C1A1A02037791).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00448.
Additional experimental data, figures, and digital CV simulation results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bard A. J.; Fox M. A. Artificial Photosynthesis: Solar Splitting of Water to Hydrogen and Oxygen. Acc. Chem. Res. 1995, 28, 141–145. 10.1021/ar00051a007. [DOI] [Google Scholar]
- Kanan M. W.; Nocera D. G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- Walter M. G.; Warren E. L.; McKone J. R.; Boettcher S. W.; Mi Q.; Santori E. A.; Lewis N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Mario A. M.; Alpuche-Aviles A.; López J. R.; Rondinini S.; Bard A. J. Screening of Oxygen Evolution Electrocatalysts by Scanning Electrochemical Microscopy Using a Tip Shielding Approach. Anal. Chem. 2008, 80, 4055–4064. 10.1021/ac8001287. [DOI] [PubMed] [Google Scholar]
- Du P.; Eisenberg R. Catalysts Made of Earth-Abundant Elements (Co, Ni, Fe) for Water Splitting: Recent Progress and Future Challenges. Energy Environ. Sci. 2012, 5, 6012–6021. 10.1039/c2ee03250c. [DOI] [Google Scholar]
- Kärkäs M. D.; Verho O.; Johnston E. V.; Åkermark B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. 10.1021/cr400572f. [DOI] [PubMed] [Google Scholar]
- Asraf M. A.; Younus H. A.; Yusubov M.; Verpoort F. Earth-Abundant Metal Complexes as Catalysts for Water Oxidation; is it Homogeneous or Heterogeneous?. Catal. Sci. Technol. 2015, 5, 4901–4925. 10.1039/C5CY01251A. [DOI] [Google Scholar]
- Sun Y.; Gao S.; Lei F.; Liu J.; Liang L.; Xie Y. Atomically-Thin Non-Layered Cobalt Oxide Porous Sheets for Highly Efficient Oxygen-Evolving Electrocatalysts. Chem. Sci. 2014, 5, 3976–3982. 10.1039/C4SC00565A. [DOI] [Google Scholar]
- Rosser T. E.; Gross M. A.; Lai Y.-H.; Reisner E. Precious-Metal Free Photoelectrochemical Water Splitting with Immobilized Molecular Ni and Fe Redox Catalysts. Chem. Sci. 2016, 7, 4024–4035. 10.1039/C5SC04863J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran J.; Zhang J.; Yu J.; Jaroniec M.; Qiao S. Z. Earth-Abundant Cocatalysts for Semiconductor-Based Photocatalytic Water Splitting. Chem. Soc. Rev. 2014, 43, 7787. 10.1039/C3CS60425J. [DOI] [PubMed] [Google Scholar]
- Pijpersa J. J. H.; Winklerc M. T.; Surendranatha Y.; Buonassisic T.; Nocera D. G. Light-Induced Water Oxidation at Silicon Electrodes Functionalized with a Cobalt Oxygen-Evolving Catalyst. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10056–10061. 10.1073/pnas.1106545108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguzzi A.; Fan F.-R. F.; Vertova A.; Rondinini S.; Bard A. J. Dynamic potential–pH Diagrams Application to Electrocatalysts for Water Oxidation. Chem. Sci. 2012, 3, 217–229. 10.1039/C1SC00516B. [DOI] [Google Scholar]
- Seabold J. S.; Choi K.-S. Effect of a Cobalt-Based Oxygen Evolution Catalyst on the Stability and the Selectivity of Photo-Oxidation Reactions of a WO3 Photoanode. Chem. Mater. 2011, 23, 1105–1112. 10.1021/cm1019469. [DOI] [Google Scholar]
- Abdi F. F.; Han L.; Smets A. H. M.; Zeman M.; Dam B.; van de Krol R. Efficient Solar Water Splitting by Enhanced Charge Separation in a Bismuth Vanadate-Silicon Tandem Photoelectrode. Nat. Commun. 2013, 2195 10.1038/ncomms3195. [DOI] [PubMed] [Google Scholar]
- Lyons M. E. G.; Brandon M. P. A Comparative Study of the Oxygen Evolution Reaction on Oxidised Nickel, Cobalt and Iron Electrodes in Base. J. Electroanal. Chem. 2010, 641, 119–130. 10.1016/j.jelechem.2009.11.024. [DOI] [Google Scholar]
- Trotochaud L.; Ranney J. K.; Williams K. N.; Boettcher S. W. Solution-Cast Metal Oxide Thin Film Electrocatalysts for Oxygen Evolution. J. Am. Chem. Soc. 2012, 134, 17253–17261. 10.1021/ja307507a. [DOI] [PubMed] [Google Scholar]
- Subbaraman R.; Tripkovic D.; Chang K.-C.; Strmcnik D.; Paulikas A. P.; Hirunsit P.; Chan M.; Greeley J.; Stamenkovic V.; Markovic N. M. Trends in Activity for the Water Electrolyser Reactions on 3d M(Ni,Co,Fe,Mn) Hydr(oxy)oxide Catalysts. Nat. Mater. 2012, 11, 550–557. 10.1038/nmat3313. [DOI] [PubMed] [Google Scholar]
- Louie M. W.; Bell A. T. An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen. J. Am. Chem. Soc. 2013, 135, 12329–12337. 10.1021/ja405351s. [DOI] [PubMed] [Google Scholar]
- Kanan M. W.; Surendranath Y.; Nocera D. G. Cobalt–Phosphate Oxygen-Evolving Compound. Chem. Soc. Rev. 2009, 38, 109–114. 10.1039/B802885K. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Hamel J. A.; Sung K.; Jarvi T. D.; Esswein A. J.; Pijpers J. J. H.; Nocera D. G. Wireless Solar Water Splitting Using Silicon-Based Semiconductors and Earth-Abundant Catalysts. Science 2011, 334, 645–648. 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- Zhong D. K.; Sun J.; Inumaru H.; Gamelin D. R. Solar Water Oxidation by Composite Catalyst/α-Fe2O3 Photoanodes. J. Am. Chem. Soc. 2009, 131, 6086–6087. 10.1021/ja9016478. [DOI] [PubMed] [Google Scholar]
- Zhong D. K.; Cornuz M.; Sivula K.; Grätzel M.; Gamelin D. R. Photo-Assisted Electrodeposition of Cobalt–Phosphate (Co-Pi) Catalyst on Hematite Photoanodes for Solar Water Oxidation. Energy Environ. Sci. 2011, 4, 1759–1764. 10.1039/c1ee01034d. [DOI] [Google Scholar]
- Cong Y.; Park H. S.; Wang S.; Dang H. X.; Fan F.-R. F.; Mullins C. B.; Bard A. J. Synthesis of Ta3N5 Nanotube Arrays Modified with Electrocatalysts for Photoelectrochemical Water Oxidation. J. Phys. Chem. C 2012, 116, 14541–14550. 10.1021/jp304340a. [DOI] [Google Scholar]
- Wang Z.; Qi Y.; Ding C.; Fan D.; Liu G.; Zhao Y.; Li C. Insight into the Charge Transfer in Particulate Ta3N5 photoanode with High Photoelectrochemical Performance. Chem. Sci. 2016, 7, 4391–4399. 10.1039/C6SC00245E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong D. K.; Choi S.; Gamelin D. R. Near-Complete Suppression of Surface Recombination in Solar Photoelectrolysis by “Co-Pi” Catalyst-Modified W:BiVO4. J. Am. Chem. Soc. 2011, 133, 18370–18377. 10.1021/ja207348x. [DOI] [PubMed] [Google Scholar]
- Pilli S. K.; Furtak T. E.; Brown L. D.; Deutsch T. G.; Turner J. A.; Herring A. M. Cobalt-Phosphate (Co-Pi) Catalyst Modified Mo-doped BiVO4 Photoelectrodes for Solar Water Oxidation. Energy Environ. Sci. 2011, 4, 5028–5034. 10.1039/c1ee02444b. [DOI] [Google Scholar]
- Zhang M.; Zhang M.-T.; Hou C.; Ke Z.-F.; Lu T.-B. Homogeneous Electrocatalytic Water Oxidation at Neutral pH by a Robust Macrocyclic Nickel(II) Complex. Angew. Chem., Int. Ed. 2014, 53, 13042–13048. 10.1002/anie.201406983. [DOI] [PubMed] [Google Scholar]
- Han Y.; Wu Y.; Lai W.; Cao R. Electrocatalytic Water Oxidation by a Water-Soluble Nickel Porphyrin Complex at Neutral pH with Low Overpotential. Inorg. Chem. 2015, 54, 5604–5613. 10.1021/acs.inorgchem.5b00924. [DOI] [PubMed] [Google Scholar]
- Nakato Y.; Ueda K.; Yano H.; Tsubomura H. Effect of Microscopic Discontinuity of Metal Overlayers on the Photovoltages in Metal-Coated Semiconductor-Liquid Junction Photoelectrochemical Cells for Efficient Solar Energy Conversion. J. Phys. Chem. 1988, 92, 2316–2324. 10.1021/j100319a043. [DOI] [Google Scholar]
- Ping Y.; Goddard W. A. III; Galli G. A. Energetics and Solvation Effects at the Photoanode/Catalyst Interface: Ohmic Contact versus Schottky Barrier. J. Am. Chem. Soc. 2015, 137, 5264–5267. 10.1021/jacs.5b00798. [DOI] [PubMed] [Google Scholar]
- Nakibli Y.; Kalisman P.; Amirav L. Less Is More: The Case of Metal Cocatalysts. J. Phys. Chem. Lett. 2015, 6, 2265–2268. 10.1021/acs.jpclett.5b00872. [DOI] [PubMed] [Google Scholar]
- Trotochaud L.; Mills T. J.; Boettcher S. W. An Optocatalytic Model for Semiconductor-Catalyst Water-Splitting Photoelectrodes Based on In Situ Optical Measurements on Operational Catalysts. J. Phys. Chem. Lett. 2013, 4, 931–935. 10.1021/jz4002604. [DOI] [PubMed] [Google Scholar]
- McKone J. R.; Lewis N. S.; Gray H. B. Will Solar-Driven Water-Splitting Devices See the Light of Day?. Chem. Mater. 2014, 26, 407–414. 10.1021/cm4021518. [DOI] [Google Scholar]
- Dincă M.; Surendranath Y.; Nocera D. G. Nickel-Borate Oxygen-Evolving Catalyst that Functions under Benign Conditions. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10337–10341. 10.1073/pnas.1001859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazri G.; Corrigan D. A.; Maheswari S. P. Angle-Resolved Infrared Spectroelectrochemistry. 1. An In situ Study of Thin-Film Nickel Oxide Electrodes. Langmuir 1989, 5, 17–22. 10.1021/la00085a004. [DOI] [Google Scholar]
- Miller F. A.; Wilkins C. A. Infrared Spectra and Characteristic Frequencies of Inorganic Ions. Anal. Chem. 1952, 24, 1253–1294. 10.1021/ac60068a007. [DOI] [Google Scholar]
- Skoog D. A.; Holler F. J.; Nieman T. A.. Principles of Instrumental Analysis, 5th ed.; Saunders College Pub.: Philadelphia, 1998; p 412. [Google Scholar]
- Li Y.; Zhao C. Iron-Doped Nickel Phosphate as Synergistic Electrocatalyst for Water Oxidation. Chem. Mater. 2016, 28, 5659–5666. 10.1021/acs.chemmater.6b01522. [DOI] [Google Scholar]
- Lampert C. M. In Large Area Chromogenics: Materials and Devices for Transmittance Control; Lampert C. M., Granqvist C. G., Eds.; SPIE Optical Engineering Press: Bellingham, WA, 1989; Vol. IS 4, p 414. [Google Scholar]
- Sialvi M. Z.; Mortimer R. J.; Wilcox G. D.; Teridi A. M.; Varley T. S.; Wijayantha K. G. U.; Kirk C. A. Electrochromic and Colorimetric Properties of Nickel(II) Oxide Thin Films Prepared by Aerosol-Assisted Chemical Vapor Deposition. ACS Appl. Mater. Interfaces 2013, 5, 5675–5682. 10.1021/am401025v. [DOI] [PubMed] [Google Scholar]
- Arvia A. J.; Posadas D. In Standard Potentials in Aqueous Solution; Bard A. J., Parsons R., Jordan J., Eds.; CRC Press: New York, 1985; Chapter 12, p 329. [Google Scholar]
- Vanýsek P.Ionic Conductivity and Diffusion at Infinite Dilution. In CRC Handbook of Chemistry and Physics; Lide D. R., Ed.; CRC Press: Boca Raton, FL, 2005; pp 5–93. [Google Scholar]
- Ye H.; Lee J.; Jang J. S.; Bard A. J. Rapid Screening of BiVO4-Based Photocatalysts by Scanning Electrochemical Microscopy (SECM) and Studies of Their Photoelectrochemical Properties. J. Phys. Chem. C 2010, 114, 13322–13328. 10.1021/jp104343b. [DOI] [Google Scholar]
- Ye H.; Park H. S.; Bard A. J. Screening of Electrocatalysts for Photoelectrochemical Water Oxidation on W-Doped BiVO4 Photocatalysts by Scanning Electrochemical Microscopy. J. Phys. Chem. C 2011, 115, 12464–12470. 10.1021/jp200852c. [DOI] [Google Scholar]
- Ferrell R. T.; Himmelblau D. M. Diffusion Coefficients of Nitrogen and Oxygen in water. J. Chem. Eng. Data 1967, 12, 111–115. 10.1021/je60032a036. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Method: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, 2001; p 231. [Google Scholar]
- Surendranath Y.; Dincă M.; Nocera D. G. Electrolyte-Dependent Electrosynthesis and Activity of Cobalt-Based Water Oxidation Catalysts. J. Am. Chem. Soc. 2009, 131, 2615–2620. 10.1021/ja807769r. [DOI] [PubMed] [Google Scholar]
- Shinagawa T.; Takanabe K. Electrocatalytic Hydrogen Evolution under Densely Buffered Neutral pH Conditions. J. Phys. Chem. C 2015, 119, 20453–20458. 10.1021/acs.jpcc.5b05295. [DOI] [Google Scholar]
- Hoogvliet J. C.; van Bennekom W. P. Gold Thin-Film Electrodes: an EQCM Study of the Influence of Chromium and Titanium Adhesion Layers on the Response. Electrochim. Acta 2001, 47, 599–611. 10.1016/S0013-4686(01)00793-9. [DOI] [Google Scholar]
- Widegren J. A.; Finke R. G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A: Chem. 2003, 198, 317–341. 10.1016/S1381-1169(02)00728-8. [DOI] [Google Scholar]
- Schley N. D.; Blakemore J. D.; Subbaiyan N. K.; Incarvito C. D.; D’Souza F.; Crabtree R. H.; Brudvig G. W. Distinguishing Homogeneous from Heterogeneous Catalysis in Electrode-Driven Water Oxidation with Molecular Iridium Complexes. J. Am. Chem. Soc. 2011, 133, 10473–10481. 10.1021/ja2004522. [DOI] [PubMed] [Google Scholar]
- Crabtree R. H. Resolving Heterogeneity Problems and Impurity Artifacts in Operationally Homogeneous Transition Metal Catalysts. Chem. Rev. 2012, 112, 1536–1554. 10.1021/cr2002905. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Hong D. Homogeneous versus Heterogeneous Catalysts in Water Oxidation. Eur. J. Inorg. Chem. 2014, 2014, 645–659. 10.1002/ejic.201300684. [DOI] [Google Scholar]
- Stracke J. J.; Finke R. G. Distinguishing Homogeneous from Heterogeneous Water Oxidation Catalysis when Beginning with Polyoxometalates. ACS Catal. 2014, 4, 909–933. 10.1021/cs4011716. [DOI] [Google Scholar]
- Gerken J. B.; McAlpin J. G.; Chen J. Y. C.; Rigsby M. L.; Casey W. H.; Britt R. D.; Stahl S. S. Electrochemical Water Oxidation with Cobalt-Based Electrocatalysts from pH 0–14: The Thermodynamic Basis for Catalyst Structure, Stability, and Activity. J. Am. Chem. Soc. 2011, 133, 14431–14442. 10.1021/ja205647m. [DOI] [PubMed] [Google Scholar]
- Kim M.-S.; Kim K.-B. A Study on the Phase Transformation of Electrochemically Precipitated Nickel Hydroxides Using an Electrochemical Quartz Crystal Microbalance. J. Electrochem. Soc. 1998, 145, 507–511. 10.1149/1.1838294. [DOI] [Google Scholar]
- Corrigan D. A.; Bendert R. M. Effect of Coprecipitated Metal Ions on the Electrochemistry of Nickel Hydroxide Thin Films: Cyclic Voltammetry in 1 M KOH. J. Electrochem. Soc. 1989, 136, 723–728. 10.1149/1.2096717. [DOI] [Google Scholar]
- Bediako D. K.; Lassalle-Kaiser B.; Surendranath Y.; Yano J.; Yachandra V. K.; Nocera D. G. Structure-Activity Correlations in a Nickel-Borate Oxygen Evolution Catalyst. J. Am. Chem. Soc. 2012, 134, 6801–6809. 10.1021/ja301018q. [DOI] [PubMed] [Google Scholar]
- Kamath P. V.; Dixit M.; Indira L.; Shukla A. K.; Kumar Y. G.; Munichandraiah N. Stabilized α-Ni(OH)2 as Electrode Material for Alkaline Secondary Cells. J. Electrochem. Soc. 1994, 141, 2956–2959. 10.1149/1.2059264. [DOI] [Google Scholar]
- Oliva P.; Leonardi J.; Laurent J. F.; Delmas C.; Braconnier J. J.; Figlarz M.; Fivet F.; de Guibert D. Review of the Structure and the Electrochemistry of Nickel Hydroxides and Oxy-Hydroxides. J. Power Sources 1982, 8, 229–255. 10.1016/0378-7753(82)80057-8. [DOI] [Google Scholar]
- Bediako D. K.; Costentin C.; Jones E. C.; Nocera D. G.; Savéant J. −M. Proton-Electron Transport and Transfer in Electrocatalytic Films. Application to a Cobalt-Based O2-Evolution Catalyst. J. Am. Chem. Soc. 2013, 135, 10492–10502. 10.1021/ja403656w. [DOI] [PubMed] [Google Scholar]
- Cho S. K.; Park H. S.; Lee H. C.; Nam K. M.; Bard A. J. Metal Doping of BiVO4 by Composite Electrodeposition with Improved Photoelectrochemical Water Oxidation. J. Phys. Chem. C 2013, 117, 23048–23056. 10.1021/jp408619u. [DOI] [Google Scholar]
- Choi S. K.; Choi W.; Park H. Solar Water Oxidation Using Nickel-Borate Coupled BiVO4 Photoelectrodes. Phys. Chem. Chem. Phys. 2013, 15, 6499–6507. 10.1039/c3cp00073g. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Respinis M.; Frei H. Time-Resolved Observations of Water Oxidation Intermediates on a Cobalt Oxide Nanoparticle Catalyst. Nat. Chem. 2014, 6, 362–367. 10.1038/nchem.1874. [DOI] [PubMed] [Google Scholar]
- Ahn H. S.; Bard A. J. Surface Interrogation of CoPi Water Oxidation Catalyst by Scanning Electrochemical Microscopy. J. Am. Chem. Soc. 2015, 137, 612–615. 10.1021/ja511740h. [DOI] [PubMed] [Google Scholar]
- Riha S. C.; Klahr B. M.; Tyo E. C.; Seifert S.; Vajda S.; Pellin M. J.; Hamann T. W.; Martinson A. B. F. Atomic Layer Deposition of a Submonolayer Catalyst for the Enhanced Photoelectrochemical Performance of Water Oxidation with Hematite. ACS Nano 2013, 7, 2396–2405. 10.1021/nn305639z. [DOI] [PubMed] [Google Scholar]
- Lichterman M. F.; Shaner M. R.; Handler S. G.; Brunschwig B. S.; Gray H. B.; Lewis N. S.; Spurgeon J. M. Enhanced Stability and Activity for Water Oxidation in Alkaline Media with Bismuth Vanadate Photoelectrodes Modified with a Cobalt Oxide Catalytic Layer Produced by Atomic Layer Deposition. J. Phys. Chem. Lett. 2013, 4, 4188–4191. 10.1021/jz4022415. [DOI] [Google Scholar]
- Abdi F. F.; van de Krol R. Nature and Light Dependence of Bulk Recombination in Co-Pi-Catalyzed BiVO4 Photoanodes. J. Phys. Chem. C 2012, 116, 9398–9404. 10.1021/jp3007552. [DOI] [Google Scholar]
- Trotochaud L.; Young S. L.; Ranney J. K.; Boettcher S. W. Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. 10.1021/ja502379c. [DOI] [PubMed] [Google Scholar]
- Klaus S.; Cai Y.; Louie M. W.; Trotochaud L.; Bell A. T. Effects of Fe Electrolyte Impurities on Ni(OH)2/NiOOH Structure and Oxygen Evolution Activity. J. Phys. Chem. C 2015, 119, 7243–7254. 10.1021/acs.jpcc.5b00105. [DOI] [Google Scholar]
- Michael J. D.; Demeter E. L.; Illes S. M.; Fan Q.; Boes J. R.; Kitchin J. R. Alkaline Electrolyte and Fe Impurity Effects on the Performance and Active-Phase Structure of NiOOH Thin Films for OER Catalysis Applications. J. Phys. Chem. C 2015, 119, 11475–11481. 10.1021/acs.jpcc.5b02458. [DOI] [Google Scholar]
- Lee J.; Ye H.; Pan S.; Bard A. J. Screening of Photocatalysts by Scanning Electrochemical Microscopy. Anal. Chem. 2008, 80, 7445–7450. 10.1021/ac801142g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.